Mediated Electron Transfer at Redox Active Monolayers Part 2 : Analysis of the Chronoamperometric Response to a Potential Step Perturbation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction
Chronoamperometric Response of Surface Immobilized Redox Groups in the Absence of Solution Phase Substrate


 . The current response to the applied potential step is given by
. The current response to the applied potential step is given by




 . We can integrate eqn.6 using the initial condition t = 0 ΓA = ΓΣ ΓB = 0 to obtain
. We can integrate eqn.6 using the initial condition t = 0 ΓA = ΓΣ ΓB = 0 to obtain

 . At any time t we also note that ΓA (t) + ΓB(t) = ΓΣ and so we also obtain that
. At any time t we also note that ΓA (t) + ΓB(t) = ΓΣ and so we also obtain that



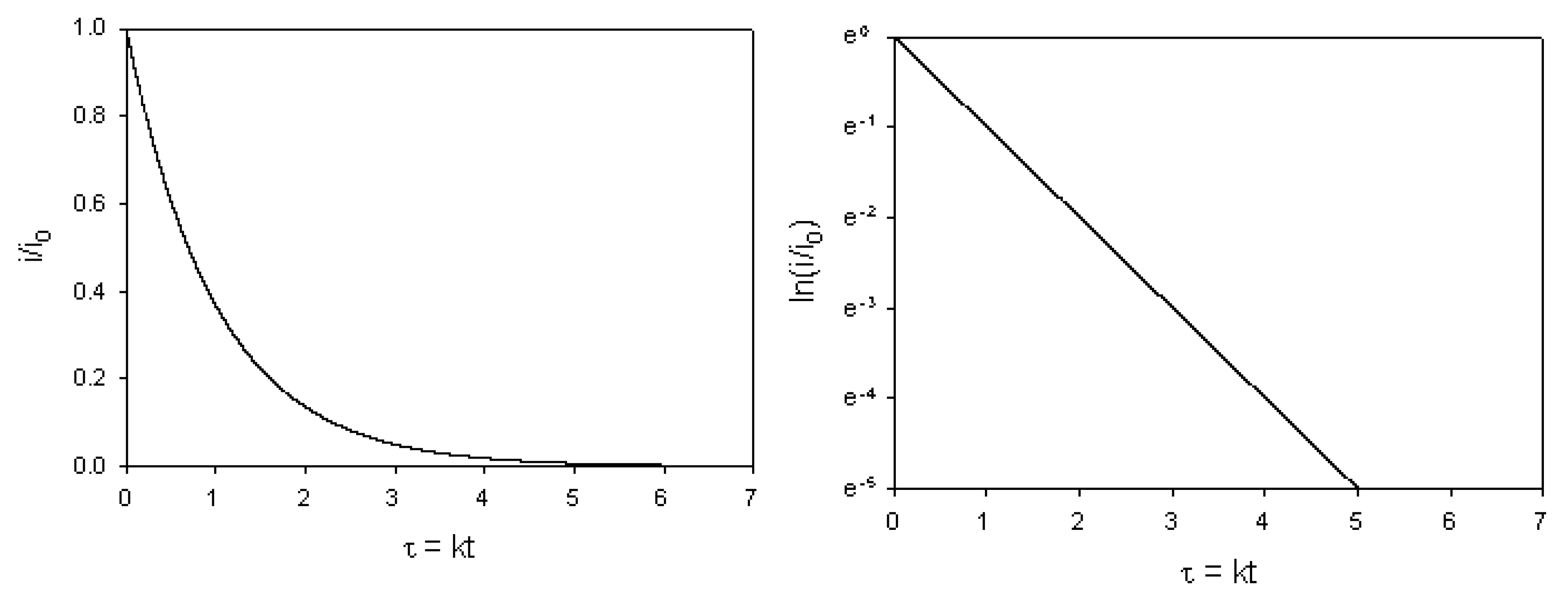
 and a slope given by
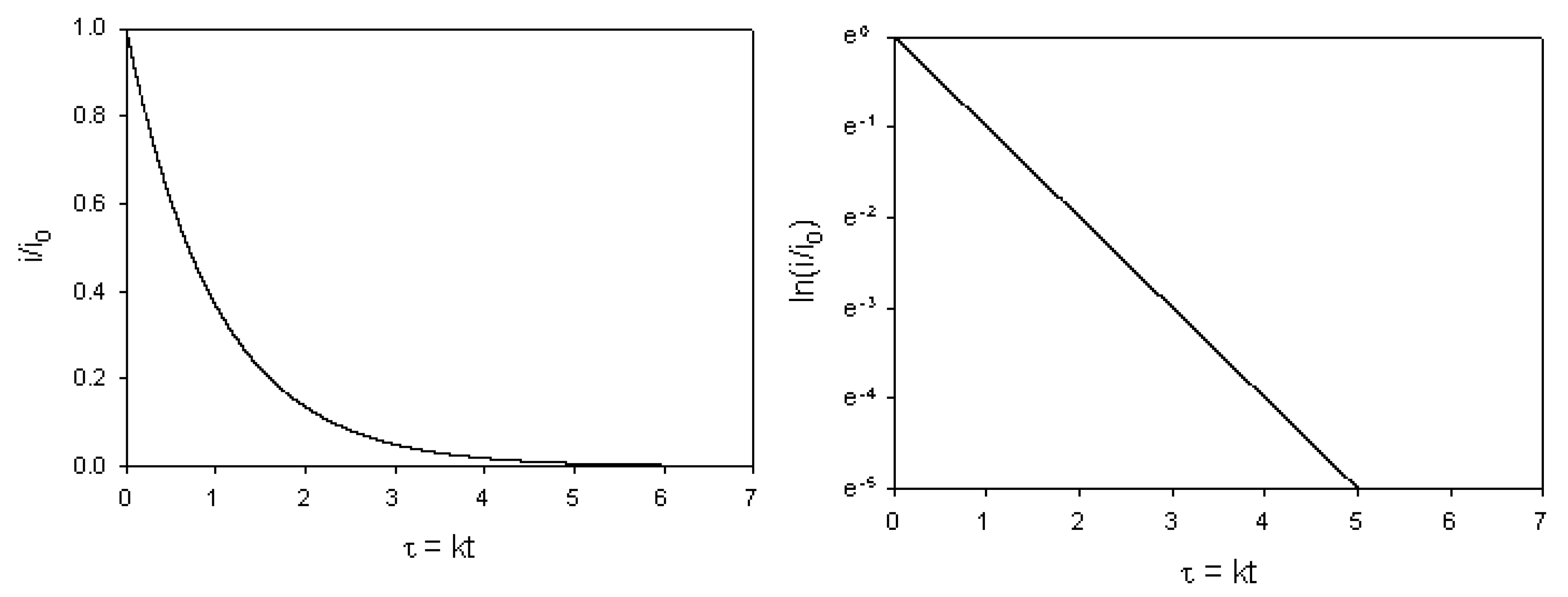
and a slope given by  . Hence the surface redox kinetics can be completely resolved using the potential step technique.
. Hence the surface redox kinetics can be completely resolved using the potential step technique.

Chronoamperometric Response of Surface Immobilized Redox Groups in the Presence of Solution Phase Substrate

Irreversible Mediated Electron Transfer with Nernstian Mediator Generation





 . Hence in normalized variables the Fick diffusion equation becomes
. Hence in normalized variables the Fick diffusion equation becomes



 .
.
 is the normalized substrate concentration in Laplace space and p denotes the Laplace parameter. The boundary conditions in Laplace space adopt the following representation
is the normalized substrate concentration in Laplace space and p denotes the Laplace parameter. The boundary conditions in Laplace space adopt the following representation


 must be finite as χ → ∞ then clearly A=0. Differentiation of eqn.25 with respect to χ and using the second relation in eqn.23 we obtain that
must be finite as χ → ∞ then clearly A=0. Differentiation of eqn.25 with respect to χ and using the second relation in eqn.23 we obtain that  . Noting that
. Noting that  , and the requirement that at χ = 0
, and the requirement that at χ = 0  , we can readily show that
, we can readily show that  . Consequently we note that
. Consequently we note that  .
.





 . We note however that the heterogeneous electron transfer rate constant k’ increases exponentially with increasing potential ξ according to the Butler-Volmer law, unless rate determining diffusive transport becomes rate limiting, whereas the composite term kHΓB → kHΓΣ as ξ → ∞. We can readily show that the current response as a function of time is given by
. We note however that the heterogeneous electron transfer rate constant k’ increases exponentially with increasing potential ξ according to the Butler-Volmer law, unless rate determining diffusive transport becomes rate limiting, whereas the composite term kHΓB → kHΓΣ as ξ → ∞. We can readily show that the current response as a function of time is given by









 and an intercept given by the reaction/diffusion parameter γ. Hence analysis of the chronoamperometric data at short times can produce kinetic information.
and an intercept given by the reaction/diffusion parameter γ. Hence analysis of the chronoamperometric data at short times can produce kinetic information.


 and
and  and so the γ parameter is potential dependent given by
and so the γ parameter is potential dependent given by  where we recall that the normalized potential is given by
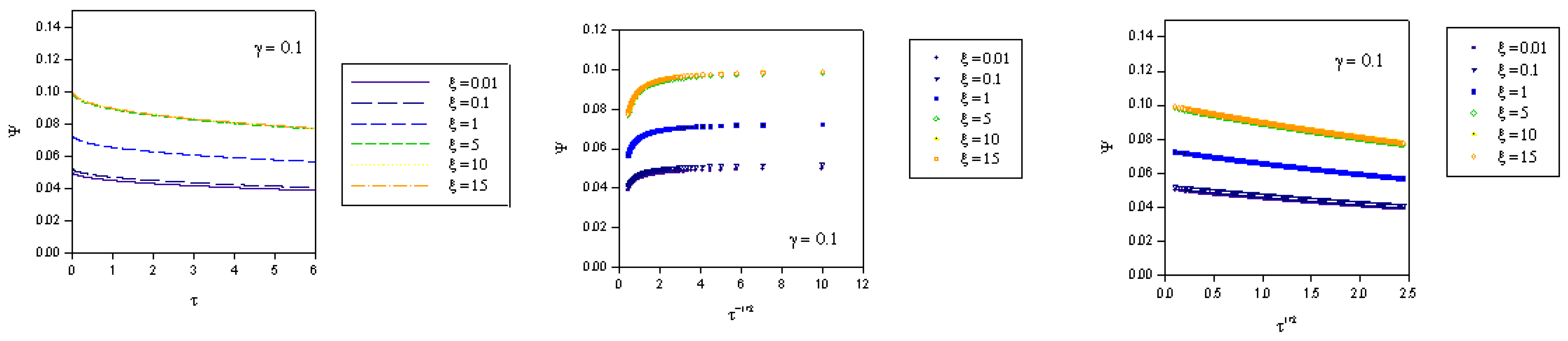
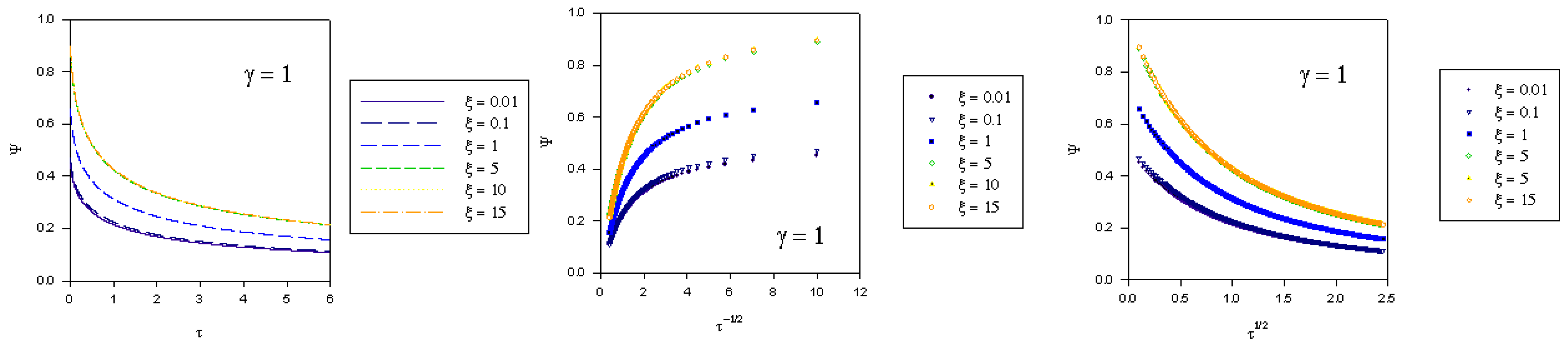
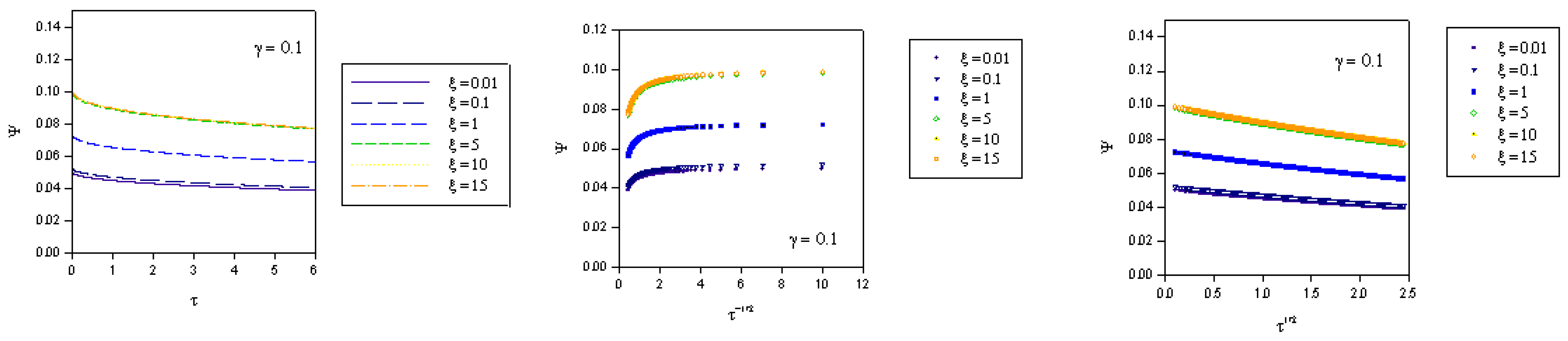
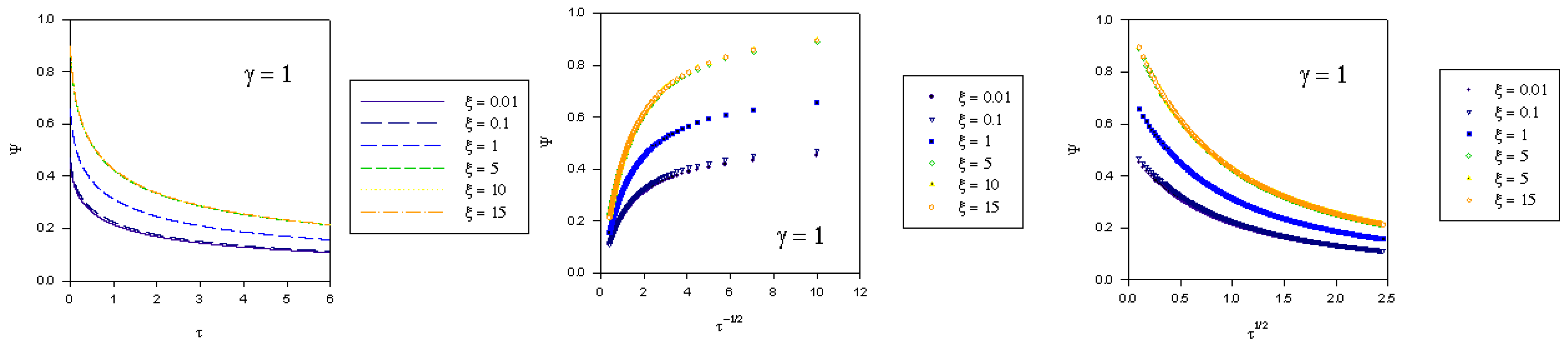
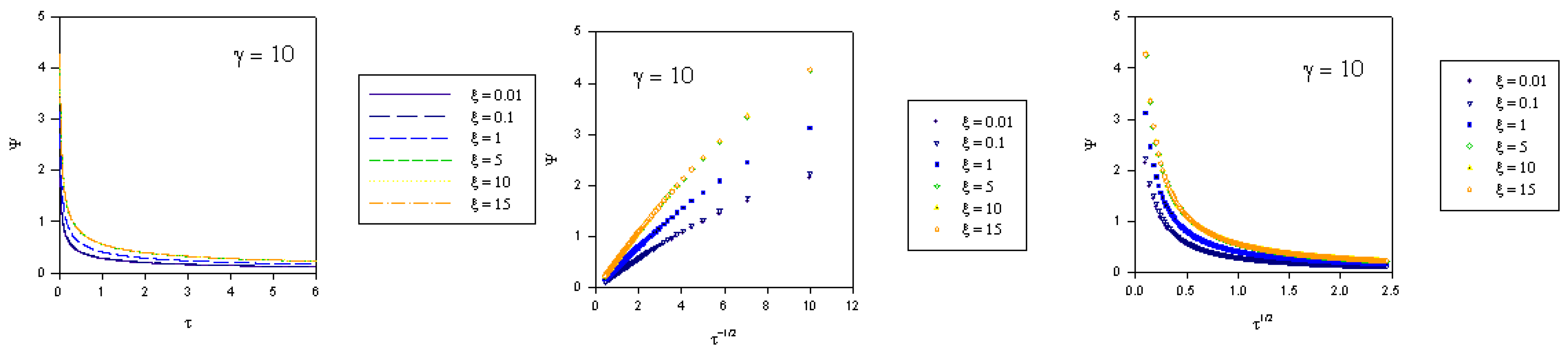
where we recall that the normalized potential is given by  . In the computations presented in figure 2, figure 3 and figure 4 the γ values quoted correspond to the quantity
. In the computations presented in figure 2, figure 3 and figure 4 the γ values quoted correspond to the quantity  .
. where fK denotes the kinetic flux arising from reaction between immobilized mediator species B and substrate S and fD represents the flux due to diffusive material transport of substrate to the site of reaction on the surface of the redox monolayer. When γ is small (as in figure 2), substrate diffusion is more rapid than heterogeneous kinetics. Hence the data when displayed in the Cottrell format will be non linear over an extensive period of the experimental timescale. Linear Cottrell behaviour will only be observed at the longest times. In contrast a plot of Ψ versus τ1/2 will exhibit good linearity over an extended timescale. This is in accord with the approximate expression presented in eqn.40 which holds when the parameter λ = γ2τ is small . If γ is small enough then λ will be small over an extended period of the normalized time τ. A well defined intercept is obtained on the Ψ axis of the plot , the magnitude of which will depend on the value of the normalized potential ξ , since we recall that
where fK denotes the kinetic flux arising from reaction between immobilized mediator species B and substrate S and fD represents the flux due to diffusive material transport of substrate to the site of reaction on the surface of the redox monolayer. When γ is small (as in figure 2), substrate diffusion is more rapid than heterogeneous kinetics. Hence the data when displayed in the Cottrell format will be non linear over an extensive period of the experimental timescale. Linear Cottrell behaviour will only be observed at the longest times. In contrast a plot of Ψ versus τ1/2 will exhibit good linearity over an extended timescale. This is in accord with the approximate expression presented in eqn.40 which holds when the parameter λ = γ2τ is small . If γ is small enough then λ will be small over an extended period of the normalized time τ. A well defined intercept is obtained on the Ψ axis of the plot , the magnitude of which will depend on the value of the normalized potential ξ , since we recall that  .
.
Irreversible Mediated Electron Transfer Coupled with Irreversible Heterogeneous Electron Transfer Kinetics


 , γ' is given by a modified form of eqn.21 and is represented as
, γ' is given by a modified form of eqn.21 and is represented as  , and the heterogeneous rate constants are of the Butler-Volmer form
, and the heterogeneous rate constants are of the Butler-Volmer form  . We have also introduced a further competition parameter ζ which relates the flux associated with mediator generation with the substrate diffusion flux. This parameter is given by
. We have also introduced a further competition parameter ζ which relates the flux associated with mediator generation with the substrate diffusion flux. This parameter is given by  .
. and the normalized time
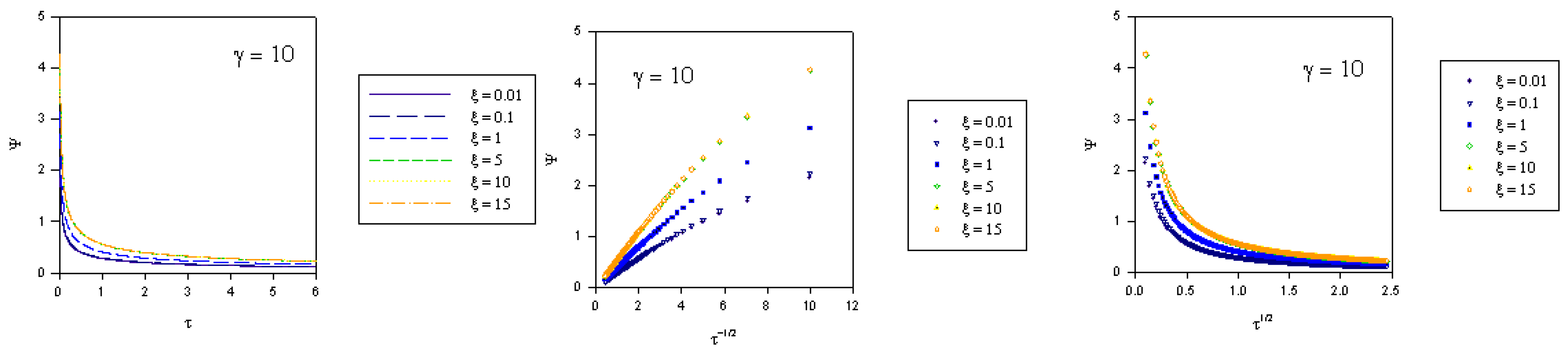
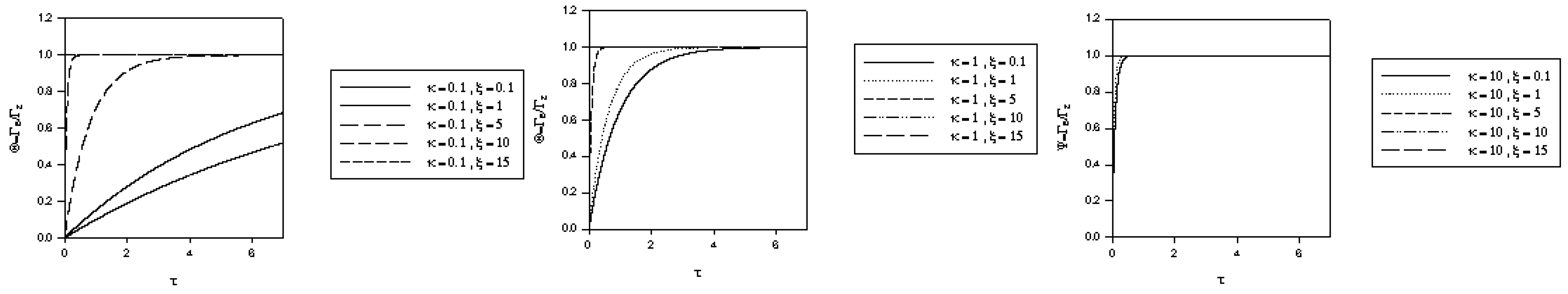
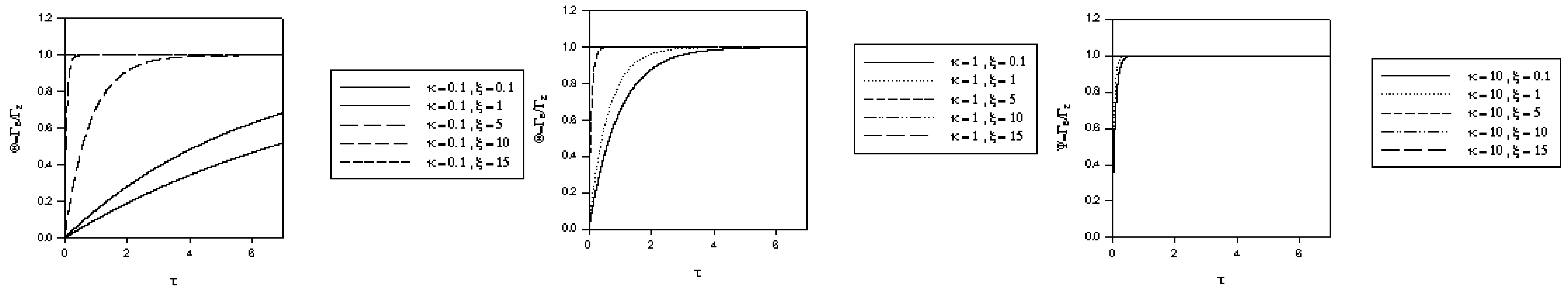
and the normalized time  . The variation of normalized surface coverage of mediator species with time, computed from eqn.46 is presented in figure 5, for various values of the competition parameter κ and the normalized potential ξ. It is clear from the latter computation that θ → 1 rather rapidly when either κ increases or ξ increases.
. The variation of normalized surface coverage of mediator species with time, computed from eqn.46 is presented in figure 5, for various values of the competition parameter κ and the normalized potential ξ. It is clear from the latter computation that θ → 1 rather rapidly when either κ increases or ξ increases. which is the same as that used in eqn.20 when Nernstian mediator generation was discussed. We can use the expression for u0 developed in our previous analysis and hence we recall from eqn.30 that
which is the same as that used in eqn.20 when Nernstian mediator generation was discussed. We can use the expression for u0 developed in our previous analysis and hence we recall from eqn.30 that  , which on application of the inverse Laplace Transformation results in the following expression for the normalized current response:
, which on application of the inverse Laplace Transformation results in the following expression for the normalized current response:

Discussion
References
- Lyons, M.E.G. Transport and kinetics in electroactive polymers. Adv. Chem. Phys. 1996, 94, 297–624. [Google Scholar]
- Gorton, L. Chemically modified electrodes for the electrocatalytic oxidation of nicotinamide coenzymes. J. Chem. Soc., Faraday Trans. I 1986, 82, 1245–1258. [Google Scholar] [CrossRef]Gorton, L.; Torstensson, A.; Jaegfeldt, H.; Johansson, G. Electrocatalytic oxidation of reduced nicotinamide coenzymes by graphite electrodes modified with an adsorbed phenoxazinium salt, medola blue. J. Electroanal. Chem. 1984, 161, 103–120. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; Fitzgerald, C.A.; Smyth, M.R. Glucose oxidation at ruthenium dioxide based electrodes. Analyst 1994, 119, 855–861. [Google Scholar] [CrossRef]Lyons, M.E.G.; Lyons, C.H.; Michas, A.; Bartlett, P.N. Heterogeneous redox catalysis at hydrated oxide layers. J. Electroanal. Chem. 1993, 351, 245–258. [Google Scholar] [CrossRef]
- The electrochemistry of alkanethiol self assembled monolayers has been the subject of huge research activity in recent years. The following citations are representative of recent publications of interest in electroanalytical chemistry. Gooding, J.J.; Hibbert, D.B. The application of alkanethiol self assembled monolayers to enzyme electrodes. Trends in Analytical Chemistry 1999, 18, 525–533. [Google Scholar] [CrossRef]Gooding, J.J.; Erokhin, P.; Losic, D.; Yang, W.; Policarpio, V.; Liu, J.; Ho, F.M.; Situmorang, M.; Hibbert, D.B.; Shapter, J.G. Parameters important in fabricating enzyme electrodes using self assembled monolayers of alkanethiols. Analytical Sciences 2001, 17, 3–9. [Google Scholar] [CrossRef] [PubMed]Gooding, J.J.; Erokhin, P.; Hibbert, D.B. Parameters important in tuning the response of monolayer enzyme electrodes fabricated using self-assembled monolayers of alkanethiols. Biosensors and Bioelectronics. 2000, 15, 229–239. [Google Scholar] [CrossRef]Alleman, K.S.; Weber, K.; Creager, S.E. Electrochemical rectification at a monolayer modified electrode. J. Phys. Chem. 1996, 100, 17050–17058. [Google Scholar] [CrossRef]Retna Raj, C.; Ohsaka, T. Electrocatalytic sensing of NADH at an in situ functionalized self assembled monolayer on a gold electrode. Electrochemistry Communications 2001, 3, 633–638. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Saveant, J.M. Heterogeneous versus homogeneous catalysis of electrochemical reactions. J. Electroanal. Chem. 1978, 93, 163–168. [Google Scholar] [CrossRef]
- Laviron, E. Electron transfer mediated by redox polymer electrodes. Theory for rotating disc voltammetry with transfer at the film/solution interface and reversible electrode coating reaction. J. Electroanal. Chem. 1982, 131, 61–75. [Google Scholar] [CrossRef]
- Aoki, K.; Tokuda, K.; Matsuda, H. Linear sweep and cyclic voltammetry for electrocatalysis at modified electrodes with very thin films. J. Electroanal. Chem. 1986, 199, 69–79. [Google Scholar] [CrossRef]
- Xie, Y.; Anson, F.C. Analysis of the cyclic voltammetric responses exhibited by electrodes modified with monolayers of catalysts in the absence and presence of substrates. J. Electroanal. Chem. 1995, 384, 145–153. [Google Scholar] [CrossRef]
- Xie, Y.; Anson, F.C. Calculation of cyclic voltammetric responses for the reduction of substrates by an inner sphere mechanism at electrode surfaces with single monolayers of catalysts. J. Electroanal. Chem. 1995, 396, 441–449. [Google Scholar] [CrossRef]
- Xie, Y.; Anson, F.C. Calculation of cyclic voltammetric responses for the catalytic reduction of substrates via intramolecular electron transfer within a catalyst/substrate complex formed on electrode surfaces. J. Electroanal. Chem. 1996, 404, 209–213. [Google Scholar] [CrossRef]
- Lyons, M.E.G. Mediated electron transfer at redox active monolayers. Sensors. 2001, 1, 215–228. [Google Scholar] [CrossRef]
- Rocklin, R.D.; Murray, R.W. Kinetics of electrocatalysis of dibromoalkyl reductions using electrodes with covalently immobilized metallotetraphenylporphyrins. J. Phys. Chem. 1981, 85, 2104–2112. [Google Scholar] [CrossRef]
- Finklea, H.O.; Liu, L.; Ravenscroft, M.S.; Punturi, S. Multiple electron tunneling paths across self-assembled monolayers of alkanethiols with attached ruthenium(II/III) redox centers. J. Phys. Chem. 1996, 100, 18852–18858. [Google Scholar] [CrossRef]Finklea, H.O. Consequences of a potential dependent transfer coefficient in ac voltammetry and in coupled electron-proton transfer for attached redox couples. J. Electroanal. Chem. 2001, 495, 79–86. [Google Scholar] [CrossRef]Finklea, H.O.; Yoon, K.; Chamberlain, E.; Allen, J.; Haddox, R. Effect of the metal on electron transfer across self-assembled monolayers. J. Phys. Chem. B. 2001, 105, 3088–3092. [Google Scholar] [CrossRef]
- Chidsey, C.E.D. Free energy and temperature dependence of electron transfer at the metal-electrolyte interface. Science 1991, 251, 919–922. [Google Scholar] [CrossRef] [PubMed]Chidsey, C.E.D.; Bertozzi, C.R.; Putvinski, T.M.; Mujsce, A.M. Coadsorption of ferrocene-terminated and unsubstituted alkanethiols on gold: electroactive self assembled monolayers. J. Am. Chem. Soc. 1990, 112, 4301–4306. [Google Scholar] [CrossRef]
- Miller, C.; Cuendet, P.; Gratzel, M. Adsorbed ω- hydroxy thiol monolayers on gold electrodes: evidence for electron tunneling to redox species in solution. J. Phys. Chem. 1991, 95, 877–886. [Google Scholar] [CrossRef]Miller, C.; Gratzel, M. Electrochemistry at ω-hydroxy thiol coated electrodes.2. Measurement of the density of electronic states distributions for several outer-sphere redox couples. J. Phys. Chem. 1991, 95, 5225–5233. [Google Scholar] [CrossRef]Becka, A.M.; Miller, C.J. Electrochemistry at ω-hydroxy thiol coated electrodes. 3. Voltage independence of the electron tunneling barrier and measurements of redox kinetics at large overpotentials. J. Phys. Chem. 1992, 96, 2657–2668. [Google Scholar] [CrossRef]
- Forster, R.J.; Faulkner, L.R. Electrochemistry of spontaneously adsorbed monolayers. Equilibrium properties and fundamental electron transfer characteristics. J. Am. Chem. Soc. 1994, 116, 5444–5452. [Google Scholar] [CrossRef]Forster, R.J.; Faulkner, L.R. Electrochemistry of spontaneously adsorbed monolayers. Effects of solvent, potential and temperature on electron transfer dynamics. J. Am. Chem. Soc. 1994, 116, 5453–5461. [Google Scholar] [CrossRef]Forster, R.J.; O’Kelly, J.P. pH modulated heterogeneous electron transfer across metal/monolayer interfaces. J. Phys. Chem. 1996, 100, 3695–3704. [Google Scholar] [CrossRef]Forster, R.J.; Keyes, T.E. Tetrazine bridged osmium dimmers: electrochemical vs photoinduced electron transfer. J. Phys. Chem. B. 2001, 105, 8829–8837. [Google Scholar] [CrossRef]
- Finklea, H.O.; Hanshew, D.D. Electron transfer kinetics in organized thiol monolayers with attached pentaammine(pyridine)ruthenium redox centers. J. Am. Chem. Soc. 1992, 114, 3173–3181. [Google Scholar] [CrossRef]
- Nahir, T.M.; Bowden, E.F. The distribution of standard rate constants for the electron transfer between thiol-modified gold electrodes and adsorbed cytochrome c. J. Electroanal. Chem. 1996, 410, 9–13. [Google Scholar] [CrossRef]Nahir, T.M.; Clark, R.A.; Bowden, E.F. Linear sweep voltammetry of irreversible electron transfer in surface confined species using the Marcus theory. Anal. Chem. 1994, 66, 2595–2598. [Google Scholar] [CrossRef]
- Marcus, R.A. Symmetry or asymmetry of kET and iSTM vs. potential curves. J. Chem. Soc., Faraday Trans. 1996, 92, 3905–3908. [Google Scholar] [CrossRef]Gosavi, S.; Gao, Y.Q.; Marcus, R.A. Temperature dependence of the electronic factor in the nonadiabatic electron transfer at metal and semiconductor electrodes. J. Electroanal. Chem. 2001, 500, 71–77. [Google Scholar] [CrossRef]Gosavi, S.; Marcus, R.A. Nonadiabatic electron transfer at metal surfaces. J. Phys. Chem. B. 2000, 104, 2067–2072. [Google Scholar] [CrossRef]Hsu, C.P. Application of the sequential formula: the electronic coupling and the distance dependence in the electron transfer of ferrocene-terminated alkanethiol monolayers. J. Electroanal. Chem. 1997, 438, 27–35. [Google Scholar] [CrossRef]Hsu, C.P.; Marcus, R.A. A sequential formula for electronic coupling in long range bridge assisted electron transfer: Formulation of theory and application to alkanethiol monolayers. J. Chem. Phys. 1997, 106, 584–598. [Google Scholar] [CrossRef]Ratner, M.A. Bridge assisted electron transfer: effective electronic coupling. J. Phys. Chem. 1990, 94, 4877–4883. [Google Scholar] [CrossRef]
- Xu, J.; Li, H-L.; Zhang, Y. Relationship between electronic tunneling coefficient and electrode potential investigated using self assembled alkanethiol monolayers on gold electrodes. J. Phys. Chem. 1993, 97, 11497–11500. [Google Scholar] [CrossRef]Arnold, S.; Feng, Z.Q.; Kakiuchi, T.; Knoll, W.; Niki, K. Investigation of the electrode reaction of cytochrome c through mixed self-assembled monolayers of alkanethiols on gold (111) surfaces. J. Electroanal. Chem. 1997, 438, 91–97. [Google Scholar] [CrossRef]Khoshtariya, D.E.; Dolidze, T.D.; Zusman, L.D.; Waldeck, D.H. Observation of the turnover between the solvent friction (overdamped) and tunneling (nonadiabatic) charge transfer mechanisms for a Au/Fe(CN)63-/4- electrode process and evidence for a freezing out of the Marcus barrier. J. Phys. Chem. A. 2001, 105, 1818–1829. [Google Scholar] [CrossRef]Yang, D.; Zi, M.; Chen, B.; Gao, Z. Separation of pinhole and tunneling electron transfer processes at self assembled polymeric monolayers on gold electrodes. J. Electroanal. Chem. 1999, 470, 114–119. [Google Scholar] [CrossRef]Kornyshev, A.A.; Kuznetsov, A.M.; Stimming, U.; Ulstrup, J. Rate processes in interfacial systems near continuous phase transitions. J. Phys. Chem. 1996, 100, 11184–11192. [Google Scholar] [CrossRef]
- Creager, S.E.; Wooster, T.T. A new way of using ac voltammetry to study redox kinetics in electroactive monolayers. Anal. Chem. 1998, 70, 4257–4263. [Google Scholar] [CrossRef]O’Connor, S.D.; Olsen, G.T.; Creager, S.E. A Nernstian electron source model for the ac voltammetric response of a reversible surface redox reaction using large amplitude ac voltages. J. Electroanal. Chem. 1999, 466, 197–202. [Google Scholar] [CrossRef]Sumner, J.J.; Creager, S.E. Redox kinetics in monolayers on electrodes: electron transfer is sluggish for ferrocene groups buried within the monolayer interior. J. Phys. Chem. B. 2001, 105, 8739–8745. [Google Scholar] [CrossRef](d) Creager, S.; Yu, C.J.; Bamdad, C.; O’Connor, S.; Maclean, T.; Lam, E.; Chong, Y.; Olsen, G.T.; Luo, J.; Gozin, M.; Kayyem, J.F. Electron transfer at electrodes through conjugated ‘molecular wire’ bridges. J. Am. Chem. Soc. 1999, 121, 1059–1064. [Google Scholar] [CrossRef]Sumner, J.J.; Creager, S.E. Topological effects in bridge mediated electron transfer between redox molecules and metal electrodes. J. Am. Chem. Soc. 2000, 122, 11914–11920. [Google Scholar] [CrossRef]Sumner, J.J.; Weber, K.S.; Hockett, L.A.; Creager, S.E. Long range heterogeneous electron transfer between ferrocene and gold mediated by ω-alkane and ω-alkyl-carboxamide bridges. J. Phys. Chem. B. 2000, 104, 7449–7454. [Google Scholar] [CrossRef]
- Brown, A.P.; Anson, F.C. Cyclic and differential pulse voltammetric behaviour of reactants confined to the electrode surface. Anal. Chem 1977, 49, 1589–1595. [Google Scholar] [CrossRef]
- Albery, W.J.; Boutelle, M.G.; Colby, P.J.; Hillman, R.A. The kinetics of electron transfer in the thionine coated electrode. J. Electroanal. Chem. 1982, 133, 135–145. [Google Scholar] [CrossRef]
- Laviron, E. A multilayer model for the study of space distributed redox modified electrodes. Part 3. Influence of the interactions between the electroactive centers in the first layer on the linear potential sweep voltammograms. J. Electroanal. Chem. 1981, 122, 37–44. [Google Scholar] [CrossRef]
- Daifuku, H.; Aoki, K.; Tokuda, K.; Matsuda, H. Electrode kinetics of surfactant polypyridine osmium and ruthenium complexes confined to tin oxide electrodes in a monomolecular layer by the Langmuir-Blodgett method. J. Electroanal. Chem. 1985, 183, 1–26. [Google Scholar] [CrossRef]
- Matsuda, H.; Aoki, K.; Tokuda, K. Theory of electrode reactions of redox couples confined to electrode surfaces at monolayer levels. Part 1. Expression of the current-potential relationship for simple redox reactions. J. Electroanal. Chem. 1987, 217, 1–13. [Google Scholar] [CrossRef]Matsuda, H.; Aoki, K.; Tokuda, K. Theory of electrode reactions of redox couples confined to electrode surfaces at monolayer levels. Part 2. Cyclic voltammetry and ac impedance measurements. J. Electroanal. Chem. 1987, 217, 15–32. [Google Scholar] [CrossRef]
- (a)Oldham, K.B.; Myland, J.C. Fundamentals of electrochemical science; Academic Press: New York, 1994; Chapter 11; pp. 413–414. [Google Scholar](b)Bard, A.J.; Faulkner, L.R. Electrochemical methods: fundamentals and applications; 2nd edition; 2001; Wiley: New York, Chapter 5; pp. 191–197. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical methods: fundamentals and applications; 2nd edition; 2001; Wiley: New York, Chapter 5; pp. 162–163. [Google Scholar]
- Spanier, J.; Oldham, K.B. An atlas of functions; 1987; Hemisphere Publishing Corporation: Washington, Chapter 41; pp. 395–403. [Google Scholar]
- Sample Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.net). Reproduction is permitted for noncommercial purposes.
Share and Cite
Lyons, M.E.G. Mediated Electron Transfer at Redox Active Monolayers Part 2 : Analysis of the Chronoamperometric Response to a Potential Step Perturbation. Sensors 2002, 2, 314-330. https://doi.org/10.3390/s20800314
Lyons MEG. Mediated Electron Transfer at Redox Active Monolayers Part 2 : Analysis of the Chronoamperometric Response to a Potential Step Perturbation. Sensors. 2002; 2(8):314-330. https://doi.org/10.3390/s20800314
Chicago/Turabian StyleLyons, Michael E.G. 2002. "Mediated Electron Transfer at Redox Active Monolayers Part 2 : Analysis of the Chronoamperometric Response to a Potential Step Perturbation" Sensors 2, no. 8: 314-330. https://doi.org/10.3390/s20800314