Electrochemical Study of Iodide in the Presence of Phenol and o-Cresol: Application to the Catalytic Determination of Phenol and o-Cresol

Abstract
:Introduction
Experimental
Reagents and solutions
Apparatus
Analytical procedure for calibration curves
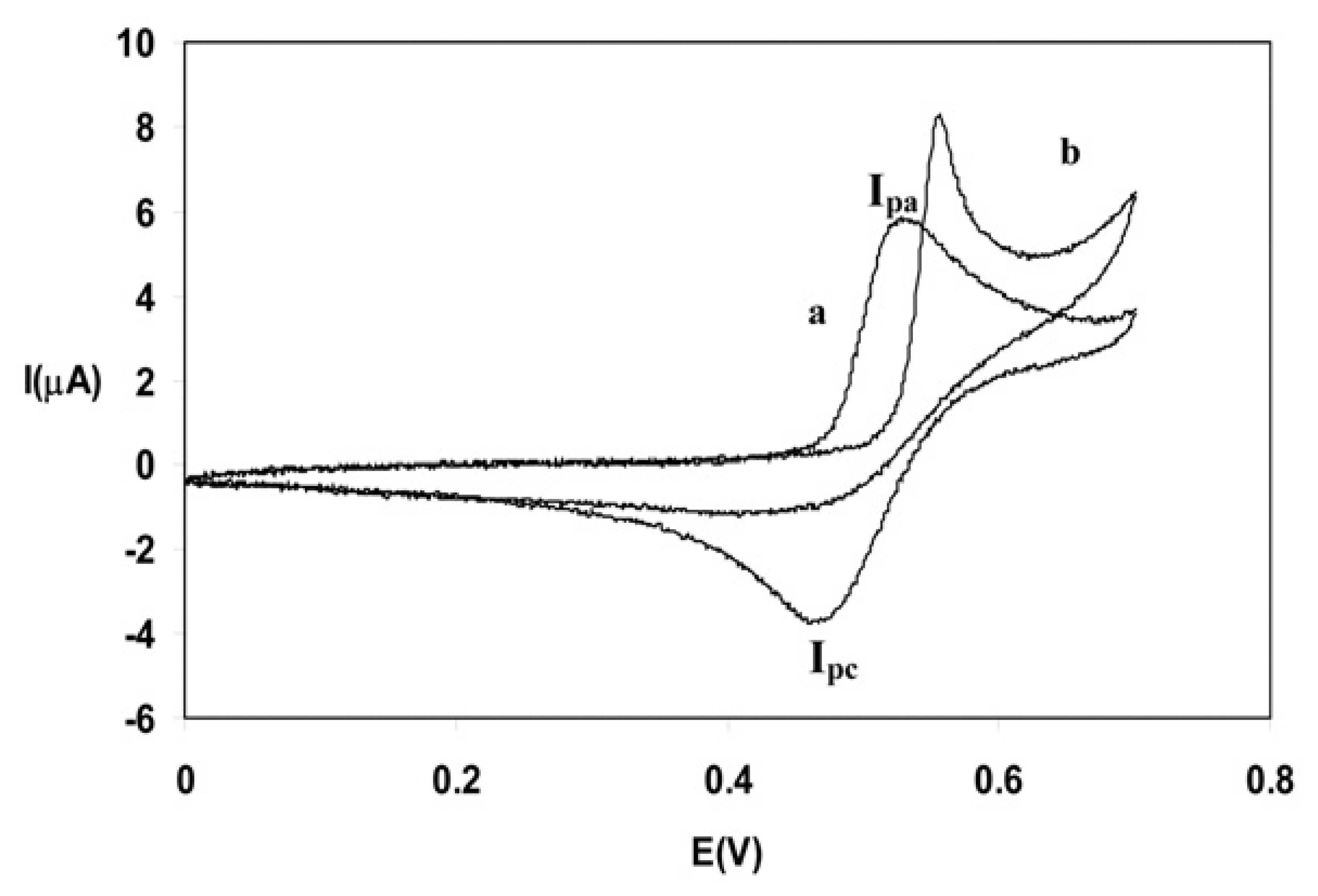
Results and discussions
Analytical aspects
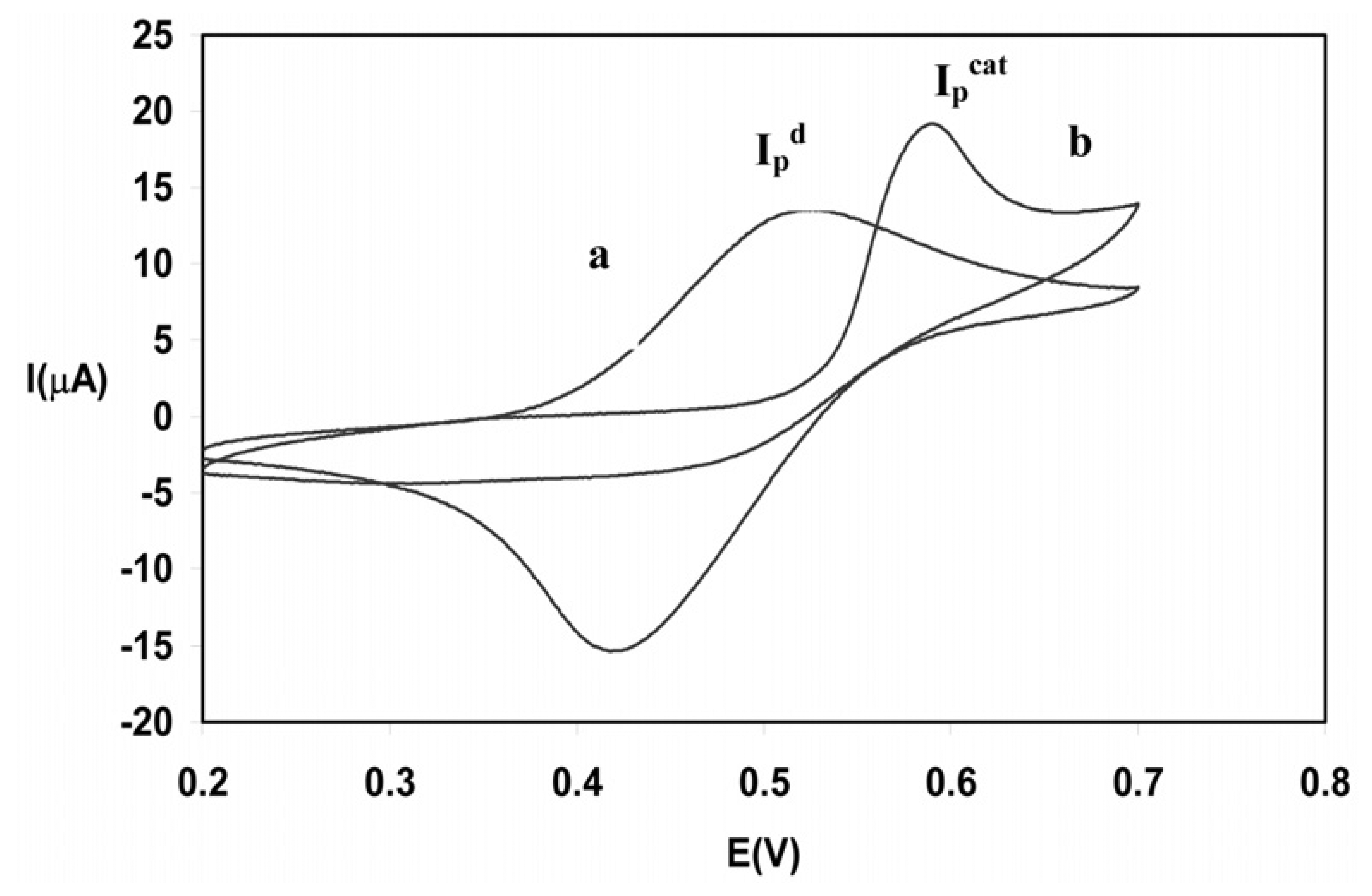
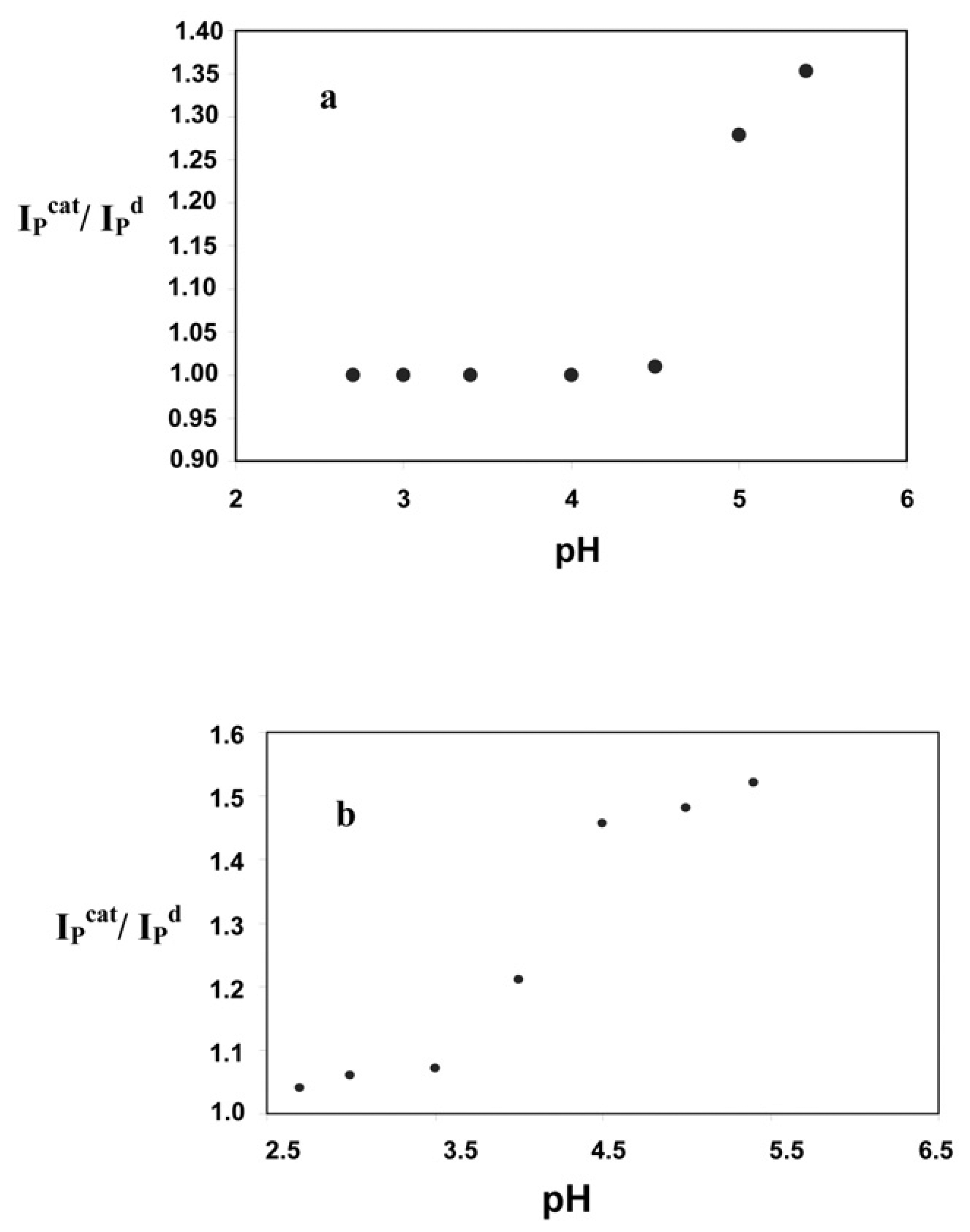
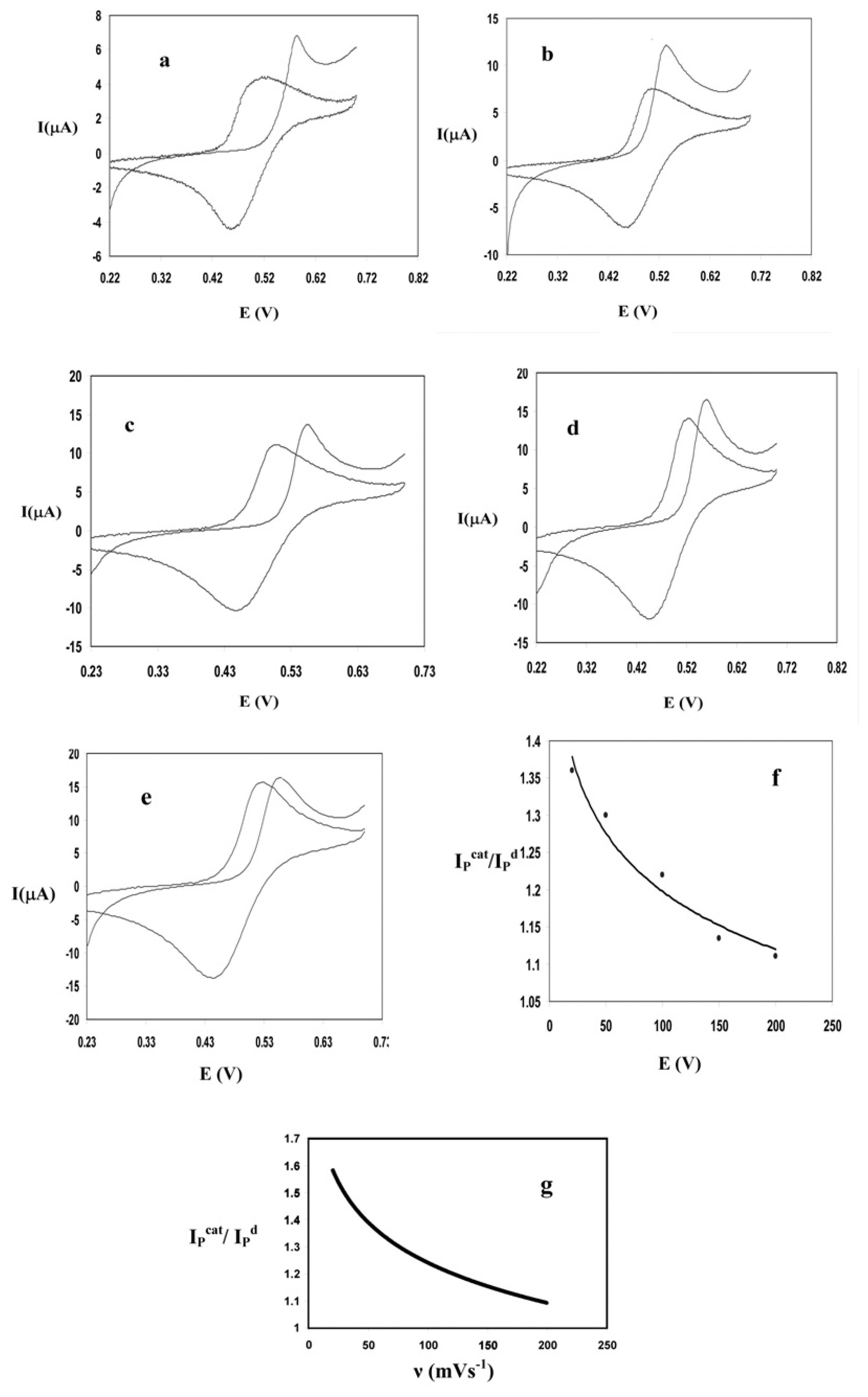
Optimization of the solution pH
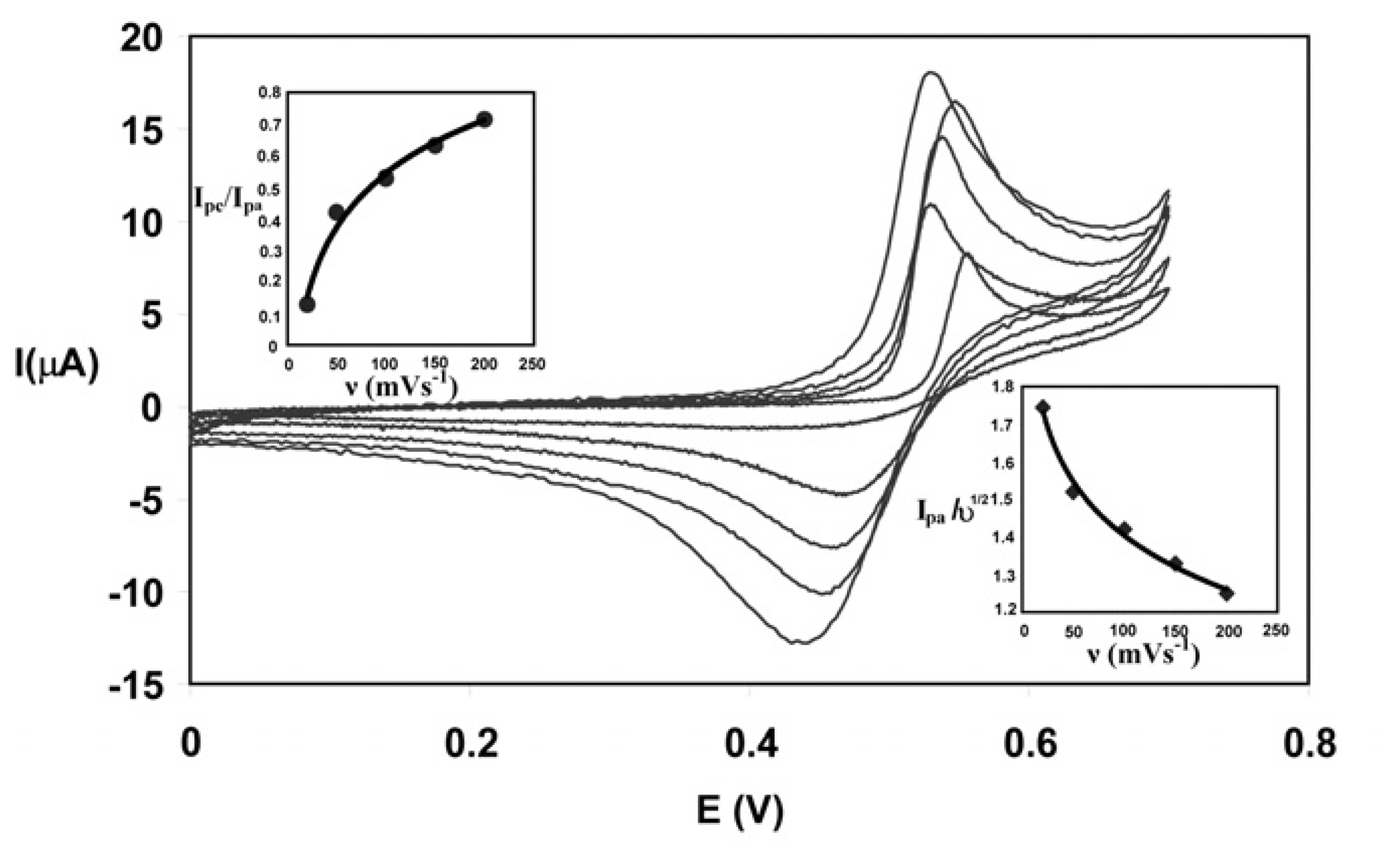
Optimization of scan rate
Optimization of sodium iodide concentration
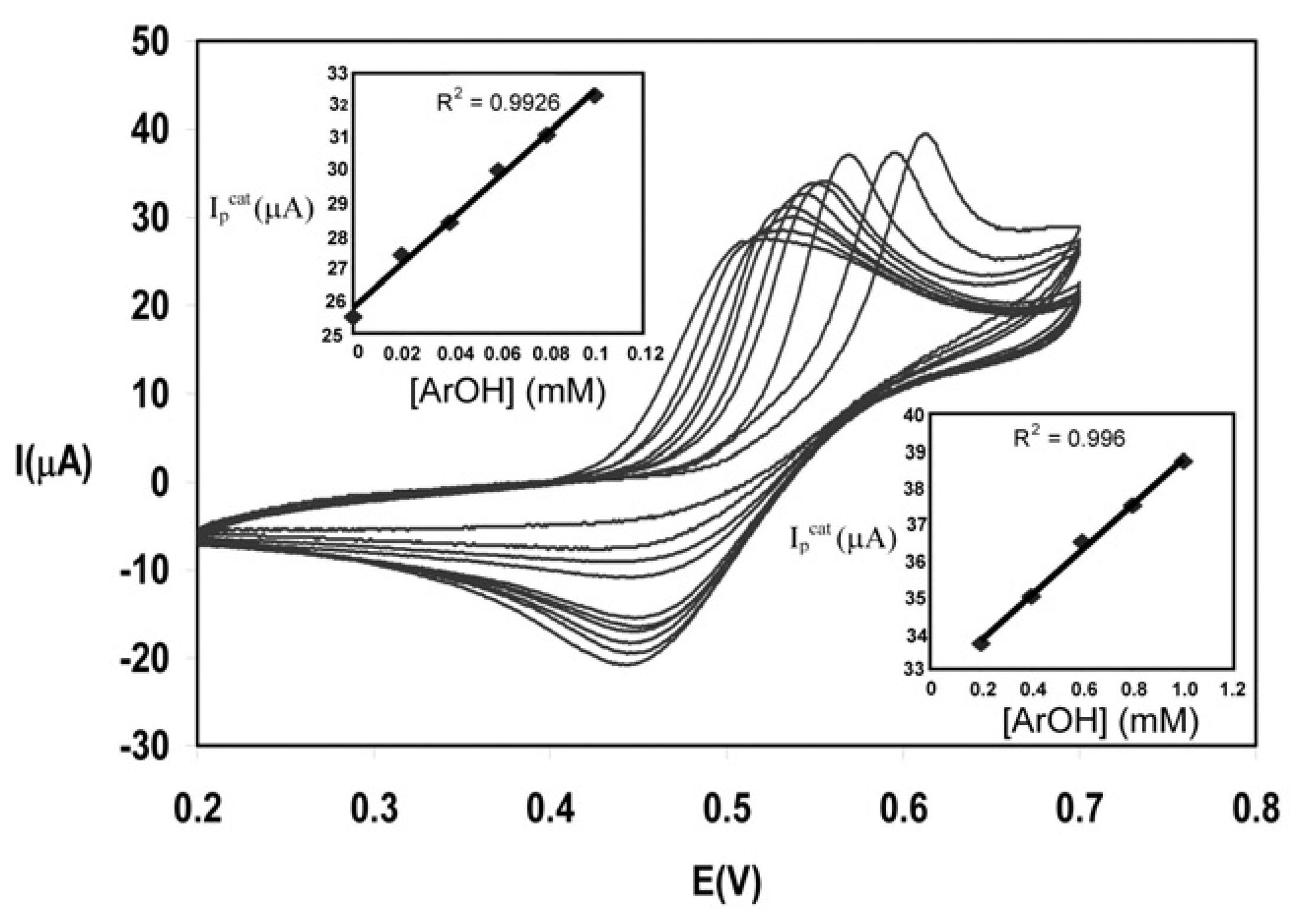
Calibration graphs
Interferences
Conclusions
Acknowledgments
References
- Patty, F.A. Industrial Hygiene and Toxicology; Vol. 2, Interscience: New York, 1963; p. p.1363. [Google Scholar]
- Kolthoff, I.M.; Elving, P.J.; Stross, F.H. Treatise on Analytical Chemistry; Vol. 2, Wiley: New York, 1971; p. p.490. [Google Scholar]
- Leithe, W. Analysis of Air Pollutants; Ann Arbor Science: Ann Arbor, MI, 1971; p. p.246. [Google Scholar]
- Leithe, W. Analysis of Organic Pollutants in Water and Waste Water; Ann Arbor Science: Ann Arbor, MI, 1972; p. p.113. [Google Scholar]
- Manahan, S.E. Fundamental of Environmental Chemistry; Lewis: Boca Raton, 1993; p. p.618. [Google Scholar]
- Morales, A.; Birkholz, D.A.; Hrudey, S.E. Analysis of pulp mill effluent contaminations in water, sediment and fish bile fatty and resin acids. Water Environ. Res. 1992, 64, 660. [Google Scholar]
- Buckee, G.K. Determination of the volatile components of beer. J. Inst. Brew. 1992, 98, 78. [Google Scholar]
- Chen, D.; Ray, A.K. Photodegradation kinetics of 4-nitrophenol in TiO2 suspension. Water Res. 1998, 32, 3223. [Google Scholar]
- Realini, P.A. Determination of priority pollutant phenols in water by HPLC. J. Chrom. Sci. 1981, 19, 124. [Google Scholar]
- Tyagi, R. Determination of substituted phenols in water by gas chromatography/mass spectroscopy after solid phase extraction. Fresenius Environ. Bull. 1995, 4, 751. [Google Scholar]
- Green, J.P.; Strierwalt, B.K.; Green, J.A.; Grizzle, P.L. Analysis of polar compound classes in SRC – II liquids-comparison of non-aqueous titrametric, i.r. spectrometric and h.p.l.c. methods. Fuel 1985, 64, 1571. [Google Scholar]
- Gonzalo, E.R.; Perez-Pavon, J.L.; Ruzicka, J.; Christinan, G.D. Flow injection analysis determination of phenols in kerosene and naphtha by membrane extraction preconcentration. Anal. Chim. Acta 1992, 259, 37. [Google Scholar]
- Heemken, O.P.; Theobald, N.; Wenclawiak, B.W. Comparison of ASE and SFE with soxhlet, sonication and mathematic saponification extraction for the determination of organic micropollutants in marine particulate matter. Anal. Chem. 1997, 69, 2171. [Google Scholar]
- Snyder, J.L.; Grob, R.L.; McNally, M.E.; Oostdyk, T.S. Comparison of supercritical fluid extraction with classical sonication and soxhlet extractions for selected pesticides. Anal. Chem. 1992, 64, 1940. [Google Scholar]
- Lompart, M.P.; Lorenzo, R.A.; Cela, R. Multivariate optimization of supercritical fluid derivatization and extraction of phenol in soil samples. J. Chrom. Sci. 1996, 34, 43. [Google Scholar]
- Lompart, M.P.; Lorenzo, R.A.; Cela, R. Optimization of supercritical fluid extraction of phenol and cresols in soil samples. J. Chrom A 1996, 723, 123. [Google Scholar]
- Lompart, M.P.; Lorenzo, R.A.; Cela, R.; Li, K.; Belanger, J.M.R.; Pare, J.R.J. Evaluation of supercritical fluid extraction, microwave-assisted extraction and sonication in the determination of some phenolic compounds from various soil matrics maria. J. Chrom. A 1997, 774, 243. [Google Scholar]
- Egizabal, A.; Zuloaga, O.; Etxebarria, N.; Fernandez, L.A.; Madariaga, J.M. Comparison of microwave-assisted extraction and soxhlet extraction for phenols in soil samples using experimental designs. Analyst 1998, 123, 1679. [Google Scholar]
- Asharf-Khorassani, M.; Gidanian, S.; Yamini, Y. Effect of pressure, temperature, modifier, modifier concentration, and sample matrix on the supercritical fluid extraction efficiency of different phenolic compounds. J. Chrom. Sci. 1995, 33, 658. [Google Scholar]
- Santos, F.J.; Jauregui, O.; Pinto, F.J.; Galceran, M.T. Experimental design approach for the optimization of supercritical fluid extraction of chlorophenols from pollutant soils. J. Chrom. A 1998, 823, 249. [Google Scholar]
- Deng, Q.; Gue, Y.; Dong, S. Cyro-hydrogel for the construction of a tyrosinase-based biosensor. Anal. Chim. Acta 1996, 319, 71. [Google Scholar]
- Gorton, L. Carbon paste electrodes modified with enzymes, tissues, and cells. Electroanal 1995, 7, 23. [Google Scholar]
- Sampath, S.; Lev, O. Membrane-free, rhodium-modified, methyl silicate-graphite amperometric biosensor. J. Electroanal. Chem. 1997, 426, 141. [Google Scholar]
- Wang, R.; Narang, U.; Prasad, P.N.; Bright, F.V. Affinity of antifluorescein antibodies encapsulated within a transparent sol-gel glass. Anal. Chem. 1993, 65, 2671. [Google Scholar]
- Dong, S.; Guo, Y. Organic phase enzyme electrode operated in water-free solvents. Anal. Chem. 1994, 66, 3895. [Google Scholar]
- Schubert, F.; Saini, S.; Turner, A.P.F.; Scheller, F. Organic phase enzyme electrodes for the determination of hydrogen peroxide and phenol. Sens. Actuators B. 1992, 7, 408. [Google Scholar]
- Conner, M.P.; Sanchez, J.; Wang, J.; Smyth, M.R.; Mannino, S. Silicone-grease-based immobilisation method for the preparation of enzyme electrodes. Analyst 1989, 114, 1427. [Google Scholar]
- Hedenburg, J.F.; Freiser, H. Anodic voltammetry of phenols. Anal. Chem. 1953, 25, 1355. [Google Scholar]
- Smyth, M.R.; Smyth, W.F. Voltammetric methods for the determination of foreign organic compounds of biological significance. A review. Analyst 1978, 103, 529. [Google Scholar]
- Kalcher, K. Chemically modified carbon paste electrodes in voltammetric analysis. Electroanal. 1990, 2, 419. [Google Scholar]
- Nematollahi, D.; Hesari, M. Electrochemical study of iodide in the presence of barbituric acid. Application to the catalytic determination of barbituric acid. J. Anal. Chem. 2001, 56, 1109. [Google Scholar]
- Nematollahi, D.; Hesari, M. Electrochemical study of iodide in the presence of barbituric acid. Application to coulometric titration of barbituric acid. Microchemical J. 2001, 70, 7. [Google Scholar]
- Golabi, S.M.; Nematollahi, D. Electrochemical study of catechol and some of 3-substituted catechols in the presence of 4-hydroxycoumarin. Application to electro-organic synthesis of new coumestan derivatives. J. Electroanal. Chem. 1997, 420, 127. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; New York; Wiley, 1980; p. p.452. [Google Scholar]
- Nematollahi, D.; Afkhami, A.; Zolfigol, M.A.; Akaberi, N. Electrochemical iodination and bromination of dibenzoylmethan. Bull. Electrochem. 2000, 16, 89. [Google Scholar]
- Wang, J.; Fang, L.; Lopez, D. Amperometric biosensor for phenol based on a tyrosinase-graphite epoxy biocomposite. Analyst 1994, 119, 455. [Google Scholar]
- Stanca, S.E.; Popescu, I.C.; Oniciu, L. Biosensors for phenol derivatives using biochemical signal amplification. Talanta 2003, 61, 501. [Google Scholar]
- Kim, H.; Chang, S.; Shim, Y. Cyclodextrin modified screen printed graphite electrodes for detection of phenols. Bull. Korean Chem. Soc. 2002, 23, 427. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Theoretical DL (M) | Experimental DL (M) | SD Relative | Recovery |
|---|---|---|---|---|
| Phenol | 1.16×10-5 | 1.125×10-5 | 1.06 | 97 % |
| o-Cresol | 4.56×10-5 | 4.201×10-5 | 1.44 | 95 % |
© 2004 by MDPI ( http://www.mdpi.net). Reproduction is permitted for non-commercial purposes.
Share and Cite
Fotouhi, L.; Ganjavi, M.; Nematollahi, D. Electrochemical Study of Iodide in the Presence of Phenol and o-Cresol: Application to the Catalytic Determination of Phenol and o-Cresol. Sensors 2004, 4, 170-180. https://doi.org/10.3390/s41100170
Fotouhi L, Ganjavi M, Nematollahi D. Electrochemical Study of Iodide in the Presence of Phenol and o-Cresol: Application to the Catalytic Determination of Phenol and o-Cresol. Sensors. 2004; 4(11):170-180. https://doi.org/10.3390/s41100170
Chicago/Turabian StyleFotouhi, Lida, Mahsa Ganjavi, and Davood Nematollahi. 2004. "Electrochemical Study of Iodide in the Presence of Phenol and o-Cresol: Application to the Catalytic Determination of Phenol and o-Cresol" Sensors 4, no. 11: 170-180. https://doi.org/10.3390/s41100170