Introduction
The chemistry of molecularly designed electrode surfaces has been a major aspect of interfacial electrochemistry for over twenty years. ‘Tailormade’ electrode surfaces at the molecular level provide much added value to the unique feature inherent in electrochemistry where, as recently stated by Murray [
1] , ‘…one can employ the electrode potential as a source or sink of pure, uncomplicated electrons of flexibly chosen free energy…’ . This feature of electrochemistry is readily apparent when the reaction is Nernstian or reversible. However many redox reactions of practical importance are irreversible. They exhibit finite kinetics at the electrode surface, an activation energy barrier at the interface must be overcome, significant overpotential must be applied to make the reaction proceed at a finite rate, and the rate of product formation, and indeed its identity, will depend in a marked manner on the nature of the electrode surface. Hence the quest to understand and control electrochemical reaction kinetics and catalysis has been the motivation of research in physical electrochemistry for decades. The science of electrocatalysis was enhanced in the 1970’s when electrochemically reactive molecular materials were attached to electrode surfaces using either a systematic synthetic strategy or via controlled adsorption. The seminal papers of Lane and Hubbard [
2], as well as the early review articles authored by Murray [
3] and by Albery and Hillman [
4] well illustrate the latter approach. Covalent attachment of redox active groups was accomplished via specific chemical coupling strategies such as surface silanization, the use of cyanuric chloride, or in the case of carbon electrodes, direct reaction of the redox moiety with acidic and carbonyl functionalities on the electrode surface. Surface modification via adsorption resulting in stable modified surfaces, is well illustrated in the work of Gorton and co-workers [
5] who developed viable amperometric biosensors for NADH using redox dye monolayers.
Redox mediation is simple in concept. In this process surface immobilized sites may be activated electrochemically via application of a voltage to the support electrode. The latter sites may then oxidise or reduce other redox agents located in the solution phase adjacent to the immobilized layer, for which the direct oxidation or reduction at the electrode surface is inhibited, either because of intrinsically slow heterogeneous electron transfer kinetics or because close approach of the soluble redox species to the electrode is prevented.
Although the initial focus of studies in the field of chemically modified electrodes was in the area of molecularly modified surfaces of monolayer thickness, interest in the latter configurations for electrocatalytic purposes decreased when it was realized that significant catalatytic advantage could only be obtained if electrodes were modified with many monolayers of redox active sites. The attractions of a three dimensional network of catalytically active sites localized next to a support electrode surface, compared with the corresponding two dimensional situation characteristic of a monolayer were clear, and much investigation was made on the fundamental properties of electrode surfaces modified with electroactive polymer thin films [
6]. Multilayer polymer films were not without their problematic aspects with respect to mediated electrocatalysis however. The current response reflecting the net reaction rate was found to depend not only on the heterogeneous cross exchange kinetics involving the dispersed mediator species and the substrate, and the diffusion of substrate in the adjacent solution to the redox site (the latter being pertinent also to monolayer systems), but also rate limitation could involve electron percolation or hopping between adjacent redox sites in the polymer matrix (described via a diffusion/migration mechanism), and partition and diffusion of reactant species through solvent containing pores in the polymer matrix. Hence, although catalytic efficiency could be enhanced via the adoption of a multilayer strategy, it could be reduced if charge percolation and/or substrate transport processes proved to be kinetically significant. Furthermore, a full physicochemical characterization of immobilized polymer films using the standard spectroscopic tools of modern chemistry proved elusive. The complex physical chemistry underlying mediated electrocatalysis in polymer modified electrode systems has been elucidated by Andrieux and Saveant [
7] and by Albery and Hillman [
8].
However the concept that a molecular electrode surface could be developed, whose reactivity with respect to a target reactant could be reasonably predicted, was never fully disregarded, and, more recently, metallic electrodes coated with adsorbed redox active monolayers which are generated via self assembly mechanisms [
9] have been the subject of considerable attention. An organized self assembled monolayer (SAM) is a single layer of molecules on a substrate surface in which the molecules exhibit a high degree of orientation, molecular order and packing. In the self assembly method, the monolayer spontaneously forms upon exposure of the substrate to a solution containing the molecules. It is clear that successful self assembly requires a relatively strong bond between the substrate and a moiety or atom in the molecule, and an additional lateral interaction between molecules in the monolayer. The strength of the head group- substrate bonds, the lateral interactions and the density of packing result in good monolayer stability. A specific focus has been on ordered monolayers fabricated from alkane- thiols with pendant redox active centers such as Ru(NH
3)
52+/3+ or ferrocene/ferricinium. Such well defined systems have been used to examine current theories of interfacial electron transfer [
10], but have also been used as model well defined systems, to increase our understanding of the structure of electrified interfaces [
11], and to develop novel enzyme based biosensors [
12]. Indeed the construction of modified enzyme systems covalently attached to electrode surfaces has led to a new generation of sophisticated molecular biorecognition systems [
13].
Mediated electron transfer of solution phase species at electrode surfaces containing immobilized redox species can be examined experimentally using a number of electrochemical techniques. The technique of rotating disc voltammetry is most often applied, since in principle, the processes of solute transport in solution and solute reaction kinetics may be cleanly separated, by conducting voltammetric experiments over a range of electrode rotation speeds. In the present paper, we discuss the theoretical analysis of mediated electrocatalysis of a solution soluble substrate at an electrode surface containing an immobilized monolayer of redox active molecules. The analysis will be cast in the context of rotating disc voltammetry and will expand on previous models developed by Andrieux and Saveant [
14], Laviron [
15] , Anson [
16] and Murray and co-workers [
17].
The paper is structured as follows. We firstly briefly discuss the thermodynamic aspects of mediated electron transfer. We then review a simple analysis of mediated electron transfer in the context of steady state rotating disc voltammetry. More detailed aspects of this analysis have been presented in the first paper of this series [
18]. Our attention will in the main be focused on an analysis of the catalytic efficiency expected for redox active monolayers assuming that the heterogeneous cross exchange process can be described in terms of simple bimolecular kinetics, so that the mediator/reactant interaction is of the simple outer sphere type. We then proceed to describe mediated electron transfer at immobilized monolayers in the context of a generalised Koutecky-Levich model, and point out the pitfalls of such an approach in the analysis of experimental data. Finally, the Koutecky-Levich approach is then compared with a more sophisticated analysis, more in keeping with reported experimental observations, in which the solution phase substrate forms a complex or adduct with the surface immobilized redox species. In this circumstance the substrate/redox site reaction can be described in terms of Michaelis-Menten kinetics. We shall derive theoretical expressions for the normalised steady state current response for both the generalised Koutecky-Levich and Michaelis-Menten models and present kinetic case diagrams for each model. We also describe a simple method of experimental data manipulation which will serve to distinguish between the two approaches. In the present paper we focus on an analysis of the steady state current response. We do not consider the transient response such as that obtained using potential step chronoamperometry, which is more complex, and was the subject of the proceeding paper in this series [
19].
Mediated Electrocatalysis Via the Outer-Sphere Bimolecular Mechanism
In this section we discuss the most simple description of mediated electron transfer via a surface immobilized redox couple. A more complete analysis has been presented in the first paper of the present series [
18]. This analysis will prove to be useful when we develop more sophisticated approaches in later sections. Three possible rate determining steps are possible. The first is the heterogeneous electron transfer at the electrode surface generating the mediator species B. The generation of B can be described either by the Nernst equation, or by the Butler-Volmer equation. In simple models proposed previously the former was assumed. This may well be a good approximation . The second involves the bimolecular reaction between the substrate species S and the mediator species B. The cross exchange reaction involving B and S is quantified via a second order rate constant k
H (units: cm
3mol
-1s
-1). The third involves the logistics of substrate supply to the electrode surface, which is described by diffusion. The rate of substrate diffusive transport to the active site is described via a diffusion rate constant
kD (units: cms
-1). The latter is given by
, where D
S represents the diffusion coefficient of the substrate species in solution and δ is the diffusion layer thickness. Typically
δ ≅ 10
−3 cm and D
S ≅ 10
−6 cm2 s−1, hence k
D ≅ 10
−3 cms−1.
The net flux (units: mol cm
-2s
-1) is given by:
where Γ
B denotes the surface coverage of mediator species B (units: mol cm
-2) and s
0 is the surface concentration of substrate. We can use the Nernst equation to derive a relation between the mediator coverage and the total surface coverage G
S as follows:
where Γ
Σ = Γ
A + Γ
B and
ξ is a normalised potential given by:
where E
0A/B represents the standard redox potential for the immobilized redox couple. The quasi steady state approximation may be used to determine s
0 to obtain:
where
s∞ denotes the bulk substrate concentration.
The net steady state flux is therefore given by:
We can introduce a non dimensional flux (or current) given as the ratio of the net flux to the diffusive flux
fD = kDs∞ , the latter corresponding to the diffusion controlled reaction of the substrate species S at an unmodified electrode surface:
From eqn.7, eqn.10 and eqn.11 we can readily obtain an expression for the current/potential curve expressed in non dimensional format:
where we introduce a parameter
which relates the rate of the surface cross exchange reaction to that of substrate diffusion in solution and the potential dependence is governed by the function
. It is readily shown that eqn.12 may be written in the following form:
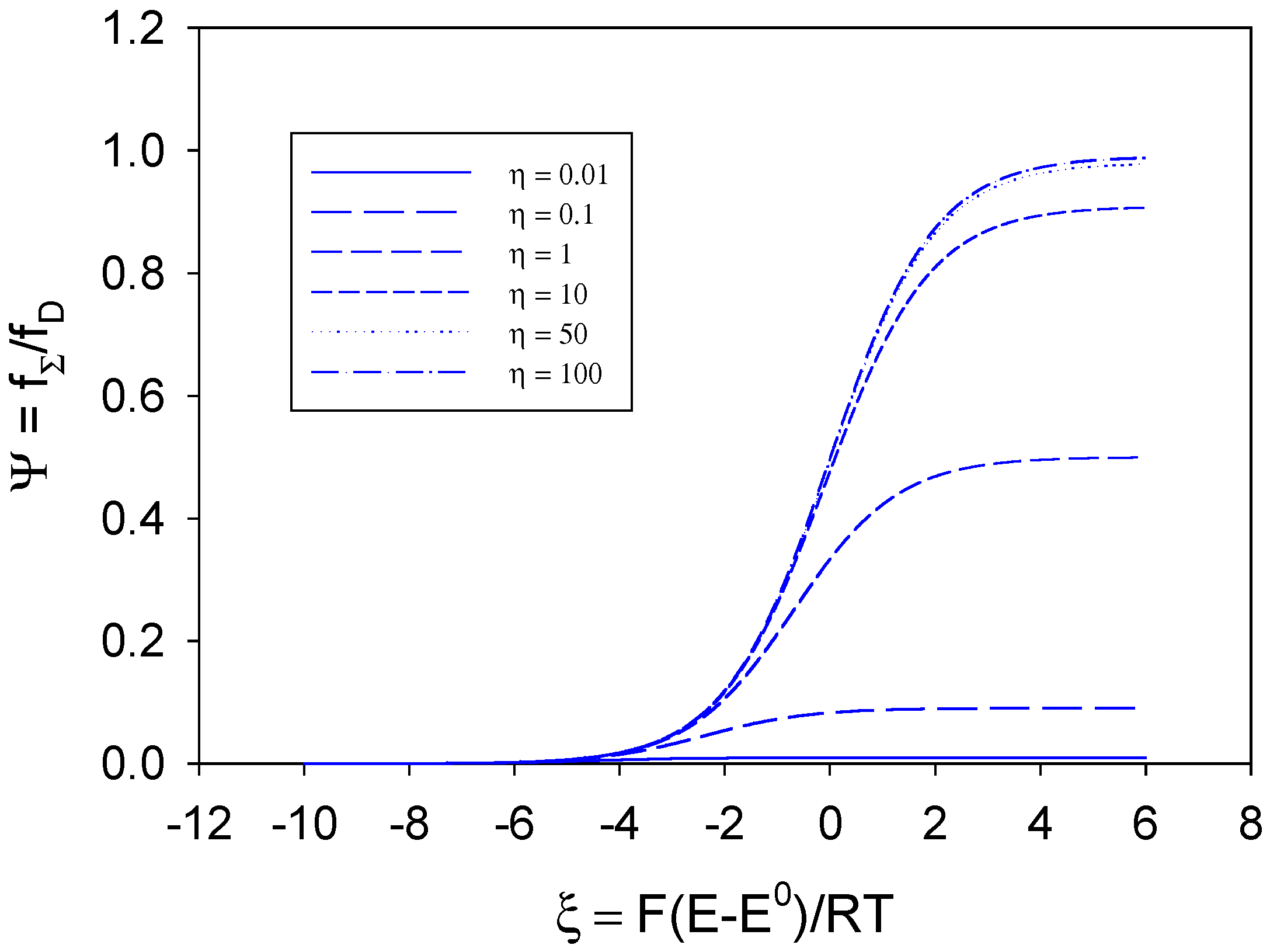
he normalised voltammograms corresponding to various values of the competition parameter
η are illustrated in
figure 1. We note that when
η is large then
kHΓ
Σ >>
kD and the rate of the cross exchange reaction between B and S is not rate determining. The shape of the normalised voltammogram tends to that characteristic of a simple diffusion controlled process and eqn.13 reduces to
. A limiting current plateau given by Ψ
L ≅ 1 or
fΣ,L ≅
kDs∞ is obtained when the normalised potential
ξ is large. In contrast when
η is small
kHΓ
Σ <<
kD, substrate diffusion to the active sites is fast and the net flux is controlled by the bimolecular kinetics of the cross exchange reaction. Under such circumstances assuming that
η−1 >> 1 we obtain
![Sensors 02 00473 i053]()
and for large values of the normalised potential a limiting current response is given by Ψ
L ≅
η or
fΣ,L ≅
kHΓ
Σs∞ reflecting pure kinetic control. We also note from
figure 1 that the half wave potential is a strong function of the competition parameter
η , and becomes less positive as the latter parameter increases.
We note that the ratio of the substrate reaction flux f
Σ at the monolayer modified electrode to the substrate diffusion flux f
D given by eqn.13 is also a direct measure of the catalytic efficiency of the monolayer modified electrode under conditions where the heterogeneous cross exchange reaction is of an outer-sphere bimolecular type. If we assume that the normalized potential ξ is large then the surface coverage of active mediator species Γ
B = Γ
S and eqn.13 takes the form:
We will now show that the maximum catalytic efficiency cannot be greater than unity for a monolayer modified electrode. The bimolecular rate constant for the mediator/substrate reaction at the monolayer can be estimated from the Marcus relation [
20]:
where k
AB, k
SP represent the corresponding homogeneous solution phase bimolecular self exchange rate constants for the A/B and S/P redox couples, K is the equilibrium constant defined in eqn.5 and G is a constant, usually close to unity and is given by [
20] :
where Z denotes a characteristic collision frequency between reactants. Typically,
Z ≅ 10
13 s−1,
kAB ≅
kSP ≅ 10
7M −1s−1 = 10
10cm3mol−1 s−1 (the
maximum value of the latter rate constants will be the diffusion controlled value which typically has the value 10
11 M
-1s
-1) . Assuming that n = 1 and T = 298K, then if we set ΔE
0 = 0.2 V we can compute that
K ≅ 2400,
G ≅ 0.58 and hence the heterogeneous cross exchange rate constant is estimated from eqn.15 and eqn.16 to be
kH ≅ 4 ×10
8M −1s−1. Since the surface coverage of active sites corresponding to a monolayer is Γ
Σ ≅ 10
−10 mol cm−2, then, for a bulk substrate concentration of ca. 1 mM = 10
-6 mol cm
-3, the heterogeneous cross exchange flux is given by
fH =
kH Γ
Σs∞ ≅4×10
−5 mol cm−2 s−1. This value should be compared with the substrate diffusion flux to the unmodified electrode
fD =
kDs∞ ≅ 10
−9 mol cm−2 s−1. Hence under the circumstances considered we see that
fD <<
fH, and from eqn.14 we see that the catalytic efficiency will approach unity. As the ratio
![Sensors 02 00473 i054]()
gets larger we note from eqn.14 that the catalytic efficiency
![Sensors 02 00473 i055]()
. Hence for the simple scenario outlined here (Nernstian mediator generation, irreversible cross exchange reaction), it is clear that under all conditions the catalytic efficiency of a monolayer modified electrode will not exceed that obtained for direct mass transport controlled substrate reaction at the unmodified electrode surface. This fact was pointed out by Andrieux and Saveant [
14] some time ago.
A similar conclusion pertains if a more complete analysis is performed. If we assume that mediator generation is kinetically irreversible and obeys the Butler-Volmer equation, and that the cross exchange reaction has a large equilibrium constant, then simplification of the general steady state expression obtained in eqn.9 of the first paper of the present series [
18] results in the assignment that the net reaction flux at the monolayer coated electrode is:
To compute the catalytic efficiency we compare the latter reaction flux with that obtained for the substrate at an unmodified electrode surface:
The latter expression was obtained using a steady state analysis and considering the rate of reactant diffusion and reaction at a bare electrode surface [
21]. We have introduced potential dependent heterogeneous electrochemical rate constants for both mediator generation ( k’
E) and direct substrate reaction (k’
S) . Both quantities are given by Butler-Volmer expressions:
In the latter expressions β
E, β
S denote the symmetry factors for the mediator generation process and the direct substrate reaction respectively, and
,
are the standard rate constants for mediator generation and direct substrate reaction [
22]. The catalytic efficiency is given by the ratio
. Now, assuming a single step mechanism for mediator generation on the surface and direct substrate reaction, the symmetry factors will both be in the range 0 – 1, and if both of the activation energy barriers are symmetric then to a good approximation we can set
βE ≅ βS ≅ β ≅ 0.5. From eqn.17-eqn.19 we obtain after some manipulation:
where we note that
and
. Also the parameter Φ is given by
where we define
,
, and
. We now use the relationship:
to note that
to obtain:
An expression for the catalytic efficiency of the monolayer modified electrode may now be obtained using eqn.20 and eqn.23:
where we have introduced the normalized potential ξ as
. Hence we note that the catalytic efficiency in general depends on the applied potential (via the ξ term), the equilibrium constant K for the heterogeneous cross exchange reaction between surface immobilized mediator and solution phase substrate, and on the kinetic flux terms describing mediator generation, heterogeneous cross exchange, direct substrate reaction and finally, on the flux for substrate diffusion to the monolayer.
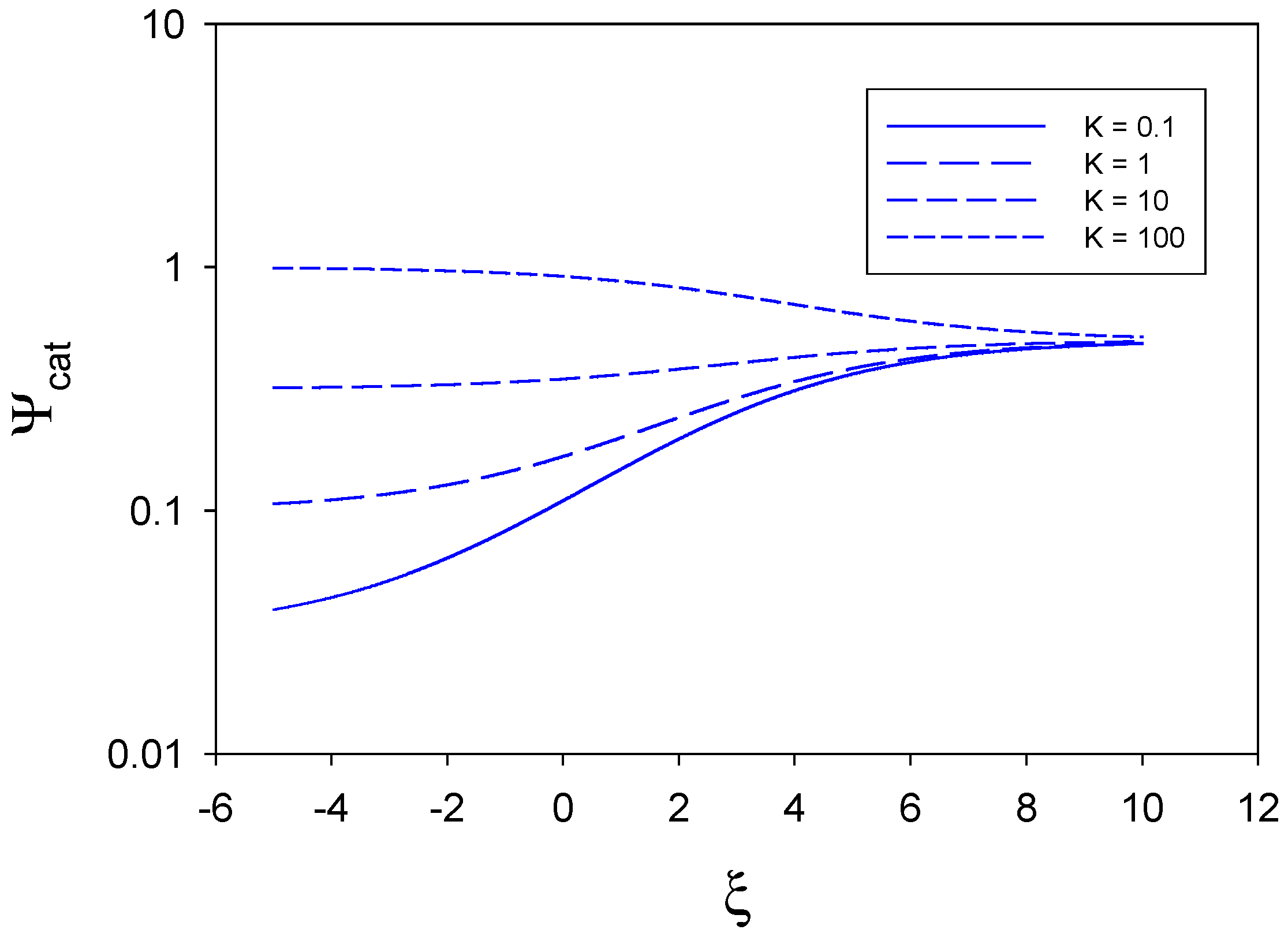
The results contained within eqn.24 may be better appreciated by resorting to a graphical analysis. In
figure 2,
figure 3 and
figure 4 we examine the manner in which the catalytic efficiency varies with normalized potential ξ for various values of the equilibrium constant K and for specific limiting kinetic cases determined by the values of the characteristic flux terms. In all computations we assume that the symmetry factor β = 0.5, which is a reasonable assumption. In all cases we have set 0.1 ≤
K ≤ 100
and − 5 ≤
ξ ≤ 10 which are realistic ranges.
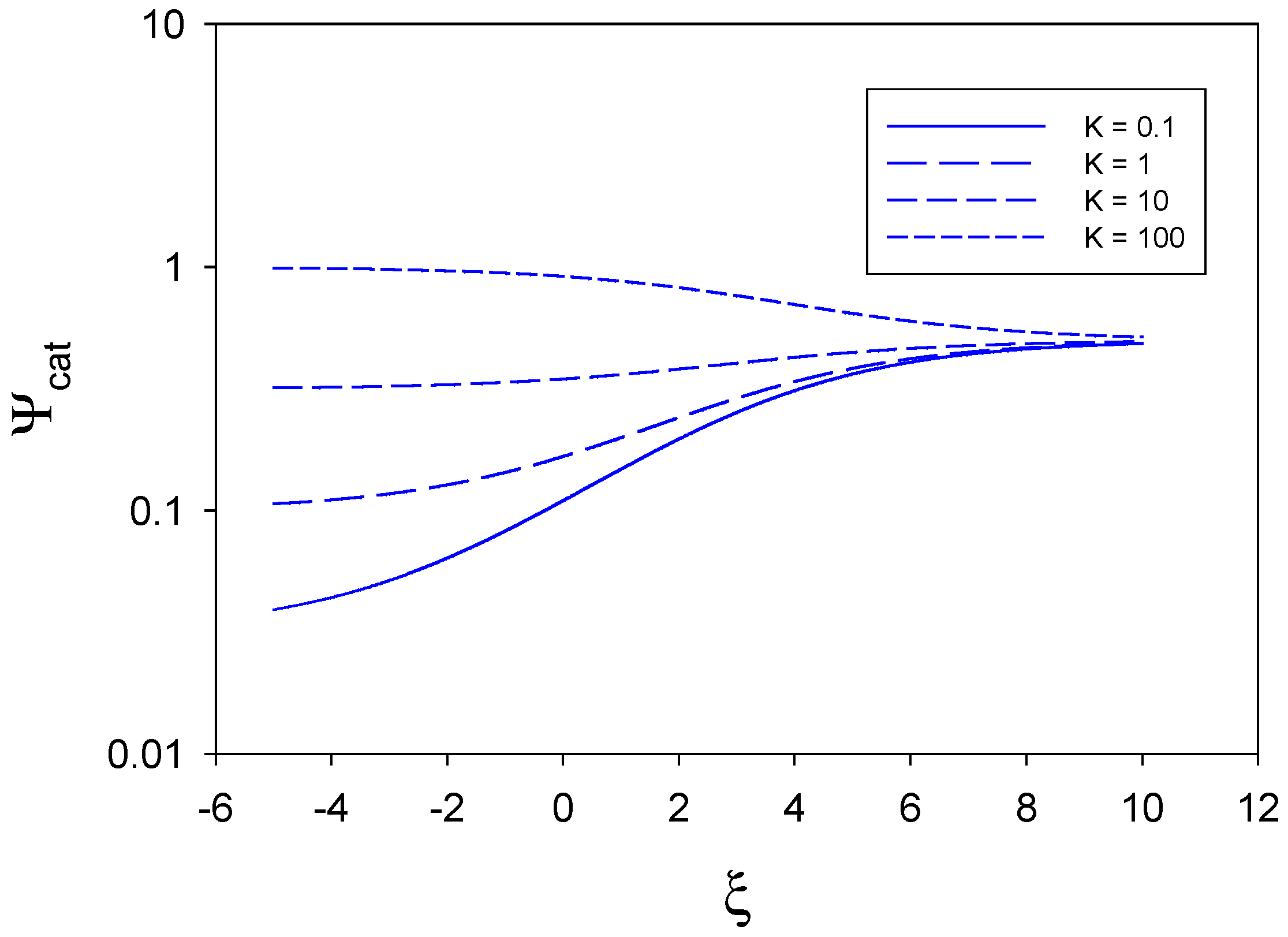
In
figure 2 the catalytic efficiency is plotted as a function of normalized potential for the situation where
,
and
. Hence we note that
, and so we consider the situation where mediator generation and heterogeneous kinetics are slow, the direct reaction of substrate at the unmodified electrode surface is facile and substrate diffusion to the redox monolayer is facile. When the equilibrium constant for the cross exchange reaction is small (K = 0.1) the catalytic activity rises rapidly with increasing electrode potential which reflects the increasingly facile generation of bound catalytically active sites on the electrode surface. This behaviour is also seen in the curves corresponding to K = 1 and K = 10. In contrast, when the equilibrium constant is large (K = 100), the catalytic activity is seen to decrease slightly with increasing electrode potential from an initial value of unity to a limiting value close to 0.8, at large ξ , which is also attained by the other curves corresponding to lower K values.
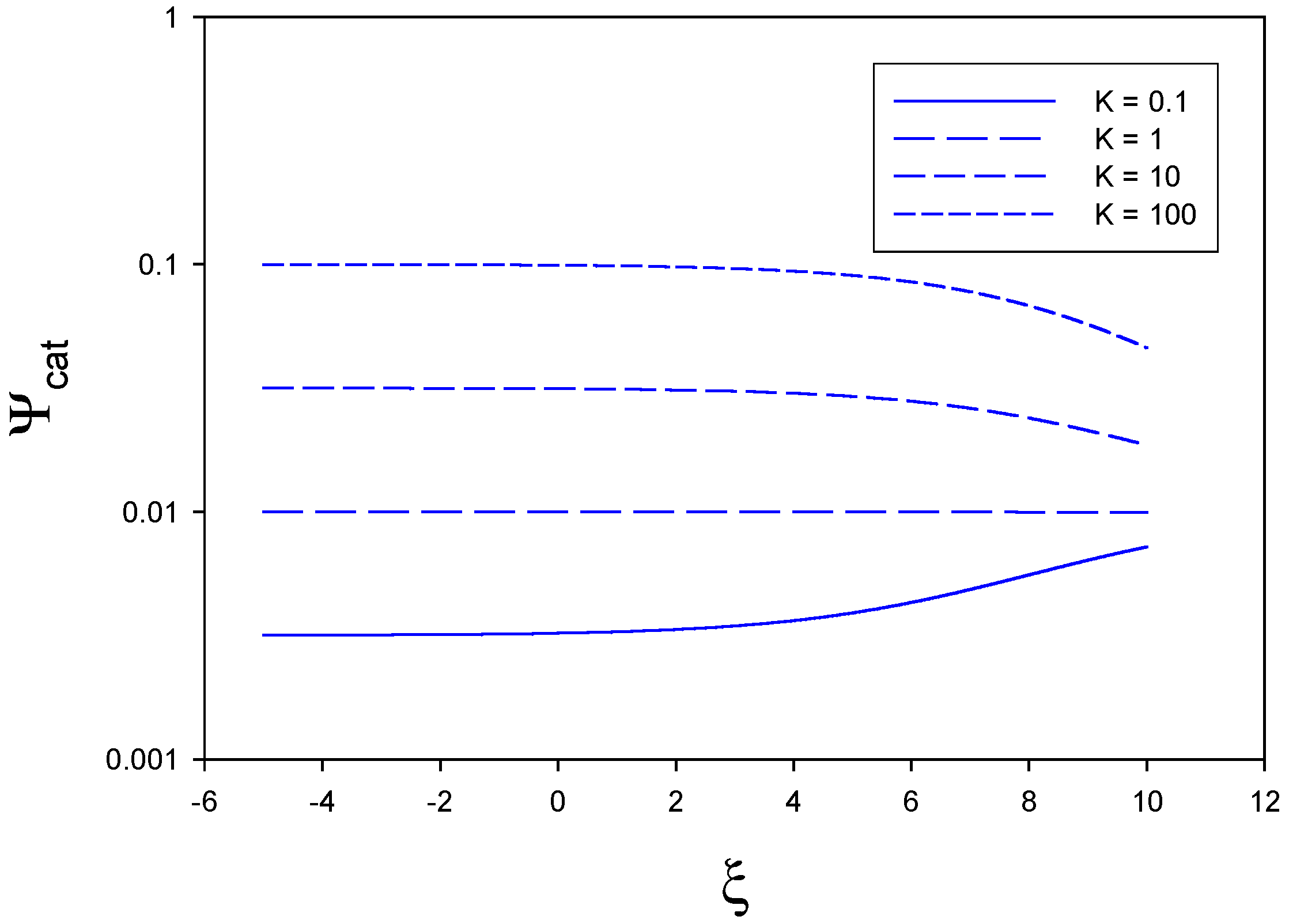
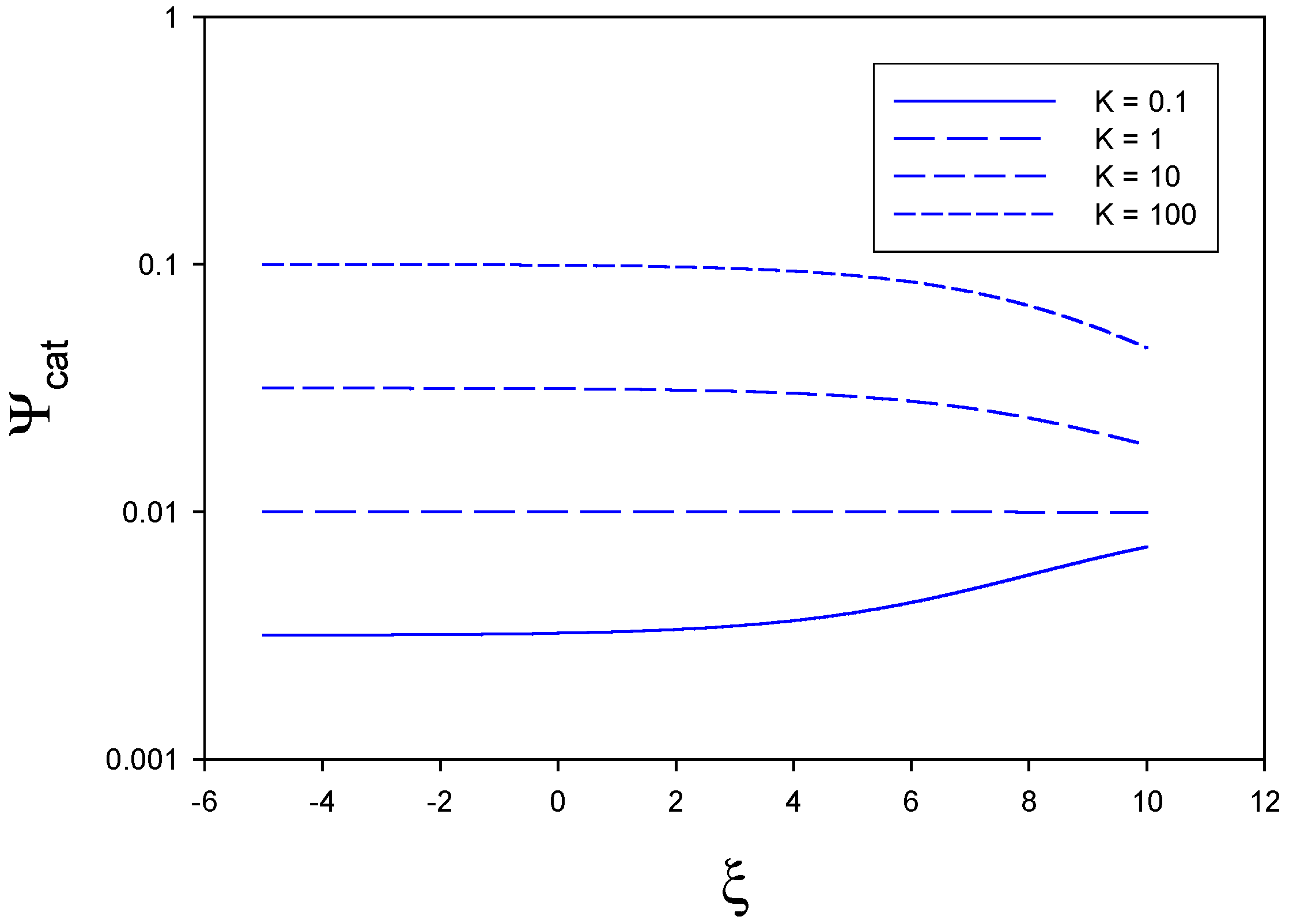
In
figure 3 we illustrate the situation where mediator generation is slow and rate determining. Here we have set
,
and . Hence we examine the situation where
. For all assumed values of the equilibrium constant the catalytic efficiency is low and less than 0.1, When K is small (0.1) the catalytic efficiency rises slightly with applied electrode potential. In contrast, for larger values of K the catalytic efficiency remains constant (at a low value) over much of the potential range and decreases slightly at elevated values of normalized potential.
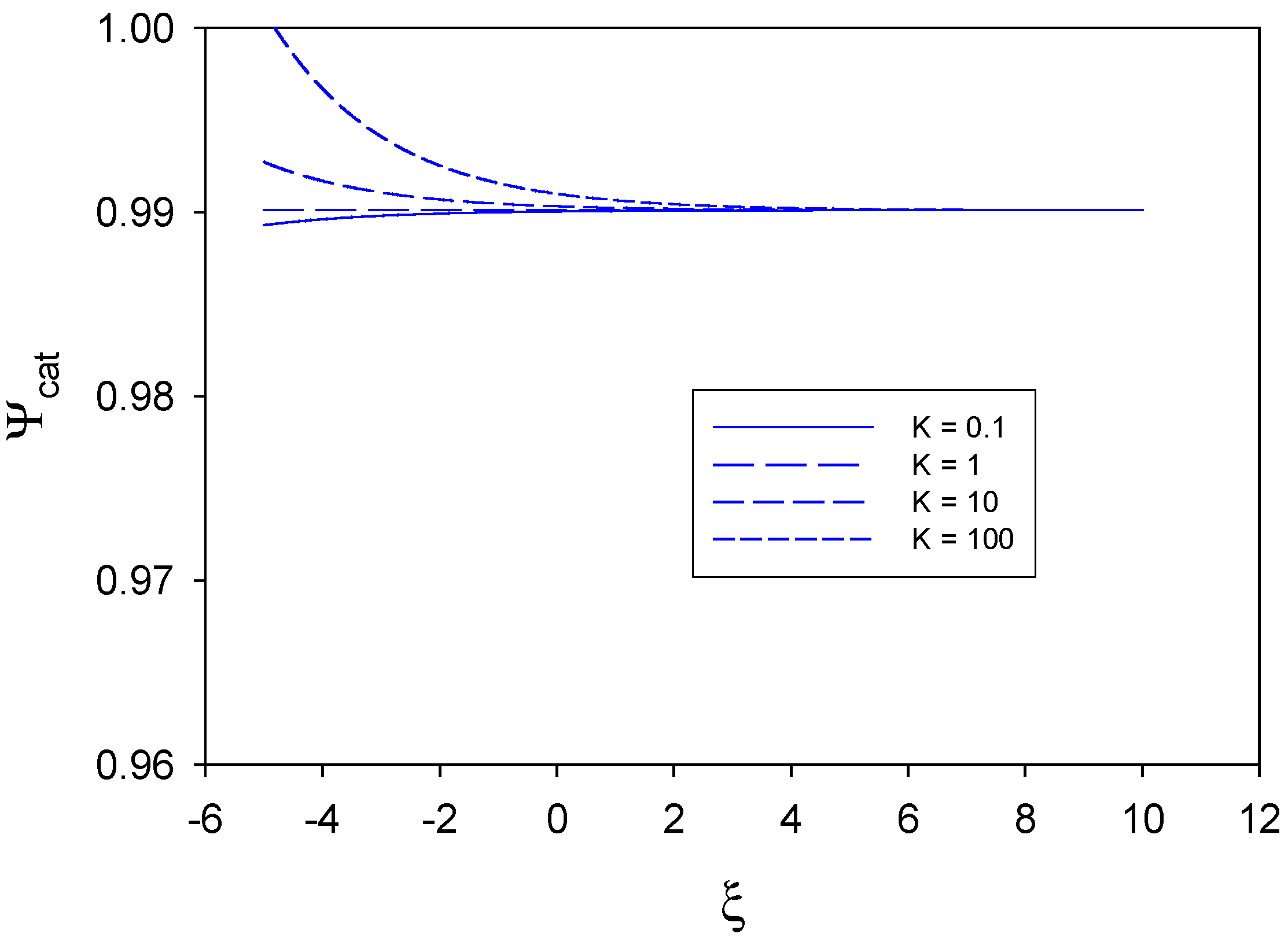
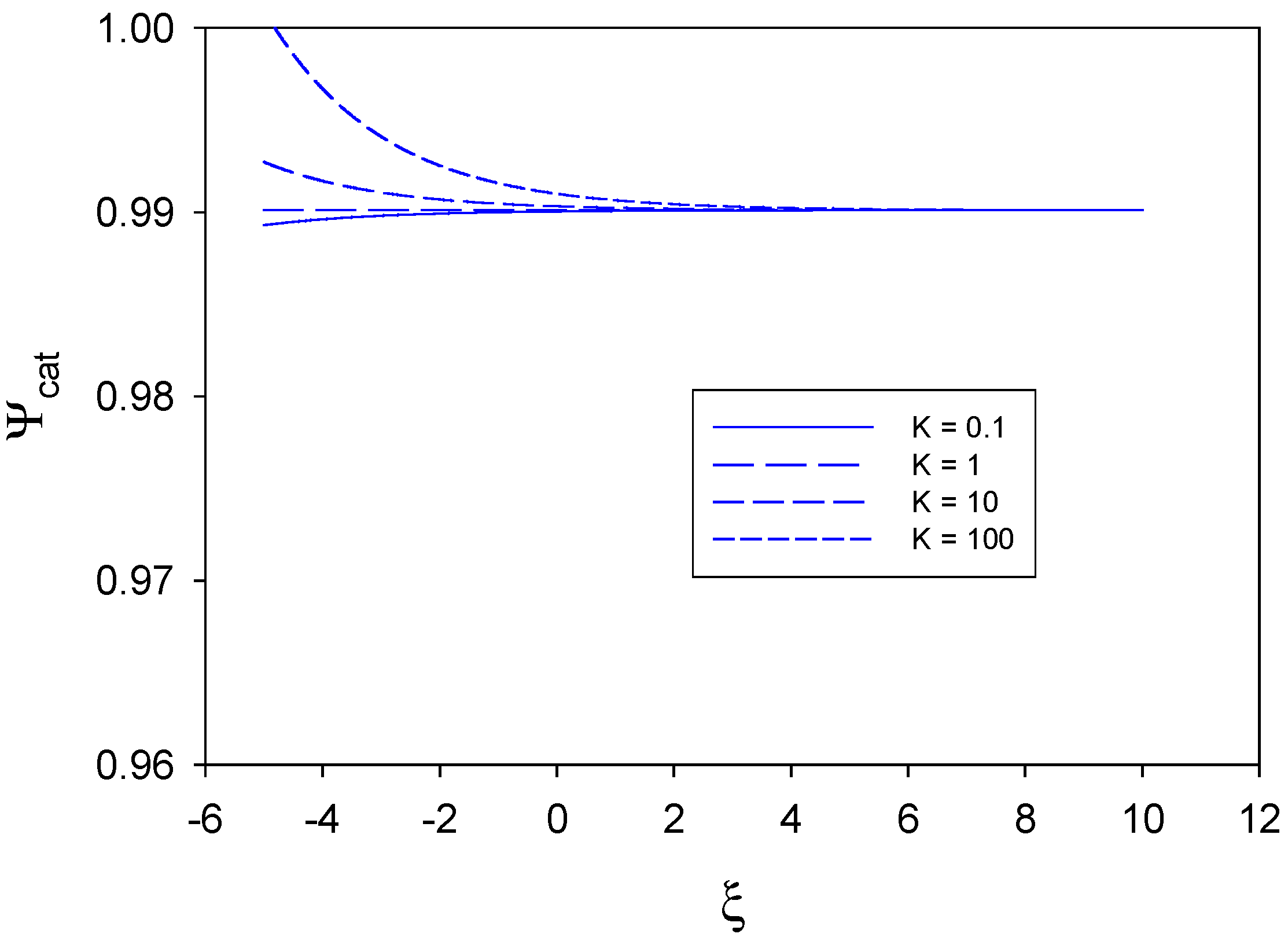
In
figure 4 we examine the situation where substrate diffusion to the redox monolayer is slow and rate determining, and where mediator generation and direct substrate reaction at the unmodified electrode surface are greater than the heterogeneous cross exchange reaction kinetics between the immobilized mediator and the substrate species. We have assumed that
,
,
, and so
. For all values of the equilibrium constant K (0.1-100) the catalytic efficiency is very large and close to unity.
We finish this section of the paper by drawing attention to a method of data analysis much used in the literature. In many situations the technique of rotating disc voltammetry is used to probe the processes of mediated electron transfer between surface immobilized redox couples and solution phase reactant species, since the matter transport of the latter is well defined using the rotating disc configuration. Indeed the diffusive rate constant k
D for the solution phase substrate species is well characterized at the rotating disc due to the fact that the diffusion layer thickness δ is a well defined function of electrode rotation speed. We note that using the Nernst diffusion layer approximation the diffusive rate constant k
D is given by
where D
S denotes the diffusion coefficient of the substrate in solution and δ is the diffusion layer thickness [
23]. For the rotating disc electrode it is well established [
24] that the diffusion layer thickness is a well defined function of electrode rotation speed
, where υ denotes the kinematic viscosity of the solution.
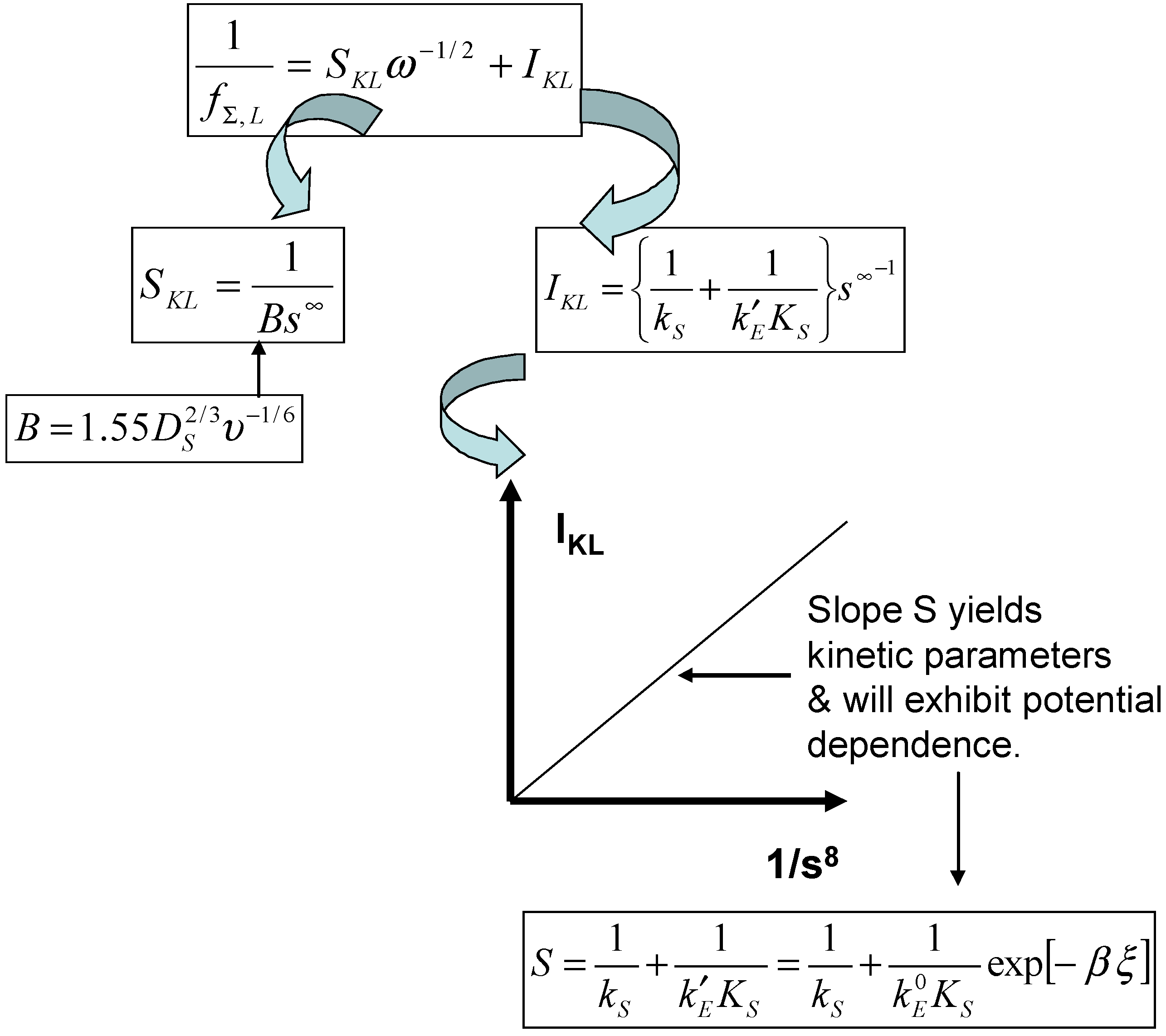
Returning to eqn.10 we note that when the normalized potential ξ is large the limiting flux is given by:
and the consecutive processes of diffusive transport and heterogeneous cross exchange kinetics are cleanly separated. From eqn.25 we can readily derive the experimentally useful Koutecky-Levich equation:
Thus a plot of inverse limiting flux versus inverse root of the rotation speed is linear with a slope
and an intercept
from which the heterogeneous rate constant for the cross exchange reaction can be derived. The expression presented in eqn.26 has been used many times in the early literature which focused on chemically modified electrode surfaces of monolayer thickness. In many cases the Koutecky-Levich expression will also apply to mediated electrocatalysis at polymer coated electrodes of significant thickness [
25]. However it has not been generally realized that the Koutecky-Levich equation must be applied with care, and that is some circumstances its use will provide an erroneous result, especially if an adduct species is formed between the immobilized mediator and the solution phase substrate .
As a consequence, in the next section of the paper, we examine the generalized Koutecky-Levich mechanism for mediated electrocatalysis at a redox active monolayer and then examine extended versions of the basic equation presented in eqn.25, which take specifically into consideration the precise nature of the interaction between the substrate in solution and the active site molecule.
A Generalised Koutecky-Levich Analysis of Mediated Electron Transfer at Immobilized Monolayers.
It is not often realised that the Koutecky-Levich analysis is strictly applicable only to first order surface reactions. Levich clearly states this limitation in his seminal monograph on physicochemical hydrodynamics [
26]. In this section of the paper we extend the standard Koutecky-Levich analysis and assume a mechanism of the following type:
Here we assume that all processes are first order, any of which can be rate determining. We have mass transport of the substrate to the electrode surface (quantified by a rate constant k
D as before), pre-activation of the substrate to form an activated substrate S
* (quantified by the rate constants k
S and k
-S) via processes such as adsorption, binding or dissociation, and finally electron transfer between S* and the electrode surface described by the heterogeneous electrochemical rate constant k’
E which will be potential dependent. In this model we specifically introduce a first order activation process and include the possibility that electron transfer between the active species and the underlying electrode may determine the net rate.
We apply the quasi steady state approximation to the reaction scheme above and obtain:
We can readily show that:
and also that:
and so the net flux is given by:
We invert the latter expression to obtain:
where we have introduced the equilibrium constant
. Again all possible rate limiting processes are cleanly separated: activation, electron transfer and mass transport. In fact:
for any sequence of consecutive first order processes it is possible, using the quasi steady state approximation, to obtain a separation of the pertinent rate limiting steps if an expression for the reciprocal of the net flux at steady state is developed.
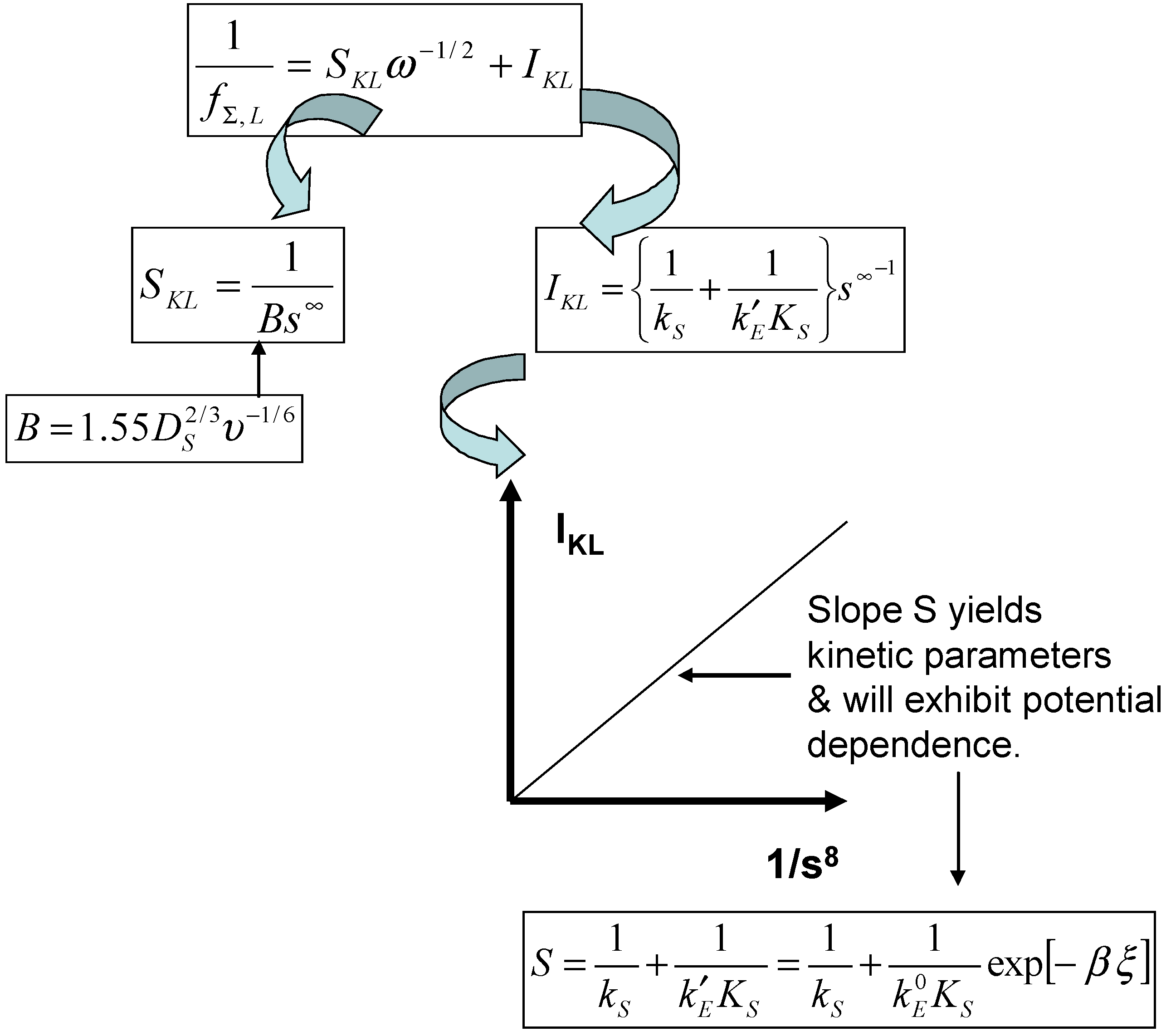
Noting that the diffusive rate constant is proportional to the square root of the electrode rotation speed for a rotating disc electrode geometry, eqn.31 can readily be transformed into an expression of the Koutecky-Levich type as outlined previously in eqn.26. Hence a plot of reciprocal flux (or indeed reciprocal current) versus inverse square root of the rotation speed is linear with a slope
where
, and an intercept given by
. The general experimental approach is outlined schematically in
figure 5.
If a Koutecky-Levich type mechanism is operative a plot of the Koutecky-Levich intercept versus inverse substrate concentration should be linear and pass through the origin as presented in
figure 5. The slope of the latter diagnostic plot should contain the kinetic parameters for the activation process and should also depend on the applied electrode potential via the k’
E term. We also note from eqn.30 that the steady state current response will be
always directly proportional to the bulk concentration of substrate. The latter predictions are easy to check experimentally.
Again it proves useful to derive an expression for the normalised flux Ψ as defined previously in eqn.11. We can readily show that eqn.30 may be transformed into the following non-dimensional form:
where we have introduced the competition parameters:
which gives the ratio of the rate of the surface reaction to that of substrate transport in solution and
which compares the rate of surface electron transfer with the rate of de-activation. The latter parameter will be strongly potential dependent.
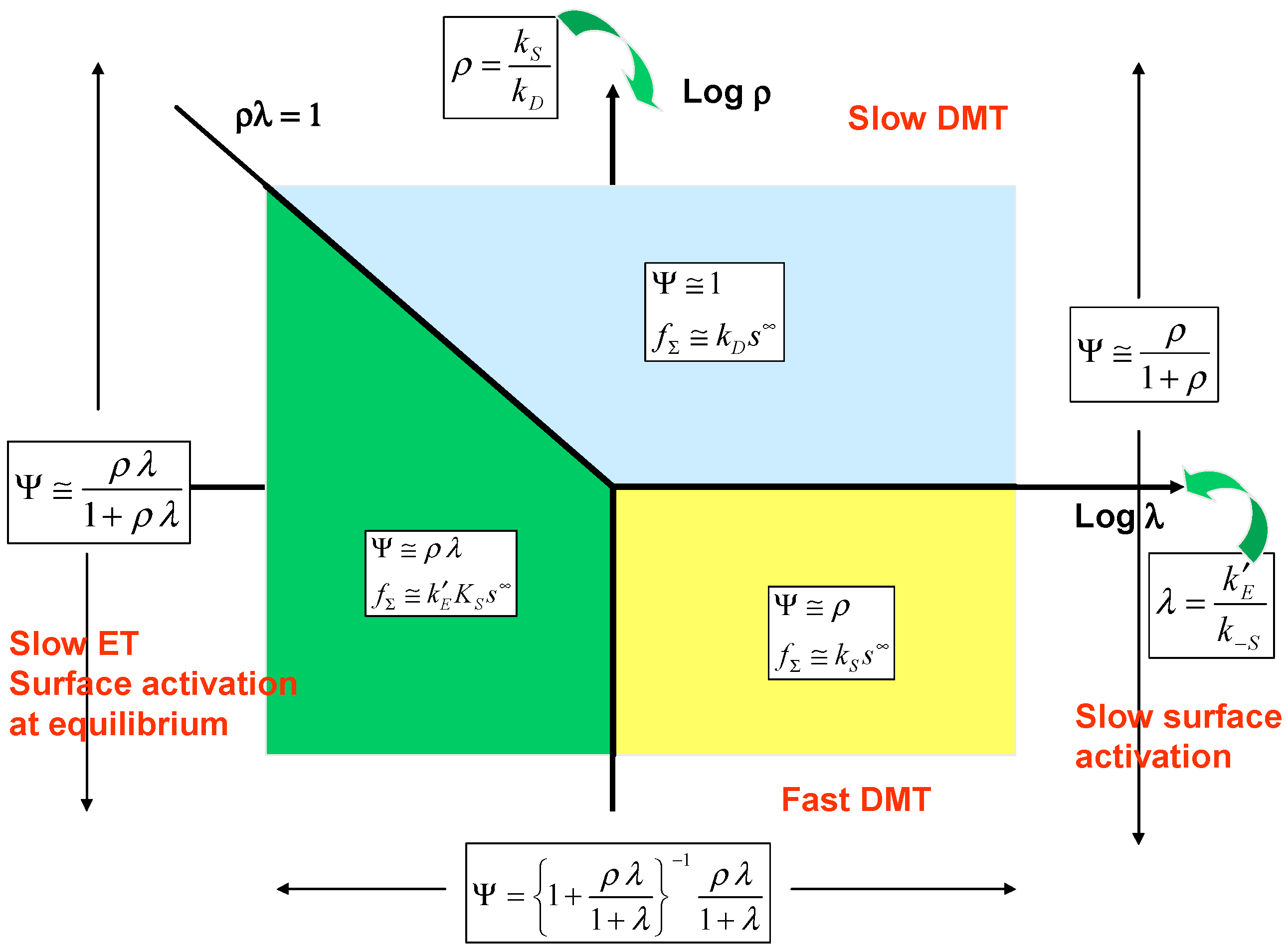
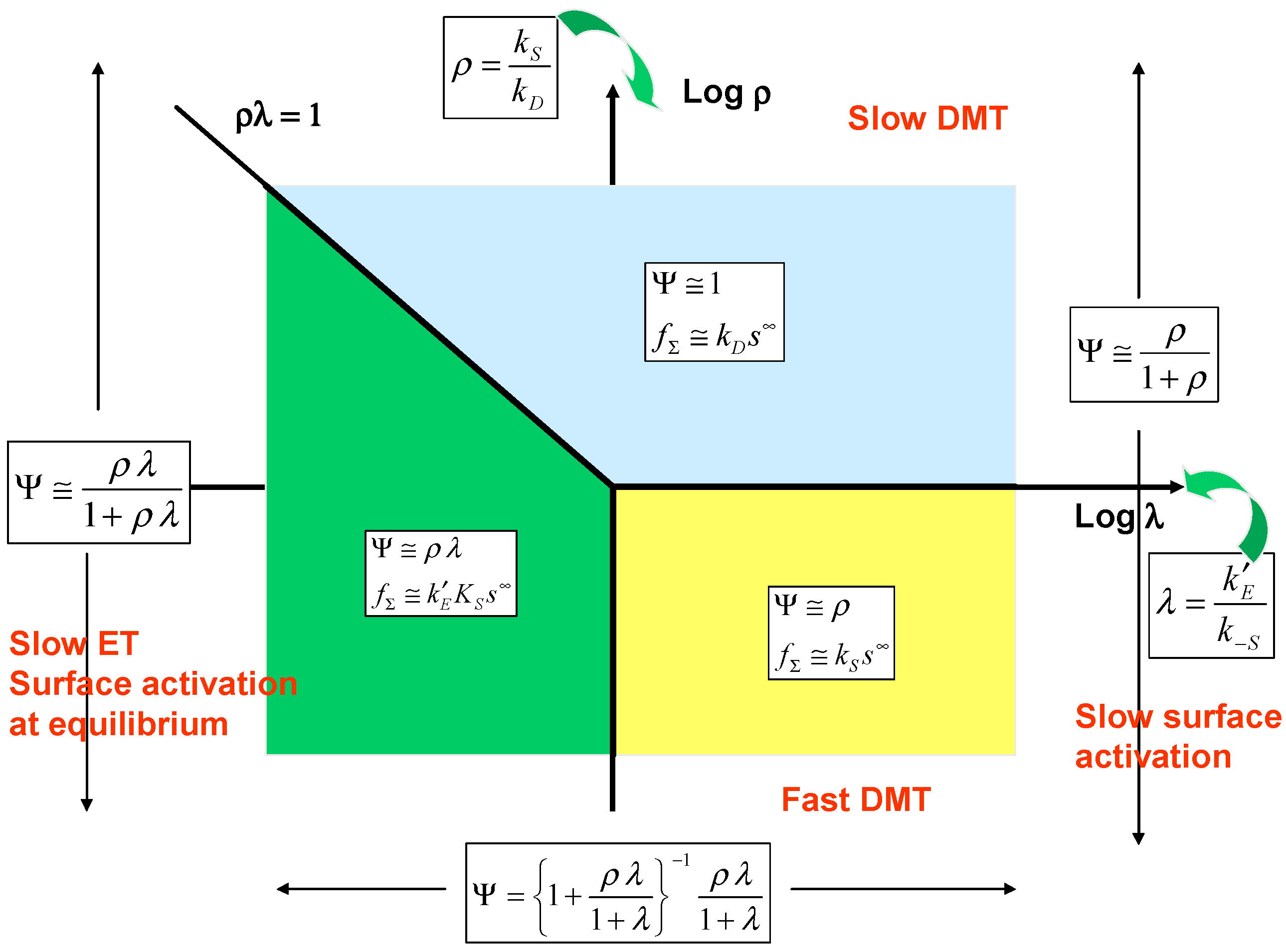
Examination of eqn.32 suggests that the parameters ρ and λ are suitable axes for a kinetic case diagram. Furthermore simplified expressions for the normalised flux may be obtained depending on the magnitude of λ and on the product ρλ . The kinetic case diagram is illustrated in
figure 6. Eqn.32 adopts the following limiting form when the parameter λ << 1:
whereas when λ >> 1 we note that:
Now taking the case when λ << 1 we obtain two distinct rate limiting situations depending on the magnitude of the product ρλ . Firstly when ρλ < 1 then eqn.33 reduces to:
or in terms of the net flux:
In this situation surface activation is at equilibrium and surface electron transfer is slow and rate determining. The flux will be potential dependent via a Tafel relationship because of the exponential dependence of the electrochemical rate constant on applied potential. We label this situation as case I. On the other hand when ρλ > 1 eqn.33 reduces to:
and the flux is given by its mass transfer controlled value since:
The net flux will be independent of potential. We label this situation case II. In the kinetic case diagram the line ρλ = 1 will separate cases I and II. Now turning to the case when λ >> 1 we examine eqn.34 and consider two situations which depend on the magnitude of the parameter ρ . Firstly, when ρ << 1 eqn.34 reduces to:
and the net flux is given by:
Here the surface activation step is slow and rate determining and both electron transfer and substrate diffusion are fast. This situation is designated case III and will be located in the lower right hand quadrant of the kinetic case diagram. On the other hand when ρ >> 1 eqn.34 reduces to Ψ ≅ 1 again and kinetic case II is regained.
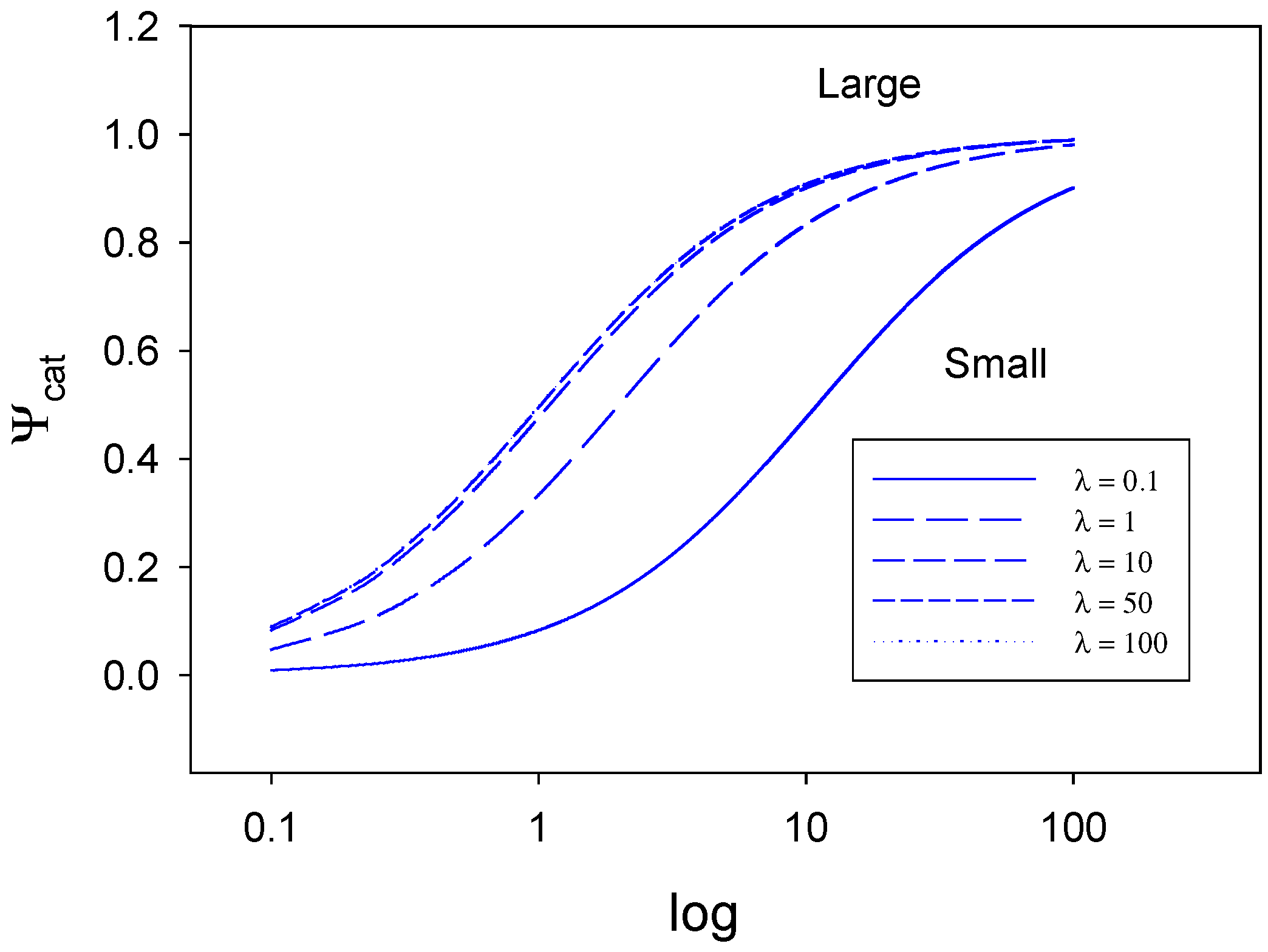
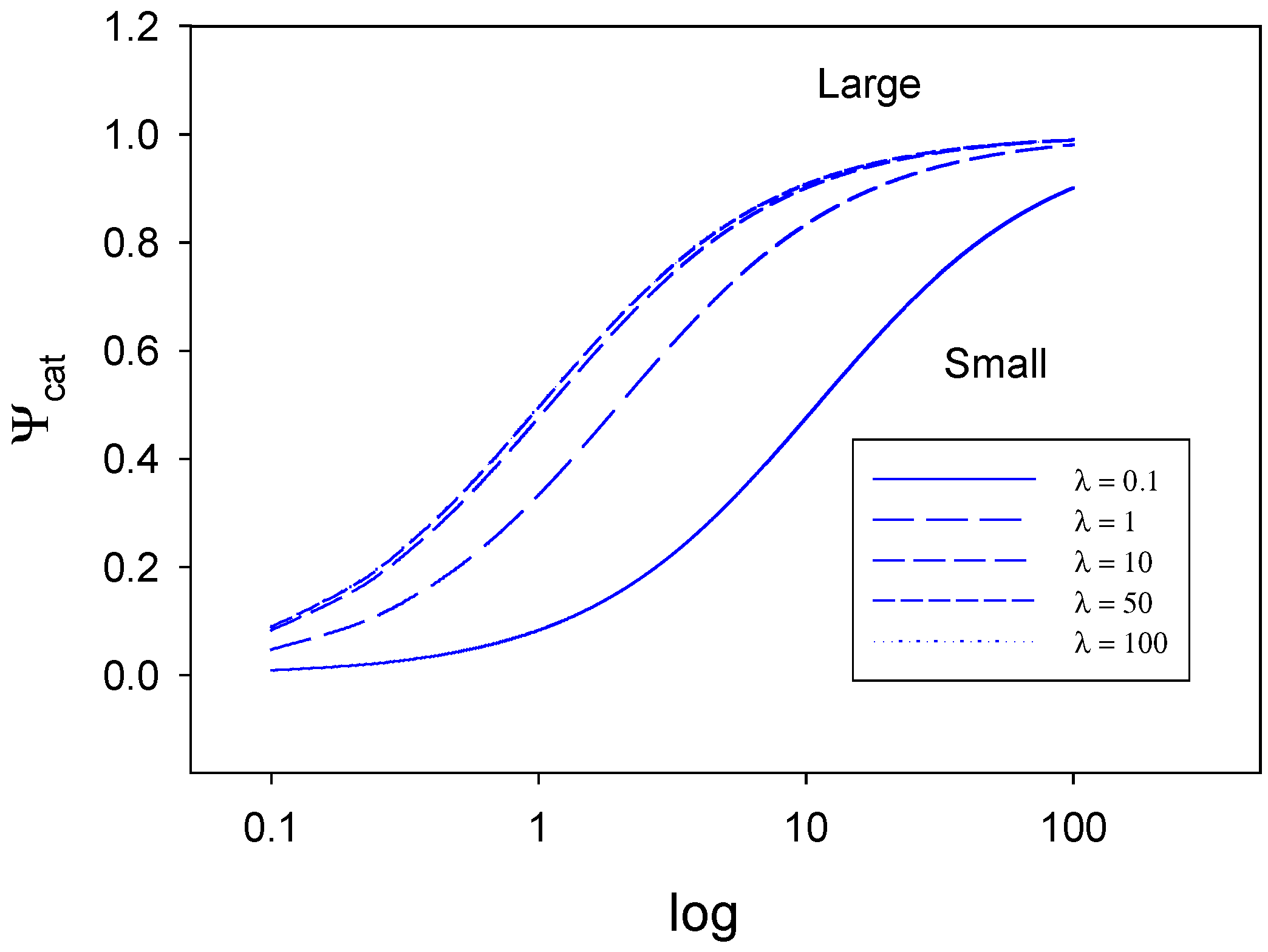
In
figure 7 and
figure 8 we plot eqn.32 for various values of λ and ρ . In
figure 7 we indicate the way that the normalized catalytic current varies with the parameter ρ over the range 0.1 -100, for various values of the parameter λ .When the parameter ρ is small the normalised current Ψ will adopt numerical values considerably less than the mass transfer controlled value of unity over the entire range of λ values examined. When both ρ and λ are small the rate is determined by slow electron transfer (case I) When λ is small the normalized flux only increases slowly with increasing ρ. This is to be expected since the potential applied to the electrode to transform the activated S* species to product will be small. In contrast when λ is large, we begin in a case III situation corresponding to slow surface activation in which the generation of the active species S* is rate determining, and we note that the normalized flux increases much more rapidly with increasing ρ value. This is brought about by the fact that the system is now subjected to a far larger electrochemical driving force via the applied potential. As ρ gets larger Ψ gradually attains the value characteristic of diffusion control over the range of λ values examined (0.1 – 100).
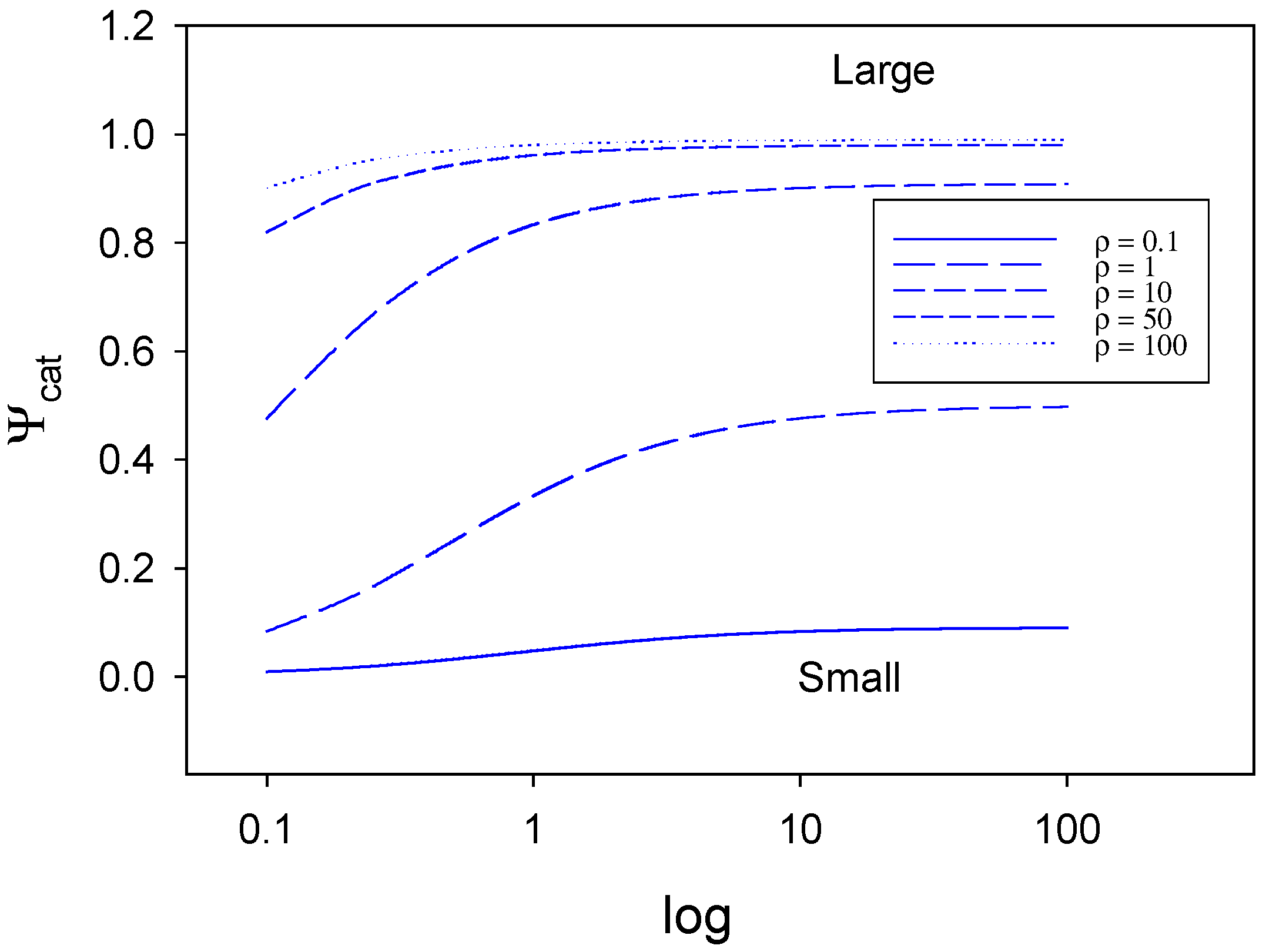
In
figure 8 we show the way that the normalized catalytic current varies with the parameter λ over the range 0.1 – 100 for various values of the ρ parameter. When ρ is small at a value of 0.1 say, the normalized catalytic current remains at a low value over all of the range of λ values examined. This will correspond either to the case of slow electron transfer involving the active surface species S* (case I) valid when λ is small, or case III, when λ is large, corresponding to slow surface activation in which the active species S* is generated. Also for any given value of ρ the normalized current Ψ attains a constant value once values of λ > 20 are attained. In contrast when ρ admits large values (50, 100) the value of the catalytic current is close to unity over much of the range of λ values examined in the computation. This can be understood by examination of the case diagram in
figure 6, where case II, the region defining diffusion control is valid for a wide range of λ values when ρ is large. The drop in normalized current when λ gets less than unity observed in
figure 8 corresponds to the changeover from rate determining case II to kinetic case I involving slow reaction of active species S* to form product which will be potential dependent.
A Precursor/Successor and Complex Formation/Dissociation Model
In the previous sections of the paper we developed models in which the steady state current response varies in a linear manner with the bulk concentration of the substrate species in solution. This result will pertain when either simple bimolecular reaction between substrate and immobilized mediator species is assumed, or if an activated complex involving the substrate species undergoes surface electron transfer to yield product. In many experimental situations a more complex relationship between the steady state amperometric current response and the bulk substrate concentration is observed. A typical current versus concentration response profile might exhibit a first order relationship at low substrate concentrations and display a current response independent of substrate concentration at high values of substrate concentration. At intermediate values of the latter a non linear response pertains. This type of behavior is of quite wide applicability. Some specific examples may be quoted. The work of Albery and Bartlett [
27] on the direct oxidation of the biologically important co-factor NADH and the enzyme catalysed oxidation of glucose at conducting organic salt electrodes such as TTF.TCNQ (TTF = tetrathiafulvalene, TCNQ = tetracyanoquinodimethane) and NMP.TCNQ (NMP = N-methylphenazinium ion) and that reported by Gorton and co-workers [
28] on NADH oxidation at redox dye modified electrodes specifically presents characteristic “dog leg” or biphasic current/concentration plots obtained from batch amperometric experiments using rotating disc electrodes. Organic electro-oxidation at conductive hydrated metal oxide surfaces provides another example [
29] . In more recent publications biphasic kinetic data are reported for glucose oxidation at thin electropolymerized poly(phenol) films containing immobilized glucose oxidase [
30] and at alkanethiol self assembled monolayers containing adsorbed glucose oxidase [
31] , and for both NADH [
32] and ascorbate [
33] oxidation at poly(aniline)/poly(vinylsulfonate) composite modified electrodes and ascorbate oxidation at an electrode modified by a Tosflex ionomer film containing ferricyanide [
34].
Biphasic kinetics may be readily explained using a Michaelis-Mentin mechanism [
35] which is already well established in enzyme kinetics [
36]. The key aspect is that the substrate will form a distinct
precursor complex with the immobilized mediator species which can then transform via intramolecular electron transfer into a
successor complex which will subsequently dissociate to regenerate the surface bound pre-catalyst species and the product. Hence key kinetic parameters in the mechanism will quantify the binding reaction to form the precursor complex, and the dissociation rate of the successor complex.
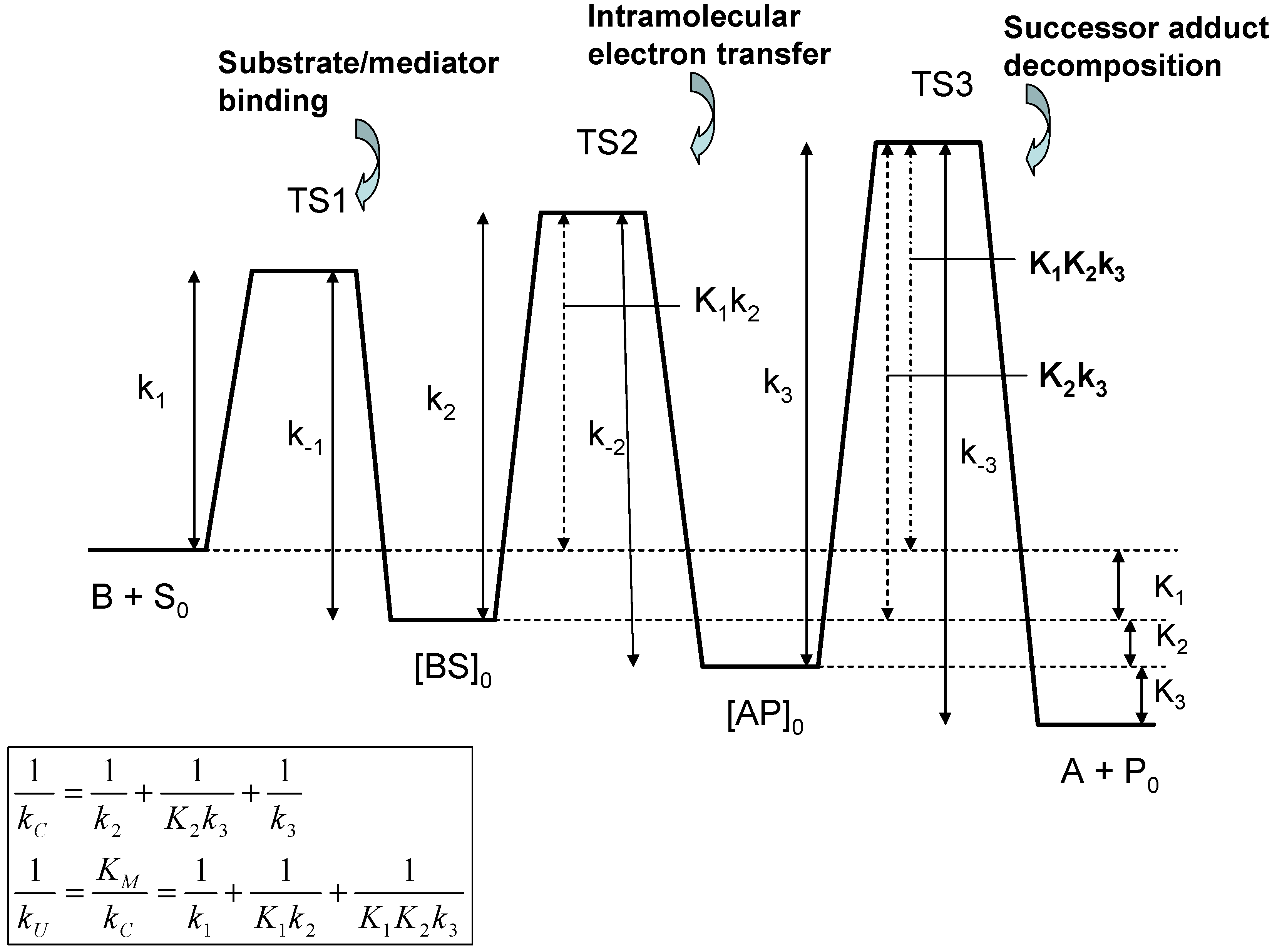
We consider the following reaction sequence:
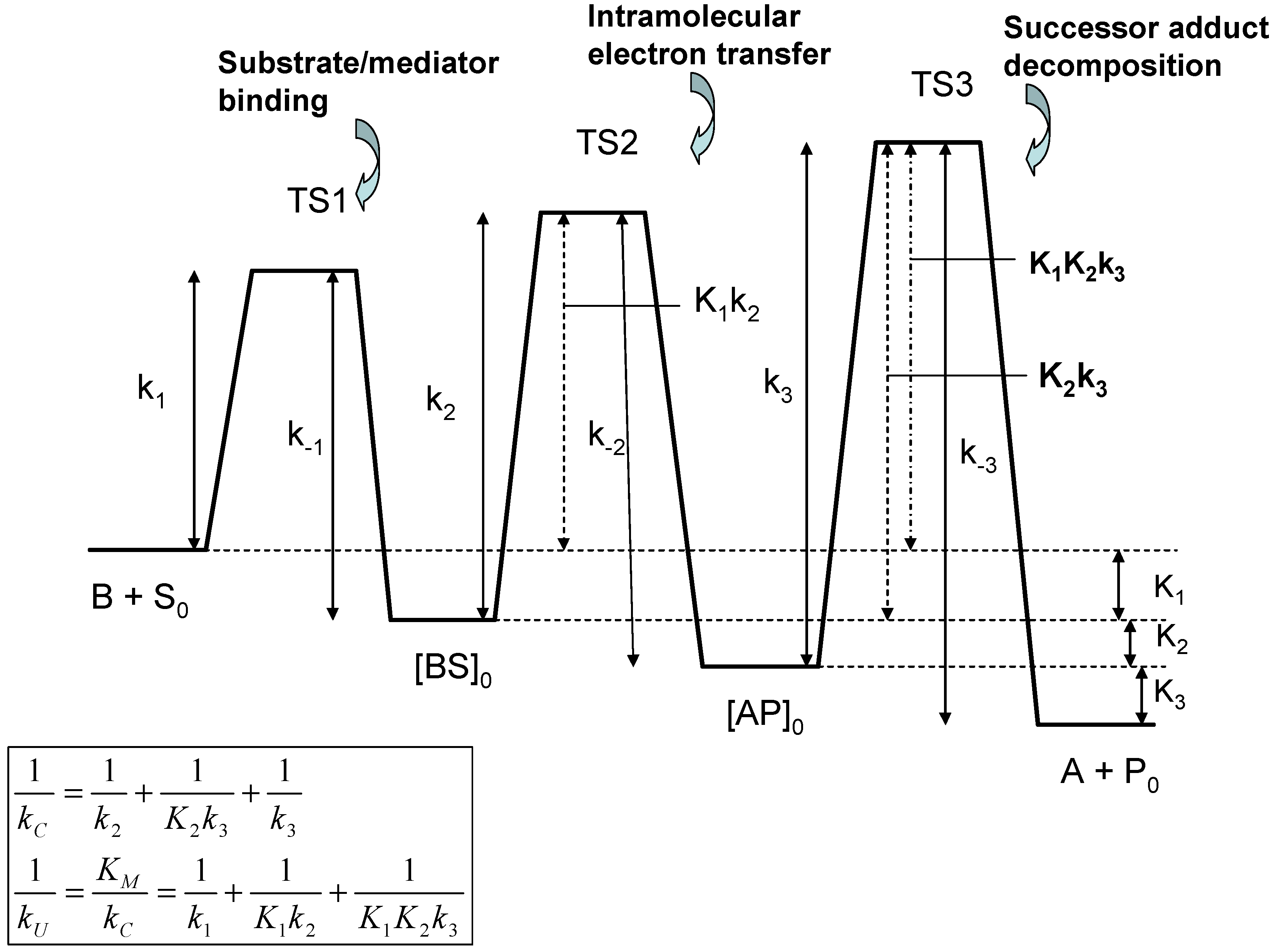
In the latter reaction sequence S, P denote the bulk reactant and product species, whereas S0 and P0 denote the corresponding species at the monolayer/solution interface. We will assume that the product is absent from the bulk solution initially and so product inhibition can be neglected. As before, kD represents the diffusive rate constant of reactant (units: cm s-1). Also kE is a first order (unit: s-1) heterogeneous electrochemical rate constant for generation of the active form of the mediator species B within the monolayer . We note that k1 denotes the bimolecular rate constant (unit : cm3 mol-1 s-1) for precursor adduct formation involving the mediator and the substrate species, and k-1 is the corresponding first order rate constant for precursor dissociation (unit: s-1) to reform mediator and substrate. Furthermore, k2 and k-2 are first order rate constants for the intramolecular electron transfer reaction which transforms the precursor complex BS to the successor complex AP, and k3 is a first order rate constant for dissociation of the successor complex to form the pre-catalyst and the product. All of the latter rate constants have units of s-1. We can assume that successor dissociation is irreversible if product inhibition can be neglected.
We again apply the quasi-steady state approximation to the latter reaction sequence to obtain an expression for the net reaction flux f
Σ (unit: mol cm
-2 s
-1):
The total surface coverage is given by:
A useful expression for the steady state reaction flux can be obtained if expressions for the unknown quantities s0, ΓA, ΓB, ΓBS and ΓAP can be determined. Since there are five unknowns and six defining expressions (presented in eqn.42 and eqn.43) the latter aim can be readily accomplished.
If we initially neglect the diffusion of the substrate species to the monolayer surface, we can readily show after some algebra that the inverse steady state reaction flux is given by :
Eqn.44 consists of three terms, each of which reflects a possible rate limiting situation. The first term on the rhs describes the electrode kinetics for the A/B transformation within the monolayer; the second reflects unsaturated binding kinetics and the third reflects adduct transformation processes to generate products. In the latter expression we have followed the approach of Albery and Knowles [
37] and introduce the Michaelis constant K
M and the catalytic rate constant k
C in terms of the fundamental rate constants for the consecutive steps in the reaction sequence as follows:
where we have written
. The significance of these equations have been discussed by Albery and Knowles [
37]. Note that the quantity
which has the dimensions of a bimolecular rate constant, provides a measure of the rate of capture of the substrate species by the immobilized catalyst B to form the adduct BS. The terms Michaelis constant and catalytic rate constant are well established in the field of enzyme kinetics. The Michaelis constant K
M provides a measure of the binding affinity or adduct formation ability of the substrate species for the immobilized mediator. Alternatively, it defines the maximum value of the substrate concentration for which the catalytic kinetics are first order with respect to substrate concentration. The catalytic rate constant k
C is a first order rate constant, quantifying the rate of decomposition of the surface adduct species to form product. We note from eqn.45 that both k
U and k
C are composite quantities, and may be considered to be
internal parameters connected with processes occurring within the monolayer region, as opposed to
external parameters which include the electrochemical rate constant k
E and the diffusive rate constant k
D. Both quantities, when expressed in reciprocal format, consist of three separated terms. Considering the k
C component terms first, we note that
corresponds to slow rate determining intramolecular electron transfer involving the transformation of the precursor adduct BS to the successor adduct AP. The
term corresponds to the case where the precursor/successor adduct transformation is at a pre-equilibrium followed by a slow rate determining decomposition of the successor adduct to form products. Finally the
term corresponds to slow rate determining successor adduct decomposition. The smallest of these terms will be the major contributor to the net catalytic rate constant k
C. Similarly, if we examine the k
U component terms we note that the
term reflects rate determining adduct formation involving the bimolecular reaction between S and B. The second and third terms
or involve either single (K
1) or multiple (K
1K
2) pre-equilibria followed by slow intramolecular electron transfer between the adducts BS and AP within the monolayer (k
2) or slow successor adduct dissociation (k
3). A schematic free energy profile illustrating the free energy differences associated with each of the possible rate limiting steps associated with the internal parameters is presented in
figure 9.
It is clear from eqn.44 that by neglecting substrate diffusion effects in solution , we can obtain a transparent expression in which the important kinetically limiting steps may be cleanly separated from one another.
Our analysis to date has neglected the external process of substrate diffusion to the monolayer. However in experiments mediated electrocatalysis is probed using rotating disc voltammetry and the surface concentration of substrate s
0 will differ from that of its bulk value s
`. This simple observation has a marked consequence for the expression for the net reaction flux. Noting that the substrate surface and bulk concentrations s
0 and s
` are related via:
Substituting eqn.46 into eqn.44 results in the more complete expression:
We use the following result
in eqn.46 to obtain:
We can readily recast eqn.48 into the following useful form:
where we have rescaled the unsaturated rate constant factor to transform into a pseudo first order rate constant via
. Eqn.49 describes the net reaction flux when substrate diffusion in the solution to the monolayer surface is taken into account. The important point to note from eqn.49 is that the net reaction flux appears on both sides of the expression and so each of the components of the expression are not separated in a clear manner using the strategy of deriving an expression for the inverse of the net reaction flux. The term which causes the complication is
fΣ/
kDs∞. If the net flux f
Σ becomes close to the limit imposed by substrate transport through the solution diffusion layer given by
fD=
kDs∞, then the latter diffusive transport results in the immobilized receptor species being less saturated than one would expect from the value of the bulk concentration s
`. This effect is described by the 1 −
fΣ/
kDs∞ term. We can see that its presence reduces the significance of the mediator generation kinetics, and the unimolecular decomposition of surface adducts to form products. If we can assume that
fΣ/
kDs∞ << 1 then 1 −
fΣ/
kDs∞ ≅ 1 and eqn.49 reduces to:
and we note that all the possible rate limiting terms are cleanly separated. This expression has been often utilized in the literature in conjunction with the Koutecky-Levich equation to analyse data obtained via use of the rotating disc electrode. It is important to note that the expression will only be valid when the net current (or reaction flux) is much less that the diffusion controlled value. The assumption implicit in the use of eqn.50 is that the monolayer surface and hence the immobilized mediator site is exposed to the bulk concentration of the substrate species. If , on the other hand, as is often the case, the experimentally observed current is the same order of magnitude as the mass transport limiting value, there will be a considerable concentration gradient in the Nernst diffusion layer and the surface and bulk concentration of substrate will differ considerably. The effect of this concentration gradient is that the immobilized mediator will be exposed to lower values of substrate than expected, and this will lead to the prediction that the surface bound mediator species will be more efficient than it is in reality. Hence the full expression presented in eqn.49 must be used in data analysis if the experimentally observed steady state current response in a sensor system approaches that of the diffusion controlled value. This very fundamental point has not been realized in much of the data analysis presented in the literature.
We can develop a variant of eqn.49 which is similar to the Hanes equation used in the analysis of enzyme based systems. We can show that:
which can be subsequently simplified to:
In the latter expression k
ME represents the effective electrochemical rate constant (unit: cms
-1) for the monolayer modified electrode at low substrate concentration , and K
ME denotes the Michaelis constant for the monolayer modified electrode. Both of these quantities are total parameters in the sense that they are composite quantities not only reflecting the binding and kinetics within the monolayer but also include the external variables related to substrate diffusion in the solution and mediator generation via an external applied potential. We can readily show that:
We note that KME should exhibit a potential dependence via the kE term.
We can transform the Hanes type expression presented in eqn.51 into a quadratic equation in the net reaction flux to obtain:
which has the solution:
We can follow the previous sections of this paper and introduce a normalized flux or effective catalytic efficiency of the modified electrode as
![Sensors 02 00473 i001]()
and from eqn.55 obtain the following dimensionless master expression:
where we have introduced
and we note that
The first parameter in eqn.57 compares the electrochemical rate constant for the modified electrode with the diffusive rate constant of the substrate, and the second compares the bulk substrate concentration with the Michaelis constant of the modified electrode system.
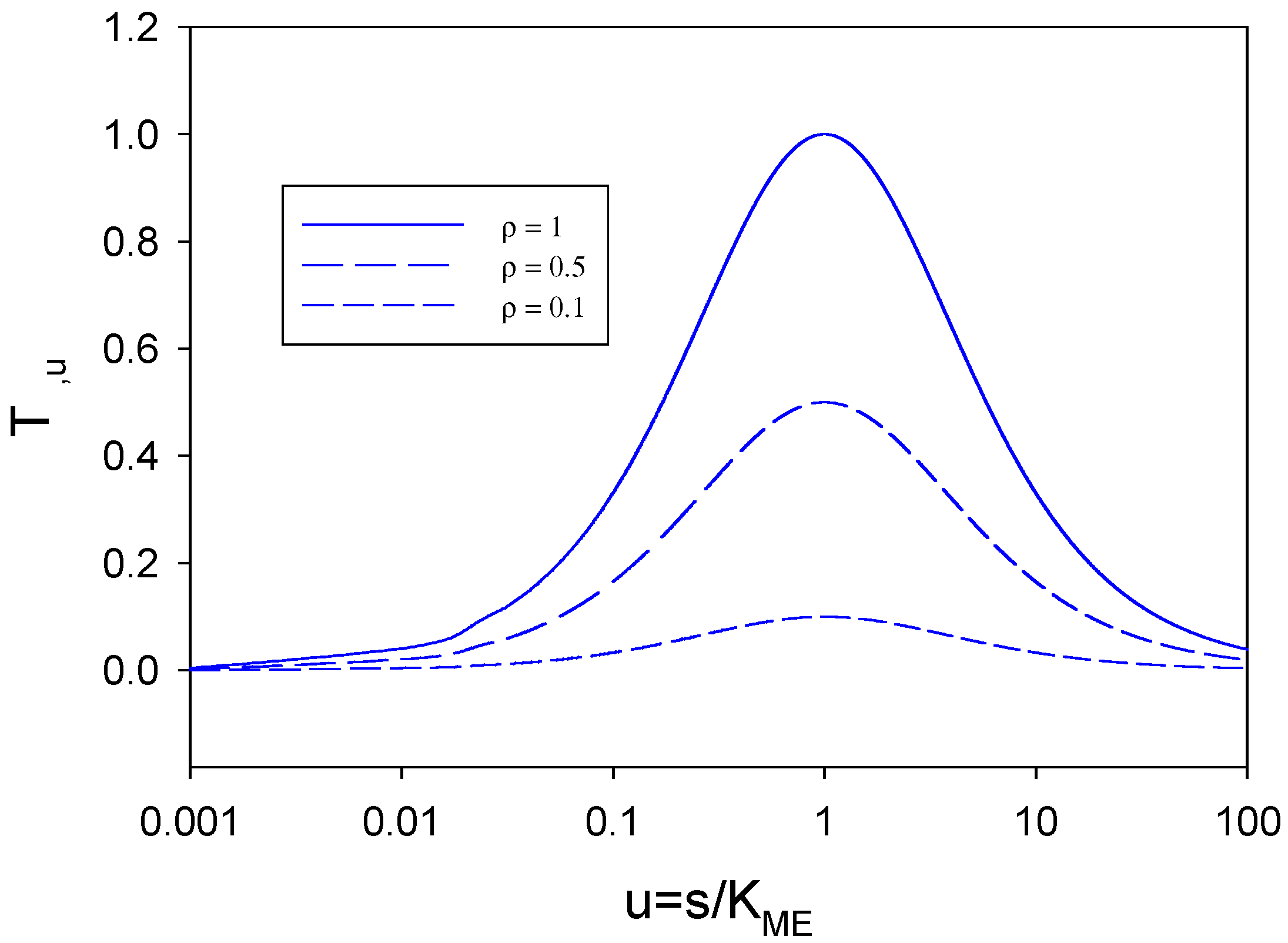
We now examine the term T
ρ,u defined in eqn.58. We firstly note that 0 ≤
ρ ≤ 1. We can plot the T term versus u for typical values of ρ . This is done in
figure 10 for three values of ρ (1, 0.5 and 0.1). We note from these plots that when u is very small (u < 0.1) or very large (u > 10) the T - function approaches zero. This will be so for all values of ρ.
If T is small then for all ρ we have
![Sensors 02 00473 i005]()
. Specifically if u is large then for u >> 1 we note (1+
u)
2 ≅
u2 and so
![Sensors 02 00473 i007]()
and eqn.56 for the normalized flux or catalytic efficiency reduces to:
in the limit of large u where s
` >> K
ME . We can now show using eqn. 59 that the net reaction flux when s
` >> K
ME is given by:
Recalling from eqn.53 the definition of K
ME in terms of the more fundamental kinetic and binding parameters we obtain:
The pertinent parameter in eqn.61 is the ratio
kE/kC , which compares the rate of mediator generation (via the k
E term) with the rate of adduct decomposition (via the k
C term). Firstly if
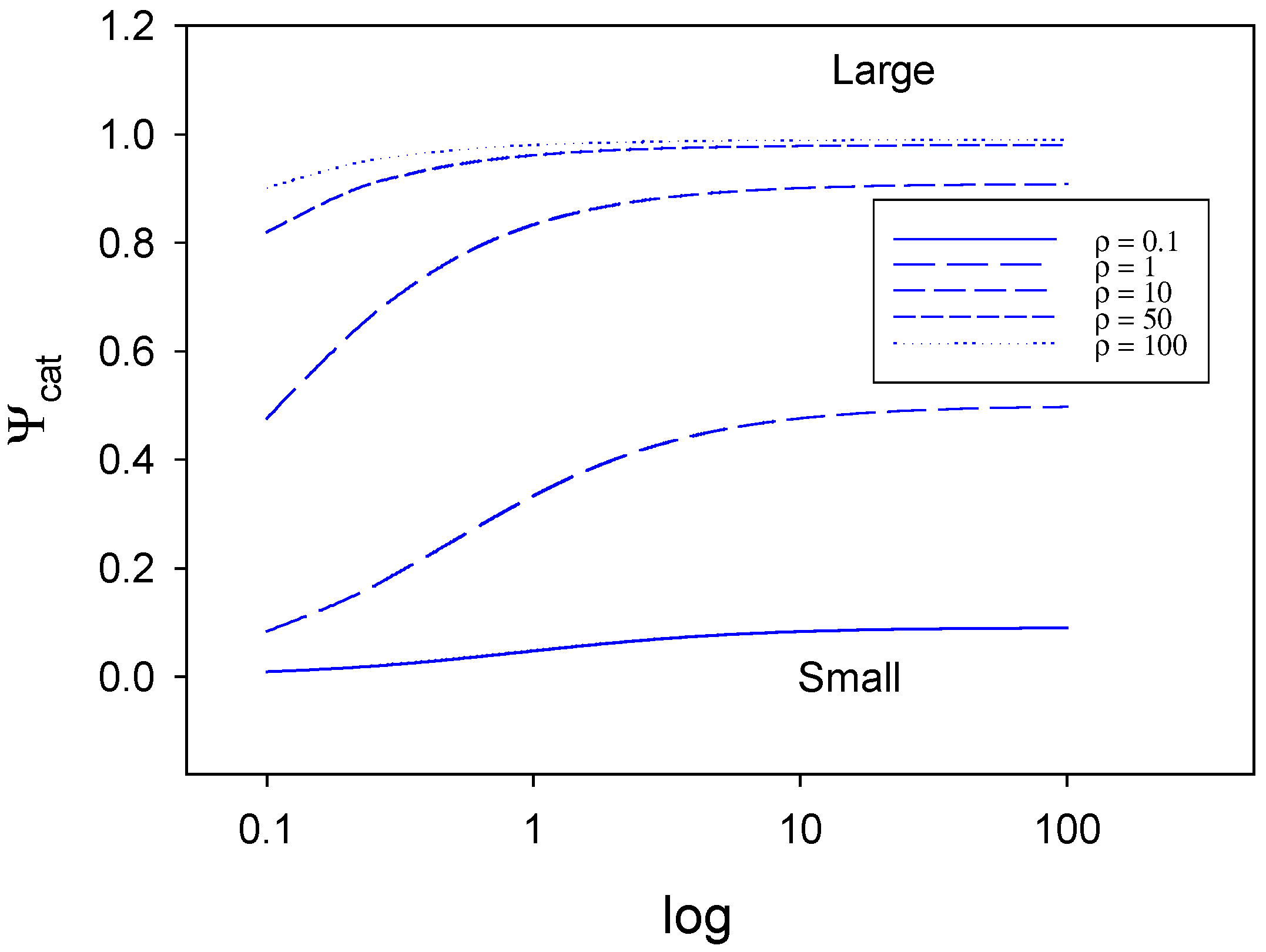
kE/kC << 1 then we have the situation where mediator generation is slow and rate limiting and surface adduct decomposition is rapid. In this particular situation eqn.61 reduces to:
which we will label case III. On the other hand, if
kE/kC >> 1 then the process of mediator generation will be rapid and the surface adduct decomposition to form products will be slow and rate determining. In this circumstance (case IV) we obtain the following limiting expression for the net reaction flux:
We note that both for case III and case IV we are concerned with the limit where s
` >> K
ME. In both cases the net reaction rate is independent of the bulk substrate concentration s
` and depends directly on the total surface coverage of the mediator species in the monolayer. For case III the net rate should be potential dependent via the Tafel relation because of the k
E term. For case IV no such potential dependence is expected.
We now focus attention on the situation where the bulk substrate concentration is much less than the Michaelis constant of the modified electrode, when s
` << K
ME. When u is small u << 1 or 1/u >> 1 and so (1 +
u)
2≅ 1. Under these circumstances eqn.56 for the normalized flux reduces to:
Now since 0 ≤
ρ ≤ 1 and u << 1 then the term 4ρu will be small. Under these conditions (1 − 4
ρ u)
1/2 ≅ 1 − 2
ρ u and eqn.64 reduces to:
Again, this is a useful limiting expression, valid when s
∞ << K
ME . Noting the definition of ρ we can show that:
If we recall the definition of the modified electrode rate constant provided in eqn.53 we can show that
Again this is a useful expression. The pertinent parameter here is the ratio
![Sensors 02 00473 i012]()
which represents the balance between substrate diffusion in the diffusion layer and chemical reaction involving the surface bound adducts. Two limiting cases may be enumerated. First, when
![Sensors 02 00473 i013]()
we have the situation of slow rate determining mass transport of substrate in the solution to the modified electrode surface and fast unsaturated kinetics involving substrate and mediator species. Under such conditions eqn.67 reduces to:
which we label case I. Here the steady state current response will vary in a linear manner with bulk concentration of the substrate. Second, when
![Sensors 02 00473 i014]()
, we have the case of facile material transport and slow rate determining chemical reaction between immobilized mediator and substrate. Here eqn.67 reduces to:
which we denote as case II. Again, the steady state current response will vary in a linear manner with bulk substrate concentration. We can differentiate between cases I and II by noting that in the former, the current is independent of surface coverage of redox mediator whereas in the latter, it depends linearly on Γ
Σ via the k’
U term.
Development of a Kinetic Case Diagram
It is desirable to derive a relationship between the overall quantity K
ME and the more fundamental Michaelis constant K
M. We recall that important parameters which define rate limiting situations are
![Sensors 02 00473 i016]()
and
![Sensors 02 00473 i017]()
. Using the latter definitions and the expressions presented in eqn.53 we can show, after some algebra that the Michaelis constant for the modified electrode is related to the Michaelis constant describing the adduct formation process via:
We can define a further normalized variable which quantifies the degree of unsaturation of the system and introduce
![Sensors 02 00473 i019]()
. We can relate the normalized concentration variable
![Sensors 02 00473 i020]()
introduced earlier to the latter quantity via:
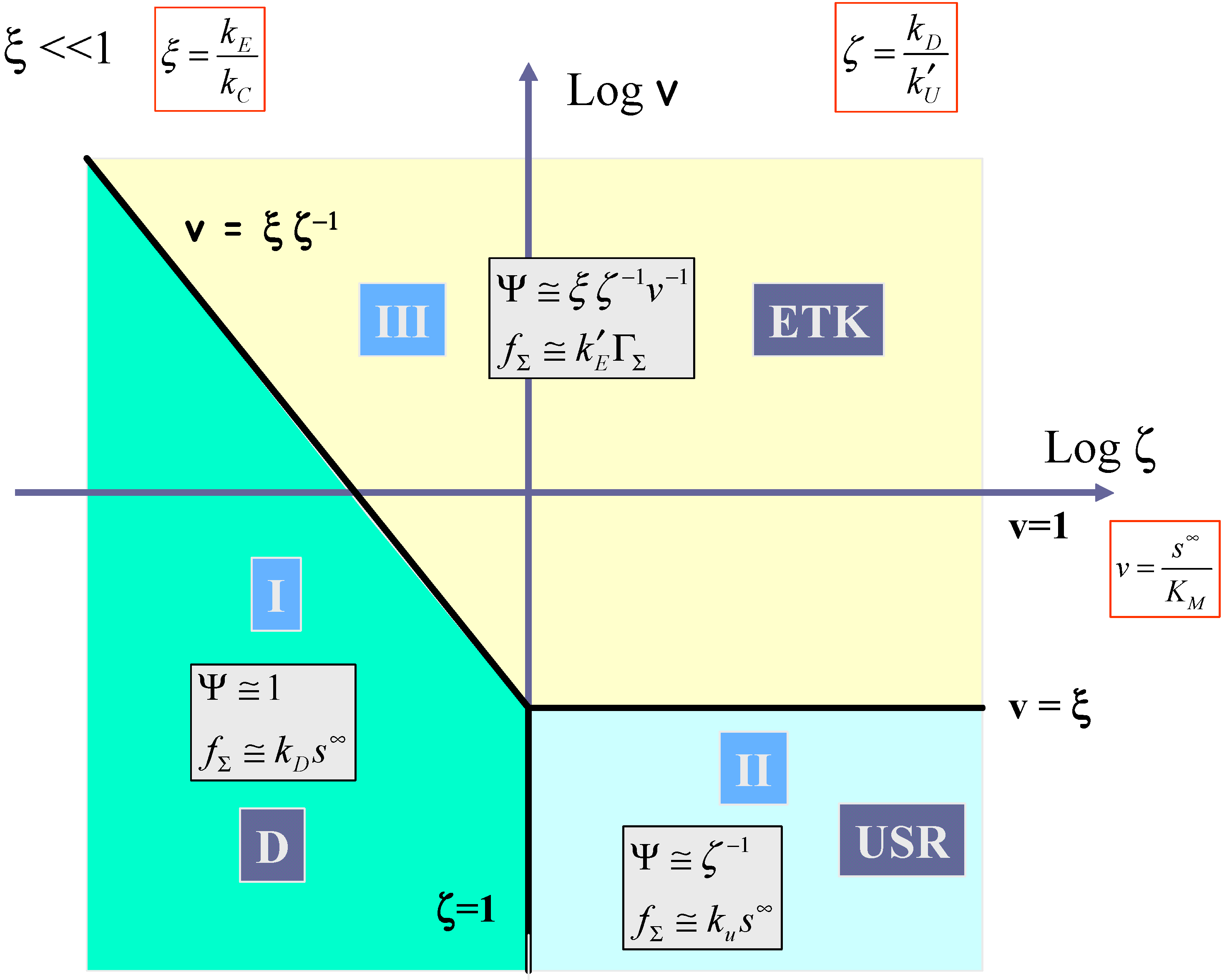
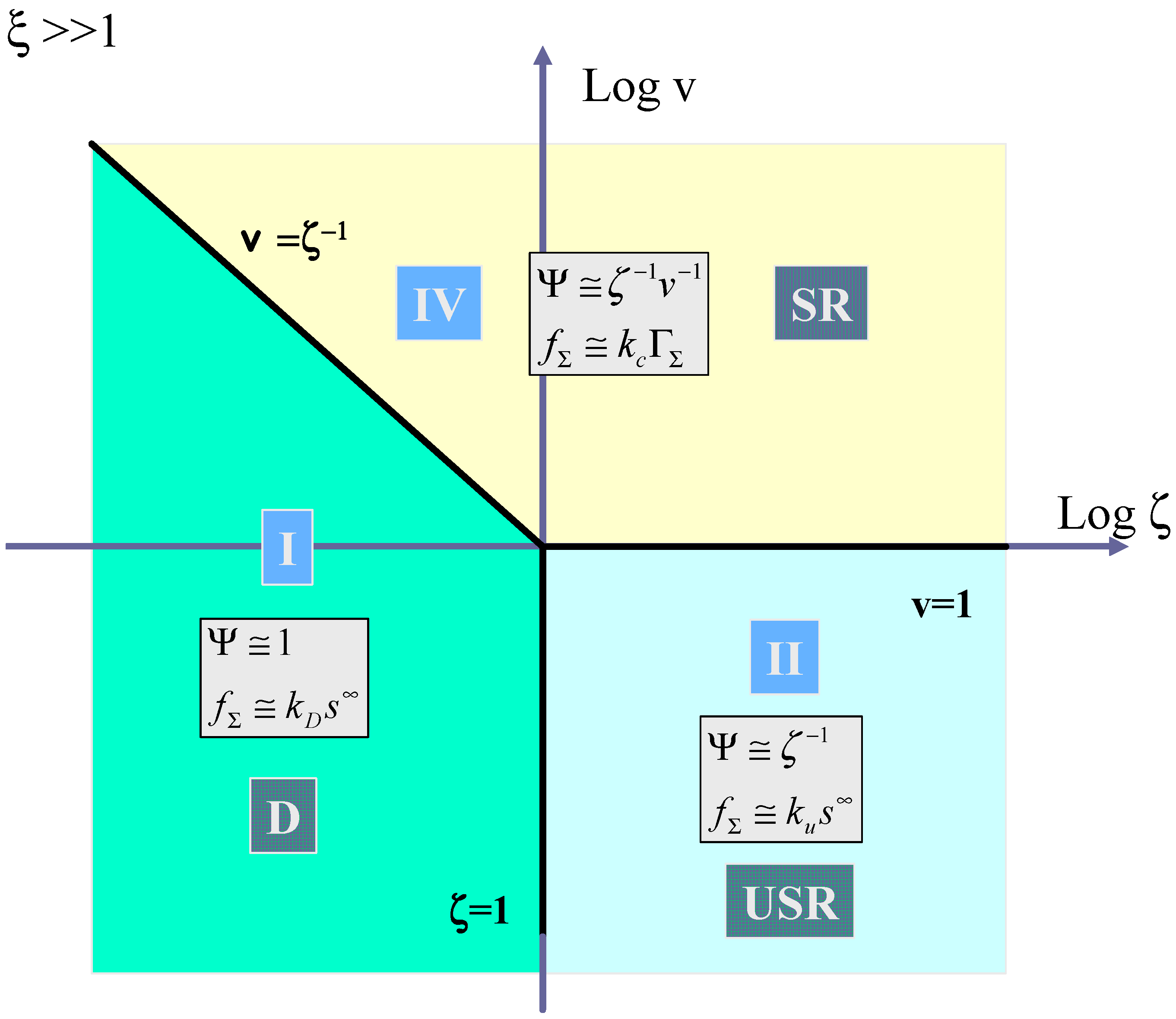
Hence the three pertinent parameters which will define a kinetic case diagram are
ζ , v, and
ξ . We can, if we so wish, construct a three dimensional case diagram involving the latter parameters. However instead, to aid visualization, we construct the two dimensional analogue, where we present plots of log v versus log
ζ for the limiting cases of
ξ << 1 and
ξ >> 1. We choose log
ζ as the abscissa and log v as the ordinate in these diagrams. The case diagrams are presented in
figure 11 and
figure 12.
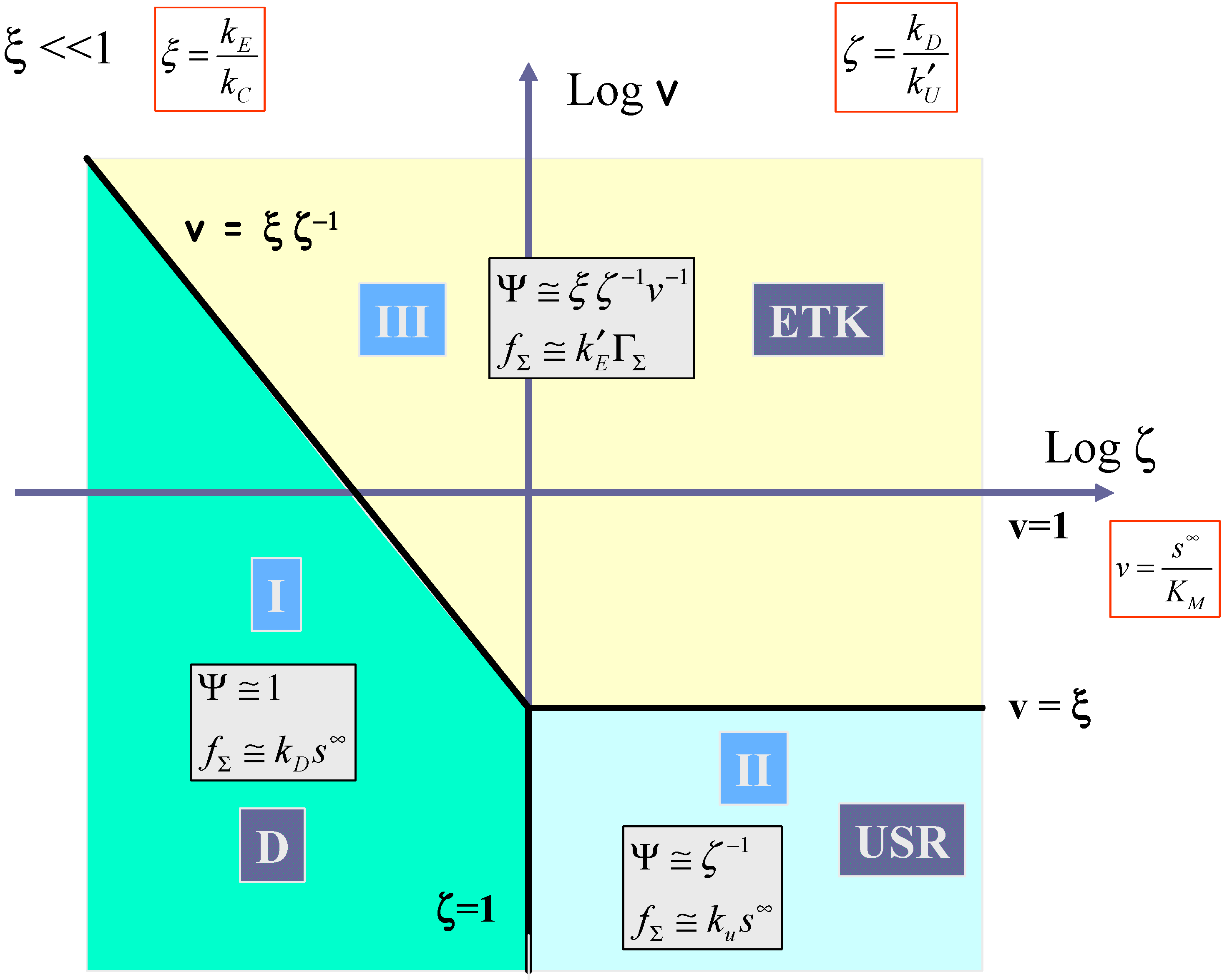
In
figure 11 the case where the kinetics of mediator generation is much slower than the decomposition of the surface adducts (ξ << 1) is outlined. Here cases I, II and III must be considered. Kinetic case I corresponding to diffusive control of the substrate to the redox monolayer (which we label D) is located between the vertical boundary
ζ = 1 which separates case I and case II, and the line
v =
ξ ζ−1, which defines the boundary between case I and case III. The expression for the net reaction flux is given by eqn.68, and we note that the flux will be first order in bulk substrate concentration and independent of the surface coverage of the surface immobilized mediator. Kinetic case II located in the lower right hand section of the case diagram, is bounded to the north by the horizontal line v = ξ and at the western edge by the vertical line ζ = 1. As previously noted case II corresponds to slow rate determining chemical reaction between surface mediator and substrate (which we label USR). In normalized form the flux is given by Ψ ≅
ζ−1, which simplifies to the expression presented in eqn.69. Here the flux will be first order both in substrate concentration and mediator coverage. Case III labeled ETK corresponds to the situation where mediator generation is slow and rate limiting. The normalized flux is approximately given by
![Sensors 02 00473 i022]()
which can be shown to reduce to eqn.62. Here the flux is independent of the bulk substrate concentration and is first order with respect to mediator surface coverage. It will also be potential dependent. Case III is bounded by the line
![Sensors 02 00473 i023]()
and by the horizontal line v = ξ.
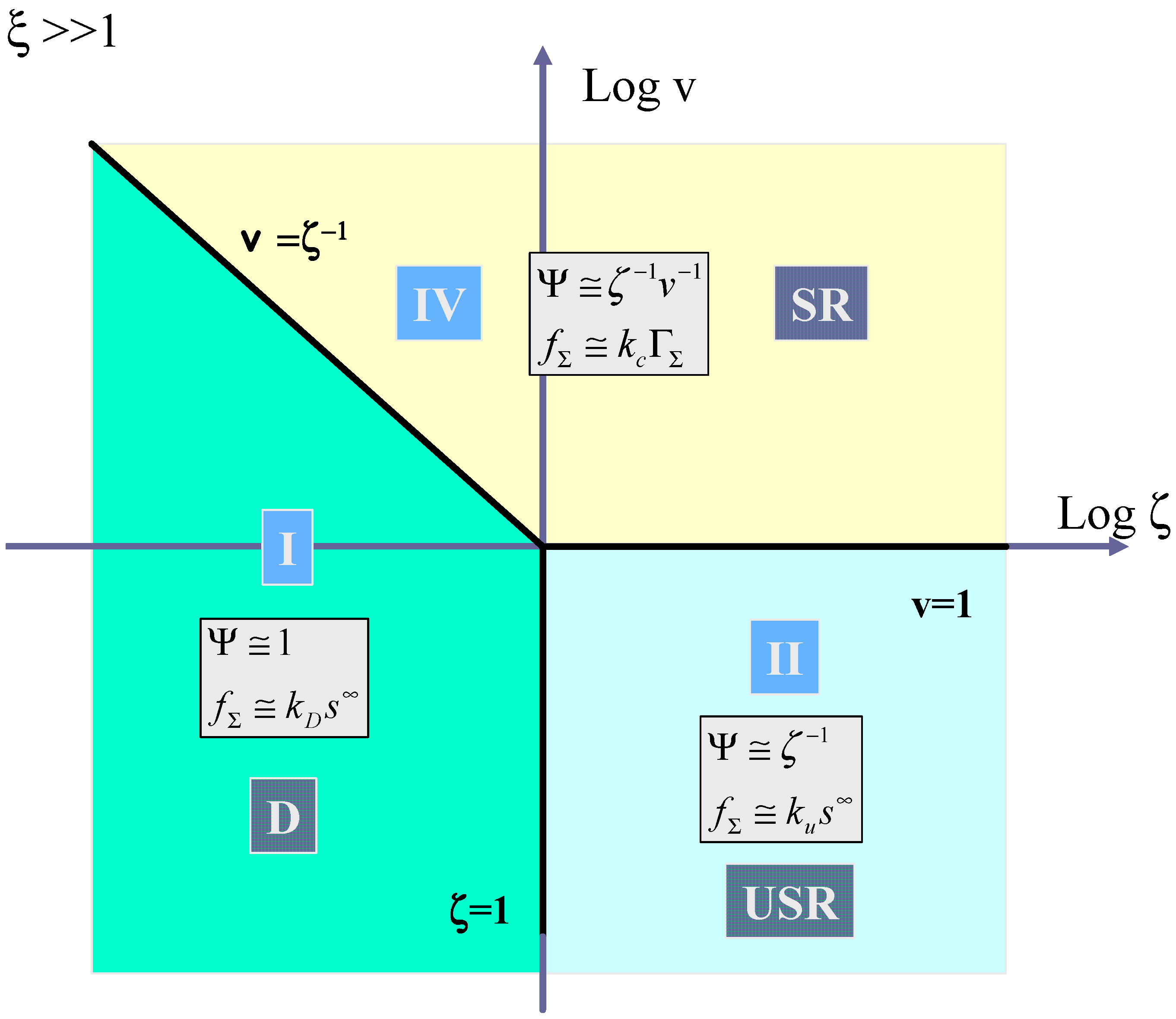
We now turn to
figure 12 which shows the kinetic case diagram for the situation where ξ >> 1. Here mediator generation will be much more rapid than surface adduct decomposition. Three distinct cases are discerned in this slice of the three dimensional case diagram. Again case I (labeled D) corresponding to diffusive substrate transport is located on the left hand side of the diagram bounded by the line
![Sensors 02 00473 i024]()
and the vertical line ζ = 1. Case II (labeled USR) corresponding to unsaturated reaction kinetics, is located in the lower right hand quadrant of the case diagram and is bounded to the west by the line ζ = 1 and to the north by the line v = 1. Finally case IV corresponding to saturated reaction kinetics (labeled SR) is located in the region bounded by the line v = 1 and the line
v =
ζ−1. Here the normalized flux is given by
![Sensors 02 00473 i025]()
which reduces to eqn.63.
We summarise the expressions for the limiting flux for each of the four possible kinetic cases in
table 1 and also delineate the conditions under which each limiting case pertains.

 and for large values of the normalised potential a limiting current response is given by ΨL ≅ η or fΣ,L ≅ kHΓΣs∞ reflecting pure kinetic control. We also note from figure 1 that the half wave potential is a strong function of the competition parameter η , and becomes less positive as the latter parameter increases.
and for large values of the normalised potential a limiting current response is given by ΨL ≅ η or fΣ,L ≅ kHΓΣs∞ reflecting pure kinetic control. We also note from figure 1 that the half wave potential is a strong function of the competition parameter η , and becomes less positive as the latter parameter increases. gets larger we note from eqn.14 that the catalytic efficiency
gets larger we note from eqn.14 that the catalytic efficiency  . Hence for the simple scenario outlined here (Nernstian mediator generation, irreversible cross exchange reaction), it is clear that under all conditions the catalytic efficiency of a monolayer modified electrode will not exceed that obtained for direct mass transport controlled substrate reaction at the unmodified electrode surface. This fact was pointed out by Andrieux and Saveant [14] some time ago.
. Hence for the simple scenario outlined here (Nernstian mediator generation, irreversible cross exchange reaction), it is clear that under all conditions the catalytic efficiency of a monolayer modified electrode will not exceed that obtained for direct mass transport controlled substrate reaction at the unmodified electrode surface. This fact was pointed out by Andrieux and Saveant [14] some time ago. and from eqn.55 obtain the following dimensionless master expression:
and from eqn.55 obtain the following dimensionless master expression:



 . Specifically if u is large then for u >> 1 we note (1+u)2 ≅ u2 and so
. Specifically if u is large then for u >> 1 we note (1+u)2 ≅ u2 and so  and eqn.56 for the normalized flux or catalytic efficiency reduces to:
and eqn.56 for the normalized flux or catalytic efficiency reduces to:





 which represents the balance between substrate diffusion in the diffusion layer and chemical reaction involving the surface bound adducts. Two limiting cases may be enumerated. First, when
which represents the balance between substrate diffusion in the diffusion layer and chemical reaction involving the surface bound adducts. Two limiting cases may be enumerated. First, when  we have the situation of slow rate determining mass transport of substrate in the solution to the modified electrode surface and fast unsaturated kinetics involving substrate and mediator species. Under such conditions eqn.67 reduces to:
we have the situation of slow rate determining mass transport of substrate in the solution to the modified electrode surface and fast unsaturated kinetics involving substrate and mediator species. Under such conditions eqn.67 reduces to:
 , we have the case of facile material transport and slow rate determining chemical reaction between immobilized mediator and substrate. Here eqn.67 reduces to:
, we have the case of facile material transport and slow rate determining chemical reaction between immobilized mediator and substrate. Here eqn.67 reduces to:

 and
and  . Using the latter definitions and the expressions presented in eqn.53 we can show, after some algebra that the Michaelis constant for the modified electrode is related to the Michaelis constant describing the adduct formation process via:
. Using the latter definitions and the expressions presented in eqn.53 we can show, after some algebra that the Michaelis constant for the modified electrode is related to the Michaelis constant describing the adduct formation process via:

 . We can relate the normalized concentration variable
. We can relate the normalized concentration variable  introduced earlier to the latter quantity via:
introduced earlier to the latter quantity via:

 which can be shown to reduce to eqn.62. Here the flux is independent of the bulk substrate concentration and is first order with respect to mediator surface coverage. It will also be potential dependent. Case III is bounded by the line
which can be shown to reduce to eqn.62. Here the flux is independent of the bulk substrate concentration and is first order with respect to mediator surface coverage. It will also be potential dependent. Case III is bounded by the line  and by the horizontal line v = ξ.
and by the horizontal line v = ξ. and the vertical line ζ = 1. Case II (labeled USR) corresponding to unsaturated reaction kinetics, is located in the lower right hand quadrant of the case diagram and is bounded to the west by the line ζ = 1 and to the north by the line v = 1. Finally case IV corresponding to saturated reaction kinetics (labeled SR) is located in the region bounded by the line v = 1 and the line v = ζ−1. Here the normalized flux is given by
and the vertical line ζ = 1. Case II (labeled USR) corresponding to unsaturated reaction kinetics, is located in the lower right hand quadrant of the case diagram and is bounded to the west by the line ζ = 1 and to the north by the line v = 1. Finally case IV corresponding to saturated reaction kinetics (labeled SR) is located in the region bounded by the line v = 1 and the line v = ζ−1. Here the normalized flux is given by  which reduces to eqn.63.
which reduces to eqn.63.




 . Now if we recall the definitions of kME and KME presented in eqn.53 we can readily show that
. Now if we recall the definitions of kME and KME presented in eqn.53 we can readily show that  and kMEKME = kCΓΣ. Hence eqn.75 reduces to the expression for the net reaction flux used by Gorton and co-workers, but recast in the terminology adopted in the present paper:
and kMEKME = kCΓΣ. Hence eqn.75 reduces to the expression for the net reaction flux used by Gorton and co-workers, but recast in the terminology adopted in the present paper:


 . We let
. We let  . Clearly T ≤ 1. We also set
. Clearly T ≤ 1. We also set  . Hence eqn.76 transforms to
. Hence eqn.76 transforms to  and eqn.52 reduces to
and eqn.52 reduces to  . Hence the flux ratio is given by:
. Hence the flux ratio is given by:

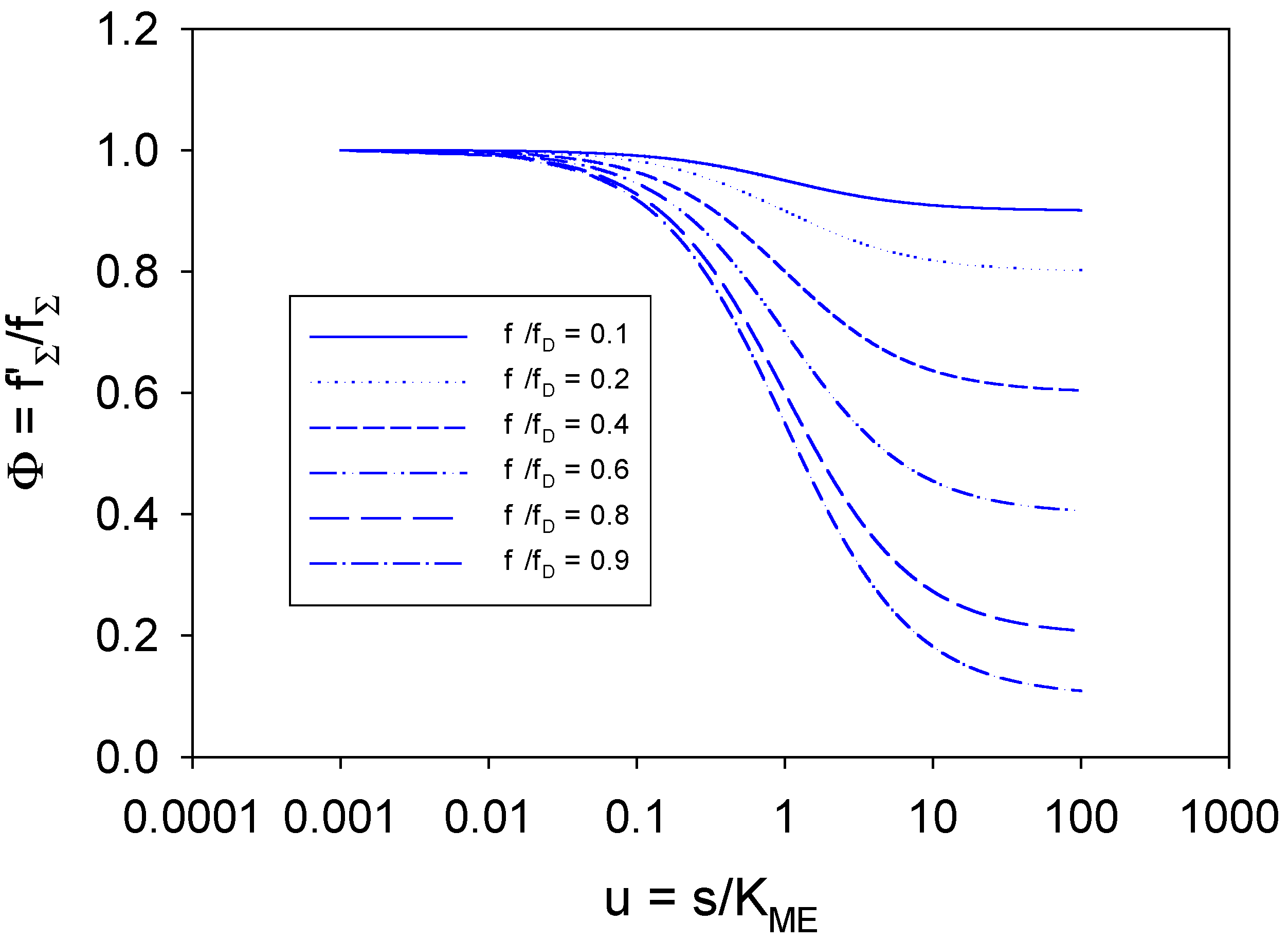
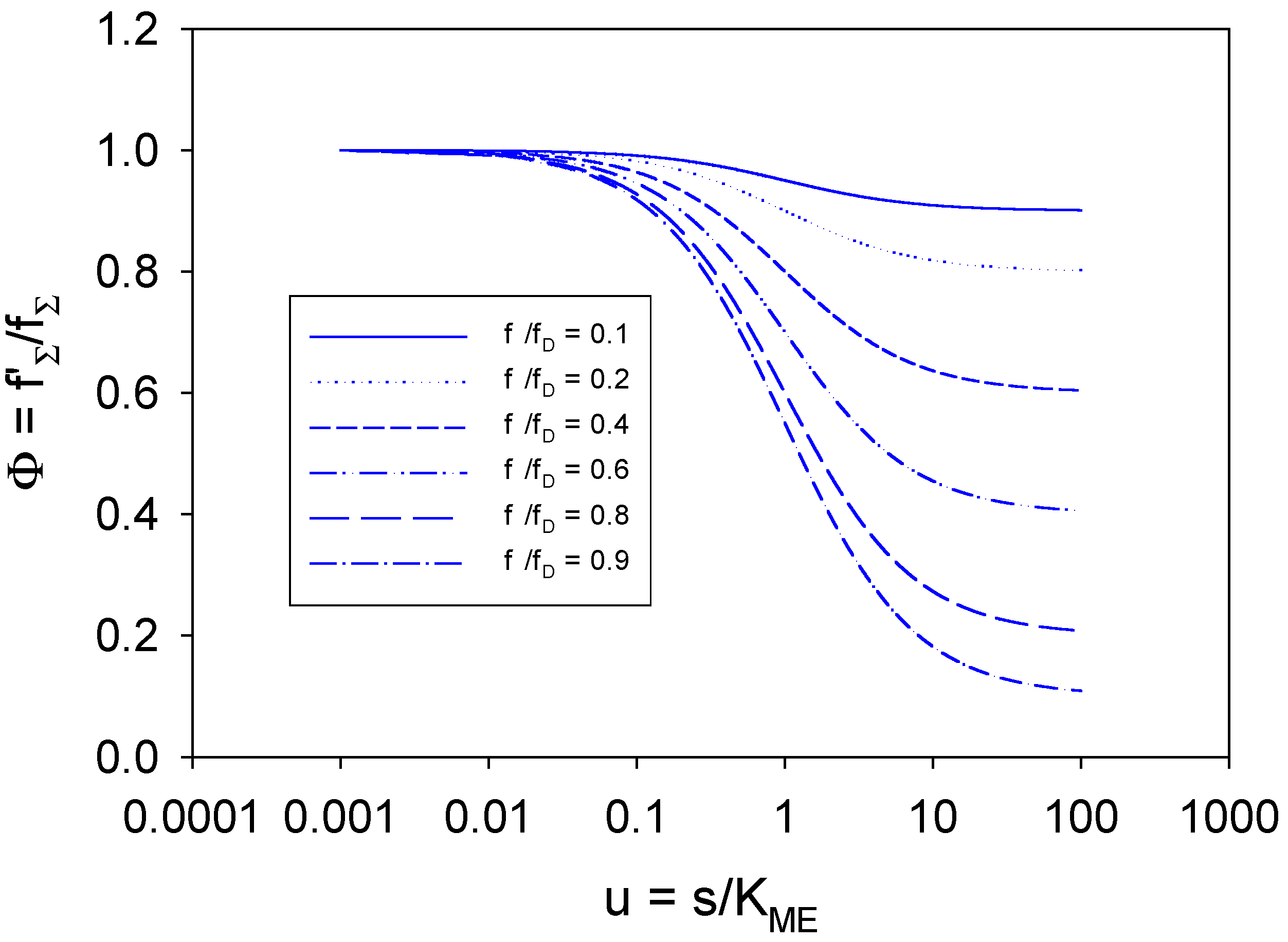
 in the range 0.1-0.9.
in the range 0.1-0.9. . When u << 1 then Φ ≅ 1 + Tu ≅ 1 when Tu << 1. Under these circum-stances the Gorton approximation will be valid and the observed current will be much less than the mass transport controlled value. We note from figure 13 that the Φ function deviates appreciably from unity and decreases considerably as the saturation parameter u increases in value. The deviation is more marked for larger values of the ratio
. When u << 1 then Φ ≅ 1 + Tu ≅ 1 when Tu << 1. Under these circum-stances the Gorton approximation will be valid and the observed current will be much less than the mass transport controlled value. We note from figure 13 that the Φ function deviates appreciably from unity and decreases considerably as the saturation parameter u increases in value. The deviation is more marked for larger values of the ratio  and the magnitude of the T factor will determine the flux function Φ . Under these conditions the Gorton expression for the reaction flux will not be a good representation to use for the analysis of experimental data. When u is very close to unity
and the magnitude of the T factor will determine the flux function Φ . Under these conditions the Gorton expression for the reaction flux will not be a good representation to use for the analysis of experimental data. When u is very close to unity  . Now we note that the flux ratio function Φ deviates from unity when the parameter u >> 1. We can show that this condition reduces to
. Now we note that the flux ratio function Φ deviates from unity when the parameter u >> 1. We can show that this condition reduces to  . As previously noted the Gorton analysis neglects concentration polarization effects and using this analysis will result in the computation of artificially high values of the Michaelis constant KM for substrate binding and for the catalytic rate constant kC for adduct decomposition. In the Gorton analysis the implicit assumption is that the redox mediator is exposed to the bulk concentration of the substrate (refer to eqn.73), and neglects the significant concentration gradient of substrate which can arise if the flux approaches that characteristic of the mass transport controlled value.
. As previously noted the Gorton analysis neglects concentration polarization effects and using this analysis will result in the computation of artificially high values of the Michaelis constant KM for substrate binding and for the catalytic rate constant kC for adduct decomposition. In the Gorton analysis the implicit assumption is that the redox mediator is exposed to the bulk concentration of the substrate (refer to eqn.73), and neglects the significant concentration gradient of substrate which can arise if the flux approaches that characteristic of the mass transport controlled value.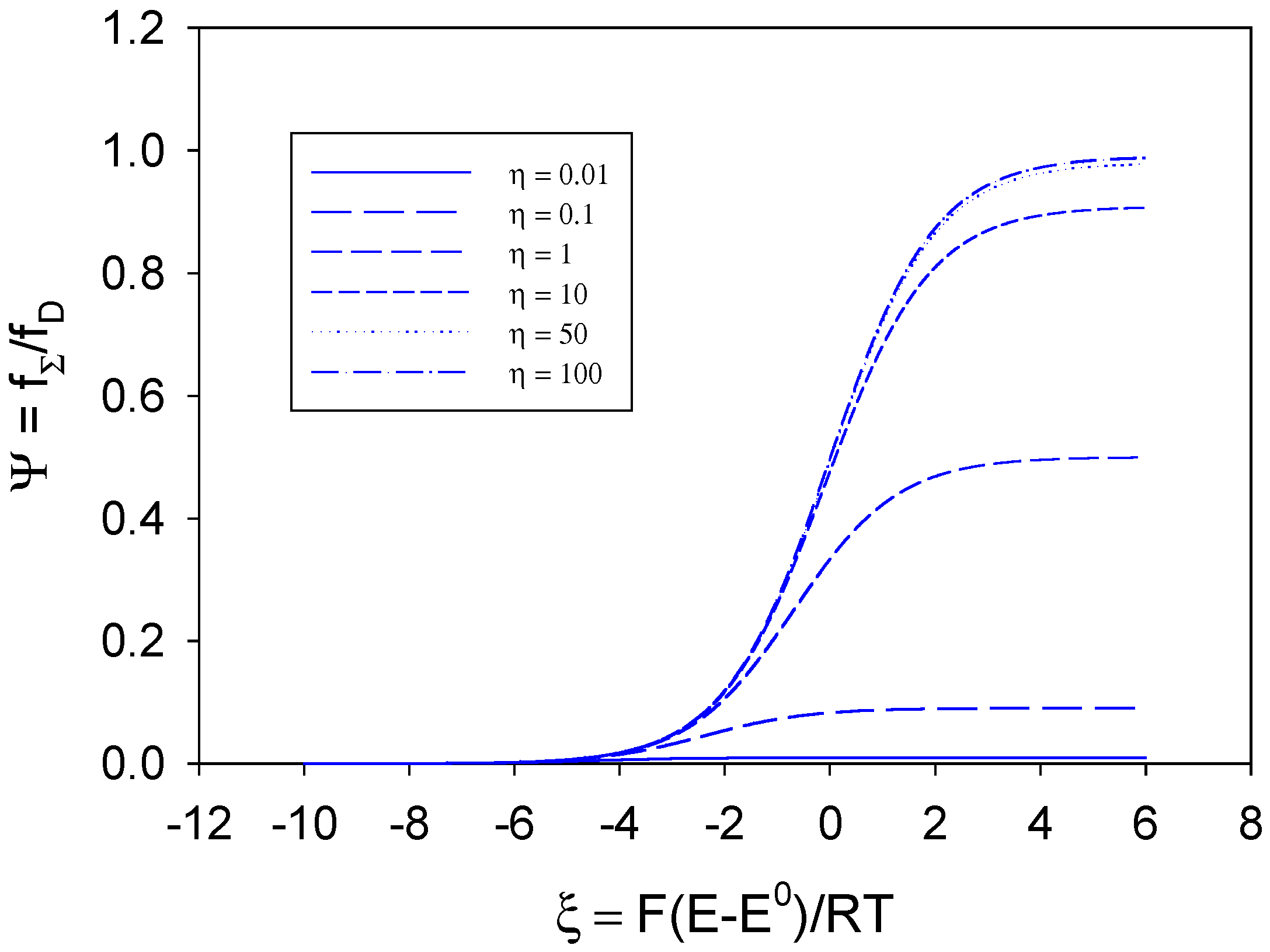
 with u.
with u.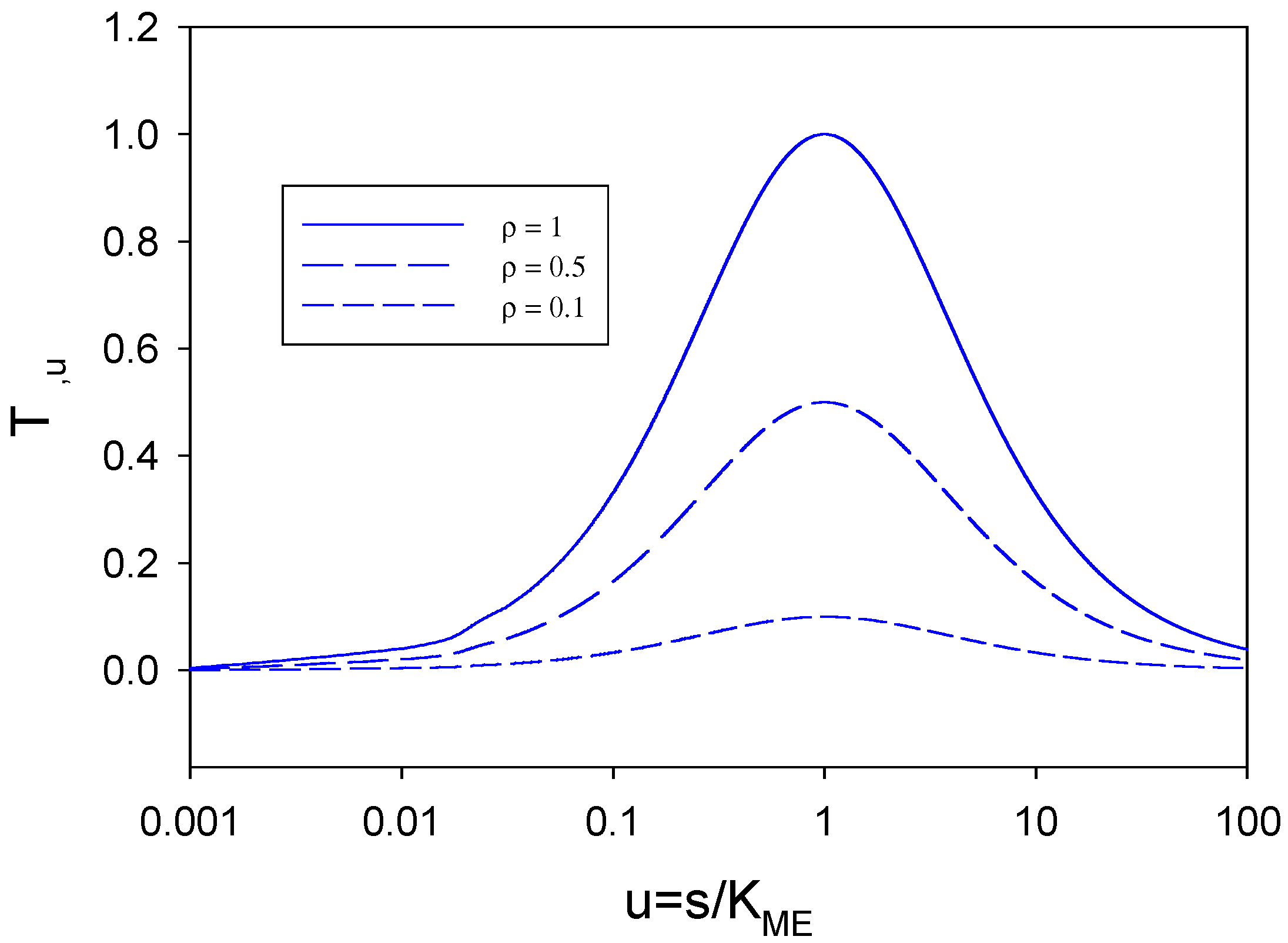

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




