Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells



1
Department of Chemistry and Drug Technologies, Sapienza University of Rome P.le Aldo Moro 5, 00185 Rome, Italy
2
Department of Biochemistry and Structural Biology, Lund University, P.O. Box 124, 221 00 Lund, Sweden
*
Author to whom correspondence should be addressed.
Sensors 2018, 18(5), 1319; https://doi.org/10.3390/s18051319
Submission received: 7 March 2018
/
Revised: 16 April 2018
/
Accepted: 19 April 2018
/
Published: 24 April 2018
(This article belongs to the Special Issue I3S 2017 Selected Papers)
Abstract
:Dehydrogenase based bioelectrocatalysis has been increasingly exploited in recent years in order to develop new bioelectrochemical devices, such as biosensors and biofuel cells, with improved performances. In some cases, dehydrogeases are able to directly exchange electrons with an appropriately designed electrode surface, without the need for an added redox mediator, allowing bioelectrocatalysis based on a direct electron transfer process. In this review we briefly describe the electron transfer mechanism of dehydrogenase enzymes and some of the characteristics required for bioelectrocatalysis reactions via a direct electron transfer mechanism. Special attention is given to cellobiose dehydrogenase and fructose dehydrogenase, which showed efficient direct electron transfer reactions. An overview of the most recent biosensors and biofuel cells based on the two dehydrogenases will be presented. The various strategies to prepare modified electrodes in order to improve the electron transfer properties of the device will be carefully investigated and all analytical parameters will be presented, discussed and compared.
1. Introduction
Dehydrogenase enzymes (E.C. 1.1.1.-1.1.2) belong to the group of oxidoreductases, which oxidise a substrate by reducing an electron acceptor. Hundreds of dehydrogenases occur in nature and most of them are NAD(P) (nicotinamide adenine dinucleotide (phosphate), acting as electron acceptor/donor) dependent [1]. In this case the cofactor is not covalently bound to the enzyme but it must be present in the reaction medium to activate the biocatalytic function of the enzyme itself, by acting as a carrier of two electrons and one proton. By contrast, all other dehydrogenases require for their activity a cofactor tightly bound to the protein, which can be either FAD (flavin adenine dinucleotide), FMN (flavine mononucleotide), PQQ (pyrroloquinoline quinone) [2] or Moco (molybdopterin) [3,4]. These enzymes are also called pyridine nucleotide-independent dehydrogenases, as they do not use NAD+ or NADP+ (nicotinamide adenine dinucleotide phosphate) as electron acceptor [1].
Dehydrogenases can therefore be divided into four main classes, depending on the type of cofactor: (i) NAD+/NADP+ dependent; (ii) flavin (FAD or FMN) dependent; (iii) PQQ dependent; and (iv) Moco dependent enzymes.
The thermodynamic redox potential of NAD+/NADH is −0.52 V versus Ag|AgClsat at pH 7 [5,6], determined by using potentiometric titration [7]. The NADH cofactor itself is not a useful redox electron shuttle in bioelectrochemistry, because of a high electrochemical overpotential, required to drive the electron transfer reaction and lack of chemical reversibility for the NADH/NAD+ redox process and because of the interfering adsorption of the cofactor itself and reaction products at electrode surfaces [5]. For these reasons, NAD+ dependent dehydrogenase based biosensors were realised by the introduction of various redox mediators [8,9,10] or of diaphorase (NADH:acceptor oxidoreductase), a redox enzyme, which catalyses the oxidation of NADH with the concomitant reduction of a redox mediator. Several 2nd generation biosensors were described by using the two-enzyme cascade NAD(P)-dependent dehydrogenase/diaphorase [11,12,13,14].
The FAD, FMN PQQ, Moco cofactors show thermodynamic redox potentials in the potential window of about −0.26 and +0.14 V vs Ag|AgClsat at pH 7, respectively [9,15], which are less negative than that for the NAD+/NADH couple. In general biosensors based on FAD-, FMN-, PQQ- and Moco-dependent dehydrogenases require the use of redox mediators to shuttle the electrons between the active site and the electrode [16,17,18,19]. Many examples of 2nd generations biosensors by employing different mediators were reported in the literature based on FAD dependent [20,21,22] and PQQ-dependent dehydrogenases [23,24,25,26,27,28].
Several dehydrogenases harbouring FAD, FMN, PQQ and Moco as cofactor can contain an additional heme prosthetic group [29]. Therefore they can in turn be divided into two categories: flavoproteins/flavohemoproteins, quinoproteins/quinohemoproteins, molybdopterin/ molybdopterinhemoproteins respectively, depending on whether they contain the heme group or not. In the case of also containing a heme the FAD, FMN, PQQ or Moco containing domain may be connected via the heme containing cytochrome domain to an electrode and thus the cytochrome domain acts as “a built in mediator” [30].
Such (FAD, FMN, PQQ, or Moco dependent) dehydrogenases have been investigated as potential enzymes that can undergo direct electron transfer (DET) with electrodes [29]. Recent studies indicate that efficient DET is hard to obtain in the absence of the additional heme group (quinohemoprotein and flavohemoprotein). The catalytic FAD, FMN, PQQ, or Moco containing dehydrogenase domain is structurally connected in various ways to the heme containing cytochrome domain thereby allowing smooth electron transfer from the catalytic site via the heme to the external electron acceptor (electrode). However, the connection between the two domains may be very sensitive to a series of parameters such as pH, ionic strength, concentration of specific cations, determining the distance between the two domains and thus the rate of the internal electron transfer.
Due to their superior electron transferability, such heme-containing, multicofactor dehydrogenases, consisting of a FAD-, FMN-, PQQ-, or Moco-harbouring catalytic subunit/domain and a heme-containing electron transfer subunit/domain, were recently shown to undergo DET with several different electrode surfaces [31,32,33,34,35,36,37,38,39,40,41,42] as initially was pointed out by Ikeda et al. already in the early 1990s [30,43].
In this review we focus our attention on two dehydrogenases that have been extensively discussed with data reported in the literature for their ability to undergo efficient DET: cellobiose dehydrogenase (CDH) and fructose dehydrogenase (FDH). CDH and FDH are gaining increased attention in the last few years as catalysts for the oxidation of lactose/glucose and fructose in several biosensing and biofuel cells (BFCs) applications [44,45,46,47,48,49,50,51,52,53].
In the following sections, the DET mechanism of the two dehydrogenases is described and an analytical comparison of the electrochemical performances of various CDH and FDH based biosensors and BFCs are reported. For all electrochemical systems investigated, information is given on the type of electrode modification performed, electrode assembly, electrode platform and device stability. Analytical parameters such as LOD, sensitivity, dynamic linear range for biosensing applications and OCV and power for BFC applications are also reported.
2. Direct Electron Transfer of Dehydrogenases
The electronic coupling between redox enzymes and electrodes for the development of analytical devices such as biosensors and BFCs can be realised according to three mechanisms, which in turn are denoted 1st, 2nd and 3rd generation biosensors [29]: (i) electroactivity of the substrate or product of the enzymatic reaction (1st generation); (ii) mediated electron transfer (MET) with the use of redox mediators, small electroactive molecules (freely diffusible or bound to side chains of flexible redox polymers), which are able to shuttle the electrons between the enzyme active site and the electrode (2nd generation); (iii) DET between the redox centre of the enzyme and the electrode surface (3rd generation). Figure 1 shows a schematic representation of the three mechanisms of electronic communications between enzyme and electrode. Commercial biosensors are mostly based on either 1st or 2nd generation biosensors [29], however, recent progress in bioelectrochemistry in combination with bioengineering of enzymes have shown that 3rd generation biosensors have now reached the market [54].
In particular, when discussing the 2nd generation sensors we should distinguish between a freely soluble monomeric mediator (e.g., Fe(CN)63−/4−, quinones) and an immobilised mediator in the form of for example, a redox polymer equipped with Os3+/2+-complexes, quinones, or other redox compounds that can be co-immobilised with the enzyme making reagentless biosensors possible. For biosensor development, the 3rd generation format based on DET mechanism shows important advantages compared to MET, 2nd generation biosensors with a soluble or polymer bound mediator: first of all the absence of a mediator allows a higher selectivity, because the biosensor can operate at a potential closer to the formal potential (E°’) of the redox enzyme, thus reducing possible interfering reactions. Moreover, both soluble and polymer bound mediators may also mediate unspecific reactions and the lack of a reagent in the reaction sequence makes the device easier to realise. However, as mentioned adsorbed/immobilised mediators allow the making of reagentless biosensors possible (no freely diffusing mediator in solution), which is an obvious advantage compared to other 2nd generation biosensors that rely on the addition of a mediator to the sensing solution [55,56]. This type of bound mediators can drive catalytic processes also at low overpotentials by modifying the molecular structure, thus tuning the E°’ of the redox polymer [57]. Finally, 3rd generation biosensors based on DET offer a wide range of possibility to modulate the desired properties of the analytical device using protein modification either through gene or protein engineering techniques [58] or new interfacial technologies making use of nanomaterials (e.g., metal nanoparticles and carbon based nanomaterials) [59].
Also for development of BFCs, DET possesses some important advantages over MET [60]. The use of redox mediators usually leads to voltage losses arising from the potential difference between the active site of the enzyme and the mediator. Mediators for anodic reactions show redox potentials at least 90–200 mV higher than those of the oxidative redox enzymes, thus decreasing the potential difference between the BFC anode and the cathode. Nevertheless, highly efficient BFCs obtained also with lower potential differences are reported in the literature [61]. Moreover, a DET design allows several simplifications in the construction of sugar/O2 BFCs based on oxygen insensitive dehydrogenases, as no membranes and no separate compartments are necessary, thus facilitating the miniaturisation process. One of the main drawbacks of using DET communication in constructing BFCs is the difficulty to electrically connect a sufficient amount of enzyme molecules, which can limit the efficiency and the power output of the devices [60].
Although DET presents favourable characteristics for both biosensors and BFCs, only a few groups of enzymes are found to be capable of interacting directly with an electrode via a DET mechanism. This is because the redox centres of the enzymes are often buried within the protein structure in combination with a lack of an electron transfer pathway connecting the active site with the protein surface [60,62,63,64,65,66,67,68,69,70,71]. An efficient DET mechanism has been demonstrated for a restricted number of enzymes [60,63,64,66,67], such as multicopper blue oxidases (laccases and bilirubin oxidases) [72,73,74], hydrogenases [64,70,71,74,75], peroxidases [29,76,77] and a few dehydrogenases, all containing metallocentres and/or one or more heme groups [63,65,66,67,78]. As for the dehydrogenases, DET communication has been reported also for a limited number of heme containing enzymes, similar in their multicofactor composition (FAD, FMN, PQQ, or Moco in combination with heme) to dehydrogenase enzymes. However, these enzymes exhibit a medium-high reactions rate with molecular oxygen and are therefore denoted oxidases rather than dehydrogenases. These are reported in Table 1. All of them are FAD, FMN, PQQ, or Moco containing heme proteins [32,45,47,48,79]. Some even containing Fe-S clusters such as membrane bound succinate dehydrogenase (complex II in the respiratory chain), which shows DET properties both in its isolated “native” state [80] as well as in its truncated form for which the heme containing part was removed [81]. Among them, CDH and FDH are the most widely investigated for biosensing and BFCs applications and for this reason they will be treated separately below.
3. Cellobiose Dehydrogenase
Cellobiose dehydrogenase (CDH; E.C 1.1.99.18) is an extracellular enzyme, belonging to the oxidoreductase group, secreted by wood-degrading, phytopatogenic and saprotrophic fungi within the phyla of Basidiomycota and Ascomycota [93,94]. In 1974, Westermark and Eriksson reported for the first time about the enzymatic activity in Trametes versicolor and Phanaerochaete chrysosporium observing the oxidation of guiacol while adding cellobiose [95]. During the last 40 years, a lot of efforts have been addressed to elucidate the full electron transfer mechanism, enzyme catalytic activity towards several substrates (cellobiose, lactose, glucose, etc.), enzyme structure and bioengineering pathways to adapt the enzyme to some commercial/industrial purposes [44,83].
CDH belongs to the flavohemoprotein family, which includes also mandelate dehydrogenase, fumarate dehydrogenase, bacterial cytochrome P-450, nitric oxidase synthase and flavocytochrome b2 [93]. Unlike these enzymes, CDH is the only extracellular one. It contains two subunits/domains: subunit I is the catalytic dehydrogenase domain (DHCDH) similar to the DH domain of other oxidoreductases belonging to the glucose-methanol-choline (GMC) oxidoreductase superfamily with a flavin adenine dinucleotide (FAD) co-factor non-covalently bound to the enzyme structure; subunit II is the cytochrome domain (CYTCDH), which contains a heme b and in bioelectrochemistry acts as “a built-in mediator” by facilitating the shuttling of the electrons to an electrode [96,97]. Both subunits are connected through a flexible linker responsible for the modulation of the rate of the internal electron transfer (IET) reaction by varying the experimental conditions such as changing the pH, ionic strength or the concentration of divalent cations [98,99,100]. The oxidation of the natural substrate cellobiose fully reduces FAD in a 2e−/2H+ process and the electrons are sequentially transferred one by one through the IET pathway to the CYTCDH resulting in reduced heme b, which in turn finally donates the electrons to the electrode [101,102].
Based on the phyla of fungi secreting CDH, it is possible to divide them into two classes: class I and class II, from Basidiomycota and Ascomycota, respectively. The CDHs belonging to class I exhibit a shorter enzyme sequence and a lower molecular weight (c.a. 80 kDa) compared to those of class II as well as a highly conserved linker region, an acidic working pH (3.5–4.5) and a poor oxidizing capacity towards glucose and other monosaccharides. Conversely, class II CDHs are able to oxidise both disaccharides and monosaccharides, exhibit a more complex and longer enzyme sequence (including also a cellulose binding module), a higher molecular weight (c.a. 115 kDa) and some have a neutral working pH (7.4) [45,103].
The crystal structure of the separated domains of CDH has been reported for the first time by the group of Divne et al. for Phanerochaete chrysosporium (PcCDH) considering the CYTCDH domain (PDB 1D7C) and DHCDH domain (1KDG) separately [104,105], as shown in Figure 2. The crystal structure of CYTCDH showed a sandwich fold of two antiparallel β sheets with an exposed heme b cofactor, like in other cytochromes. The heme b is hexacoordinated so that the propionate residues are sufficiently exposed and affecting the internal electron transfer (IET). Only one crystal structure is available for the whole enzyme, extracted from Neurospora crassa and published by the same group recently [106]. The natural electron acceptor to CDH, lytic polysaccharide monooxygenase (LPMO) was only recently discovered [107] revealing the central role of CDH in the degradation of lignocellulose in nature [108,109].
The electron transfer reaction between the DHCDH domain and an electrode can occur mainly according to two different routes depending on the absence (DET based reaction) or the presence of a redox mediator (MET based reaction) with a suitable redox potential [82], as schematised in Figure 3.
In the presence of any electron acceptor, an aldose is oxidised at the C1 position (only the β-D-anomer is a substrate for CDH) into its corresponding lactone and concurrently FAD in the active site of the DHCDH is fully reduced to FADH2-DHCDH:
The reoxidation of FADH2-DHCDH can follow two different reaction mechanisms:
(1) The electrons are transferred from FADH2 to the electrode directly if a 2e−, 2H+ acceptor is employed, such as a quinone (Q) or an equivalent aromatic redox compound [82]:
The reduced quinone, QH2, is then reoxidized at the electrode at an applied potential (Eapp) higher than the E°’ of the Q/QH2 redox couple,
(2) the electrons are transferred from FADH2 to the electrode directly and sequentially if a 1e−, non-H+ acceptor, is employed, such as an Os3+-complex (Os3+) [83]:
The Os2+ ions formed are reoxidized at the electrode if Eapp is higher than the E°’ of the Os3+/2+ redox couple, :
In the absence of a mediator the electrons can be transferred from FADH2-DHCDH to the CYTCDH sequentially in an internal electron transfer process (IET)
The reoxidation of the reduced enzyme can be summarised as follows:
This step is followed by a first electron transfer (DET) step to the electrode, which is immediately followed by a second IET step delivering the second electron from the DHCDH to the CYTCDH:
Finally, the second electron is delivered to the electrode:
Various CDHs have been extensively utilised over the past twenty years to develop lactose/glucose biosensors and enzymatic fuel cells [44,82,83], which are carefully reviewed in the following two sections, highlighting the results achieved in terms of sensitivity, selectivity, linear range, stability, operational voltage and maximal power output.
3.1. Cellobiose Dehydrogenase Based Biosensors
As mentioned above, CDH can be classified into two groups: class I CDH, only catalysing cellodextrins and lactose efficiently and class II, which can catalyse not only cellodextrins and lactose but may also the oxidation of monosaccharides like glucose and other low molecular weight dextrins such as maltose and so forth, although the turnover number is higher for cellodextrins and lactose than for example, for glucose. Therefore in this review the most recent applications of CDH based biosensors for lactose and glucose will be reported separately (Table 2).
3.1.1. CDH Based Lactose Biosensors
Before the discovery of the DET properties of CDH biosensors for lactose were typically based on combining lactase, hydrolysing lactose into glucose and galactose, with either glucose oxidase (GOx) or galactose oxidase [116,117,118,119]. Novel electrochemical biosensors for lactose detection based on class I CDHs were initially based on simple adsorption onto plain graphite electrodes [96] or covalent immobilisation on premade screen printed carbon based electrodes [110,120] used with success for lactose detection in the dairy industry [119,121]. Also screen printed electrodes (SPEs) have been considered by Safina et al. to develop portable lactose biosensors based on two different class I CDHs, T. villosa CDH and P. sordida CDH [110]. They used two different SPEs: modified with multiwalled carbon nanotubes (MWCNTs) and unmodified graphite. Two different immobilisation methods have been exploited, cross-linking the enzyme on top of the electrode with glutaraldehyde or poly(ethyleneglycol)deglycidyl ether (PEGDGE), which showed different sensitivities and stabilities. After optimisation of the working conditions of the biosensors, they were able to detect lactose in a concentration range between 0.5–200 μM and 0.5–100 μM, with TvCDH and PsCDH, respectively, with a LOD of 250 nM for both. The developed biosensors were successfully tested for the determination of lactose in dairy (milk with different percentages of fat, lactose-free milk and yogurt) with a good reproducibility (RSD = 1.5–2.2%). Improved performance of such lactose biosensors was developed by Tasca and co-workers [111,122]. Most of the work was focused on the electrode modification in order to achieve efficient DET through the electrostatic interactions between the negatively recharged PsCDH and the positively charged electrode surface due to the presence of –NH2 groups. In particular, single walled carbon nanotubes (SWNCTs) were surface modified with aryl diazonium salts of p-phenylenediamine (NH2-PD) and deposited on top of a glassy carbon (GC) electrode [123]. The PsCDH/NH2-PD/SWCNTs-GC based lactose biosensor showed very efficient DET and exhibited an extraordinary high current density of 500 μA cm−2 in a 5 mM lactose solution at pH 3.5. The biosensor exhibited a detection limit for lactose of 0.5 μM, a large linear range from 1 to 150 μM lactose and a high sensitivity (476.8 nA μM−1 cm−2). It showed also a fast response time (4 s), good reproducibility (RSD = 1.75%) and good stability (half-life 12 days).
In another approach, Tavahodi and co-workers developed a lactose biosensor based on PsCDH immobilised onto positively charged polyethyleneimine (PEI) modified gold nanoparticles (AuNPs) [101]. In this work, they synthesised PEI@AuNPs by using PEI as reducing agent for Au(III) and as stabiliser for the NPs. Next, the PEI@AuNPs were drop-cast on top of solid planar Au electrodes creating a favourable environment for the correct orientation of PsCDH, due to the strong electrostatic interactions. The heterogeneous electron-transfer (ET) rate (ks) for the redox reaction of immobilised PsCDH at the modified electrodes was calculated based on the Laviron theory [124] and was found to be 39.6 ± 2.5 s−1. The proposed lactose biosensor exhibits a good long term stability as well as a high and reproducible sensitivity to lactose, viz. 3.93 µA mM−1, with a response time less than 5 s and a linear range from 1 to 100 µM.
Other nanomaterials for example, palladium (PdNPs) or platinum nanoparticles (PtNPs) in combination with MWCNTs or nanohybrids have been used to enhance the lactose biosensor sensitivity and stability in recent years. In particular, Bozorgzadeh et al. developed a biosensor based on DET of PcCDH immobilised onto PtNPs or PdNPs decorated MWCNTs [112]. The prepared nanohybrids, PtNPs–MWCNTs and PdNPs–MWCNTs, were cast on the surface of spectrographic graphite electrodes and then PcCDH was adsorbed on the modified layer. DET between PcCDH and the modified nanostructured electrodes was studied using flow injection amperometry and cyclic voltammetry. The maximum current responses (Imax) and the apparent Michaelis–Menten constants (KMapp) for the different PcCDH modified electrodes were calculated by fitting the data to the Michaelis–Menten equation and compared. The sensitivity towards lactose was 3.07 and 3.28 µA mM−1 at the PcCDH/PtNPs–MWCNTs/SPGE and PcCDH/PdNPs–MWCNTs/SPGE electrodes, respectively, which were higher than those measured at the PcCDH/MWCNTs/SPGE (2.60 µA mM−1) and PcCDH/SPGE (0.92 µA mM−1).
More recently, Bollella et al. evaluated the influence of an ordered electrode nanostructuration on the sensitivity of a lactose biosensor based on the immobilisation of a class II CDH, Corynascus thermophilus CDH (CtCDH) [113]. An efficient DET between CtCDH and the novel gold electrode platform was achieved by covalently linking “green” AuNPs and AgNPs (obtained through a green synthetic pathway using quercetin as reducing agent at room temperature [125]) modified with a dithiol based self-assembled monolayer, consisting of biphenyl-4,4′-dithiol (BPDT). The apparent kS of CtCDH was calculated to be 21.5 ± 0.8 s−1 and 10.3 ± 0.7 s−1, for the AuNPs/BPDT/AuE and the AgNPs/BPDT/AuE platforms, respectively. The modified electrodes were successively used to develop a biosensor for lactose detection. However, the CtCDH/AuNPs/BPDT/AuE based biosensor showed the best analytical performances with an excellent stability, a detection limit of 3 μM, a linear range between 5 and 400 μM and a sensitivity of 27.5 ± 2.5 μA cm−2 mM−1.
3.1.2. CDH Based Glucose Biosensors
The discovery of class II CDHs also revealed that several of these were also good candidates for making 3rd generation glucose biosensors [103], in contrast to other glucose oxidising dehydrogenases, such as various FAD-glucose dehydrogenases (FAD-GDH) [20], PQQ-glucose dehydrogenase (PQQ-GDH) [27] and GOx that do not show any DET properties [126,127]. Several Class II CDHs were investigated for their catalytic properties and pH profiles [103] and among these especially the following two CDHs, Myriococcum thermophilum CDH (MtCDH) [128,129] and CtCDH [53,114,115] have been intensively studied as possible candidates for glucose biosensor making as well as for biofuel cells anodes.
In 2009 the very first paper appeared on the possibility to make 3rd generation glucose sensors based on Class II CDH [130]. In 2011 a glucose biosensors based on DET of CtCDH was published by Tasca et al. [114]. CtCDH was immobilised onto oxidatively shortened SWCNTs through cross-linking the enzyme with PEGDGE. By drop-casting SWCNTs onto the electrode, it was possible to observe a higher sensitivity (222 nA mM−1 cm−2) compared to a bare graphite electrode and with a linear range between 0.1 and 30 mM and a LOD of 0.05 mM, which covers the whole concentration range relevant for the direct determination of the blood glucose level. With respect to its linear range and detection limit the 3rd generation glucose biosensor might be suited for monitoring the glucose concentration in human blood.
Furthermore, Zafar and co-workers reported on the immobilisation of CtCDH on top of carbon screen-printed electrodes (SPCEs), in order to develop portable DET based glucose biosensor [115]. The enzyme was immobilised on two different commercially available SPCEs, carboxyl-functionalised single-walled carbon nanotubes (SPCE–SWCNTs) and multiwalled carbon nanotubes (SPCE–MWCNTs), by simple physical adsorption or a combination of adsorption followed by cross-linking using with PEGDGE Mn = 400 or glutaraldehyde (GA). The SPCE–MWCNTs/CtCDH/PEGDGE electrode showed the highest sensitivity, a wide linear range (0.025–30 mM) with an LOD of 0.01 mM. Moreover, this biosensor exhibited a high stability by losing only 10% of its initial signal after continuously injecting 50 mM glucose for 7 h.
3.2. CDH Based Biofuel Cells
It is known that the development of enzymatic fuel cells (EFCs) recently attracts a growing interest because of the possibility to be used as implantable electric power sources for self-contained biodevices as they can be significantly miniaturised and are also able to operate under human physiological conditions [60,131,132]. Most of the efforts have been addressed to find out the “ideal” sugar oxidising enzyme, able to oxidise both α- and β-D-glucose preferably more than once, insensitive to oxygen and possibly also able to show efficient DET with the enzyme modified electrode [52]. Class II CDHs fulfil some of the aforementioned parameters concerning the “ideal” sugar oxidising enzyme. In this section, the main findings related to EFCs based on DET of class II CDHs as glucose/O2 EFCs are summarised (Table 3).
In 2008 Coman et al. published the very first report on a glucose/O2 BFC based on DET for both the anode and cathode. In this BFC Dichomera saubinetii CDH and Trametes hirsuta laccase (ThLac) were both immobilised by simply drop-casting on top of a graphite electrode [133]. The optimum pH of the DET reaction was found close to pH 5, similar to the previously reported DET-optimum pH for class II MtCDH, being in this case coupled with ThL, which shows also an acidic optimal pH. The biocatalytic limiting current values of the bioanode vary considerably depending on the sugar, in the order: lactose > cellobiose > glucose. Nevertheless, the EFC exhibited good operational and stability parameters with an open-circuit voltage of 0.73 V, a maximum power density of 5 µWcm−2 at 0.5 V and an estimated half-life of more than 38 h in an air-saturated 0.1 M citrate–phosphate buffer at pH 4.5 containing 5 mM glucose. The work was very useful as a proof of concept of the possibility to couple such enzymes through DET based reactions, however, they do not work at human physiological conditions.
Later the same research groups reported on an EFC based on the combination of CtCDH and Myrothecium verrucaria bilirubin oxidase (MvBOx) both working nicely at human physiological pH, again simply drop-casting the enzymes on top of graphite electrodes [134]. The mediator-less and membrane-less EFC was tested in phosphate buffer and human serum and showed an open-circuit voltage of 0.62 and 0.58 V and a maximum power density of ca. 3 and 4 µW cm−2 at 0.37 and 0.19 V of cell voltage, respectively.
Wang et al. reported on the fabrication and characterisation of a gold-nanoparticle (AuNPs)-based mediatorless sugar/O2 EFC operating in neutral sugar-containing buffers and human physiological fluids, such as blood and plasma [53]. In this paper CtCDH and MvBOx were used for making the anode and the cathode, respectively. After a detailed characterisation of each bioelement separately, the CtCDH/AuNPs-based bioanode and the MvBOx/AuNPs-based biocathode were combined into a functional EFC. The following characteristics of the mediator- and membrane-less miniaturised EFC were obtained: an open-circuit voltage of 0.68 V, a maximum power density of 15 µW cm−2 at a cell voltage of 0.52 V in phosphate buffer and an open-circuit voltage of 0.65 V and a maximum power density of 3 µW cm−2 at a cell voltage of 0.45 V, in human blood. The estimated half-lives of the biodevices were found to be >12, <8 and <2 h in sugar-containing buffer, human plasma and blood, respectively.
More recently, the dimensions of the above reported electrodes were scaled down in order to obtain an EFC operating in basal human lachrymal liquid, which produces enough electrical energy for powering modern low-power electronic devices [50]. Indeed, the following characteristics of miniature glucose/O2 biodevices operating in human tears were registered: 0.57 V open-circuit voltage, about 1 µW cm−2 maximum power density at a cell voltage of 0.5 V and more than 20 h operational half-life. Theoretical calculations regarding the maximum recoverable electrical energy can be extracted from the biofuel and the biooxidant, glucose and molecular oxygen, each readily available in human lachrymal liquid.
Similarly, the miniaturised EFC has been used also in sweat and saliva to be able to power small electronic devices for biomedical in-situ monitoring, which require a low power output [135]. In this work thin gold wires were used as electrodes, modified according to the protocol reported by Wang et al. [53]. The following characteristics of miniature glucose/O2 biodevices were registered in human sweat and saliva, respectively: 580 and 560 mV open-circuit voltage, 0.26 and 0.1 µW cm−2 power density at a cell voltage of 0.5 V, with up to ten times higher power output at 0.2 V. When saliva was collected after meal ingestion, roughly a two-fold increase in power output was obtained, with a further two-fold increase by addition of 500 µM glucose. Likewise, the power generated in sweat at 0.5 V increased two-fold by addition of 500 µM glucose.
In a similar approach, Krikstolaityte et al. developed a glucose/O2 EFC based on the DET of another class II CDH, Humicola insolens CDH (HiCDH), immobilised on positively charged thiol capped AuNPs with N-(6-mercapto)hexylpyridinium (MHP), while the biocathode was still based on MvBOx/AuNPs modified electrode [51]. In this paper, the results achieved in terms of power output were slightly lower compared to the results of the paper reported previously: (i) 5 mM glucose-open-circuit voltage (OCV) of 0.65 V and the maximal power density of 4.77 µW cm−2 at an operating voltage of 0.50 V; (ii) 10 mM lactose-OCV of 0.67 V and the maximal power density of 8.64 µW cm−2 at an operating voltage of 0.50 V. Conversely, the stability parameters resulted to be higher, indeed the half-life operation times of the EFC were estimated to be at least 13 and 44 h in air saturated PBS containing 5 mM glucose and 10 mM lactose, respectively.
4. Fructose Dehydrogenase
Fructose dehydrogenase (FDH; EC 1.1.99.11) is a membrane-bound oxidoreductase found among bacteria belonging to the acetic acid bacteria (AAB) family, a group of gram-negative bacteria able to oxidise various sugars and ethanol with the production of acetic acid during the fermentation process [65]. Among the AAB family, only two genera exhibit oxidizing activity towards D-fructose producing 5-keto-D-fructose, Acetobacter and Gluconobacter [136]. In particular, Acetobacter can oxidise also ethanol to carbon dioxide and water through Krebs cycle enzymes, whereas Gluconobacter does not completely oxidise ethanol due to lack of some Krebs cycle enzymes.
FDH belongs to the flavohemoproteins family. It is a heterotrimeric membrane-bound enzyme complex with a molecular mass of 146.4 kDa, consisting of three subunits: subunit I (DHFDH), which is the catalytic domain, with a covalently bound FAD cofactor, where D-(-)-fructose is involved in a 2H+/2e− oxidation to 5-keto-D-fructose; subunit II (CYTFDH), which acts as a built-in electron acceptor with three heme c moieties covalently bound to the enzyme scaffold and two of them are involved in the stepwise electron transfer pathway; subunit III, which is not involved in the electron transfer but plays a key role for the enzyme complex stability [48,79,84,137,138].
Unfortunately, the crystal structure of FDH is not available yet, because the enzyme is a membrane bound protein with a high molecular weight (ca. 146 kDa). In the last few years a lot of efforts have been addressed considering new crystallisation methods to solve the problem with membrane bound enzymes. The crystal structure would be a fundamental finding to clarify the electron transfer mechanism of this enzyme with particular attention on the co-factors involved.
The ET pathway for FDH immobilised on the electrode surface and in the absence of any competing e− acceptor, is similar as was outlined for CDH above and is assumed to occur as follows (Figure 4) [139]:
- (1)
- D-(-)-fructose is oxidised to 5-keto-D-(-)-fructose involving 2e−/2H+ with the concomitant reduction of FAD to FADH2;
- (2)
- FADH2 is sequentially reoxidized in two separate 1 ET steps. In the first FADH2 is partially reoxidized to FADH· through the IET pathway between the DHFDH and CYTFDH domains, whereby one of the three heme c moieties (heme c1) is reduced. Next, the electron is transferred from heme c1 to a second heme c (heme c2) of the two hemes involved in the ET pathway and then to a final electron acceptor, which is the electrode when FDH is adsorbed onto the electrode surface;
- (3)
- FADH· is finally reoxidized to FAD by heme c1 and the electron is then transferred to heme c2 (which gives the second internal electron transfer (IET) step), which in turn is reoxidized by the electrode whereby FDH is returned to its fully oxidised state.
Conversely, in the presence of an electron acceptor such as quinone (Q), the reoxidation of FADH2-DHFDH can be accomplished by a 2e−/2H+ reduction of Q to QH2, with a consequently reoxidation of QH2 at the electrode surface. Alternatively, in the case of a monoelectronic and non-H+ acceptor—for example, an Os3+-complex—the electrons are sequentially transferred from the FADH2-DHFDH to Os3+-complex. Moreover, a second ET from the FADH2-DHFDH to a second Os3+ complex will fully re-oxidise the DHFDH with the formation of two Os2+ complexes at the electrode surface [85,140,141].
Over the past twenty years FDH has been used to develop DET-based fructose biosensors and enzymatic fuel cells less extensively than CDH, probably due to the more restricted application in food technology compared to that in medicine. Glucose, unlike fructose, is an essential energy source in many living organisms and glucose biofuel cells have found applications in powering implantable bioelectronic devices used for diagnose and biomedical applications [70,142]. Nevertheless, the most significant electrochemical platforms will carefully reviewed in following sections.
4.1. FDH Based Biosensors
In the earliest article on a fructose biosensor based on DET of FDH, Ikeda et al. reported on the immobilisation of FDH onto a carbon paste electrode [79]. The modified electrode showed an electrocatalytical wave starting at EONSET = 0 V versus Ag|AgClsat and rising up to approximately 2.5 µA at 0.7 V versus Ag|AgClsat. The oxidation of D-fructose occurred at the catalytic dehydrogenase domain, from which the electrons were transferred to the second subunit, the cytochrome domain containing the heme c:s and finally shuttling the electrons to the electrode [143,144,145,146]. The KM and Imax values of this electrode were determined as 8.0 ± 1 mM and 1.4 ± 0.4 µA, respectively. Moreover, the FDH biosensor exhibited a linear range between 0.2 and 30 mM.
Afterwards, other papers have been published, where the authors mistakenly believed that the cofactor of the catalytic dehydrogenase domain was PQQ instead of FAD. For example, Parellada et al. reported on the immobilisation of FDH onto polyethyleneimine (PEI) modified carbon paste electrodes [147]. The enzyme was introduced into the carbon paste matrix and the addition of PEI allows an mediatorless anodic current to be achieved at 400 mV versus Ag|AgClsat after injections of fructose in the FIA system. Operating at 400 mV, the analytical characteristics of the sensor were: limit of detection 75 µM (S/N 3), linearity up to l0 mM, sensitivity 385 µA mM−1 cm−2, complete specificity for fructose and operational stability of 10 h. Yabuki and co-workers immobilised FDH onto AuNPs modified GC electrodes showing a catalytic current proportional to a concentration of D-fructose up to 0.5 mM [148]. However, the long-term stability of the biosensor was not very good, as the response decreased to 50%, 20% and 10% of the initial current after 1 h, 1 day and 10 days, respectively.
Besides the papers mentioned above, FDH has also been employed by co-immobilising the enzyme with several mediators, like coenzyme Q-6, tetrathiafulvalene (TTF) and tetracyanoquinodimethane (TCNQ), hexacyanoferrate (III) or ferrocene and dihydroxyphenols [145,149,150,151].
Only more recently, several research groups claimed the presence of FAD as co-factor instead of PQQ in subunit I of the enzyme complex, as supposed by the previous works. After that, all researchers referred to FDH as a heterotrimeric flavoprotein-cytochrome c complex.
Tominaga and co-workers reported on the development of a novel platform based on FDH directly adsorbed onto CNTs synthesised on a platinum plate by the chemical vapour deposition (CVD) method using iron nanoparticles derived from ferritin [152]. The so modified electrode showed a well-defined electrocatalytical wave from −0.15 V (vs. Ag|AgClsat), which was close to the E°’ of one of the heme c’s functioning as prosthetic group of FDH. Moreover, the calibration curve exhibited a linear relationship between current density and fructose concentration up to ca. 40 mM. The Michaelis-Menten constant (KM) was found to be 11 ± 1 mM.
In 2014, Nazaruk et al. used liquid crystalline cubic phase (LCP) and CNTs for the immobilisation of FDH in order to enhance the DET of FDH, exploiting the possibility to retain native conformation and bioactivity of FDH [153]. The calibration curve of FDH/LPC/SWCNTs/GC electrode showed a linear relationship between current density and fructose concentration up to 10 mM. The Michaelis-Menten constant (KM) was found to be 11 ± 1 mM. The biosensor was tested for fructose detection in fruit juices by using both cyclic voltammetry and amperometry at a constant applied potential. Moreover, the same modified electrode has been used also as bioanode in combination with Trametes hirsuta laccase (ThLac) to realise an EFC, of which the performance is reviewed in the next section.
More recently, Šakinytė et al. reported on the immobilisation of FDH onto thermally reduced graphene oxide (TRGO) obtaining three different fractions, namely TRGO1, TRGO2 and TRGO3 [154]. All three TRGO fractions were applicable for design of amperometric fructose biosensors acting on DET principles. The achieved high values of the sensitivity are in the same order of magnitude as for other fructose biosensors based on synergistic mediated processes. The electrode based on TRGO1 exhibited the highest sensitivity of 14.5 μA mM−1 cm−2, with a LOD of 0.7 mM. In this regard, the functional groups present in the TRGO1 fraction are able not only to orient FDH properly but they can also participate in the electron transfer reactions. In this case, the surface area of the electrode material in bioelectrocatalysis based on DET does not play any crucial role in the effectivity of the ET.
Siepenkoetter and co-workers studied the effect of nanoporous gold with different pores sizes on the bioelectrocatalytical behaviour of FDH in order to develop a highly sensitive and selective biosensor for the detection of fructose. In fact, they synthesised nanoporous gold with pore diameters of 9, 18, 42 and 62 nm by using a de-alloying method [155]. From the preliminary results, the 42 nm-nanoporous gold electrode showed the best performance in terms of catalytic current. Moreover, the so prepared electrode was modified following two different methods: self-assembling monolayer (SAM) of 3-mercaptopropionic acid (3-MPA) and a mixed layer based on 3-MPA SAM with the electrodeposition of 2-carboxy-6-naphthoyl diazonium salt. Finally, FDH was cross-linked onto the modified electrode using carbodiimide (CMC) in a manner that enables DET to occur. The latter modified electrode exhibited the highest electrocatalytical current. So far the response of the biosensor correlated very well with the results obtained with a spectrophotometric commercially available enzymatic kit. The biosensor demonstrated rapid response times (less than 5 s), with a linear range of 0.05–0.3 mM D-fructose, a sensitivity of 3.7 µA cm−2 µM and a limit of detection of 1.2 µM, with high selectivity. The biosensor is a promising alternative to established analytical measurements for the detection of fructose.
The main analytical parameters of the reported fructose biosensors based on DET of FDH are summarised in Table 4.
4.2. FDH Based Biofuel Cells
In early research into FDH based BFCs, Kamitaka et al. [47] realised a one-compartment biofuel cell, in which FDH from Gluconobacter sp. and laccase from Trametes sp. (TsLac) were used as DET-type bioelectrocatalysts in the two-electron oxidation of D-fructose and the four-electron reduction of O2 as fuels, respectively. In particular, FDH was drop-cast on top of carbon paper (CP) electrodes modified with Ketjen black (KB) particles, which resulted in a tight immobilisation because of the strong π-π stacking interactions. On the other hand, TsLac was immobilised onto carbon aerogel (CG) particles with an average pore size of 22 nm, modified on CP electrodes. Both elements resulted in an efficient bioelectrocatalysis, with a maximum current density of 4 mA cm−2. The FDH adsorbed KB-modified CP electrodes and the TsLac-adsorbed CG-modified CP electrodes were combined to construct a one-compartment BFC without separators with an open-circuit voltage of 790 mV, a maximum current density of 2.8 mA cm−2 and a maximum power density of 850 mW cm−2 at 410 mV of the cell voltage under stirring.
A similar fructose/O2 biofuel cell based on DET was realised by Murata et al. [156] because of the large surface area of the electrodes it was possible to increase the current density during the bioelectrocatalysis. AuNPs were synthesised according to a citrate protocol and successively directly drop-cast to immobilise MvBOx, for the biocathode and modified through a SAM reaction with mercaptoethanol (ME) to have a positive influence on the immobilisation of FDH, for the bioanode. The BOx-adsorbed AuNPs/CP and FDH-adsorbed ME-AuNPs/CP electrodes were combined to construct a mediatorless BFC without separator. The following characteristics of the mediator-, separator- and membrane-less BFC were obtained: a maximum current density of 2.6 mA cm−2, a maximum power density of 0.66 mW cm−2 at 360 mV of the cell voltage in quiescent solution while under stirring, a maximum current density of 4.9 mA cm−2 and a maximum power density of 0.87 mW cm−2 at an operating voltage of 300 mV.
More recently, Haneda et al. prepared a totally flexible, sheet shaped BFC by using a carbon fabric (CF) as the flexible, conductive base for the enzyme electrodes. The CF strips were modified with MWCNTs and FDH for the oxidation of fructose and with Ketjien black (KB) and MvBOx for the reduction of oxygen. The FDH anode strip and the BOx cathode strip were stacked with a hydrogel film of agarose, which retains the electrolyte solution and fuel (fructose). This assembly provides a stand-alone, sheet-shaped power source that can be bent without loss of output power. The open-circuit voltage of the cell was found to be 0.7 V with a maximum power density reaching 550 µW cm−2 at 0.4 V. It is interesting to note that this device could be repeatedly bent up to a 44° angle without significant loss of output power, while bending in excess of this value would cause fracture of the modified electrodes [157].
In a similar approach, Miyake et al. [158] described a layered BFC constructed by laminating enzyme-modified carbon fabric (CF) strips and hydrogel film containing electrolyte and fuel. The hydrogel sheets ensure ion-conduction between anode/cathode fabrics and also serve as the fuel tank to eliminate the necessity of packaging. This device resulted in having a better performance compared to that previously developed by Haneda et al. because of the different and improved hydrogel. Using agarose resulted in a thick layer but really weak and not resistive to continuous bending stress. Conversely, Miyake and co-workers used a heavy-duty “double network (DN) hydrogel,” resulting in a very flexible, thinner BFC (~1 mm thickness). The pre-modification of CF with CNTs was effective to improve the performance of both bioelements, with a maximum power of 0.64 mW at 1.21 V and an open circuit voltage of 2.09 V.
In 2014, Nazaruk et al. [153] used monoolein or phytantriol liquid crystalline cubic phase and CNTs for the immobilisation of FDH. Entrapment within the cubic phase prevents the guest protein from chemical and physical degradation, thus facilitating retention of its native conformation and bioactivity. The mesophase environment was therefore found appropriate for retaining FDH close to the electrode surface. Phytantriol was used as the cubic phase component in case of measurements carried out in biological fluids containing hydrolysing enzymes. The enzymatic fuel cell based on FDH in the cubic phase film at the anode and Cerrena unicolor C-139 laccase at the biocathode showed open circuit potential of 703 ± 10 mV in the presence of 40 mM fructose and power density of 850 µW cm−2 at 250 mV under continuous flow of O2.
In the same year, So and co-workers tried to improve the performances of a DET-type fructose/O2 BFC with a substrate modified biocathode. The hypothesis was that when the electrode is modified with adsorbed bilirubin, a substrate for MvBOx, the enzyme would attractively interact with the modified compound in such an orientation that the electron-accepting T1 site faces the electrode surface. The orientation seems to be convenient for the DET-type bioelectrocatalysis. The authors also attempted to create a high performance bioanode optimising the conditions for adsorbing FDH on carbon cryogel (CCG) electrodes, a mesoporous carbon material. Finally, a one-compartment DET-type BFC without separators was realised, an open-circuit voltage of 0.79 V and a maximum power density of 2.6 mW cm−2 at 0.46 V were achieved without stirring and under air atmospheric conditions [49].
In the most recent paper for a fructose/O2 BFC, Kizling and co-workers [159] employed an eco-friendly cellulose/polypyrrole composite to immobilise FDH in order to obtain a bioanode working at a more negative potential thus affecting the operational parameters of the BFC. The modified bioanode was combined with a ThLac based biocathode. In this case, laccase was immobilised onto naphthoquinone-modified CNTs. This work was aiming at increasing the capacitance of both electrodes in order to possibly use the final device as a biosupercapacitor. The BFC with the GCE cathode covered with laccase adsorbed on naphthylated MWCNTs exhibits improved parameters with an open circuit voltage of 0.76 V and a maximum power density of 1.6 mWcm−2 at cell voltage of 0.33 V.
Table 5 summarises the most important characteristics of the mentioned fructose/O2 BFCs based on DET of FDH in the bioanode.
5. Enzyme Engineering to Enhance DET for Future Biosensors and BFC Developments
One of the most promising approaches to improve bioelectrocatalytic reactions based on DET of dehydrogenases is the production of mutated or modified enzymes through the use of genetic engineering and recombinant technology, both of which have opened the doors to widespread exploitations of new dehydrogenases for sensing devices with desired electrochemical properties [160,161,162]. In particular, engineered enzymes enabled to realise DET-based biodevices with enhanced sensitivity and wider linear range and with extended stability, as shown in the papers referred to below.
Sode and co-workers performed a series of mutations on PQQ-glucose dehydrogenase extracted from different bacteria and realised glucose biosensors with improved performances. In one of the earlier reports, they used engineered Escherichia coli PQQ-glucose dehydrogenase, where a histidine residue, His775, was exchanged for aspartate (Asp). This mutant exhibited a 25-fold higher KM, thus showing an extended linear range for the glucose biosensor (3–70 mM) compared to the colorimetric test (0.5–30 mM) [163]. In another work they fused the cytochrome domain of PQQ-alcohol dehydrogenase to the C-terminal of PQQ-glucose dehydrogenase from Acinetobacter calcoaceticus and the constructed fused protein showed very good electron transfer properties from the heme to the electrode, allowing the construction of a glucose biosensor with higher sensitivity [164].
Yuhashi et al. [163] demonstrated that the use of suitable mutants can improve the stability of BFCs. They constructed a BFC employing an engineered PQQ-glucose dehydrogenase, the SER415Cys mutant, from Acinetobacter calcoaceticus as the bioanode enzyme and BOx as the biocathode enzyme. The lifetime of the system was greatly enhanced up to 152 h, more than six times than that of the BFC employing the wild-type enzyme. Recently a FAD-dependent glucose dehydrogenase was fused to a natural minimal cytochrome domain in its C-terminus to achieve DET [165] revealing that through protein engineering DET properties of the enzyme can be obtained.
As for FAD-dependent CDH and FDH, we have reported several investigations, where the enzymes have been shown to undergo internal ET to promote direct bioelectrocatalysis but also in these cases enzyme engineering has been utilised to improve the selectivity, performance and stability of the bioelectrodes for both biosensors and BFCs, as illustrated in the following examples.
Both CtCDH and HiCDH have been altered by a single cysteine to tyrosine substitution in position 291 and 285 of the active sites of CtCDH and HiCDH, respectively. The engineering modification resulted in improved kinetic constants for glucose and a concomitant decrease in activity for several disaccharides, including maltose, with a consequent higher glucose sensitivity and reduced maltose affinity of the biosensor [166,167,168].
Hibino et al. [169] constructed a variant of FDH that lacks 143 amino acid residues. The downsized engineered protein caused an increase in the surface concentration of the electrochemically effective enzyme molecules and an improvement in the heterogeneous electron transfer kinetics. This study is the first trial of the deletion of such a long region that includes one of the prosthetic groups to enhance the direct electrocatalysis of DET-based devices such as biosensor and BFCs. Drastic increases in current densities and sensitivity for lactose could also be obtained by deglycosylating Phanerochaete chrysosporium (PcCDH) and Ceriporiopsis subvermispora (CsCDH) as a result of both increasing ks as well as the number of enzyme molecules on the electrode surface [170].
Although the electrochemical properties of many dehydrogenases have been improved by protein engineering, the coupling of such engineered enzyme with various nanomaterial based modified electrodes still represents an important tool to facilitate the electron transfer between enzyme and electrode.
Nanomaterial-facilitated protein DET has been extensively studied in recent years in order to explore the fundamental insights in the effects of nanomaterial properties on protein DET behaviours. Research aims to design and fabricate novel nanocomposites with desirable properties to enhance protein DET capacity for the development of high-performance bioelectrochemical devices. However, the further enhancement of the DET capacity necessary to develop high-performance biosensors and BFCs for practical applications remains a great challenge.
6. Conclusions
Direct electron transfer between dehydrogenase and electrode is a very promising approach to obtain high performance biosensors and BFCs. Unfortunately, only a restricted number of dehydrogenases show the phenomenon of DET on electrode surfaces.
Although many efforts have been carried out to elucidate the direct electrocatalysis of dehydrogenases, the mechanism of the DET is not thoroughly understood yet. There are some questions that need to be clarified: (i) what is the exact role of the enzyme prosthetic group in the electrocatalysis and in the electron transfer process; (ii) what is the influence of the nature of the electrode material and the structure of the electrode surface; (iii) the relationship between the distance separating the active site of the enzyme and the electrode surface and the efficiency of the electrocatalysis.
Despite these difficulties, researchers are exploiting two strategies in order to improve the DET efficiency of dehydrogenases: the design and development of new nanomaterial-based electrode platforms and of novel surface immobilisation methods and the synthesis of new engineered redox proteins/enzymes, achieved through gene and protein engineering techniques, resulting in a manipulation of their redox potentials and an increase in electron-transfer efficiency.
The use of both nanomaterials and engineered dehydrogenases with desired properties may led to a broad spectrum of practical applications ranging from biosensors to BFCs with improved performances.
Another important advantage of the direct electrocatalysis of dehydrogenases is the possibility of miniaturisation of the sensing elements and the development of implantable devices. However, further research into improving the stability and performance are still needed to improve sensitivity, operational lifetimes and open circuit potentials of the DET-dehydrogenase-based electrochemical devices.
Acknowledgments
The authors thank the following agencies for financial support; LG: The Swedish Research Council (project 2014-5908).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cardosi, M.; Liu, Z. Amperometric glucose sensors for whole blood measurement based on dehydrogenase enzymes. In Dehydrogenases; Canuto, R.A., Ed.; InTechOpen: London, UK, 2012; pp. 319–354. [Google Scholar]
- Anthony, C.; Ghosh, M. The structure and function of the PQQ-containing quinoprotein dehydrogenases. Prog. Biophys. Mol. Biol. 1998, 69, 1–21. [Google Scholar] [CrossRef]
- Bray, R.C. Molybdenum-containing enzymes. J. Less Common Met. 1977, 54, 527–536. [Google Scholar] [CrossRef]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Gorton, L.; Domínguez, E. Electrochemistry of NAD(P)+/NAD(P)H. Encyclopedia of Electrochemistry; Bard, A.J., Stratmann, M., Wilson, G.S., Eds.; Bioelectrochemistry, Wiley-VCH: Weinheim, Germany, 2002; Volume 9, pp. 67–143. [Google Scholar]
- Chenault, H.K.; Whitesides, G.M. Regeneration of nicotinamide cofactors for use in organic-synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.M. Oxidation-Reduction Potentials of Organic Systems; Robert E. Krieger Publishing Company: Huntington, NY, USA, 1972. [Google Scholar]
- Gorton, L.; Domínguez, E. Electrocatalytic oxidation of NAD(P)H at mediator-modified electrodes. Rev. Mol. Biotechnol. 2002, 82, 371–392. [Google Scholar] [CrossRef]
- Gorton, L. Carbon-paste electrodes modified with enzymes, tissues and cells. Electroanalysis 1995, 7, 23–45. [Google Scholar] [CrossRef]
- Gorton, L.; Csöregi, E.; Domínguez, E.; Emnéus, J.; Jönsson-Pettersson, G.; Marko-Varga, G.; Persson, B. Selective detection in flow-analysis based on the combination of immobilized enzymes and chemically modified electrodes. Anal. Chim. Acta 1991, 250, 203–248. [Google Scholar] [CrossRef]
- Antiochia, R.; Lavagnini, I. Alcohol biosensor based on the immobilization of meldola blue and alcohol dehydrogenase into a carbon nanotube paste electrode. Anal. Lett. 2006, 39, 1643–1655. [Google Scholar] [CrossRef]
- Antiochia, R.; Gorton, L. Development of a carbon nanotube paste electrode osmium polymer-mediated biosensor for determination of glucose in alcoholic beverages. Biosens. Bioelectron. 2007, 22, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, O.; Shleev, S.; Gayda, G.; Demkiv, O.; Gonchar, M.; Gorton, L.; Csöregi, E.; Nistor, M. Bi-enzyme biosensor based on NAD+-and glutathione-dependent recombinant formaldehyde dehydrogenase and diaphorase for formaldehyde assay. Sens. Actuators B Chem. 2007, 125, 1–9. [Google Scholar] [CrossRef]
- Antiochia, R.; Palleschi, G. Atri-enzyme electrode probe for the sequential determination of fructose and glucose in the same sample. Anal. Lett. 1997, 30, 683–697. [Google Scholar] [CrossRef]
- Klinman, J.P.; Dove, J.E. Novel Cofactors; Gulf Professional Publishing: Houston, TX, USA, 2001; Volume 58. [Google Scholar]
- Ferri, S.; Kojima, K.; Sode, K. Review of glucose oxidases and glucose dehydrogenases: A bird’s eye view of glucose sensing enzymes. J. Diabetes Sci. Technol. 2011, 5, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Heller, A. Electron-conducting redox hydrogels: Design, characteristics and synthesis. Curr. Opin. Chem. Biol. 2006, 10, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Hämmerle, M.; Olsthoorn, A.J.J.; Schuhmann, W.; Schmidt, H.-L.; Duine, J.A.; Heller, A. High current density “wired” quinoprotein glucose dehydrogenase electrode. Anal. Chem. 1993, 65, 238–241. [Google Scholar] [CrossRef]
- Zafar, M.N.; Beden, N.; Leech, D.; Sygmund, C.; Ludwig, R.; Gorton, L. Characterization of different FAD-dependent glucose dehydrogenases for possible use in glucose-based biosensors and biofuel cells. Anal. Bioanal. Chem. 2012, 402, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Kojima, S.; Kano, K.; Ikeda, T.; Sato, M.; Sanada, H.; Omura, H. Novel FAD-dependent glucose dehydrogenase for a dioxygen-insensitive glucose biosensor. Biosci. Biotechnol. Biochem. 2006, 70, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Antiochia, R.; Vinci, G.; Gorton, L. Rapid and direct determination of fructose in food: A new osmium-polymer mediated biosensor. Food Chem. 2013, 140, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Laurinavicius, V.; Kurtinaitiene, B.; Liauksminas, V.; Ramanavicius, A.; Meskys, R.; Rudomanskis, R.; Skotheim, T.; Boguslavsky, L. Oxygen insensitive glucose biosensor based on PQQ-dependent glucose dehydrogenase. Anal. Lett. 1999, 32, 299–316. [Google Scholar] [CrossRef]
- Tseng, T.-F.; Yang, Y.-L.; Lou, S.-L. Chromium Hexacyanoferrate Modified Biosensor Based on PQQ-Dependent Glucose Dehydrogenase. In Proceedings of the 29th Annual International Conference of the Engineering in Medicine and Biology Society, EMBS 2007, Lyon, France, 22–26 August 2007; pp. 2681–2684. [Google Scholar]
- Razumiene, J.; Cirbaite, E.; Razumas, V.; Laurinavicius, V. New mediators for biosensors based on PQQ-dependent alcohol dehydrogenases. Sens. Actuators B Chem. 2015, 207, 1019–1025. [Google Scholar] [CrossRef]
- Laurinavicius, V.; Razumiene, J.; Ramanavicius, A.; Ryabov, A.D. Wiring of PQQ-dehydrogenases. Biosens. Bioelectron. 2004, 20, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wollenberger, U.; Scheller, F.W. PQQ as redox shuttle for quinoprotein glucose dehydrogenase. Biol. Chem. 1998, 379, 1207–1211. [Google Scholar] [PubMed]
- Pinyou, P.; Ruff, A.; Pöller, S.; Ma, S.; Ludwig, R.; Schuhmann, W. Design of an Os complex modified hydrogel with optimized redox potential for biosensors and biofuel cells. Chem.-A Eur. J. 2016, 22, 5319–5326. [Google Scholar] [CrossRef] [PubMed]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.; Ruzgas, T.; Gazaryan, I. Direct electron transfer between heme-containing enzymes and electrodes as basis for third generation biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar] [CrossRef]
- Ikeda, T.; Kobayashi, D.; Matsushita, F.; Sagara, T.; Niki, K. Bioelectrocatalysis at electrodes coated with alcohol-dehydrogenase, a quinohemoprotein with heme c serving as a built-in mediator. J. Electroanal. Chem. 1993, 361, 221–228. [Google Scholar] [CrossRef]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef] [PubMed]
- Karyakin, A.A. Principles of direct (mediator free) bioelectrocatalysis. Bioelectrochemistry 2012, 88, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Matsumura, H.; Ishida, T.; Yoshida, M.; Igarashi, K.; Samejima, M.; Ohno, H.; Nakamura, N. pH-dependent electron transfer reaction and direct bioelectrocatalysis of the quinohemoprotein pyranose dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 477, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Gorton, L. Direct electrochemistry of heme multicofactor-containing enzymes on alkanethiol-modified gold electrodes. Bioelectrochemistry 2005, 66, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Ruzgas, T.; Gorton, L. Direct electron transfer of heme- and molybdopterin cofactor-containing chicken liver sulfite oxidase on alkanethiol-modified gold electrodes. Anal. Chem. 2003, 75, 4841–4850. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Shipovskov, S.; Gorton, L. Bioelectrocatalytic detection of theophylline at theophylline oxidase electrodes. Biosens. Bioelectron. 2007, 22, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Sosna, M.; Bonamore, A.; Gorton, L.; Boffi, A.; Ferapontova, E.E. Direct electrochemistry and Os-polymer-mediated bioelectrocatalysis of NADH oxidation by Escherichia coli flavohemoglobin at graphite electrodes. Biosens. Bioelectron. 2013, 42, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Christenson, A.; Dock, E.; Gorton, L.; Ruzgas, T. Direct heterogeneous electron transfer of theophylline oxidase. Biosens. Bioelectron. 2004, 20, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.J.; McElhaney, A.E.; Feng, C.J.; Enemark, J.H.; Armstrong, F.A. Avoltammetric study of interdomain electron transfer within sulfite oxidase. J. Am. Chem. Soc. 2002, 124, 11612–11613. [Google Scholar] [CrossRef] [PubMed]
- Frasca, S.; Rojas, O.; Salewski, J.; Neumann, B.; Stiba, K.; Weidinger, I.M.; Tiersch, B.; Leimkühler, S.; Koetz, J.; Wollenberger, U. Human sulfite oxidase electrochemistry on gold nanoparticles modified electrode. Bioelectrochemistry 2012, 87, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Smutok, O.; Gayda, G.; Gonchar, M.; Schuhmann, W. A novel L-lactate-selective biosensor based on flavocytochrome b2 from methylotrophic yeast Hansenula polymorpha. Biosens. Bioelectron. 2005, 20, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Aguey-Zinsou, K.F.; Bernhardt, P.V.; Kappler, U.; McEwan, A.G. Direct electrochemistry of a bacterial sulfite dehydrogenase. J. Am. Chem. Soc. 2003, 125, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Miyaoka, S.; Matsushita, F.; Kobayashi, D.; Senda, M. Direct bioelectrocatalysis at metal and carbon electrodes modified with adsorbed D-gluconate dehydrogenase or adsorbed alcohol-dehydrogenase from bacterial-membranes. Chem. Lett. 1992, 21, 847–850. [Google Scholar] [CrossRef]
- Ludwig, R.; Harreither, W.; Tasca, F.; Gorton, L. Cellobiose dehydrogenase: A versatile catalyst for electrochemical applications. ChemPhysChem 2010, 11, 2674–2697. [Google Scholar] [CrossRef] [PubMed]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Nöll, G. Comparison of direct and mediated electron transfer for cellobiose dehydrogenase from Phanerochaete sordida. Anal. Chem. 2009, 81, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Yakushi, T.; Matsushita, K.; Kitazumi, Y.; Shirai, O.; Kano, K. The electron transfer pathway in direct electrochemical communication of fructose dehydrogenase with electrodes. Electrochem. Commun. 2014, 38, 28–31. [Google Scholar] [CrossRef]
- Kamitaka, Y.; Tsujimura, S.; Setoyama, N.; Kajino, T.; Kano, K. Fructose/dioxygen biofuel cell based on direct electron transfer-type bioelectrocatalysis. Phys. Chem. Chem. Phys. 2007, 9, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Kano, K. Bioelectrocatalysis-based application of quinoproteins and quinoprotein-containing bacterial cells in biosensors and biofuel cells. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2003, 1647, 121–126. [Google Scholar] [CrossRef]
- So, K.; Kawai, S.; Hamano, Y.; Kitazumi, Y.; Shirai, O.; Hibi, M.; Ogawa, J.; Kano, K. Improvement of a direct electron transfer-type fructose/dioxygen biofuel cell with a substrate-modified biocathode. Phys. Chem. Chem. Phys. 2014, 16, 4823–4829. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Andoralov, V.; Blum, Z.; Sotres, J.; Suyatin, D.B.; Ruzgas, T.; Arnebrant, T.; Shleev, S. Biofuel cell as a power source for electronic contact lenses. Biosens. Bioelectron. 2012, 37, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Krikstolaityte, V.; Lamberg, P.; Toscano, M.; Silow, M.; Eicher-Lorka, O.; Ramanavicius, A.; Niaura, G.; Abariute, L.; Ruzgas, T.; Shleev, S. Mediatorless carbohydrate/oxygen biofuel cells with improved cellobiose dehydrogenase based bioanode. Fuel Cells 2014, 14, 792–800. [Google Scholar] [CrossRef]
- Shleev, S. Quo vadis, implanted fuel cell? ChemPlusChem 2017, 82, 522–539. [Google Scholar] [CrossRef]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef] [PubMed]
- DirectSens Biosensors. Available online: http://www.directsens.com/ (accessed on 3 April 2018).
- Heller, A. Electrical wiring of redox enzymes. Acc. Chem. Res. 1990, 23, 128–134. [Google Scholar] [CrossRef]
- Ruff, A. Redox polymers in bioelectrochemistry: Common playgrounds and novel concepts. Curr. Opin. Electrochem. 2017, 5, 66–73. [Google Scholar] [CrossRef]
- Milton, R.D.; Wang, T.; Knoche, K.L.; Minteer, S.D. Tailoring biointerfaces for electrocatalysis. Langmuir 2016, 32, 2291–2301. [Google Scholar] [CrossRef] [PubMed]
- Prabhulkar, S.; Tian, H.; Wang, X.; Zhu, J.-J.; Li, C.-Z. Engineered proteins: Redox properties and their applications. Antioxid. Redox Signal. 2012, 17, 1796–1822. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Mathias, J.P.; Seto, C.T. Molecular self-assembly and nanochemistry: A chemical strategy for the synthesis of nanostructures. Science 1991, 254, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Blum, Z.; Shleev, S. Direct electron transfer based enzymatic fuel cells. Electrochim. Acta 2012, 82, 191–202. [Google Scholar] [CrossRef]
- Flexer, V.; Mano, N. Wired pyrroloquinoline quinone soluble glucose dehydrogenase enzyme electrodes operating at unprecedented low redox potential. Anal. Chem. 2014, 86, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, S.; Hartwich, G.; Schulte, A.; Schuhmann, W. Amperometric Enzyme Sensors based on Direct and Mediated Electron Transfer. In Electrochemistry of Nucleic Acids and Proteins: Towards Electrochemical Sensors for Genomics and Proteomics; Palecek, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 599–655. [Google Scholar]
- Ferapontova, E.E.; Shleev, S.; Ruzgas, T.; Stoica, L.; Christenson, A.; Tkac, J.; Yaropolov, A.I.; Gorton, L. Direct Electrochemistry of Proteins and Enzymes. In Electrochemistry of Nucleic Acids and Proteins: Towards Electrochemical Sensors for Genomics and Proteomics; Palecek, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 517–598. [Google Scholar]
- Leger, C.; Bertrand, P. Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghindilis, A.L.; Atanasov, P.; Wilkins, E. Enzyme-catalyzed direct electron transfer: Fundamentals and analytical applications. Electroanalysis 1997, 9, 661–674. [Google Scholar] [CrossRef]
- Wollenberger, U.; Spricigo, R.; Leimkühler, S.; Schröder, K. Protein electrodes with direct electrochemical communication. In Biosensing for the 21st Century; Renneberg, R., Lisdat, F., Eds.; Springer: Berlin, Germany, 2008; Volume 109, pp. 19–64. [Google Scholar]
- Wollenberger, U. Third Generation Biosensors—Integrating Recognition and Transduction in Electrochemical Sensors; Elsevier Science B.V.: Amsterdam, The Netherlands, 2005; pp. 65–130. [Google Scholar]
- Zhang, W.J.; Li, G.X. Third-generation biosensors based on the direct electron transfer of proteins. Anal. Sci. 2004, 20, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent advances on developing 3rd generation enzyme electrode for biosensor applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef] [PubMed]
- De Poulpiquet, A.; Ciaccafava, A.; Lojou, E. New trends in enzyme immobilization at nanostructured interfaces for efficient electrocatalysis in biofuel cells. Electrochim. Acta 2014, 126, 104–114. [Google Scholar] [CrossRef]
- de Poulpiquet, A.; Ranava, D.; Monsalve, K.; Giudici-Orticoni, M.-T.; Lojou, E. Biohydrogen for a new generation of H2/O2 biofuel cells: A sustainable energy perspective. ChemElectroChem 2014, 1, 1724–1750. [Google Scholar] [CrossRef]
- Shleev, S.; Jarosz-Wilkolazka, A.; Khalunina, A.; Morozova, O.; Yaropolov, A.; Ruzgas, T.; Gorton, L. Direct electron transfer reactions of laccases from different origins on carbon electrodes. Bioelectrochemistry 2005, 67, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Shleev, S.; Tkac, J.; Christenson, A.; Ruzgas, T.; Yaropolov, A.I.; Whittaker, J.W.; Gorton, L. Direct electron transfer between copper-containing proteins and electrodes. Biosens. Bioelectron. 2005, 20, 2517–2554. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Ruzgas, T.; Csöregi, E.; Emnéus, J.; Gorton, L.; Marko-Varga, G. Peroxidase-modified electrodes: Fundamentals and application. Anal. Chim. Acta 1996, 330, 123–138. [Google Scholar] [CrossRef]
- Christenson, A.; Dimcheva, N.; Ferapontova, E.E.; Gorton, L.; Ruzgas, T.; Stoica, L.; Shleev, S.; Yaropolov, A.L.; Haltrich, D.; Thorneley, R.N.F.; Aust, S.D. Direct electron transfer between ligninolytic redox enzymes and electrodes. Electroanalysis 2004, 16, 1074–1092. [Google Scholar] [CrossRef]
- Guo, L.-H.; Allen, H.; Hill, O. Direct electrochemistry of proteins and enzymes. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 1991; Volume 36, pp. 341–375. [Google Scholar]
- Ikeda, T.; Matsushita, F.; Senda, M. Amperometric fructose sensor based on direct bioelectrocatalysis. Biosens. Bioelectron. 1991, 6, 299–304. [Google Scholar] [CrossRef]
- Christenson, A.; Gustavsson, T.; Gorton, L.; Hägerhäll, C. Direct and mediated electron transfer between intact succinate:quinone oxidoreductase from Bacillus subtilis and a surface modified gold electrode reveals redox state-dependent conformational changes. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Sucheta, A.; Ackrell, B.A.; Armstrong, F.A. Electrocatalytic voltammetry of succinate dehydrogenase: Direct quantification of the catalytic properties of a complex electron-transport enzyme. J. Am. Chem. Soc. 1996, 118, 5031–5038. [Google Scholar] [CrossRef]
- Bollella, P.; Ludwig, R.; Gorton, L. Cellobiose dehydrogenase: Insights on the nanostructuration of electrodes for improved development of biosensors and biofuel cells. Appl. Mater. Today 2017, 9, 319–332. [Google Scholar] [CrossRef]
- Ludwig, R.; Ortiz, R.; Schulz, C.; Harreither, W.; Sygmund, C.; Gorton, L. Cellobiose dehydrogenase modified electrodes: Advances by materials science and biochemical engineering. Anal. Bioanal. Chem. 2013, 405, 3637–3658. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Nishina, A.; Hamano, Y.; Kano, K.; Shiraishi, S. Electrochemical reaction of fructose dehydrogenase on carbon cryogel electrodes with controlled pore sizes. Electrochem. Commun. 2010, 12, 446–449. [Google Scholar] [CrossRef]
- Xia, H.-q.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Interaction between D-fructose dehydrogenase and methoxy-substituent-functionalized carbon surface to increase productive orientations. Electrochim. Acta 2016, 218, 41–46. [Google Scholar] [CrossRef]
- Treu, B.L.; Minteer, S.D. Isolation and purification of PQQ-dependent lactate dehydrogenase from Gluconobacter and use for direct electron transfer at carbon and gold electrodes. Bioelectrochemistry 2008, 74, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Ramanavicius, A.; Habermüller, K.; Csöregi, E.; Laurinavicius, V.; Schuhmann, W. Polypyrrole-entrapped quinohemoprotein alcohol dehydrogenase. Evidence for direct electron transfer via conducting-polymer chains. Anal. Chem. 1999, 71, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Miyaoka, S.; Miki, K. Enzyme-catalysed electrochemical oxidation of D-gluconate at electrodes coated with D-gluconate dehydrogenase, a membrane-bound flavohemoprotein. J. Electroanal. Chem. 1993, 352, 267–278. [Google Scholar] [CrossRef]
- Shiota, M.; Yamazaki, T.; Yoshimatsu, K.; Kojima, K.; Tsugawa, W.; Ferri, S.; Sode, K. An Fe-S cluster in the conserved Cys-rich region in the catalytic subunit of FAD-dependent dehydrogenase complexes. Bioelectrochemistry 2016, 112, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Tuurala, S.; Lau, C.; Atanassov, P.; Smolander, M.; Minteer, S.D. Characterization and stability study of immobilized PQQ-dependent aldose dehydrogenase bioanodes. Electroanalysis 2012, 24, 229–238. [Google Scholar] [CrossRef]
- Treu, B.L.; Sokic-Lazic, D.; Minteer, S. Bioelectrocatalysis of pyruvate with PQQ-dependent pyruvate dehydrogenase. ECS Trans. 2010, 25, 1–11. [Google Scholar]
- Xu, S.; Minteer, S.D. Investigating the impact of multi-heme pyrroloquinoline quinone-aldehyde dehydrogenase orientation on direct bioelectrocatalysis via site specific enzyme immobilization. ACS Catal. 2013, 3, 1756–1763. [Google Scholar] [CrossRef]
- Zamocky, M.; Ludwig, R.; Peterbauer, C.; Hallberg, B.; Divne, C.; Nicholls, P.; Haltrich, D. Cellobiose dehydrogenase-a flavocytochrome from wood-degrading, phytopathogenic and saprotropic fungi. Curr. Protein Pept. Sci. 2006, 7, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Sutzl, L.; Laurent, C.V.F.P.; Abrera, A.T.; Schutz, G.; Ludwig, R.; Haltrich, D. Multiplicity of enzymatic functions in the CAZy AA3 family. Appl. Microbiol. Biotechnol. 2018, 102, 2477–2492. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, G.; Johansson, G.; Pettersson, G. Acritical review of cellobiose dehydrogenases. J. Biotechnol. 2000, 78, 93–113. [Google Scholar] [CrossRef]
- Stoica, L.; Ludwig, R.; Haltrich, D.; Gorton, L. Third-generation biosensor for lactose based on newly discovered cellobiose dehydrogenase. Anal. Chem. 2006, 78, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.D.; Aust, S.D. Cellobiose dehydrogenase—An extracellular fungal flavocytochrome. Enzym. Microb. Technol. 2001, 28, 129–138. [Google Scholar] [CrossRef]
- Schulz, C.; Ludwig, R.; Micheelsen, P.O.; Silow, M.; Toscano, M.D.; Gorton, L. Enhancement of enzymatic activity and catalytic current of cellobiose dehydrogenase by calcium ions. Electrochem. Commun. 2012, 17, 71–74. [Google Scholar] [CrossRef]
- Kielb, P.; Sezer, M.; Katz, S.; Lopez, F.; Schulz, C.; Gorton, L.; Ludwig, R.; Wollenberger, U.; Zebger, I.; Weidinger, I.M. Spectroscopic observation of calcium-induced reorientation of cellobiose dehydrogenase immobilized on electrodes and its effect on electrocatalytic activity. ChemPhysChem 2015, 16, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Zahma, K.; Schulz, C.; Sygmund, C.; Gorton, L.; Ludwig, R. Inter-domain electron transfer in cellobiose dehydrogenase: Modulation by pH and divalent cations. FEBS J. 2015, 282, 3136–3148. [Google Scholar] [CrossRef] [PubMed]
- Tavahodi, M.; Ortiz, R.; Schulz, C.; Ekhtiari, A.; Ludwig, R.; Haghighi, B.; Gorton, L. Direct electron transfer of cellobiose dehydrogenase on positively charged polyethyleneimine gold nanoparticles. ChemPlusChem 2017, 82, 546–552. [Google Scholar] [CrossRef]
- Igarashi, K.; Yoshida, M.; Matsumura, H.; Nakamura, N.; Ohno, H.; Samejima, M.; Nishino, T. Electron transfer chain reaction of the extracellular flavocytochrome cellobiose dehydrogenase from the basidiomycete Phanerochaete chrysosporium. FEBS J. 2005, 272, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Harreither, W.; Sygmund, C.; Augustin, M.; Narciso, M.; Rabinovich, M.L.; Gorton, L.; Haltrich, D.; Ludwig, R. Catalytic properties and classification of cellobiose dehydrogenases from ascomycetes. Appl. Environ. Microbiol. 2011, 77, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.M.; Henriksson, G.; Pettersson, G.; Divne, C. Crystal structure of the flavoprotein domain of the extracellular flavocytochrome cellobiose dehydrogenase. J. Mol. Biol. 2002, 315, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.M.; Bergfors, T.; Bäckbro, K.; Pettersson, G.; Henriksson, G.; Divne, C. A new scaffold for binding haem in the cytochrome domain of the extracellular flavocytochrome cellobiose dehydrogenase. Structure 2000, 8, 79–88. [Google Scholar] [CrossRef]
- Tan, T.-C.; Kracher, D.; Gandini, R.; Sygmund, C.; Kittl, R.; Haltrich, D.; Hallberg, B.M.; Ludwig, R.; Divne, C. Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat. Commun. 2015, 6, 7542. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. AC4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmayr, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 2016, 352, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Ludwig, R. Cellobiose dehydrogenase: An essential enzyme for lignocellulose degradation in nature—A review/Cellobiosedehydrogenase: Ein essentielles Enzym für den Lignozelluloseabbau in der Natur-Eine Übersicht. Bodenkultur 2016, 67, 145–163. [Google Scholar] [CrossRef]
- Safina, G.; Ludwig, R.; Gorton, L. Asimple and sensitive method for lactose detection based on direct electron transfer between immobilised cellobiose dehydrogenase and screen-printed carbon electrodes. Electrochim. Acta 2010, 55, 7690–7695. [Google Scholar] [CrossRef]
- Tasca, F.; Harreither, W.; Ludwig, R.; Gooding, J.J.; Gorton, L. Cellobiose dehydrogenase aryl diazonium modified single walled carbon nanotubes: Enhanced direct electron transfer through a positively charged surface. Anal. Chem. 2011, 83, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Bozorgzadeh, S.; Hamidi, H.; Ortiz, R.; Ludwig, R.; Gorton, L. Direct electron transfer of Phanerochaete chrysosporium cellobiose dehydrogenase at platinum and palladium nanoparticles decorated carbon nanotubes modified electrodes. Phys. Chem. Chem. Phys. 2015, 17, 24157–24165. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Mazzei, F.; Favero, G.; Fusco, G.; Ludwig, R.; Gorton, L.; Antiochia, R. Improved DET communication between cellobiose dehydrogenase and a gold electrode modified with a rigid self-assembled monolayer and green metal nanoparticles: The role of an ordered nanostructuration. Biosens. Bioelectron. 2017, 88, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Tasca, F.; Zafar, M.N.; Harreither, W.; Nöll, G.; Ludwig, R.; Gorton, L. Athird generation glucose biosensor based on cellobiose dehydrogenase from Corynascus thermophilus and single-walled carbon nanotubes. Analyst 2011, 136, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.N.; Safina, G.; Ludwig, R.; Gorton, L. Characteristics of third-generation glucose biosensors based on Corynascus thermophilus cellobiose dehydrogenase immobilized on commercially available screen-printed electrodes working under physiological conditions. Anal. Biochem. 2012, 425, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ammam, M.; Fransaer, J. Two-enzyme lactose biosensor based on β-galactosidase and glucose oxidase deposited by AC-electrophoresis: Characteristics and performance for lactose determination in milk. Sens. Actuators B 2010, 148, 583–589. [Google Scholar] [CrossRef]
- Loğoğlu, E.; Sungur, S.; Yildiz, Y. Development of lactose biosensor based on β-galactosidase and glucose oxidase immobilized into gelatin. J. Macromol. Sci. Part A Pure Appl. Chem. 2006, 43, 525–533. [Google Scholar] [CrossRef]
- Sharma, S.K.; Kumar, A.; Chaudhary, R.; Pundir, S.; Pundir, C.S.; Sehgal, N. Lactose biosensor based on lactase and galactose oxidase immobilized in polyvinyl formal. Artif. Cells Blood Substit. Biotechnol. 2007, 35, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, D.; Ralis, E.V.; Makower, A.; Scheller, F.W. Amperometric Bi-enzyme based biosensor for the detection of lactose—Characterization and application. J. Chem. Technol. Biotechnol. 1990, 49, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Glithero, N.; Clark, C.; Gorton, L.; Schuhmann, W.; Pasco, N. At-line measurement of lactose in dairy-processing plants. Anal. Bioanal. Chem. 2013, 405, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.; Ma, S.; Ludwig, R.; Schuhmann, W.; Ruff, A. A polymer multilayer based amperometric biosensor for the detection of lactose in the presence of high concentrations of glucose. Electroanalysis 2017, 29, 154–161. [Google Scholar] [CrossRef]
- Tasca, F.; Ludwig, R.; Gorton, L.; Antiochia, R. Determination of lactose by a novel third generation biosensor based on a cellobiose dehydrogenase and aryl diazonium modified single wall carbon nanotubes electrode. Sens. Actuators B-Chem. 2013, 177, 64–69. [Google Scholar] [CrossRef]
- Ortiz, R.; Ludwig, R.; Gorton, L. Highly efficient membraneless glucose bioanode based on Corynascus thermophilus cellobiose dehydrogenase on aryl diazonium-activated single-walled carbon nanotubes. ChemElectroChem 2014, 1, 1948–1956. [Google Scholar] [CrossRef]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Bollella, P.; Schulz, C.; Favero, G.; Mazzei, F.; Ludwig, R.; Gorton, L.; Antiochia, R. Green synthesis and characterization of gold and silver nanoparticles and their application for development of a third generation lactose biosensor. Electroanalysis 2017, 29, 77–86. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Al-Lolage, F.A. There is no evidence to support literature claims of direct electron transfer (DET) for native glucose oxidase (GOx) at carbon nanotubes or graphene. J. Electroanal. Chem. 2017. [Google Scholar] [CrossRef]
- Wilson, G.S. Native glucose oxidase does not undergo direct electron transfer. Biosens. Bioelectron. 2016, 82, VII–VIII. [Google Scholar] [CrossRef] [PubMed]
- Harreither, W.; Coman, V.; Ludwig, R.; Haltrich, D.; Gorton, L. Investigation of graphite electrodes modified with cellobiose dehydrogenase from the ascomycete Myriococcum thermophilum. Electroanalysis 2007, 19, 172–180. [Google Scholar] [CrossRef]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Noll, G. Highly efficient and versatile anodes for biofuel cells based on cellobiose dehydrogenase from Myriococcum thermophilum. J. Phys. Chem. C 2008, 112, 13668–13673. [Google Scholar] [CrossRef]
- Sygmund, C.; Harreither, W.; Haltrich, D.; Gorton, L.; Ludwig, R. A new generation of glucose biosensors-engineering cellobiose dehydrogenase for increased direct electron transfer. New Biotechnol. 2009, 25, S115. [Google Scholar] [CrossRef]
- Cosnier, S.; Le Goff, A.; Holzinger, M. Towards glucose biofuel cells implanted in human body for powering artificial organs. Electrochem. Commun. 2014, 38, 19–23. [Google Scholar] [CrossRef]
- Leech, D.; Kavanagh, P.; Schuhmann, W. Enzymatic fuel cells: Recent progress. Electrochim. Acta 2012, 84, 223–234. [Google Scholar] [CrossRef]
- Coman, V.; Vaz-Domínguez, C.; Ludwig, R.; Harreither, W.; Haltrich, D.; De Lacey, A.L.; Ruzgas, T.; Gorton, L.; Shleev, S. A membrane-, mediator-, cofactor-less glucose/oxygen biofuel cell. Phys. Chem. Chem. Phys. 2008, 10, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Coman, V.; Ludwig, R.; Harreither, W.; Haltrich, D.; Gorton, L.; Ruzgas, T.; Shleev, S. Adirect electron transfer-based glucose/oxygen biofuel cell operating in human serum. Fuel Cells 2010, 10, 9–16. [Google Scholar]
- Falk, M.; Pankratov, D.; Lindh, L.; Arnebrant, T.; Shleev, S. Miniature direct electron transfer based enzymatic fuel cell operating in human sweat and saliva. Fuel Cells 2014, 14, 1050–1056. [Google Scholar] [CrossRef]
- Kawai, S.; Goda-Tsutsumi, M.; Yakushi, T.; Kano, K.; Matsushita, K. Heterologous overexpression and characterization of a flavoprotein-cytochrome c complex fructose dehydrogenase of Gluconobacter japonicus NBRC3260. Appl. Environ. Microbiol. 2013, 79, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kano, K. High current density bioelectrolysis of D-fructose at fructose dehydrogenase-adsorbed and Ketjen black-modified electrodes without a mediator. Chem. Lett. 2006, 36, 218–219. [Google Scholar] [CrossRef]
- Hamano, Y.; Tsujimura, S.; Shirai, O.; Kano, K. Micro-cubic monolithic carbon cryogel electrode for direct electron transfer reaction of fructose dehydrogenase. Bioelectrochemistry 2012, 88, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. The influence of pH and divalent/monovalent cations on the internal electron transfer (IET), enzymatic activity and structure of fructose dehydrogenase. Anal. Bioanal. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kizling, M.; Bilewicz, R. Fructose dehydrogenase electron transfer pathway in bioelectrocatalytic reactions. ChemElectroChem 2018, 5, 166–174. [Google Scholar] [CrossRef]
- Bilewicz, R.; Kizling, M.; Dzwonek, M.; Wieckowska, A. Size does matter—Mediation of electron transfer by gold clusters in bioelectrocatalysis. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Miyake, T.; Haneda, K.; Nagai, N.; Yatagawa, Y.; Onami, H.; Yoshino, S.; Abe, T.; Nishizawa, M. Enzymatic biofuel cells designed for direct power generation from biofluids in living organisms. Energy Environ. Sci. 2011, 4, 5008–5012. [Google Scholar] [CrossRef]
- Khan, G.F.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. Electrochemical behaviour of monolayer quinoprotein adsorbed on the electrode surface. J. Electroanal. Chem. 1991, 315, 263–273. [Google Scholar] [CrossRef]
- Khan, G.F.; Kobatake, E.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. Molecular interface for an activity controlled enzyme electrode and its application for the determination of fructose. Anal. Chem. 1992, 64, 1254–1258. [Google Scholar] [CrossRef]
- Khan, G.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. An amperometric biosensor for fructose using a PQQ enzyme. Sens. Actuators B Chem. 1993, 14, 673–674. [Google Scholar] [CrossRef]
- Ameyama, M.; Shinagawa, E.; Matsushita, K.; Adachi, O. D-fructose dehydrogenase of Gluconobacter industrius: Purification, characterization and application to enzymatic microdetermination of D-fructose. J. Bacteriol. 1981, 145, 814–823. [Google Scholar] [PubMed]
- Parellada, J.; Domínguez, E.; Fernández, V.M. Amperometric flow injection determination of fructose in honey with a carbon paste sensor based on fructose dehydrogenase. Anal. Chim. Acta 1996, 330, 71–77. [Google Scholar] [CrossRef]
- Yabuki, S.; Mizutani, F. D-Fructose sensing electrode based on electron transfer of D-fructose dehydrogenase at colloidal gold-enzyme modified electrode. Electroanalysis 1997, 9, 23–25. [Google Scholar] [CrossRef]
- Begum, A.; Kobatake, E.; Suzawa, T.; Ikariyama, Y.; Aizawa, M. New electrocatalytic biomolecular interface for fabricating a fructose dehydrogenase-based sensing system. Anal. Chim. Acta 1993, 280, 31–36. [Google Scholar] [CrossRef]
- Mizutani, F.; Yabuki, S. Highly-sensitive measurement of dihydroxyphenols using carbon felt electrode impregnated with fructose dehydrogenase-containing solution. Chem. Lett. 1994, 23, 1569–1572. [Google Scholar] [CrossRef]
- Kinnear, K.T.; Monbouquette, H.G. An amperometric fructose biosensor based on fructose dehydrogenase immobilized in a membrane mimetic layer on gold. Anal. Chem. 1997, 69, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Nomura, S.; Taniguchi, I. d-Fructose detection based on the direct heterogeneous electron transfer reaction of fructose dehydrogenase adsorbed onto multi-walled carbon nanotubes synthesized on platinum electrode. Biosens. Bioelectron. 2009, 24, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Nazaruk, E.; Landau, E.M.; Bilewicz, R. Membrane bound enzyme hosted in liquid crystalline cubic phase for sensing and fuel cells. Electrochim. Acta 2014, 140, 96–100. [Google Scholar] [CrossRef]
- Šakinytė, I.; Barkauskas, J.; Gaidukevič, J.; Razumienė, J. Thermally reduced graphene oxide: The study and use for reagentless amperometric D-fructose biosensors. Talanta 2015, 144, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The immobilization of fructose dehydrogenase on nanoporous gold electrodes for the detection of fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Murata, K.; Kajiya, K.; Nakamura, N.; Ohno, H. Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell. Energy Environ. Sci. 2009, 2, 1280–1285. [Google Scholar] [CrossRef]
- Haneda, K.; Yoshino, S.; Ofuji, T.; Miyake, T.; Nishizawa, M. Sheet-shaped biofuel cell constructed from enzyme-modified nanoengineered carbon fabric. Electrochim. Acta 2012, 82, 175–178. [Google Scholar] [CrossRef]
- Miyake, T.; Haneda, K.; Yoshino, S.; Nishizawa, M. Flexible, layered biofuel cells. Biosens. Bioelectron. 2013, 40, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Kizling, M.; Stolarczyk, K.; Kiat, J.S.S.; Tammela, P.; Wang, Z.; Nyholm, L.; Bilewicz, R. Pseudocapacitive polypyrrole–nanocellulose composite for sugar-air enzymatic fuel cells. Electrochem. Commun. 2015, 50, 55–59. [Google Scholar] [CrossRef]
- Holland, J.T.; Lau, C.; Brozik, S.; Atanassov, P.; Banta, S. Engineering of glucose oxidase for direct electron transfer via site-specific gold nanoparticle conjugation. J. Am. Chem. Soc. 2011, 133, 19262–19265. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kataoka, K.; Sakurai, T.; Ikeda, T.; Kano, K. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 2007, 601, 119–124. [Google Scholar] [CrossRef]
- Campàs, M.; Prieto-Simón, B.; Marty, J.-L. A Review of the Use of Genetically Engineered Enzymes in Electrochemical Biosensors. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2009; pp. 3–9. [Google Scholar]
- Yamazaki, T.; Kojima, K.; Sode, K. Extended-range glucose sensor employing engineered glucose dehydrogenases. Anal. Chem. 2000, 72, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Okuda, J.; Sode, K. PQQ glucose dehydrogenase with novel electron transfer ability. Biochem. Biophys. Res. Commun. 2004, 314, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Algov, I.; Grushka, J.; Zarivach, R.; Alfonta, L. Highly efficient flavin-adenine dinucleotide glucose dehydrogenase fused to a minimal cytochrome c domain. J. Am. Chem. Soc. 2017, 139, 17217–17220. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Rahman, M.; Zangrilli, B.; Sygmund, C.; Micheelsen, P.O.; Silow, M.; Toscano, M.D.; Ludwig, R.; Gorton, L. Engineering of cellobiose dehydrogenases for improved glucose sensitivity and reduced maltose affinity. ChemElectroChem 2017, 4, 846–855. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L.; Ludwig, R.; Antiochia, R. Athird generation glucose biosensor based on cellobiose dehydrogenase immobilized on a glassy carbon electrode decorated with electrodeposited gold nanoparticles: Characterization and application in human saliva. Sensors 2017, 17, 1912. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Fusco, G.; Stevar, D.; Gorton, L.; Ludwig, R.; Ma, S.; Boer, H.; Koivula, A.; Tortolini, C.; Favero, G. Aglucose/oxygen enzymatic fuel cell based on gold nanoparticles modified graphene screen-printed electrode. Proof-of-concept in human saliva. Sens. Actuators B Chem. 2018, 256, 921–930. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a protein-engineered variant of D-fructose dehydrogenase for direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2017, 77, 112–115. [Google Scholar] [CrossRef]
- Ortiz, R.; Matsumura, H.; Tasca, F.; Zahma, K.; Samejima, M.; Igarashi, K.; Ludwig, R.; Gorton, L. Effect of deglycosylation of cellobiose dehydrogenases on the enhancement of direct electron transfer with electrodes. Anal. Chem. 2012, 84, 10315–10323. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of electron transfer in biosensors: 1st generation (left), 2nd generation with soluble or immobilised mediator (centre) and 3rd generation (right).
Figure 1.
Schematic representation of electron transfer in biosensors: 1st generation (left), 2nd generation with soluble or immobilised mediator (centre) and 3rd generation (right).
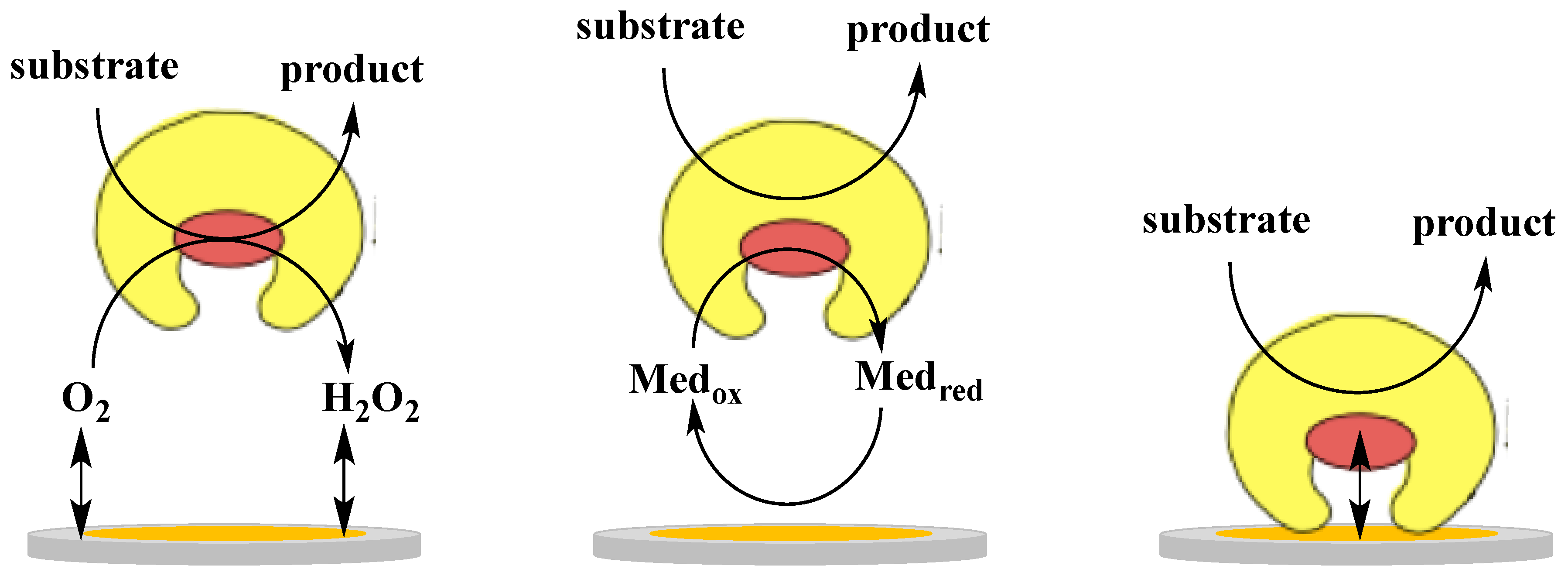
Figure 2.
Schematic representation of cellobiose dehydrogenase (CDH): DHCDH domain is shown in green with the FAD cofactor in pink; CYTCDH domain in violet with heme b cofactor in orange; the flexible linker, in blue, is responsible for the modulation of internal electron transfer (IET); all the potential glycosylation sites are shown in red.
Figure 2.
Schematic representation of cellobiose dehydrogenase (CDH): DHCDH domain is shown in green with the FAD cofactor in pink; CYTCDH domain in violet with heme b cofactor in orange; the flexible linker, in blue, is responsible for the modulation of internal electron transfer (IET); all the potential glycosylation sites are shown in red.

Figure 3.
Electron transfer pathways from the substrate through CDH to various electron acceptors. One-(1-EA) and two-electron acceptors (2-EA) can be reduced directly by FADH2 in the DHCDH. Alternatively, electrons can be transferred by IET to heme b in the CYTCDH, which works as a relay for the reduction of macromolecular electron acceptors like polysaccharide monooxygenase (PMO), cyt c or an electrode. Figure 3 is reproduced from [89] published as open-access paper in Analytical and Bioanalytical Chemistry edited by Springer-Verlag.
Figure 3.
Electron transfer pathways from the substrate through CDH to various electron acceptors. One-(1-EA) and two-electron acceptors (2-EA) can be reduced directly by FADH2 in the DHCDH. Alternatively, electrons can be transferred by IET to heme b in the CYTCDH, which works as a relay for the reduction of macromolecular electron acceptors like polysaccharide monooxygenase (PMO), cyt c or an electrode. Figure 3 is reproduced from [89] published as open-access paper in Analytical and Bioanalytical Chemistry edited by Springer-Verlag.

Figure 4.
Suggested electron transfer mechanism for fructose dehydrogenase (FDH) at the electrode surface. D-(-)-fructose is oxidised to 5-keto-D-(-)-fructose releasing 2 electrons, which are transferred one by one through FAD first, followed by two heme c working as monoelectronic acceptors. Figure 4 is reproduced from [139] published as open-access paper in Analytical and Bioanalytical Chemistry edited by Springer-Verlag.
Figure 4.
Suggested electron transfer mechanism for fructose dehydrogenase (FDH) at the electrode surface. D-(-)-fructose is oxidised to 5-keto-D-(-)-fructose releasing 2 electrons, which are transferred one by one through FAD first, followed by two heme c working as monoelectronic acceptors. Figure 4 is reproduced from [139] published as open-access paper in Analytical and Bioanalytical Chemistry edited by Springer-Verlag.


{kind=link}
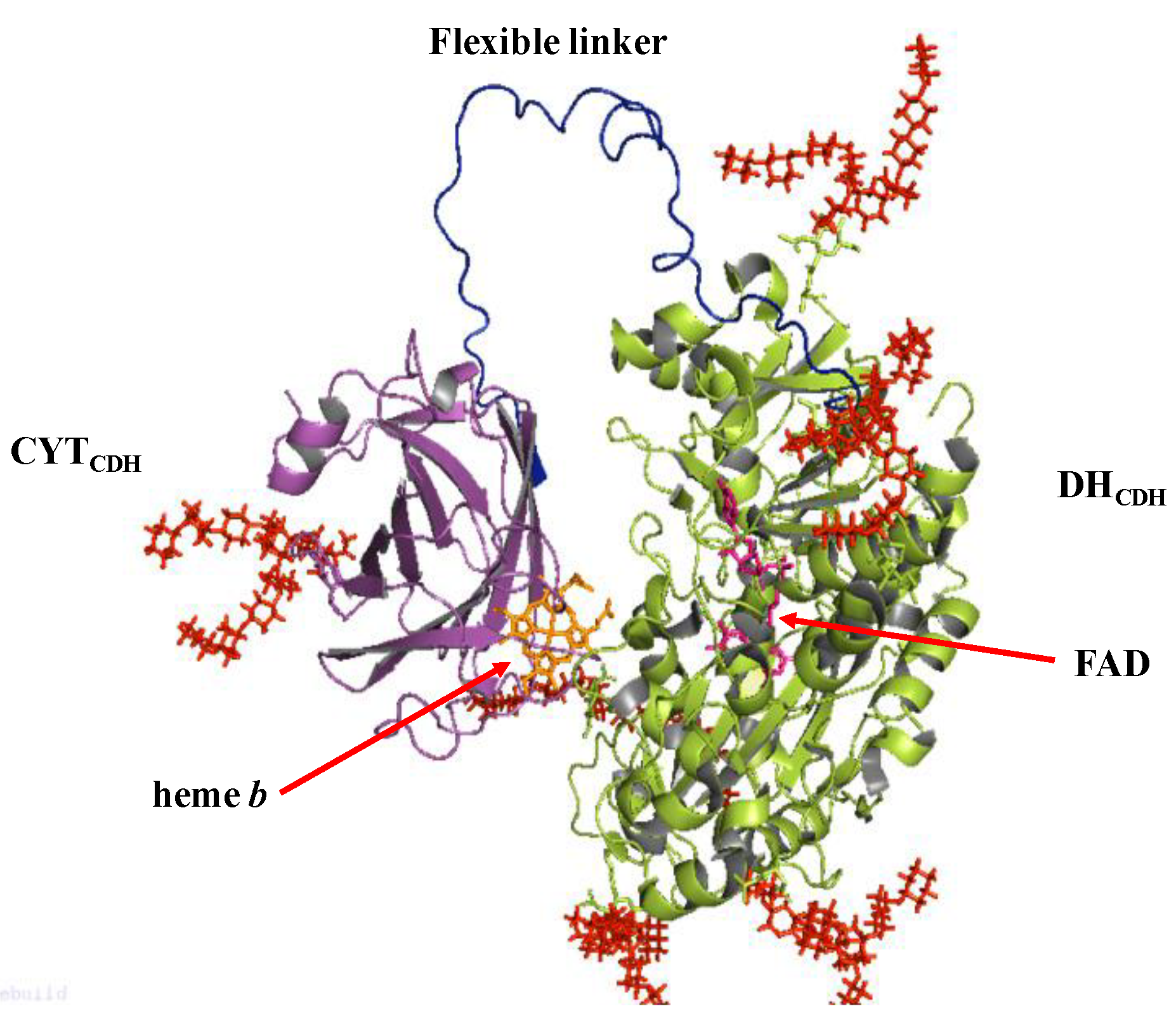{kind=link}
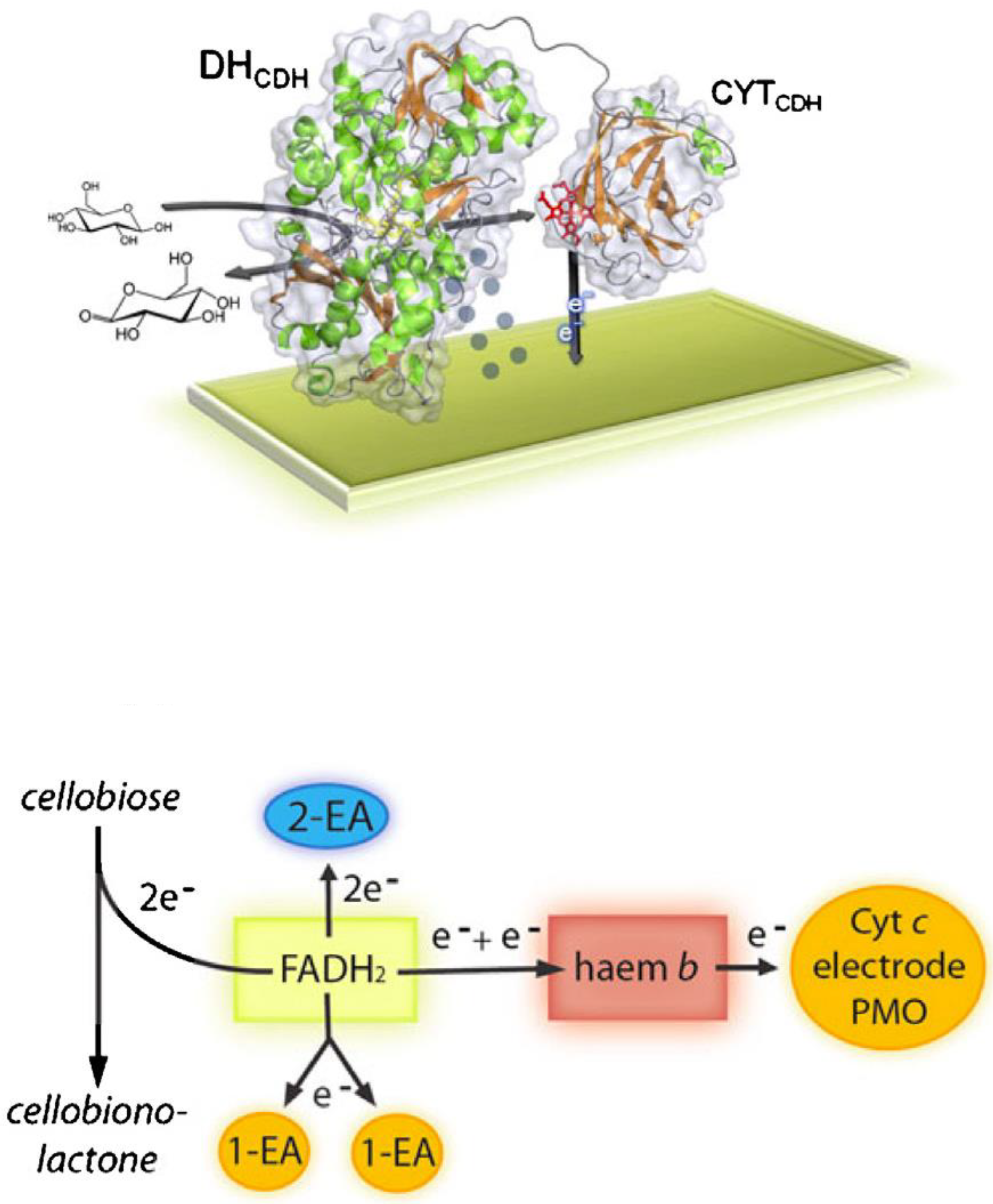{kind=link}
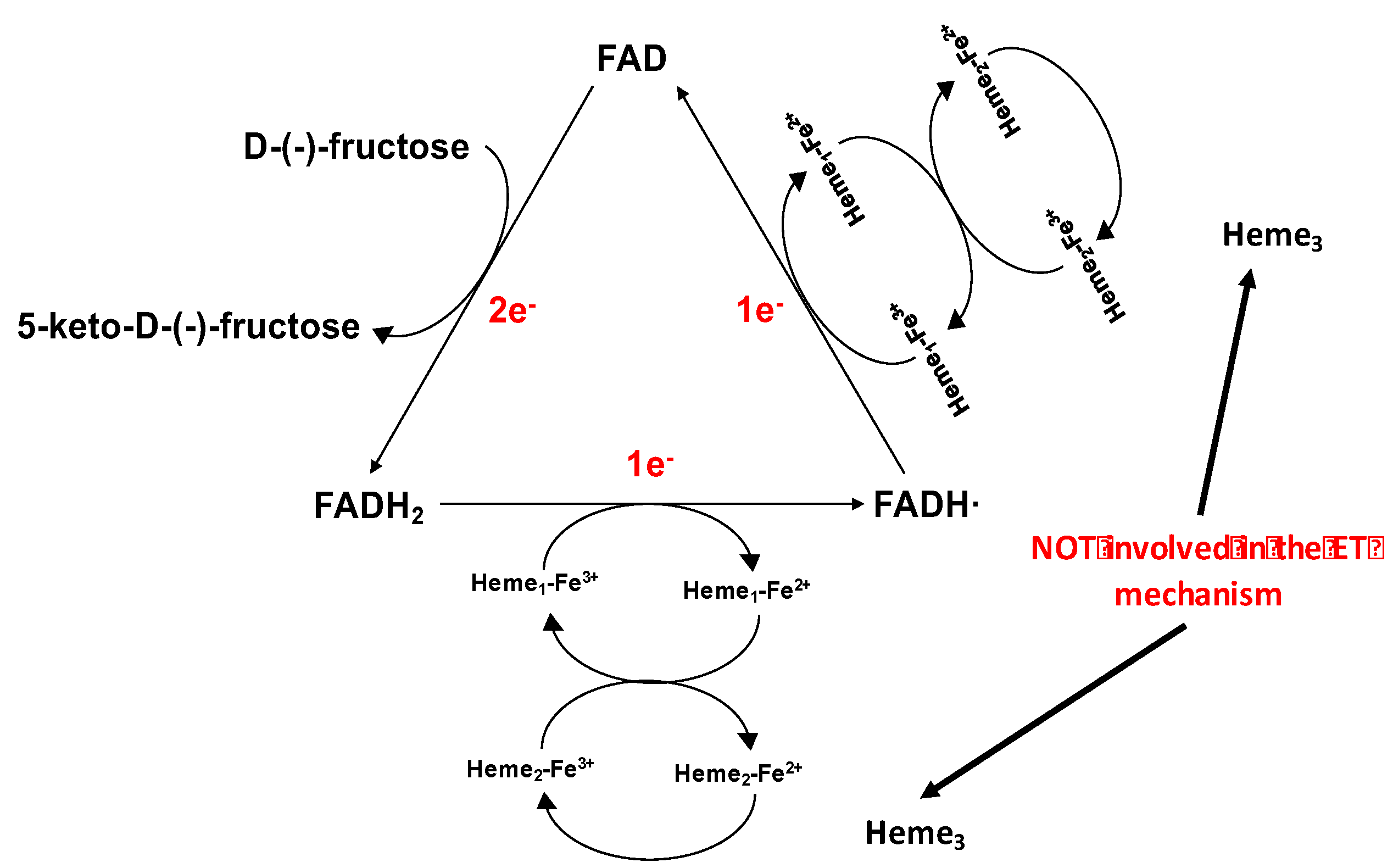{kind=link}
Table 1.
Multicofactor oxidases/dehydrogenases for which Direct Electron Transfer (DET) reactions with electrodes have been shown.
Table 1.
Multicofactor oxidases/dehydrogenases for which Direct Electron Transfer (DET) reactions with electrodes have been shown.
| Dehydrogenases | Cofactor | Substrate | Ref. |
|---|---|---|---|
| Cellobiose dehydrogenase | FAD-heme | D-glucose, cellobiose, lactose | [44,45,82,83] |
| D-Fructose dehydrogenase | FAD-heme | D-fructose | [46,49,84,85] |
| Pyranose dehydrogenase | FAD-heme | aldoses | [33] |
| Lactate dehydrogenase | PQQ-heme | lactate | [86] |
| Lactate dehydrogenase/cyt b2 | FMN-heme | lactate | [41] |
| Alcohol dehydrogenase | PQQ-heme | ethanol | [30,87] |
| Succinate dehydrogenase | FAD-Fe-S cluster-heme | succinate | [80,81] |
| D-gluconate dehydrogenase | FAD-heme-Fe-S cluster | D-gluconate | [43,88] |
| D-glucose dehydrogenase | FAD-heme-Fe-S cluster | D-glucose | [89] |
| Aldose dehydrogenase | PQQ-heme | D-glucose | [90] |
| Pyruvate dehydrogenase | PQQ-heme | pyruvate | [91] |
| Aldehyde dehydrogenase | PQQ-heme | aldehyde | [92] |
| Sulphite oxidase | Moco-heme | sulphite | [35,39] |
| Sulphite dehydrogenase | Moco-heme | sulphite | [42] |
| Theophylline oxidase | ?-heme | theophylline | [34,36,38] |
Table 2.
Lactose (upper part) and glucose biosensors (bottom part) based on DET of CDH are compared based on several analytical parameters such as linear range, LOD, sensitivity, CDH class, applied potential and stability. Abbreviations: AuE gold electrode, AuNPs gold nanoparticles, CtCDH Corynascus thermophilus CDH, GC glassy carbon electrode, MWCNTs multi-walled carbon nanotubes, NH2-PD aryl diazonium salts of p-phenylenediamine, PcCDH Phanerochaete chrysosporium CDH, PdNPs palladium nanoparticles, PEDGE poly(ethylene glycol) diglycidyl ether, PEI polyethyleneimine, PsCDH Phanerochaete sordida CDH, PtNPs platinum nanoparticles, SPE screen printed electrode, SPGE spectrographic graphite electrode, SWCNTs single-walled carbon nanotubes, TvCDH Trametes villosa CDH.
Table 2.
Lactose (upper part) and glucose biosensors (bottom part) based on DET of CDH are compared based on several analytical parameters such as linear range, LOD, sensitivity, CDH class, applied potential and stability. Abbreviations: AuE gold electrode, AuNPs gold nanoparticles, CtCDH Corynascus thermophilus CDH, GC glassy carbon electrode, MWCNTs multi-walled carbon nanotubes, NH2-PD aryl diazonium salts of p-phenylenediamine, PcCDH Phanerochaete chrysosporium CDH, PdNPs palladium nanoparticles, PEDGE poly(ethylene glycol) diglycidyl ether, PEI polyethyleneimine, PsCDH Phanerochaete sordida CDH, PtNPs platinum nanoparticles, SPE screen printed electrode, SPGE spectrographic graphite electrode, SWCNTs single-walled carbon nanotubes, TvCDH Trametes villosa CDH.
| Lactose Biosensors | |||||||
|---|---|---|---|---|---|---|---|
| Electrode Platforms | Linear Range/(µM) | LOD/(µM) | Sensitivity/(µA mM−1 cm−2) | Class | Applied Potential/V vs. Ag|AgClsat | Stability | Ref. |
| TvCDH/PEDGE/MWCNTs/SPE | 0.5–200 | 0.25 | - | I | +0.198 | 100% of initial response after 8 h | [110] |
| PsCDH/PEDGE/MWCNTs/SPE | 0.5–100 | I | |||||
| PsCDH/NH2-PD/SWCNTs-GC | 1–150 | 0.5 | 476.8 | I | +0.2 | 85% of the initial response after 50 h | [111] |
| PsCDH/ PEI@AuNPs/AuE | 1–100 | 0.3 | 196.5 | I | +0.25 | 95% of the initial response after 24 h | [101] |
| PcCDH/PtNPs–MWCNTs/SPGE | - | - | 43.5 | I | +0.29 | 75% of their initial response after 10 h | [112] |
| PcCDH/PdNPs–MWCNTs/SPGE | - | - | 46.4 | I | +0.29 | ||
| CtCDH/AuNPs/BPDT/AuE | 5–400 | 3 | 27.5 | II | +0.25 | 85% of initial response after 20 days | [113] |
| Glucose Biosensors | |||||||
| CtCDH/PEDGE/MWCNTs/GC | 0.1–30 | 0.05 | 222 | II | +0.190 | - | [114] |
| CtCDH/PEDGE/MWCNTs-SPE | 0.025–30 | 0.01 | - | II | +0.198 | 90% of initial response after 7 h | [115] |
| CtCDH/PEDGE/SWCNTs-SPE | 0.025–30 | 0.01 | II | ||||
Table 3.
DET-based lactose/glucose and glucose/oxygen enzymatic fuel cells (EFCs) are compared based on some of the operational parameters like operating conditions, open-circuit voltage (OCV), power output and operational stability. Abbreviations: AuE gold electrode, AuMWs gold microwires electrode, AuNPs gold nanoparticles, CtCDH Corynascus thermophilus CDH, DcCDH Dichomera saubinetii CDH, HiCDH Humicola insolens CDH, MHP N-(6-mercapto)hexylpyridinium, MvBOx Myrothecium verrucaria bilirubin oxidase, SPGE graphite electrode, ThLac Trametes hirsuta laccase.
Table 3.
DET-based lactose/glucose and glucose/oxygen enzymatic fuel cells (EFCs) are compared based on some of the operational parameters like operating conditions, open-circuit voltage (OCV), power output and operational stability. Abbreviations: AuE gold electrode, AuMWs gold microwires electrode, AuNPs gold nanoparticles, CtCDH Corynascus thermophilus CDH, DcCDH Dichomera saubinetii CDH, HiCDH Humicola insolens CDH, MHP N-(6-mercapto)hexylpyridinium, MvBOx Myrothecium verrucaria bilirubin oxidase, SPGE graphite electrode, ThLac Trametes hirsuta laccase.
| BFC | Conditions | OCV/(V) | Power Output/Limiting Element (l.e.) | Operational Stability | Ref. |
|---|---|---|---|---|---|
| DcCDH/ThLac SPGE-based | 100 mM citrate–phosphate air-saturated buffer, pH 4.5 containing 5 mM glucose | 0.73 | 5 µW cm−2 at 0.5 V (l.e.: anode) | Half-life > 38 h | [133] |
| CtCDH/MvBOx SPGE-based | 50 mM PBS buffer pH 7.4 containing 5 mM glucose and 150 mM NaCl | 0.62 | ~3 µW cm−2 at 0.37 V (l.e.: anode) | Half-life > 6 h | [134] |
| Human serum | 0.58 | ~4 µW cm−2 at 0.19 V (l.e. cathode) | Half-life < 2 h | ||
| CtCDH/MvBOx AuNPs/AuE-based | 50 mM PBS buffer air-saturated pH 7.4 containing 5 mM glucose and 150 mM NaCl | 0.68 | 3.3 µW cm−2 at 0.52 V (l.e. anode) | ~20% drop in 12 h of continuous operation | [53] |
| 50 mM PBS buffer air saturated pH 7.4 containing 5 mM lactose | 0.68 | 14.9 µW cm−2 at 0.52 V (l.e. anode) | Half-life > 12 h | ||
| Human blood | 0.66 | 2.8 µW cm−2 at 0.45 V (l.e. cathode) | Half-life < 3 h | ||
| Human plasma | 0.63 | 3 µW cm−2 at 0.47 V (l.e. cathode) | Half-life < 8 h | ||
| CtCDH/MvBOx AuNPs/AuE-based (contact lenses) | Human tears | 0.57 | 1 µW cm−2 at 0.5 V (l.e. cathode) | Half-life > 20 h | [50] |
| CtCDH/MvBOx AuNPs/AuMWs-based | Sweat | 0.58 | 0.26 µW cm−2 at 0.5 V (l.e. cathode) | Half-life > 10 h | [135] |
| Sweat + 500 µM glucose | 0.61 | 0.47 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva before lunch | 0.56 | 0.1 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva after lunch | 0.56 | 0.2 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva after lunch + 500 µM glucose | 0.60 | 0.46 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| HiCDH/MHP/AuNPs/AuE-based MvBOx AuNPs/AuE-based | 50 mM PBS air-saturated buffer pH 7.4 containing 5 mM glucose | 0.65 | 4.77 µW cm−2 at 0.50 V (l.e. anode) | Half-life > 13 h | [51] |
| 50 mM PBS air-saturated buffer pH 7.4 containing 10 mM lactose | 0.67 | 8.64 µW cm−2 at 0.50 V (l.e. anode) | Half-life > 44 h |
Table 4.
Fructose biosensors based on DET of FDH are compared based on several analytical parameters like linear range, LOD, sensitivity, applied potential and stability. Abbreviations: Aunanoporous gold nanoporous, AuNPs gold nanoparticles, CP carbon paste, FDH fructose dehydrogenase, GC glassy carbon electrode, LCP lipidic cubic phase, MPA 3-mercaptopropionic acid, MWCNTs multiwalled carbon nanotubes, NPD 2-carboxy-6-naphtoyl diazonium salt, PEI polyethyleneimine, SWCNTs single-walled carbon nanotubes, TRGO1 thermally reduced graphene oxide 1.
Table 4.
Fructose biosensors based on DET of FDH are compared based on several analytical parameters like linear range, LOD, sensitivity, applied potential and stability. Abbreviations: Aunanoporous gold nanoporous, AuNPs gold nanoparticles, CP carbon paste, FDH fructose dehydrogenase, GC glassy carbon electrode, LCP lipidic cubic phase, MPA 3-mercaptopropionic acid, MWCNTs multiwalled carbon nanotubes, NPD 2-carboxy-6-naphtoyl diazonium salt, PEI polyethyleneimine, SWCNTs single-walled carbon nanotubes, TRGO1 thermally reduced graphene oxide 1.
| Fructose Biosensors | |||||
|---|---|---|---|---|---|
| Electrode Platforms | Linear Range/(mM) | LOD/(mM) | Sensitivity/(µA mM−1 cm−2) | Applied Potential/V vs. Ag|AgClsat | Ref. |
| FDH/CP | 0.2–30 | - | - | +0.2 | [79] |
| FDH/PEI/CP | Up to 10 | 75 | 385 | +0.4 | [147] |
| FDH/AuNPs/GC | Up to 0.5 | - | - | +0.5 | [148] |
| FDH/MWCNTs/GC | Up to 40 | 5 | - | - | [152] |
| FDH/LCP/SWCNTs/GC | Up to 10 | - | 4 | + 0.2 | [153] |
| FDH/TRGO1/GC | 0.7–8.8 | 0.7 | 14.5 | + 0.4 | [154] |
| FDH/MPA-NPD/Aunanoporous | 0.05–0.3 | 0.0012 | 3.7 | + 0.15 | [155] |
Table 5.
DET-based fructose/O2 EFCs are compared based on some of the operational parameters like operating conditions, open-circuit voltage (OCV), power output and operational stability. Abbreviations: AuNPs gold nanoparticles, CCG carbon cryogel, cell. Cellulose, CG carbon aerogel, CP carbon paper electrodes, FDH fructose dehydrogenase, KB Ketjen black particles, LCP lipidic cubic phase, ME mercaptoethanol, MvBOx Myrothecium verrucaria bilirubine oxidase, MWCNTs multi-walled carbon nanotubes, NQ naphthoquinone, PPy polypyrrole, SWCNTs single-walled carbon nanotubes, ThLac Trametes hirsuta laccase, TsLac Trametes sp. laccase.
Table 5.
DET-based fructose/O2 EFCs are compared based on some of the operational parameters like operating conditions, open-circuit voltage (OCV), power output and operational stability. Abbreviations: AuNPs gold nanoparticles, CCG carbon cryogel, cell. Cellulose, CG carbon aerogel, CP carbon paper electrodes, FDH fructose dehydrogenase, KB Ketjen black particles, LCP lipidic cubic phase, ME mercaptoethanol, MvBOx Myrothecium verrucaria bilirubine oxidase, MWCNTs multi-walled carbon nanotubes, NQ naphthoquinone, PPy polypyrrole, SWCNTs single-walled carbon nanotubes, ThLac Trametes hirsuta laccase, TsLac Trametes sp. laccase.
| BFC | Conditions | OCV (V) | Power Output/Limiting Element | References |
|---|---|---|---|---|
| FDH/KB/CP TsLac/CG/CP | 0.1 M McIlvaine O2-satured buffer (pH 5.0) containing 200 mM fructose | 0.79 | 850 mW cm−2 at 0.41 V under stirring (l.e.: cathode) | [47] |
| FDH/ME-AuNPs/CP MvBOx/AuNPs/CP | 0.1 M acetate O2-satured buffer (pH 6.0) containing 200 mM fructose | 0.73 | 0.66 mW cm−2 at 0.36 V without stirring (l.e.: cathode) | [156] |
| 0.87 mW cm−2 at 0.3 V under stirring (l.e.: anode) | ||||
| FDH/MvBOx KB-sheet shaped electrodes | 0.15 M McIlvaine O2-satured buffer solution (pH 5.0) containing 200 mM fructose. | 0.70 | 0.55 mW cm−2 at 0.4 V (l.e.: cathode) | [157] |
| FDH/MvBOx KB-carbon strips electrodes | 0.25 M McIlvaine O2-satured buffer solution (pH 5.0) containing 500 mM fructose | 2.09 | 0.64 mW at 1.2 V (l.e.: cathode) | [158] |
| FDH/ThLac LCP-SWCNTs based GC | 0.15 M McIlvaine O2-satured buffer (pH 5.0) containing 40 mM fructose | 0.70 | 0.85 mW cm−2 at 0.25 V under stirring (l.e.: anode) | [153] |
| FDH/MvBOx CCG based electrodes | 1 M citrate O2-satured buffer (pH 5.0) containing 500 mM fructose | 0.79 | 2.6 mW cm−2 at 0.46 V (l.e: cathode) | [49] |
| FDH/cell./PPy/MWCNTs/GC ThLac/NQ/MWCNTs/GC | 0.1 M McIlvaine O2-satured buffer solution (pH 5.3) containing 100 mM fructose | 0.76 | 1.6 mW cm−2 at 0.33 V (l.e.: cathode) | [159] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bollella, P.; Gorton, L.; Antiochia, R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors 2018, 18, 1319. https://doi.org/10.3390/s18051319
AMA Style
Bollella P, Gorton L, Antiochia R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors. 2018; 18(5):1319. https://doi.org/10.3390/s18051319
Chicago/Turabian StyleBollella, Paolo, Lo Gorton, and Riccarda Antiochia. 2018. "Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells" Sensors 18, no. 5: 1319. https://doi.org/10.3390/s18051319
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.