
Molecularly Imprinted Electropolymer for a Hexameric Heme Protein with Direct Electron Transfer and Peroxide Electrocatalysis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Electrodes
2.3. Apparatus and Electrochemical Measurements
3. Results and Discussion
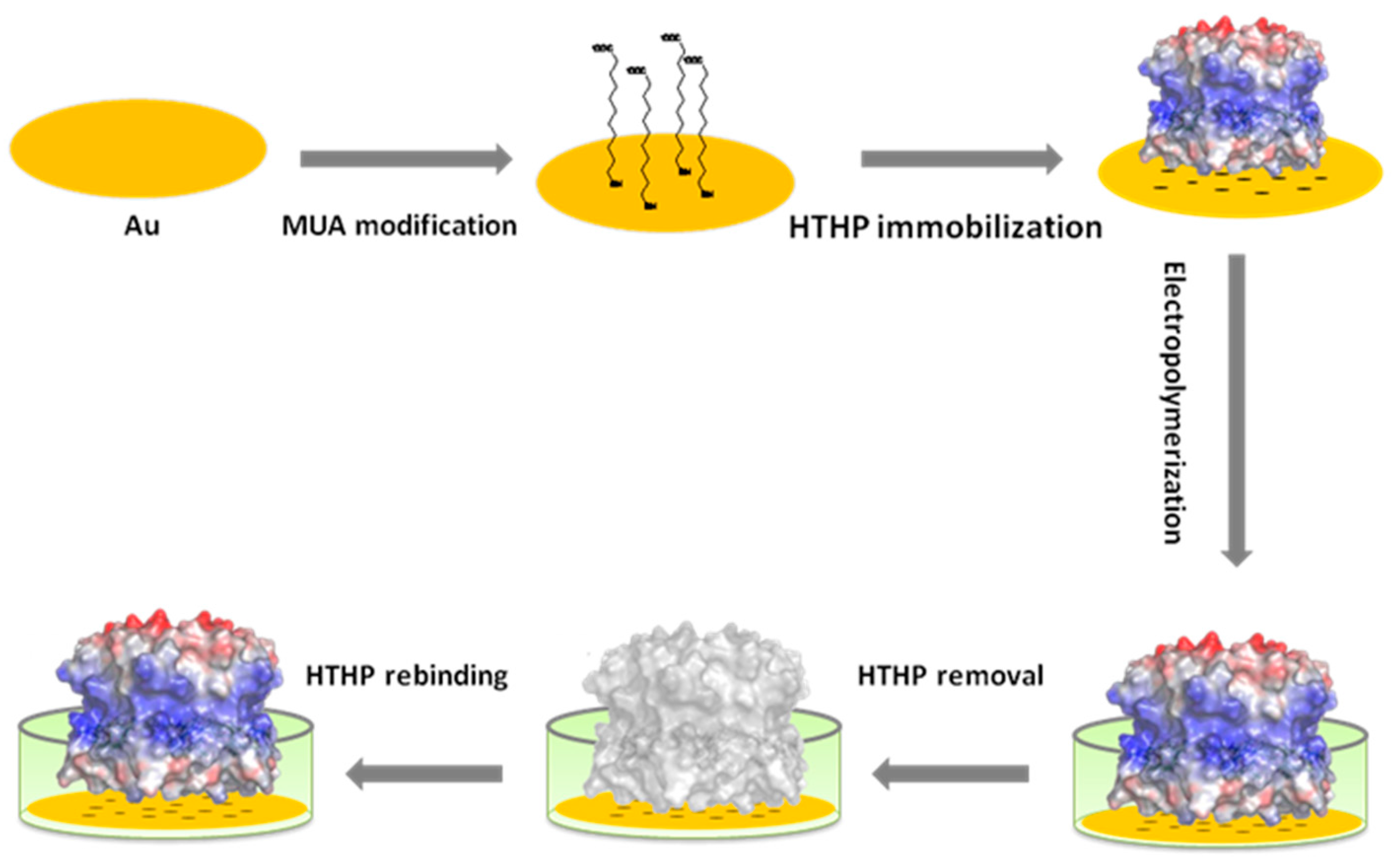
3.1. Preparation of the MIP-Modified Electrode, Template Removal and Rebinding
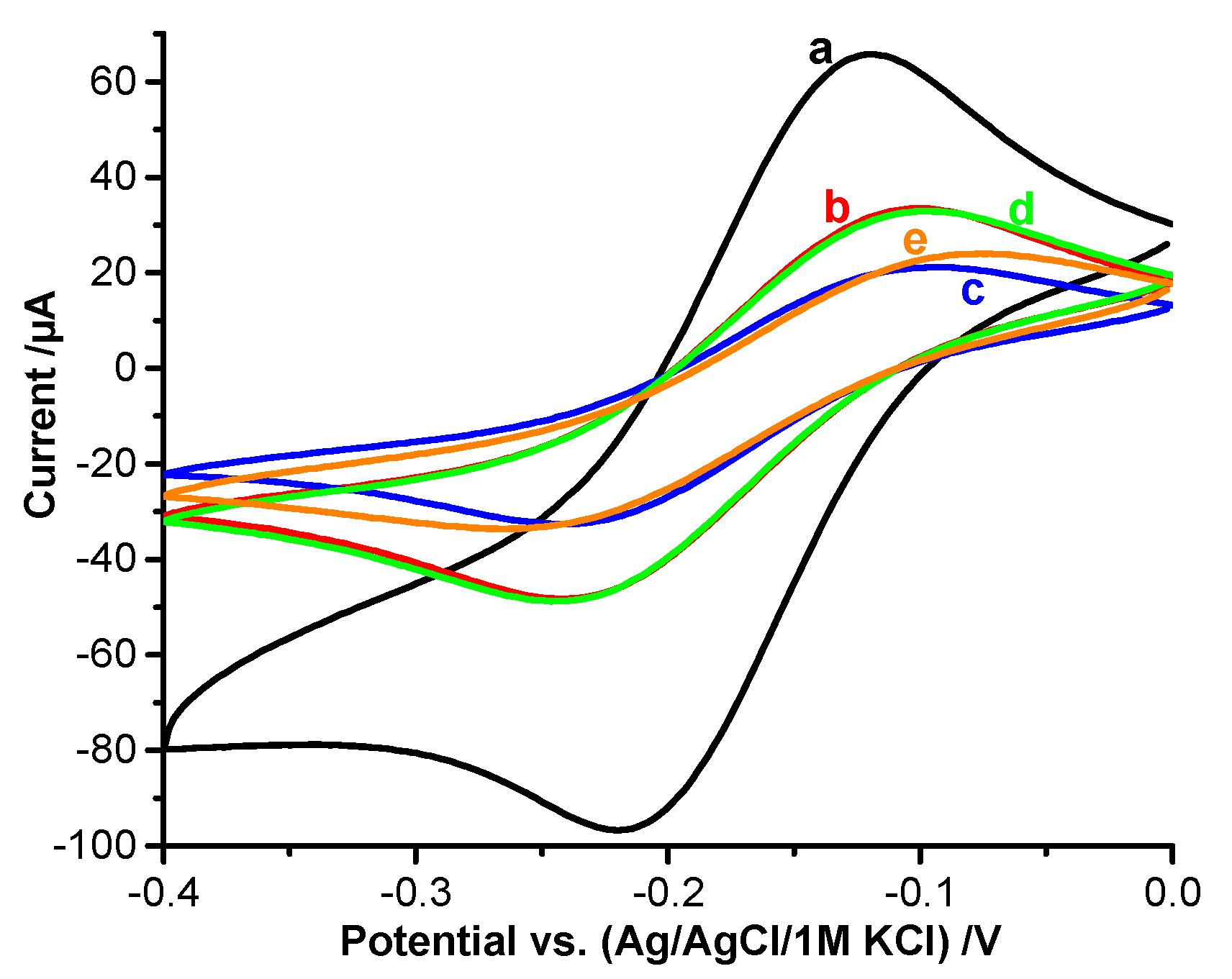
3.2. DET of HTHP Trapped in the MIP
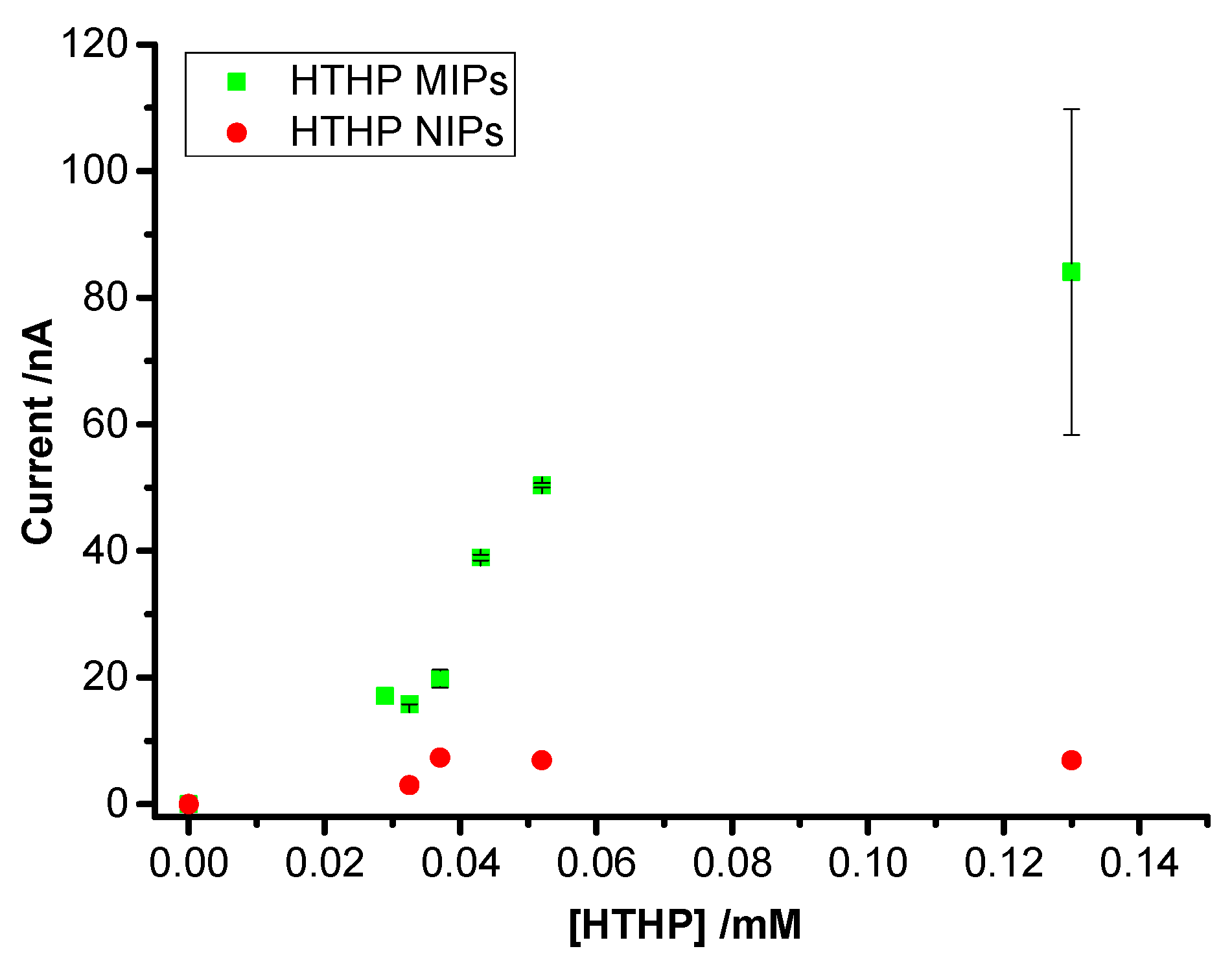
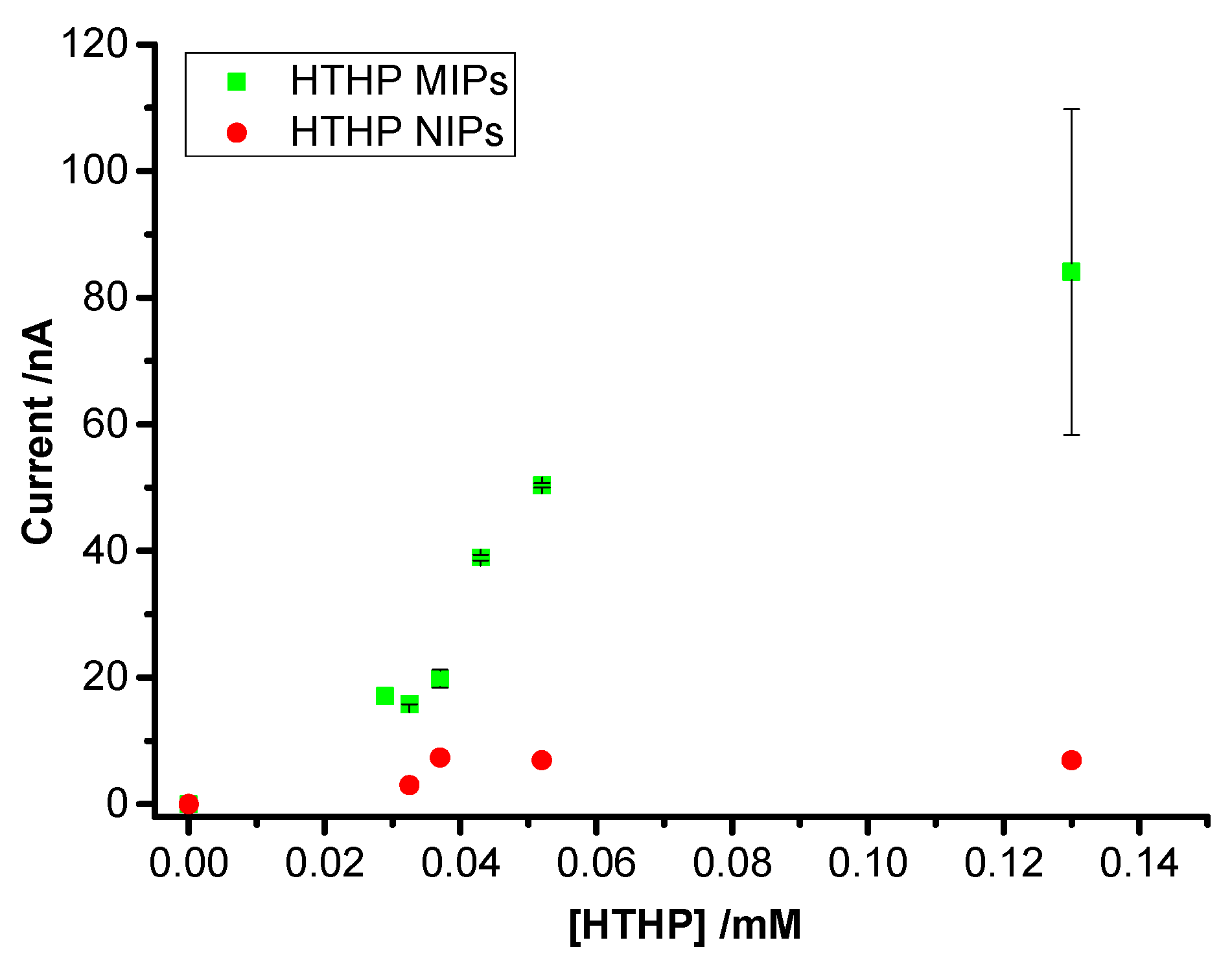
3.3. Concentration Dependence of Rebinding of HTHP to MIPs and NIPs
3.4. Binding of Cyt c to the HTHP-MIP
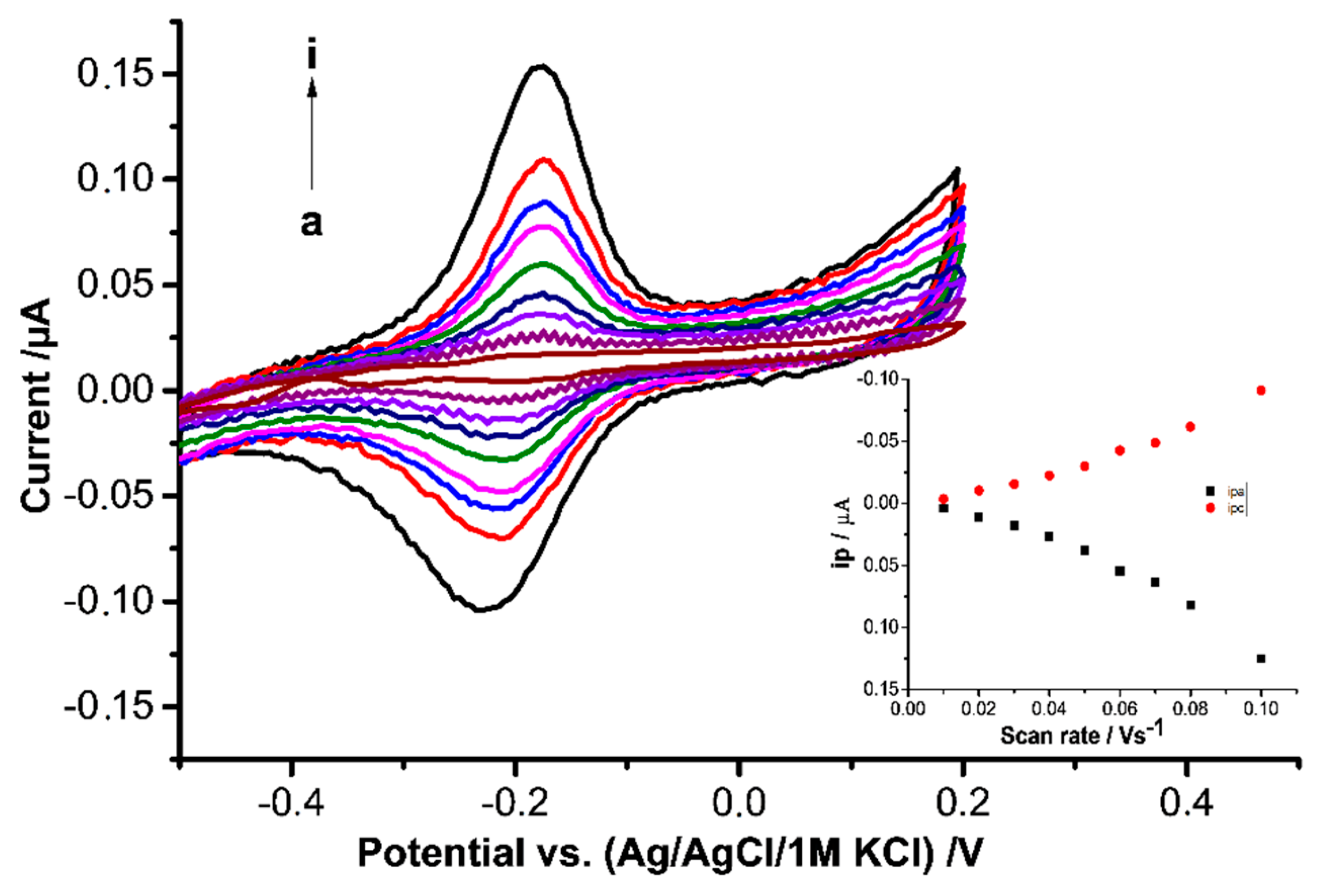
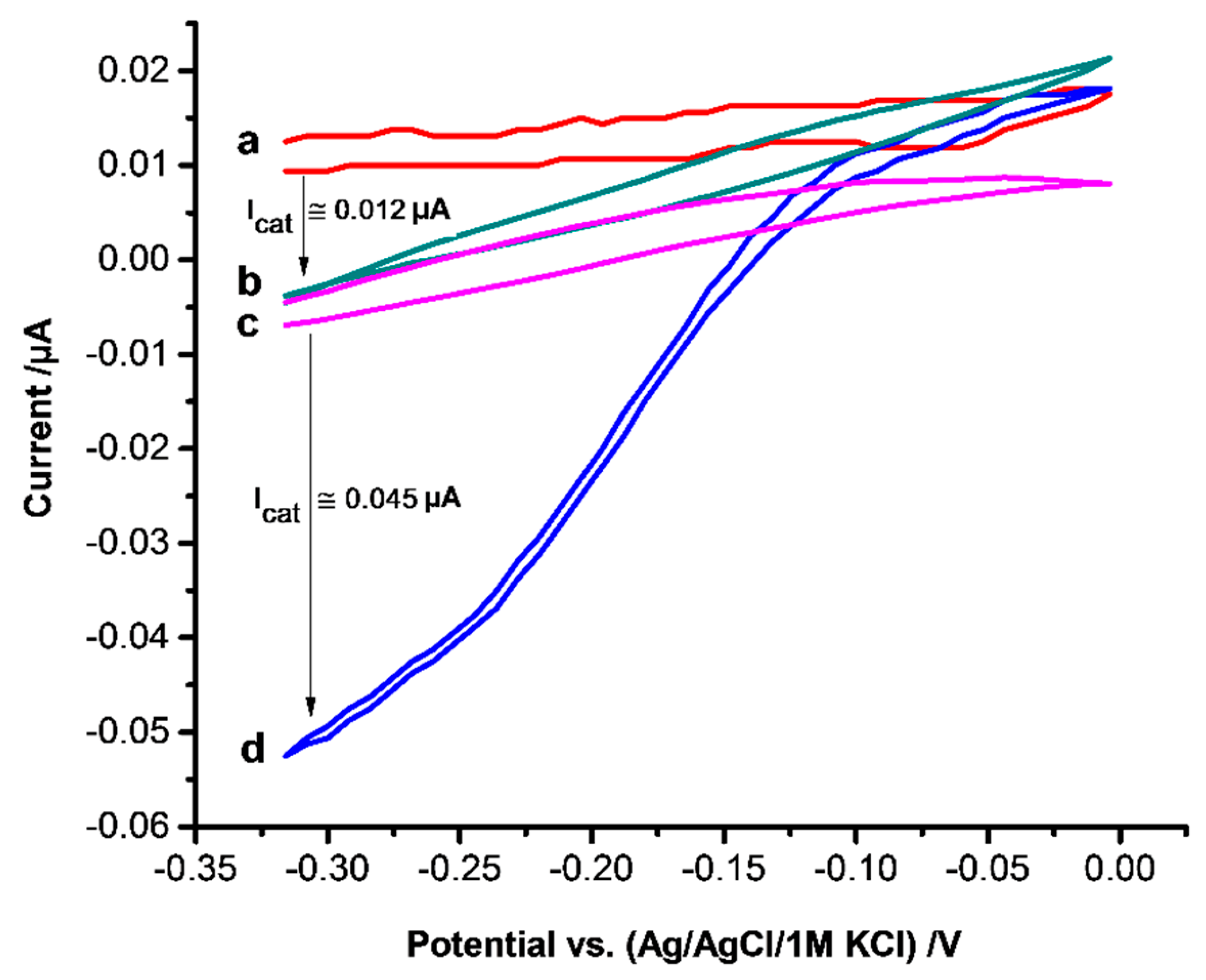
3.5. Electrocatalysis of Hydrogen Peroxide Reduction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscopy |
| Av-FITC | Avidin-fluorescein isothiocyanate |
| BSA | Bovine serum albumin |
| ConA | Concanavilin A |
| CV | Cyclic voltammetry |
| Cyt c | Cytochrome c |
| DET | Direct electron transfer |
| DPV | Differential pulse voltammetry |
| EIS | Electrochemical impedance spectroscopy |
| gp51 | Bovine leukemia virus glycoprotein gp51 |
| HTHP | Hexameric tyrosine-coordinated heme protein |
| IF | Imprinting factor |
| MIP | Molecularly imprinted polymer |
| MUA | Mercaptoundecanoic acid |
| NIP | Non-imprinted Polymer |
| PAD | Pulsed amperometric detection |
| PB | Phosphate buffer |
| QCM | Quartz crystal microbalance |
| SAM | Self-assembled monolayer |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEM | Scanning electron microscope |
| SPR | Surface plasmon resonance |
| SPS | Surface plasmon spectroscopy |
| SWV | Square wave voltammetry |
References
- Wulff, G.; Sarhan, A. Use of polymers with enzyme-analogous structures for resolution of racemates. Angew. Chem. Int. Ed. 1972, 11, 341–342. [Google Scholar]
- Shea, K.J.; Thompson, E.A.; Pandey, S.D.; Beauchamp, P.S. Template synthesis of macromolecules—Synthesis and chemistry of functionalized macroporous polydivinylbenzene. J. Am. Chem. Soc. 1980, 102, 3149–3155. [Google Scholar] [CrossRef]
- Arshady, R.; Mosbach, K. Synthesis of substrate-selective polymers by host-guest polymerization. Macromol. Chem. Phys. 1981, 182, 687–692. [Google Scholar] [CrossRef]
- Ge, Y.; Turner, A.P.F. Too large to fit? Recent developments in macromolecular imprinting. Trends Biotechnol. 2008, 26, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, M.A.E.; Allender, C.J.; Brain, K.R.; Heard, C.M. A high-throughput screening technique employing molecularly imprinted polymers as biomimetic selectors. Methodol. Surv. Bioanal. Drugs 1998, 25, 37–43. [Google Scholar]
- Hayden, O.; Haderspöck, C.; Krassnig, S.; Chena, X.; Dickert, F.L. Surface imprinting strategies for the detection of trypsin. Analyst 2006, 131, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Bossi, A.; Piletsky, S.A.; Piletska, E.V.; Righetti, P.G.; Turner, A.P.F. Surface-grafted molecularly imprinted polymers for protein recognition. Anal. Chem. 2001, 73, 5281–5286. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, Z.; Si, S. Potentiometric urea biosensor based on immobilization of urease onto molecularly imprinted TiO2 film. J. Electroanal. Chem. 2009, 635, 1–6. [Google Scholar] [CrossRef]
- Chen, Y.W.; Rick, J.; Chou, T.C. A systematic approach to forming micro-contact imprints of creatine kinase. Org. Biomol. Chem. 2009, 7, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.J.; Tong, Y.W. The effect of protein structural conformation on nanoparticle molecular imprinting of ribonuclease a using miniemulsion polymerization. Langmuir 2007, 23, 2722–2730. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Zhang, L.; Hao, Y.; Cui, X.; Liu, D.; Zhang, M.; Tang, Y. Novel polydopamine imprinting layers coated magnetic carbon nanotubes for specific separation of lysozyme from egg white. Talanta 2015, 144, 1125–1132. [Google Scholar] [CrossRef]
- Guerreiro, A.; Poma, A.; Karim, K.; Moczko, E.; Takarada, J.; de Vargas-Sansalvador, I.P.; Turner, N.; Piletska, E.; de Magalhães, C.S.; Glazova, N.; et al. Influence of surface-imprinted nanoparticles on trypsin activity. Adv. Healthc Mater. 2014, 3, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ye, J.; Bie, Z.; Liu, Z. Affinity-tunable specific recognition of glycoproteins via boronate affinity-based controllable oriented surface imprinting. Chem. Sci. 2014, 5, 1135–1140. [Google Scholar] [CrossRef]
- Kamon, Y.; Matsuura, R.; Kitayama, Y.; Ooya, T.; Takeuchi, T. Precisely controlled molecular imprinting of glutathione-s-transferase by orientated template immobilization using specific interaction with an anchored ligand on a gold substrate. Polym. Chem. 2014, 5, 4764–4771. [Google Scholar] [CrossRef]
- Erdőssy, J.; Horváth, V.; Yarman, A.; Scheller, F.W.; Gyurcsányi, R.E. Electrosynthesized molecularly imprinted polymers for protein recognition. Trends Anal. Chem. 2016. [Google Scholar] [CrossRef] [Green Version]
- Li, S.J.; Cao, S.S.; Whitcombe, M.J.; Piletsky, S.A. Size matters: Challenges in imprinting macromolecules. Prog. Polym. Sci. 2014, 39, 145–163. [Google Scholar] [CrossRef]
- Sharma, P.S.; Pietrzyk-Le, A.; D’Souza, F.; Kutner, W. Electrochemically synthesized polymers in molecular imprinting for chemical sensing. Anal. Bioanal. Chem. 2012, 402, 3177–3204. [Google Scholar] [CrossRef] [PubMed]
- Malitesta, C.; Mazzotta, E.; Picca, R.A.; Poma, A.; Chianella, I.; Piletsky, S.A. MIP sensors—The electrochemical approach. Anal. Bioanal. Chem. 2012, 402, 1827–1846. [Google Scholar] [CrossRef] [PubMed]
- Bosserdt, M.; Gajovic-Eichelmann, N.; Scheller, F.W. Modulation of direct electron transfer of cytochrome c by use of a molecularly imprinted thin film. Anal. Bioanal. Chem. 2013, 405, 6437–6444. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, J.-H.; Pippig, D.A.; Martins, B.M.; Wagener, N.; Dobbek, H. HTHP: A novel class of hexameric, tyrosine-coordinated heme proteins. J. Mol. Biol. 2007, 368, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Gajovic-Eichelmann, N.; Ehrentreich-Forster, E.; Bier, F.F. Directed immobilization of nucleic acids at ultramicroelectrodes using a novel electro-deposited polymer. Biosens. Bioelectron. 2003, 19, 417–422. [Google Scholar] [CrossRef]
- Dechtrirat, D.; Gajovic-Eichelmann, N.; Bier, F.F.; Scheller, F.W. Hybrid material for protein sensing based on electrosynthesized mip on a mannose terminated self-assembled monolayer. Adv. Funct. Mater. 2014, 24, 2233–2239. [Google Scholar] [CrossRef]
- Peng, L.; Utesch, T.; Yarman, A.; Jeoung, J.H.; Steinborn, S.; Dobbek, H.; Mroginski, M.A.; Tanne, J.; Wollenberger, U.; Scheller, F.W. Surface-tuned electron transfer and electrocatalysis of a hexameric tyrosine-coordinated heme protein (HTHP). Chemisty 2015, 21, 7596–7602. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wollenberger, U.; Kinne, M.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Fischer, A.; Scheller, F.W. Peroxygenase based sensor for aromatic compounds. Biosens. Bioelectron. 2010, 26, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.; Serna, C.; López-Tenés, M.; Moreno, M. Theoretical background for the behavior of molecules containing multiple interacting or noninteracting redox centers in any multipotential step technique and cyclic voltammetry. J. Electroanal. Chem. 2005, 576, 9–19. [Google Scholar] [CrossRef]
- Dechtrirat, D.; Jetzschmann, K.J.; Stocklein, W.F.M.; Scheller, F.W.; Gajovic-Eichelmann, N. Protein rebinding to a surface-confined imprint. Adv. Funct. Mater. 2012, 22, 5231–5237. [Google Scholar] [CrossRef]
- Bosserdt, M.; Bognár, J.; Lautner, G.; Witt, J.; Köhler, K.; Gajovic-Eichelmann, N.; Yarman, A.; Wittstock, G.; Scheller, F.W.; Gyurcsányi, R.E. Microelectrospotting as a new method for electrosynthesis of surface-imprinted polymer microarrays for protein recognition. Biosens. Bioelectron. 2015, 73, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanaviciene, A.; Ramanavicius, A. Molecularly imprinted polypyrrole-based synthetic receptor for direct detection of bovine leukemia virus glycoproteins. Biosens. Bioelectron. 2004, 20, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Kan, X.W.; Xing, Z.L.; Zhu, A.H.; Zhao, Z.; Xu, G.L.; Li, C.; Zhou, H. Molecularly imprinted polymers based electrochemical sensor for bovine hemoglobin recognition. Sens. Actuat B Chem. 2012, 168, 395–401. [Google Scholar] [CrossRef]
- Li, L.; Yang, L.L.; Xing, Z.L.; Lu, X.J.; Kan, X.W. Surface molecularly imprinted polymers-based electrochemical sensor for bovine hemoglobin recognition. Analyst 2013, 138, 6962–6968. [Google Scholar] [CrossRef] [PubMed]
- Bognar, J.; Szucs, J.; Dorko, Z.; Horvath, V.; Gyurcsanyi, R.E. Nanosphere lithography as a versatile method to generate surface-imprinted polymer films for selective protein recognition. Adv. Funct. Mater. 2013, 23, 4703–4709. [Google Scholar] [CrossRef]
- Menaker, A.; Syritski, V.; Reut, J.; Opik, A.; Horvath, V.; Gyurcsanyi, R.E. Electrosynthesized surface-imprinted conducting polymer microrods for selective protein recognition. Adv. Mater. 2009, 21, 2271–2275. [Google Scholar] [CrossRef]
- Karimian, N.; Turner, A.P.; Tiwari, A. Electrochemical evaluation of troponin T imprinted polymer receptor. Biosens. Bioelectron. 2014, 59, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Ren, L.; Zhao, H.Z.; Xu, C.J.; Zhang, L.; Yu, Y.; Wang, H.Z.; Lan, Y.C.; Roberts, M.F.; Chuang, J.H.; et al. A molecular-imprint nanosensor for ultrasensitive detection of proteins. Nat. Nanotechnol. 2010, 5, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, T.X. Surface plasmon resonance sensor chips for the recognition of bovine serum albumin via electropolymerized molecularly imprinted polymers. Chin. Chem. Lett. 2013, 24, 813–816. [Google Scholar] [CrossRef]
- Jetzschmann, K.J.; Jágerszki, G.; Dechtrirat, D.; Yarman, A.; Gajovic-Eichelmann, N.; Gilsing, H.-D.; Schulz, B.; Gyurcsányi, R.E.; Scheller, F.W. Vectorially imprinted electropolymer for acetylcholinesterase. Adv. Funct. Mater. 2015, 25, 5178–5183. [Google Scholar] [CrossRef] [Green Version]
- Cieplak, M.; Szwabinska, K.; Sosnowska, M.; Bikram, K.C.C.; Borowicz, P.; Noworyta, K.; D’Souza, F.; Kutner, W. Selective electrochemical sensing of human serum albumin by semi-covalent molecular imprinting. Biosens. Bioelectron. 2015, 74, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Clark, R.A.; Bowden, E.F.; Tarlov, M.J. Characterization of cytochrome c/alkanethiolate structures prepared by self-assembly on gold. J. Phys. Chem. 1993, 97, 6564–6572. [Google Scholar] [CrossRef]
- Davis, K.L.; Drews, B.J.; Yue, H.; Waldeck, D.H.; Knorr, K.; Clark, R.A. Electron-transfer kinetics of covalently attached cytochrome c/SAM/Au electrode assemblies. J. Phys. Chem. C 2008, 112, 6571–6576. [Google Scholar] [CrossRef]
- Csoregi, E.; Jonssonpettersson, G.; Gorton, L. Mediatorless electrocatalytic reduction of hydrogen-peroxide at graphite-electrodes chemically-modified with peroxidases. J. Biotechnol. 1993, 30, 315–337. [Google Scholar] [CrossRef]
- Presnova, G.; Grigorenko, V.; Egorov, A.; Ruzgas, T.; Lindgren, A.; Gorton, L.; Börchers, T. Direct heterogeneous electron transfer of recombinant horseradish peroxidases on gold. Faraday Discuss. 2000, 116, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, G.; Gorton, L. An electrochemical sensor for hydrogen-peroxide based on peroxidase adsorbed on a spectrographic graphite electrode. Electroanalysis 1989, 1, 465–468. [Google Scholar] [CrossRef]
- Dunford, H.B. Oxidations of iron(II)/(III) by hydrogen peroxide: From aquo to enzyme. Coord. Chem. Rev. 2002, 311, 233–234. [Google Scholar] [CrossRef]
- Yarman, A.; Nagel, T.; Gajovic-Eichelmann, N.; Fischer, A.; Wollenberger, U.; Scheller, F.W. Bioelectrocatalysis by microperoxidase-11 in a multilayer architecture of chitosan embedded gold nanoparticles. Electroanalysis 2011, 23, 611–618. [Google Scholar] [CrossRef]
- Peng, L.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. Bioelectrocatalytic properties of Agrocybe aegerita peroxygenase. Electrochim. Acta 2010, 55, 7809–7813. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | Template/Protein | IF | Detection of Binding | Ref. |
|---|---|---|---|---|
| Scopoletin | Cyt c-derived peptide | 6 | Fluorescence, SPR | [26] |
| Scopoletin | Cyt c | 2 | Fluorescence, CV (DET) | [19] |
| Scopoletin | ConA | 8.6 | QCM, SPS | [22] |
| Scopoletin | Ferritin | 13 | SPR, AFM | [27] |
| Scopoletin | HTHP | 12 | CV (DET, RM), SWV | This work |
| Pyrrole | gp51 | 9–10 | PAD (RM) | [28] |
| Pyrrole | Bovine hemoglobin | - | DPV (RM), EIS, SEM | [29] |
| Pyrrole | Bovine hemoglobin | 7.72 | DPV (RM), EIS, SEM | [30] |
| 3,4-Ethylenedioxythiophene/Poly(styrenesulphonate) | Avidin | 6.5 | QCM, AFM | [31] |
| 3,4-Ethylenedioxythiophene/Poly(styrenesulphonate) | Avidin or Av-FITC | - | Fluorescence, SEM | [32] |
| o-Phenylenediamine | Troponin T | - | CV (RM), DPV, AFM | [33] |
| Phenol | Human ferritin/Human papillomavirus | - | EIS, DPV (RM), SEM | [34] |
| 3-Aminophenylboronic acid | BSA | 2 | SPR, SEM | [35] |
| 3,4-Propylenedioxythiophene carboxylate | Acetylcholinesterase | 9.9 | Amperometry of catalysis, AFM | [36] |
| 2,2’-Bithiophene-5-carboxylic acid | Human serum albumin | 26.8 | DPV (RM), EIS, AFM | [37] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, L.; Yarman, A.; Jetzschmann, K.J.; Jeoung, J.-H.; Schad, D.; Dobbek, H.; Wollenberger, U.; Scheller, F.W. Molecularly Imprinted Electropolymer for a Hexameric Heme Protein with Direct Electron Transfer and Peroxide Electrocatalysis. Sensors 2016, 16, 272. https://doi.org/10.3390/s16030272
Peng L, Yarman A, Jetzschmann KJ, Jeoung J-H, Schad D, Dobbek H, Wollenberger U, Scheller FW. Molecularly Imprinted Electropolymer for a Hexameric Heme Protein with Direct Electron Transfer and Peroxide Electrocatalysis. Sensors. 2016; 16(3):272. https://doi.org/10.3390/s16030272
Chicago/Turabian StylePeng, Lei, Aysu Yarman, Katharina J. Jetzschmann, Jae-Hun Jeoung, Daniel Schad, Holger Dobbek, Ulla Wollenberger, and Frieder W. Scheller. 2016. "Molecularly Imprinted Electropolymer for a Hexameric Heme Protein with Direct Electron Transfer and Peroxide Electrocatalysis" Sensors 16, no. 3: 272. https://doi.org/10.3390/s16030272