An Overview of Label-free Electrochemical Protein Sensors

1
School of Materials Science, Japan Advanced Institute of Science and Technology (JAIST),1-1 Asahidai, Nomi City, Ishikawa 923-1292, Japan
2
Dept. of Chemistry, University of Western Ontario, 1151 Richmond Street, London, Ontario, N6A 5B7, Canada
3
Nanobiotechnology and Biodevice Laboratory, Dept. of Applied Physics, Graduate School of Engineering, Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan
*
Author to whom correspondence should be addressed.
Sensors 2007, 7(12), 3442-3458; https://doi.org/10.3390/s7123442
Submission received: 4 December 2007
/
Accepted: 18 December 2007
/
Published: 20 December 2007
(This article belongs to the Special Issue Utilization of Electrochemical Sensors and Biosensors in Biochemistry and Molecular Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Electrochemical-based protein sensors offer sensitivity, selectivity and reliability at a low cost, making them very attractive tools for protein detection. Although the sensors use a broad range of different chemistries, they all depend on the solid electrode surface, interactions with the target protein and the molecular recognition layer. Traditionally, redox enzymes have provided the molecular recognition elements from which target proteins have interacted with. This necessitates that the redox-active enzymes couple with electrode surfaces and usually requires the participation of added diffusional components, or assembly of the enzymes in functional chemical matrices. These complications, among many others, have seen a trend towards non-enzymatic-based electrochemical protein sensors. Several electrochemical detection approaches have been exploited. Basically, these have fallen into two categories: labeled and label-free detection systems. The former rely on a redox-active signal from a reporter molecule or a label, which changes upon the interaction of the target protein. In this review, we discuss the label-free electrochemical detection of proteins, paying particular emphasis to those that exploit intrinsic redox-active amino acids.
1. Introduction
Genetic information, imprinted on nucleic acids, has always enchanted the researchers and intrigued them into unraveling its secrets. Proteins, molecular expression of this genetic information, are at the very core of biological function. They are the centre of most pathological conditions and most disease biomarkers are proteins. Besides DNA studies, they are perhaps the subject of most intense research. Biosensors technology has emerged as one of the most promising platforms for studying proteins. Biosensors are devices that combine a biological component (a recognition layer) and a physicochemical detector component (a transducer). The transduction unit can be electrochemical, optical, piezoelectric, magnetic or calorimetric. The recognition layer can be constructed using enzymes, antibodies, cells, tissues, nucleic acids, peptide nucleic acids, and aptamers [1-4]. In this review, we discuss electrochemical sensors for protein analysis.
Electrochemical biosensors have superior properties over other existing measurement systems, because electrochemical biosensors can provide rapid, simple and low-cost on-field detection. Electrochemical measurement protocols are also suitable for mass fabrication of miniaturized devices. In fact, electrochemical biosensors have played a major role in the move towards simplified testing for point-of-care usage. Indeed, self-testing glucose strips, based on screen-printed enzyme electrodes, coupled to pocket-size amperometric meters for diabetes, have dominated the market over the past two decades [5]. There are basically four different pathways for electrochemical detection of proteins: a change in the electrochemical signal of (i) a label, which selectively binds with the target protein, (ii) electro-active amino acids of antibody or target protein, (iii) a secondary antibody-tagged probe, (iv) aptamers- and (v) an enzyme-tagged probe can be monitored [4,6,7]. In this review, we focus on the label-free electrochemical detection of proteins with particular emphasis to those that exploit intrinsic redox-active amino acids. We present recent work carried out by our group as well as work by other groups.
2. Intrinsic redox-active amino acids-based sensors: direct application
Since the middle of the 20th century, electrochemical analysis of proteins is increasingly gaining prominence [8]. From the early 1970s until today, many electro-chemists have focused on a relatively small group of proteins containing a metal center with reversible redox-activity (metalloproteins) [9]. Nowadays, the fact that most of the proteins not containing a metal center can show electrochemical activity, depending on their amino acid structure, has attracted a lot of the attention from researchers. Since polarography has been a well-established method, the first label-free electrochemistry of proteins came from mercury electrodes. Peptides and proteins containing cysteine/cystine (Cys) showed specific electrochemical signals on mercury electrodes with the help of Hg-S bond formation [10,11], reduction of disulfide groups [12], and the catalytic evolution of hydrogen in cobalt-containing solutions (Brdicka reaction) [13,14]. Hydrogen evolution was also catalyzed at highly negative potentials in the absence of transition metal ions using mercury electrodes with proteins that contained or lacked sulfur amino acids. In combination with chronopotentiometric stripping analysis, presodium catalysis resulted in a well-defined signal that enabled the detection of several important proteins [15].
In the early 1980s, Reynaud et al. [16] and Brabec et al. [17,18] showed that tyrosine (Tyr) and tryptophan (Trp) residues in proteins are electro-oxidizable at carbon electrodes. The electro-oxidation of Tyr residues involves two electron and two proton transfer with an electrode process that is similar to the oxidation of simple p-substituted phenols [16–19]. Well-developed oxidation peaks of Tyr and Trp in nM concentrations of peptides were obtained by applying voltammetric methods in combination with a sophisticated baseline correction [20].The reader is referred to a recent review by Herzog and Arrigan for a comprehensive discussion on the electrochemical strategies for label-free detection of amino acids, peptides [21]. Here-in our primary focus is on the exploitation of redox-active amino acids for protein sensing.
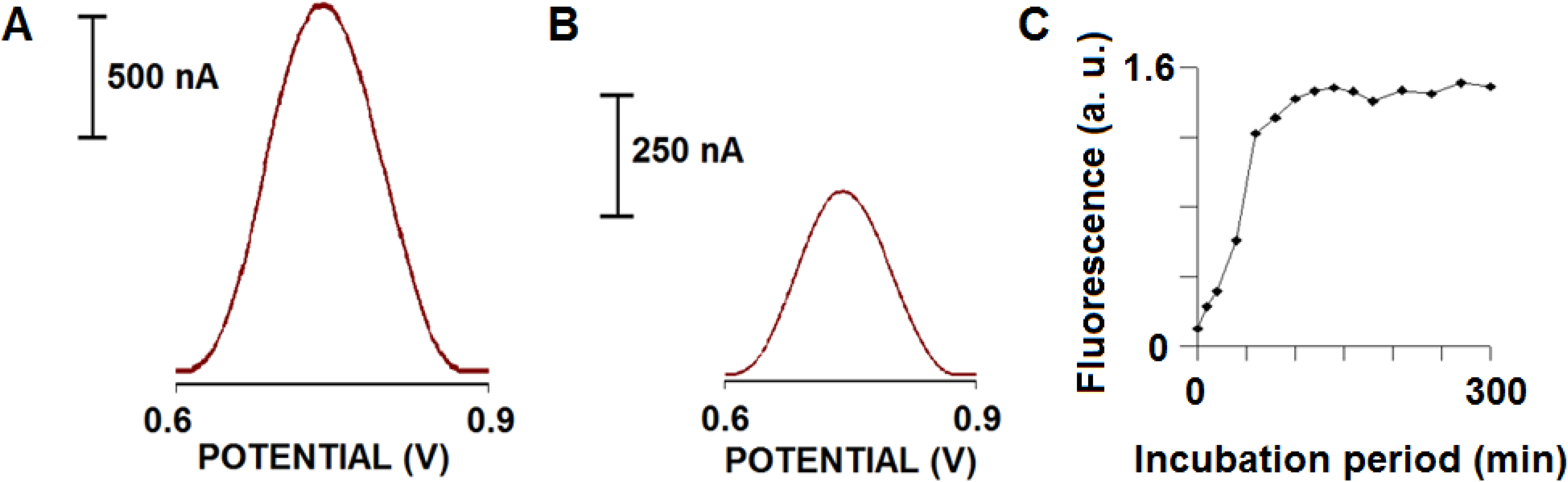
Our group successfully exploited oxidation of Tyr for detection of several biomolecules. Vestergaard at al. presented the first electrochemical detection and aggregation study of Alzheimer's amyloid beta peptides (Aβ-40 and Aβ-42) using three different voltammetric techniques at a glassy carbon electrode (GCE). The method was based on detecting changes in the oxidation signal of Tyr at various time periods during amyloid beta incubation at 37°C in Tris buffer, pH 7.4. We hypothesised that as the conformation if the peptides changed during aggregation, we should see an accompanied change in the oxidation signal of Tyr. A clear difference in the rate of aggregation was observed between the two peptides. During the study, we observed a decrease in the Try oxidation signal with increase in incubation period. The degree of aggregation was confirmed using thioflavin T label and analysed using a fluorescence spectroscopy and imaging using atomic force microscopy (AFM) [22]. The results are depicted in Fig. 1.
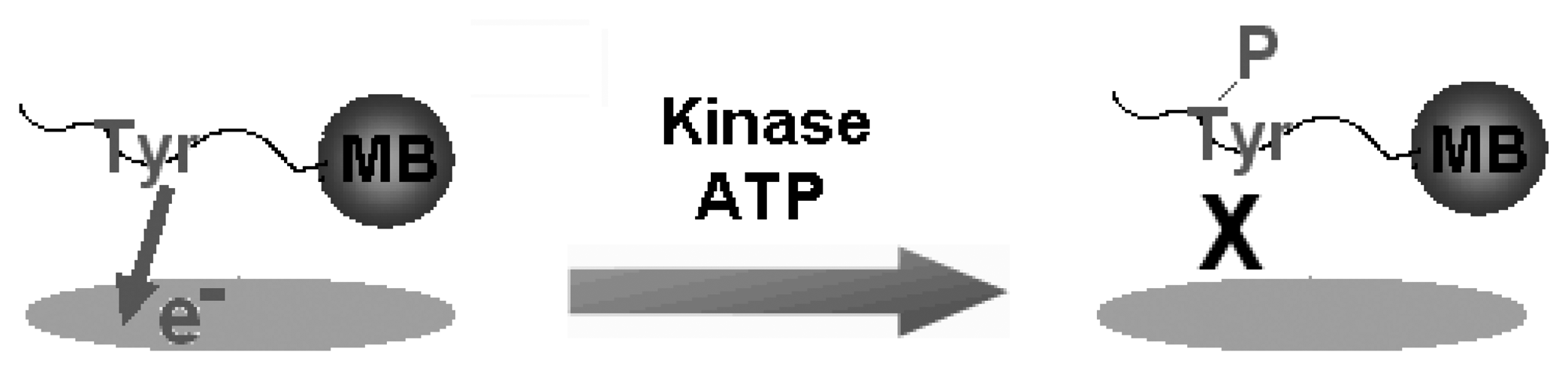
We also studied label-free electrochemical detection of phosphorylation based on the electrooxidation of Tyr in connection with differential pulse voltammetry (DPV) using a screen-printed carbon electrode (SPCE). First, we monitored the electrochemical current responses of Tyr and o-phospho-L-Tyrosine. We observed that the phosphorylation caused a significant suppression on the electro-oxidation of Tyr. We also monitored electrochemical responses of sarcoma (Src) both in the non-phosphorylated and phosphorylated forms. The procedure was very simple and we propose that label-free electrochemical in vitro detection of Tyr phosphorylation can be performed in a rapid and cost-effective format [23]. Using this principle, we detected the inhibition of Tyr phosphorylation using a small molecule. Using DPV in conjunction with multi-walled carbon nanotube-modified SPCEs, we determined the activity of c-Src non-receptor protein tyrosine kinase, p60c-Src, in combination with its highly specific substrate peptide, Raytide. Tyr kinase reactions were also performed in the presence of an inhibitor, 4-amino-5-(4-chlorophenyl)-7- (tert-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (Figure 2) [24].
Aggregation of α-synuclein has been detected based on the redox-active Tyr and Cys residues. The authors used constant current chronopotentiometric stripping analysis (CPSA) to measure hydrogen evolution (peak H) catalyzed by α-synuclein at hanging mercury drop electrodes (HMDE) and square-wave stripping voltammetry (SWSV) to measure Tyr oxidation at carbon paste electrodes (CPE). Aggregation-induced changes in peak H at HMDE were relatively large in strongly aggregated samples, suggesting that this electrochemical signal may find use in the analysis of early stages of α-synuclein aggregation. Native α-synuclein could be detected down to subnanomolar concentrations by CPSA [25]. The same group successfully detected a metallothionein from rabbit liver by CPSA in conjunction with HMDE [26], and using a phytochelatin-modified electrode, they were successful in detecting cadmium and zinc ions [27]. This highlights the versatility of proteins as recognition elements, serving not only for other macromolecules but also for small molecules such as heavy metals.
Directly capturing the possible configuration of biomolecules, and/or their involved interactions with other molecules, without a molecular recognition element is truly a remarkable progress. Although they enable quick and simple initial investigation into whether direct label-free detection is possible or not, they have a profound limitation. They cannot be used, successfully in complex sample matrices, where various protein molecules are present. Label-free protein detection is, therefore, commonly achieved by employing biomolecules with high affinity for the target protein. This ensures much improved specificity, especially when dealing with a more complex sample matrix such as urine, cerebral spinal fluid (CSF), and serum, which contains high levels of serum albumin and immunoglobulins. In this review, we will discuss antibody-based and aptamer-based electrochemical protein sensors that utilise label-free strategies.
3. Antibody-based protein detection
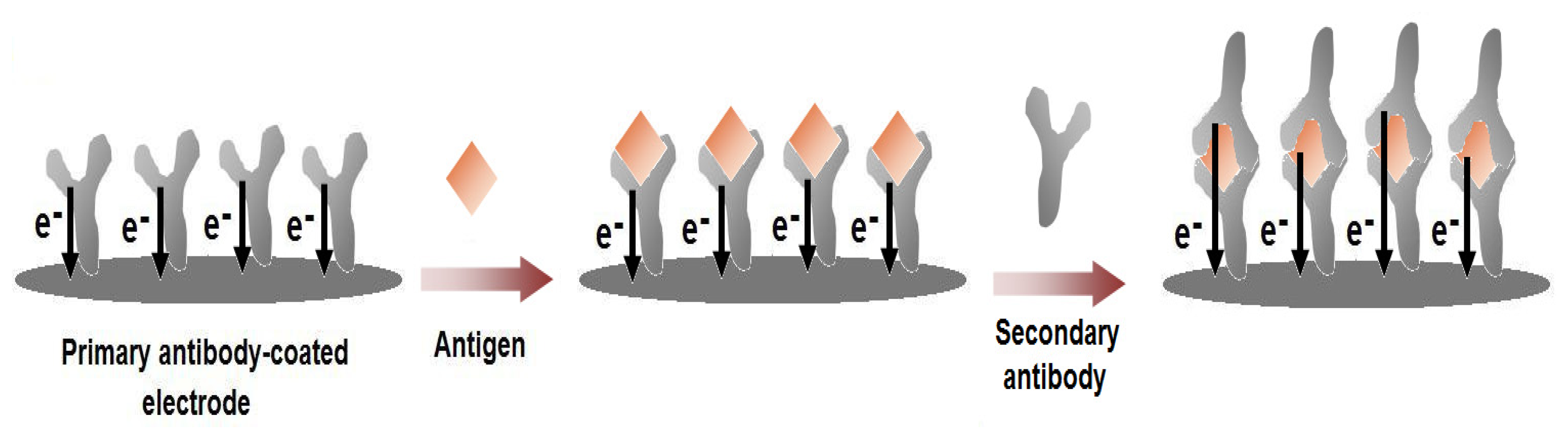
Immunosensors exploit the interaction between an antibody (Ab), synthesised in response to the target molecule, an antigen (Ag). Antibodies can be formed, when they are attached to an immunogen carrier such as serum albumin. There are two types of Abs: polyclonal and monoclonal. Polyclonal antibodies (pAb) have an affinity for the target antigen, and are directed to different binding sites, with different binding affinities. Monoclonal antibodies (mAb), on the other hand, are identical, because they are produced from one type of immune cell. They have higher sensitivity and selectivity than pAb, and are, therefore, preferred. Antibody binding sites are located at the ends of two arms (Fab units) of the Y-shaped protein. The tail end of the Y (aka Fc unit) contains species-specific structure, commonly used as an antigen for production of species-specific Abs. The antibody is used as the recognition layer in biosensor development. There exists a handful of general immunosensor formats (Figure 3) [28].
Antibody-based biosensors started to emerge in the 1970s following the work of Giaver, and Kronick and Little [29,30]. Since then, there has been an immunosensor boom, not surprising given the specificity and sensitivity of Ab:Ag interactions. Our group has developed label-free electrochemical immunosensors, mostly targeting pathologically-important biomarkers. Following successful detection of Aβ-peptide aggregation based on Tyr oxidation signal [22], Kerman and colleagues developed an immunosensor for the model hormone (human chorionic gonadotropin: hCG). Following pretreatment, a carbon paste electrode (CPE) was dipped in phosphate buffer containing EDC and NHS. After incubation at room temperature for 1 h, lysine residues of protein A were coupled onto the modified CPE. Protein A has high binding affinity for the Fc region of Ab. Monoclonal antibody for hCG (β-hCG-mAb) was immobilised on protein A-linked CPE and incubated for a specified time period. Unreacted covalent-active surface groups were subsequently passivated using ethanolamine to prevent non-specific adsorption. The immunosensor was ready for introduction of synthetic hCG and later, human urine samples from pregnant and non pregnant women as well as men. Urine samples were collected in accordance with the ethical standard of the Helsinki Declaration of 1975, as revised in 1996. The analysis was carried out using square wave voltammetry (SWV). Peak currents for both β-hCG-mAb and hCG were observed at ∼ 0.6 V (vs Ag/AgCl) (Fig. 4). Using this sensor, the limit of detection was 15 pM in synthetic hCG and 20 pM in human urine [31]. Employing the described underlying principle, we developed an immunosensor for detection of human telomerase reverse transcriptase (hTERT) in urine, using differential pulse stripping voltammetry (DPSV) in conjunction with pencil graphite electrode [32].
For label-free protein detection the use of carbon nanotubes (CNTs) has provided improved sensitivity. CNTs were first discovered in 1991 [33]. They are cylindrical graphite sheets with properties that make them potentially useful in a wide variety of applications in fields of materials science. They exhibit extraordinary strength and unique electrical properties and are used in electrochemistry for promoting electron transfer reactions with electroactive species. There are two groups of CNTs: SWCNTs and MWCNTs [34]. SWCNTs are comprised of a cylindrical graphite sheet of nanoscale diameter (∼ 1 nm) capped by hemispherical ends. MWCNTs comprise several to tens of concentric cylinders of these graphite shells with a layer spacing of 3-4°A. They have a diameter between 2-100 nm [35]. Since their first application to the study of dopamine [36], CNTs increasingly show potential in bioelectrochemistry. Over the past few years, their preparation and purification have received much attention [37,38].
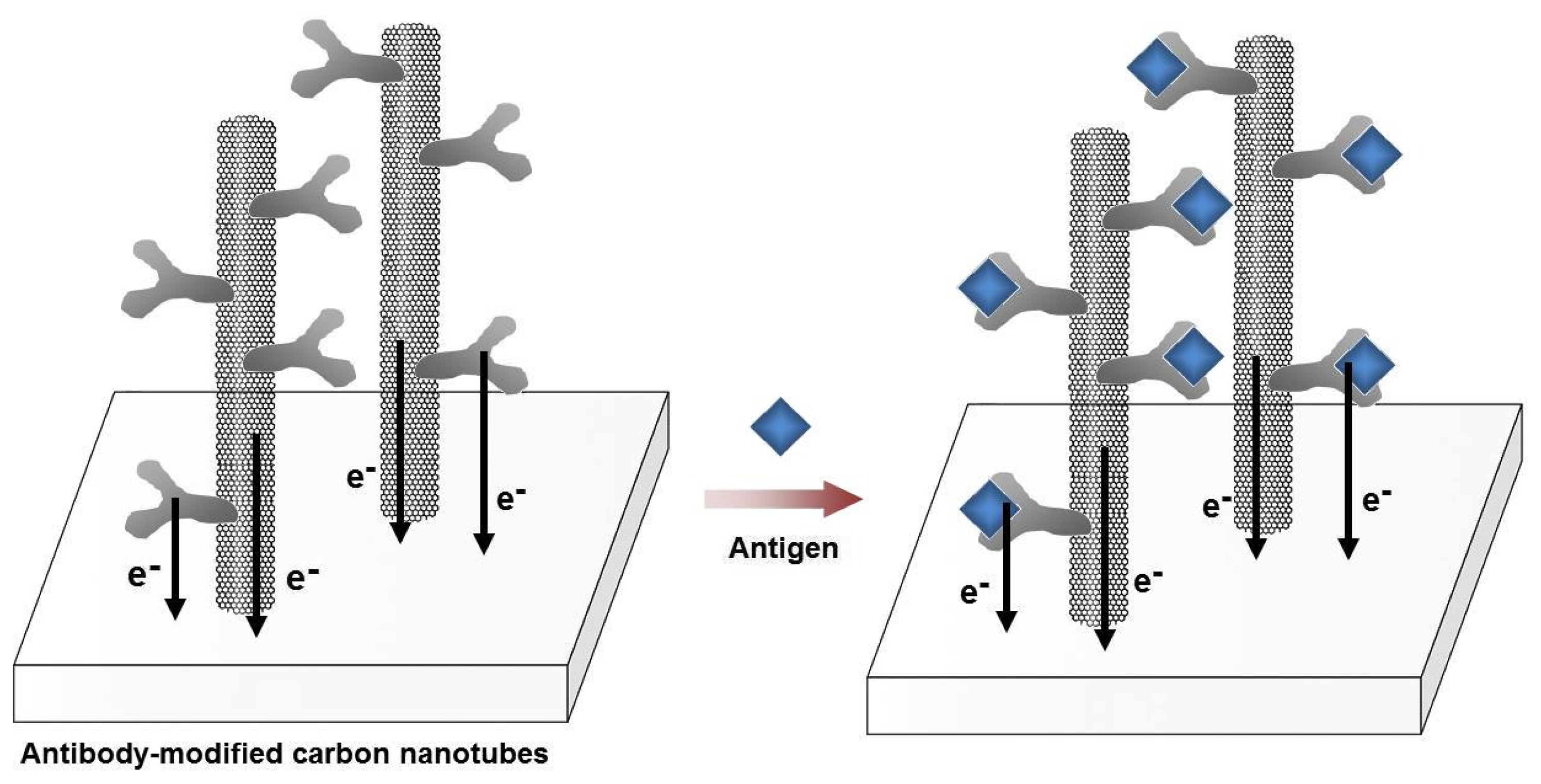
An immunosensor of IgE based on SWCNT-modified field-effect transistor (FET) was successfully developed [39]. Another label-free electrochemical immunosensor was fabricated using microelectrode arrays modified with single-walled carbon nanotubes (SWCNTs) as transducer surfaces for the detection of a cancer marker, total prostate-specific antigen (T-PSA) using DPV. The current signals, derived from the oxidation of Tyr and Trp residues, increased with the interaction between T-PSA on T-PSA-mAb covalently immobilized on SWCNTs. The selectivity of the biosensor was challenged using bovine serum albumin as the target protein. The detection limit for T-PSA was determined as 0.25 ng/mL. Since the cut-off limit of T-PSA between prostate hyperplasia and cancer is 4 ng/mL, the performance of this immunosensor is promising for further clinical applications (Fig. 5) [40].
Label-free electrochemical impedance spectroscopy (EIS) has been explored widely due to its high sensitivity for detection of small proteins such as interferon-γ (IFN-γ) [41]. EIS provides accurate mechanistic and kinetic information from repeatable adsorption and desorption measurements on the sensor transduction surface. The surface, represented in a form of an electrode, displays both resistive and capacitive properties when a small amplitude sinusoidal excitation perturbs the system at equilibrium. As a result, antibody-based biosensors in EIS are increasingly attracting interest. They allow for direct and label-free electrochemical immunosensing, potentially speeding up detection and analysis of biomarkers, without losing sensitivity.
An immunosensor was developed by immobilising anti-IFN-γ antibodies on a self-assembled monolayer (SAM) of acetylcysteine on polycrystalline Au [42]. EIS and cyclic voltammetry (CV) was employed for analysis of carcinoembryonic antigen (CEA). Antibody to CEA (Ab-CEA) was covalently attached on a glucothione monolayer-modified Au nanoparticle (NP). The resulting conjugate was immobilised on Au electrode by electro-copolymerisation with O-aminophenol, completing the sensor development. Introduction of CEA increased the electron transfer resistance of [Fe(CN)6]3-/4- redox pair [43]. Using EIS, a sensitive immunosensor was developed via covalent coupling of the antibody with functionalized AuNPs for apolipoprotein A-I (Apo A-I) detection. The hybrid AuNPs were prepared using SAM and sol–gel techniques for improved performance, ∼6-17 times higher than that fabricated by normal SAM technique did. The detection limit of the immunosensor was 50 pg mL−1 Apo A-I, two orders of magnitude lower than that of the traditional methods [44]. A label-free electrochemical impedance immunosensor for rapid detection of Escherichia coli O157:H7 was developed by immobilizing anti-E. coli antibodies onto an indium-tin oxide interdigitated array (IDA) microelectrode. Similar to Tang et al. [44], immobilization of antibodies and the binding of E. coli cells to the IDA microelectrode surface resulted in an increase in the electron-transfer resistance, directly measured with electrochemical in the presence of [Fe(CN)6]3-/4- as a redox probe [45].
4. Aptamer-based protein detection
Aptamers are synthetic oligonucleotides that can be generated to selectively bind to low-molecular weight organic and inorganic substrates and to macromolecules such as proteins, drugs, with high affinity [46-51]. A reusable label-free aptasensor for detection of small molecules using EIS was recently reported [52]. The affinity constants for aptamers are comparable to the binding constants of antibodies to antigens, i.e., it is in the micromolar to nanomolar scale [53]. Aptamer-protein interactions can be monitored using intrinsic DNA and protein oxidation signals on carbon electrodes. Recent reports on electrical/electrochemical aptasensors have highlighted the promise that aptamers hold for detection of proteins and their involved interactions. Their selection by the SELEX (systematic evolution of ligands by exponential enrichment) confers them a high affinity for their substrate. They could even substitute antibodies as primary candidates in biosensing. Their advantages over antibodies include: (i) no need for complex Ag:Ab sandwich-assay needed for labeled techniques, as modification of aptamer with label is possible for direct analysis of aptamer:substrate interaction, (ii) the synthesis of aptamers leads to highly reproducible structures of the binding ligands compared to antibodies, and (iii) chemical modification of aptamers with labels or functional groups that enable them to tether to transducers is easier than with antibodies. Still, the sensitivity of DNA and RNA to nucleases makes them more susceptible to denaturation and may require purer environments for their applications than antibodies [4, 54].
We have already discussed intrinsic redox-active amino acids in section 2. Here, we will briefly discuss electrochemical oxidation of DNA bases. Guanine and adenine are the most electro-active DNA bases because they can easily be adsorbed and oxidized on carbon electrodes. Guanine and adenine oxidation signals on carbon electrodes can be observed at around 1.0 and 1.3 V in 0.50 M acetate buffer solution at pH 4.80, respectively [55]. Guanine is the most redox-active of the DNA bases and the mechanism for its oxidation has been studied in detail [56-58]. The oxidation of guanine and adenine shows peak currents at ∼0.9 V and ∼1.2 V, respectively, depending on the pH and ionic strength of the electrolyte and the electrode material. The process was shown to have two steps, involving the total loss of four electrons and four protons. A recent review by Palecek et al. (2005) discusses the electrochemical mechanisms for the oxidation and reduction of DNA bases on carbon and mercury electrodes [59]. Monitoring the changes in these signals upon duplex formation enabled the detection of hybridization [60,61]. The electrochemical signals obtained from free adenine and guanine bases decreased on binding to their complementary thymine and cytosine bases after hybridization. Label-free interaction of DNA with drugs, metals and proteins have been monitored using the intrinsic oxidation signal of DNA by a large number of groups [62-64]. Electrocatalytic oxidation of guanine, guanosine and guanosine monophosphate has also been recently reported [65].
Rodriquez et al. [66] developed the EIS-based label-free biosensor for monitoring aptamer interactions. A biotinylated aptamer for lysozyme was linked to streptavidin-functionalised electrode, acting as an aptasensor the lysozyme protein. The same group developed a highly specific and sensitive aptasensor for detecting protein interactions using aptamer-coated magnetic beads in conjunction with chronopotentiometric stripping [67]. Using EIS, an aptamer-based sensor was developed, in an Au electrode array configuration, for the detection of IgE [68]. Thrombin was detected down to 0.1 nM First, an aptamer was self-assembled on a microfabricated thin film Au electrode. Then the protein was introduced onto the recognition surface for a binding event, monitored using EIS [69].
5. Nanotechnology and label-free protein sensors
In our recent review article, we discussed the current role and future prospects of nanosensor technology in the study of Alzheimer's disease biomarkers [70] without particular focus to the electrochemical label-free protein sensors. The reader is also referred to a recent review by Pumera and colleagues for an in-depth discussion on the main techniques and methods which use nanoscale materials for construction of electrochemical biosensors [71]. Here, we confine our review to the exploitation of nanotechnology for application to label-free protein sensing. Au and Ag NPs, have been exploited in the fabrication of localised surface plasmon resonance chips for both label and label-free protein detection [72-74] because of their unique optical properties [75-77]. These metal NPs show specific changes in their absorbance responses in the visible region of the spectrum upon binding with various molecules such as nucleic acids or proteins (Fig. 6). In electrochemistry, Au NPs have also been employed, but as labels for detection of proteins and other target molecules based on monitoring the reduction current signal of Au in HCl [78-83]. For label-free protein detection the use of carbon nanotubes (CNTs) has provided improved sensitivity. Fabregas group developed a multi-walled carbon nanotube (MWCNT)/polysulfone (PS) biocomposite membrane modified thick-film screen-printed electrochemical biosensor. The fabricated CNT/PS strips were reported to be mechanically stable, and to exhibit high electrochemical activity. Furthermore, the biocompatibility of CNT/PS composite allowed easy incorporation of the biological functional moiety of horseradish peroxidase [84], providing simplicity and robustness.
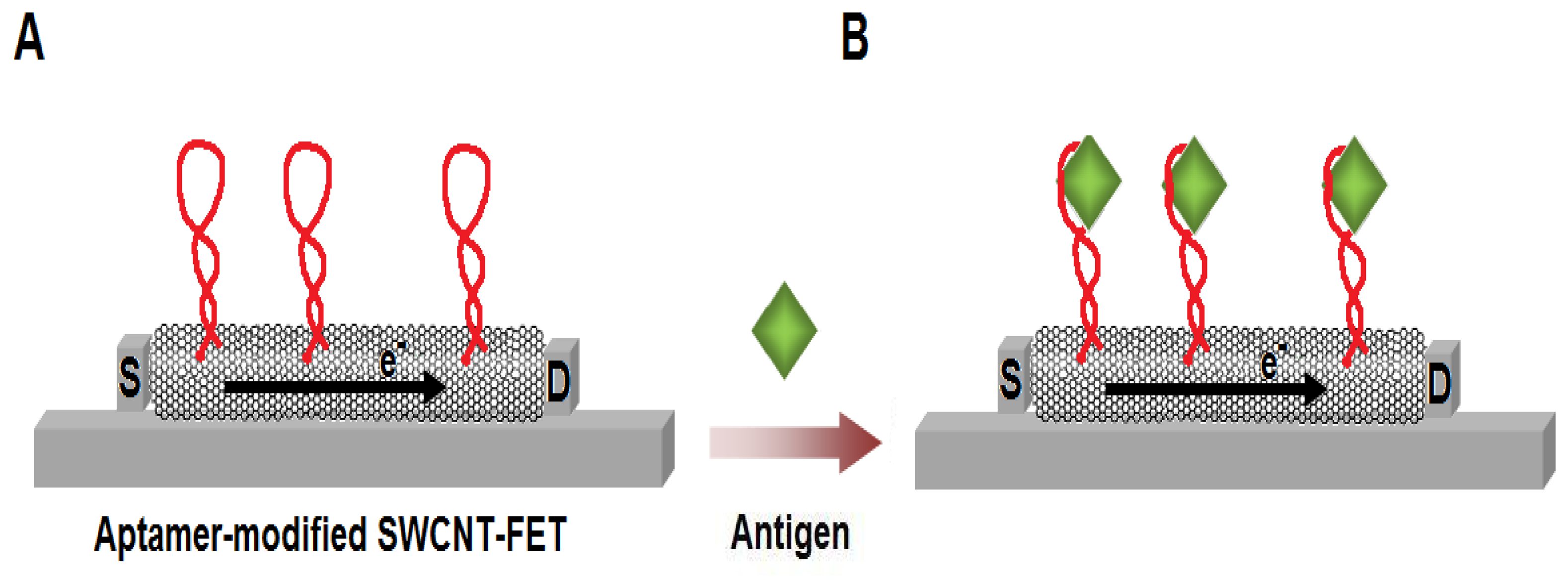
Exploiting the electrical properties of CNTs with nanotechnology, aptamers are increasingly becoming one of the most promising candidates for protein biosensors. In our group, a sensor based on aptamer-modified carbon nanotube-FET was developed for label-free detection of immunoglobulin E (IgE). Briefly, 5′-amino-modified aptamers were immobilised on CNT channels and the electrical properties monitored in real-time. Upon introduction of IgE, a sharp decrease in the source-drain current was observed (Fig. 7). After optimisation, the limit of detection was determined at 250 pM IgE [39]. Compared with performance of this aptasensor and the IgE immunosensor described earlier in the same article [39], under similar conditions, the aptasensor provided better sensitivity. So et al. [65] also utilised the real-time detection of protein using single-walled carbon nanotube (SWCNT)-FET-based aptasensors. Anti-thrombin aptamers, highly specific to serine protein thrombin were immobilized on the sidewall of a SWCNT-FET using carbodiimidazole-activated Tween 20 as the linking molecules. The binding of thrombin aptamers to SWCNT-FETs caused a rightward shift of the threshold gate voltages, presumably due to the negatively charged backbone of the DNA aptamers. While the addition of thrombin solution caused an abrupt decrease in the conductance of the thrombin aptamer immobilized SWCNT-FET [66].
6. Challenges for development of point-of-care biosensors
We will conclude by briefly touching on current and future challenges in the development and application of biosensor technology for point -of-care testing (POCT). Already there are several commercially available biosensing tools for home use such as monitoring of glucose levels for diabetic patients [5]. Most commercially available POCTs are based on immunochromatographic membrane strips (immunostrips). Examples include home-use pregnancy testing kits, influenza test strips carried out at local practitioners, and strips for cancer biomarker prostate specific antigen (PSA) [85]. Although this technology is simple and works reasonably well, the main problem is sensitivity. Most immunostrips have nanomolar level sensitivity whereas electrochemical analysis of the equivalent tests go down to picomolar concentration levels [31,34,84]. Gold NP-enhanced immunostrips and resin-based micropipette tips showed increased sensitivity for hCG and PSA [86,87]. Electrochemical analysis of the AuNP-enhanced strips gave an even higher sensitivity, showing unequivocally, the superiority of electrochemical analysis over immunostrip-based assay [88]. The challenge remains that of appropriate miniaturisation and integration.
Another current challenge is utilisation of biosensors for clinical samples, in particular, blood. Advances in sample pretreatment (extraction and purification) to counter matrix interference are needed. Detection of small and low abundant biomarkers in clinical samples is problematic mainly due to the presence of high abundant proteins such as albumin (HSA) and immunoglobulin G (IgG), accounting for at least 65% of plasma proteins [89]. Depletion of these proteins may not only facilitate detection but also biomarker discovery. Depletion should aim to provide selective removal of high abundant proteins, and concentrate low abundant components. Although commercially pre-packed columns for HSA and IgG removal are available, they lack the flexibility to accommodate issues pertaining to sample size, for example. In addition, some rely on use of pumps and other equipment to operate smoothly, limiting their use in many health centres [90,91]. In our group, we developed a simple rapid method for removal of both IgG and HSA based on affinity chromatography principles. Depletion of the proteins from serum samples substantially lowered the detection limit of PSA [92].
We are presently in the early days of the emerging technology of using label-free electrochemistry of proteins in the development of biosensors. So far, antibody-based biosensors have been intensively developed, although many improvements are required in reproducibility and sensitivity. More, there is no doubt that a growing number of aptamers and, it is to be hoped, nanomaterial-based label-free biosensors will soon be used for the diagnosis and therapeutic follow-up of diseases.
References
- Kerman, K.; Kobayashi, M.; Tamiya, E. Recent trends in electrochemical DNA biosensor technology. Meas. Sci. Tech. 2004, 15, R1–R11. [Google Scholar]
- Wang, J. Electrochemical biosensors: towards point-of-care cancer diagnostics. Biosens. Bioelectron. 2006, 21, 1887–1892. [Google Scholar]
- Kerman, K.; Vestergaard, M.; Nagatani, N.; Takamura, Y.; Tamiya, E. Electrochemical genosensor based on peptide nucleic acid-mediated PCR and asymmetric PCR techniques: electrostatic interactions with a metal cation. Anal. Chem. 2006, 8, 2182–2189. [Google Scholar]
- Bini, A; Minunni, M.; Tombelli, S.; Centi, S.; Mascini, M. Analytical performance of aptamer-based sensing for thrombin detection. Anal. Chem. 2007, 79, 3016–3019. [Google Scholar]
- Newman, J.; Turner, A. P. F. Home blood glucose biosensors: a commercial perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. [Google Scholar]
- Kerman, K.; Vestergaard, M.; Tamiya, E. Electrochemical Biosensors, Chapter: in Methods in Biotechnology; Rasooly, Ed.; Humana Press: USA, In press.
- Centi, S.; Tombelli, S.; Minnuni, M.; Mascini, M. Aptamer-based detection of plasma proteins by an electrochemical assay coupled to magnetic beads. Anal. Chem. 2007, 79, 1466–1473. [Google Scholar]
- Brezina, M.; Zuman, P. Polarography in medicine, biochemistry and pharmacy; Interscience: New York, 1958. [Google Scholar]
- Armstrong, F. A. Bioelectrochem; Wilson, G. S., Ed.; Wiley-VCH: Weinheim, 2002; pp. 11–29. [Google Scholar]
- Havran, L.; Billova, S.; Palecek, E. Electroactivity of avidin and streptavidin. Avidin signals at mercury and carbon electrodes respond to biotin binding. Electroanalysis 2004, 16, 1139–1148. [Google Scholar]
- Palecek, E.; Ostatna, V. Electroactivity of nonconjugated proteins and peptides. Towards electroanalysis of all proteins. Electroanalysis 2007, 19, 2383–2403. [Google Scholar]
- Tomschik, M.; Havran, L; Fojta, M.; Palecek, E. Constant current chronopotentiometric stripping analysis of bioactive peptides at mercury and carbon Electrodes. Electroanalysis 1998, 10, 403–409. [Google Scholar]
- Brdicka, R. Collect. Czech. Chem. Commun.; 1933; Volume 5, pp. 112–127. [Google Scholar]
- Heyrovsky, M. Early polarographic studies on proteins. Electroanalysis 2004, 16, 1067–1073. [Google Scholar]
- Tomschik, M.; Havran, L.; Palecek, E.; Heyrovsky, M. The presodium catalysis of electroreduction of hydrogen ions on mercury electrodes by metallothionein. An investigation by constant current derivative stripping chronopotentiometry. Electroanalysis 2000, 12, 274–279. [Google Scholar]
- Reynaud, J.A.; Malfoy, B.; Bere, A. The electrochemical oxidation of three proteins: RNAase A, bovine serum albumin and concanavalin A at solid electrodes. J. Electroanal. Chem. 1980, 116, 595–606. [Google Scholar]
- Brabec, V.; Schindlerova, I. Electrochemical behaviour of proteins at graphite electrodes Part III. The effect of protein adsorption. Bioelectrochem. Bioenerg. 1981, 8, 451–458. [Google Scholar]
- Brabec, V.; Mornstein, V. Electrochemical behaviour of proteins at graphite electrodes II. Electrooxidation of amino acids. Biophys. Chem. 1980, 12, 159–165. [Google Scholar]
- Reynolds, N. C., Jr.; Kissela, B. M.; Fleming, L. H. The voltammetry of neuropeptides containing L-tyrosine. Electroanalysis 1995, 7, 1177–1181. [Google Scholar]
- Cai, X.; Rivas, G.; Farias, P.A.M.; Shiraishi, H.; Wang, J.; Palecek, E. Potentiometric stripping analysis of biologically important bioactive peptides at carbon electrodes. Anal. Chim. Acta. 1996, 332, 49–57. [Google Scholar]
- Herzog, G.; Arrigan, D. W. M. Electrochemical strategies for label-free detection of amino acids, peptides and proteins. Analyst 2007, 132, 615–632. [Google Scholar]
- Vestergaard, M.; Kerman, K.; Saito, M.; Nagatani, N.; Takamura, Y.; Tamiya, E. J. Am. Chem. Soc. 2005, 127, 11892–11893.
- Kerman, K.; Vestergaard, M.; Chikae, M.; Yamamura, S.; Tamiya, E. Label-free electrochemical detection of phosphorylated and non-phosphorylated forms of peptides based on tyrosine oxidation. Electrochem. Commun. 2007, 9, 976–980. [Google Scholar]
- Kerman, K.; Vestergaard, M.; Tamiya, E. Label-free electrical sensing of small-molecule inhibition on tyrosine phosphorylation. Anal. Chem. 2007, 79, 6881–6885. [Google Scholar]
- Masařík, M.; Stobiecka, A.; Kizek, R.; Jelen, F.; Pechan, Z.; Hoyer, W.; Jovin, T.; Subramaniam, V.; Paleček, E. Sensitive electrochemical detection of native and aggregated alpha-synuclein protein involved in Parkinson's disease. Electroanalysis 2004, 16, 1172–1181. [Google Scholar]
- Kizek, R.; Trnkova, L.; Palecek, E. Determination of metallothionein at the femtomole level by constant current stripping chronopotentiometry. Anal. Chem. 2001, 73, 4801–4807. [Google Scholar]
- Adam, V.; Zehnalek, J.; Petrlova, J.; Potesil, D.; Sures, B.; Trnkova, L.; Jelen, F.; Vitecek, J.; Kizek, R. Phytochelatin modified electrode surface as a sensitive heavy-metal ion biosensor. Sensors 2005, 5, 70–84. [Google Scholar]
- Rogers, K. R. Principles of affinity-based biosensors. Mol. Biotechnol. 2000, 14, 109–129. [Google Scholar]
- Giaever, I. The antibody:antigen interaction: a visual observation. J. Immunol. 1973, 110, 1424–1426. [Google Scholar]
- Kronick, M. N.; Little, W. A. A new immunoassay based on fluorescent excitation by internal reflection spectroscopy. Proc. Natl. Acad. Sci. USA. 1974, 71, 4553–4555. [Google Scholar]
- Kerman, K.; Nagatani, N.; Chikae, M.; Yuhi, T.; Takamura, Y.; Tamiya, E. Label-free electrochemical immunoassay for the detection of human chorionic gonadotropin hormone. Anal. Chem. 2006, 78, 5612–5616. [Google Scholar]
- Takata, M.; Kerman, K.; Nagataki, N.; Konaka, H.; Namiki, M.; Tamiya, E. Label-free bioelectronic immunoassy for the detection of human telomerase reverse transcriptase in urine. J. Electroanal. Chem. 2006, 596, 109–116. [Google Scholar]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar]
- Zhao, Q.; Gan, Z.; Zhuang, Q. Electrochemical sensors based on carbon nanotubes. Electroanalysis 2002, 14, 1609–1613. [Google Scholar]
- Merkoci, A.; Alegret, S. Towards nanoanalytical chemistry: case of nanomaterial integration into [bio]sensing systems. Contributions to Science 2005, 3, 57–66. [Google Scholar]
- Britto, P. J.; Santhanam, K. S. V.; Ajayan, P. M. Carbon nanotube electrode for oxidation of dopamine. Bioelectrochem. Bioenerg. 1996, 41, 121–125. [Google Scholar]
- Zhao, W.; Song, C.H.; Pehrsson, P.E. Water-soluble and optically pH-sensitive single-walled carbon nanotubes from surface modifications. J. Am. Chem. Soc. 2002, 124, 12418–12419. [Google Scholar]
- Land, B. J.; Ruf, H. J.; Worman, J. J.; Raffaelle, R. P. Effects of alkyl amide solvents on the dispersal of single-wall carbon nanotubes. J. Phys. Chem. B 2004, 108, 17089–17095. [Google Scholar]
- Maehashi, K.; Katsura, T.; Kerman, K.; Takamura, Y.; Matsumoto, K.; Tamiya, E. Label-free protein biosensor based on aptamer-modified carbon nanotube field effect transistors. Anal. Chem. 2007, 79, 782–787. [Google Scholar]
- Okuno, J.; Mehashi, K.; Keman, K.; Takamura, Y.; Matsumoto, K.; Tamiya, E. Label-free immunosensor for prostate-specific antigen based on single-walled carbon nanotube array-modified microelectrodes. Biosens. Bioelectron. 2007, 22, 2377–2381. [Google Scholar]
- Bart, M.; Stigter, E. C. A.; Stapert, H. R.; de Jong, G. J.; van Bennekom, W. P. On response of a label-free interferon-γ immunosensor utilising electrochemical impedance spectroscopy. Biosens. Bioelectron. 2005, 21, 49–59. [Google Scholar]
- Tang, H.; Chen, J.; Nie, L.; Kuang, Y.; Yao, S. A label-free electrochemical immunassay for carcinoembryonic antigen (CEA) based on gold nanoparticles (AuNPs) and nonconductive polymer film. Biosens. Bioelectron. 2007, 22, 1061–1067. [Google Scholar]
- Zhang, S.; Huang, F.; Liu, B.; Ding, J.; Xu, X.; Kong, J. A sensitive impedance immunosensor based on functionalized gold nanoparticle–protein composite films for probing apolipoprotein A-I. Talanta 2007, 71, 874–881. [Google Scholar]
- Yang, L.; Li, Y.; Erf, G. F. Interdigitated array microelectrode-based electrochemical impedance immunosensor for detection of Escherichia coli O157:H7. Anal. Chem. 2004, 76, 1107–1113. [Google Scholar]
- Ellington, A. D.; Szostak, J. W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Wilson, D. S.; Szostak, J. W. In vitro selection of functional nucleic acids. Ann. Rev. Biochem. 1999, 68, 611–647. [Google Scholar]
- Jayasena, S. D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Cho, E. J.; Collett, J. R.; Szafranska, A. E.; Ellington, A. D. Optimization of aptamer microarray technology for multiple protein targets. Anal. Chim. Acta 2006, 564, 82–90. [Google Scholar]
- Musheev, M. U.; Krylov, S. N. Selection of aptamers by systematic evolution of ligands by exponential enrichment: Addressing the polymerase chain reaction issue. Anal. Chim. Acta 2006, 564, 91–96. [Google Scholar]
- Jenison, R. D.; Gill, S. C.; Pardi, A.; Polisky, B. High-resolution molecular discrimination by RNA. Science 1994, 263, 1425–1429. [Google Scholar]
- Jelen, F.; Fojta, M.; Palecek, E. Voltammetry of native double-stranded, denatured and degraded DNAs. J. Electroanal. Chem. 1997, 427, 49–56. [Google Scholar]
- Li, B.; Du, Y.; Wei, H.; Dong, S. Reusable, label-free electrochemical aptasensor for sensitive detection of small molecules. Chem. Commun. 2007, 3780–3783. [Google Scholar]
- Willner, I.; Zayats, M. Electronic aptamer-based sensors. Angew. Chem. Int. Ed. 2007, 46, 2–13. [Google Scholar]
- Fojta, M.; Havran, L.; Fulneckova, J.; Kubicarova, T. Adsorptive transfer stripping AC voltammetry of DNA complexes with intercalators. Electroanalysis 2000, 12, 926–934. [Google Scholar]
- Cai, X.; Rivas, G.; Farias, P.A.M.; Shiraishi, H.; Wang, J.; Palecek, E. Evaluation of different carbon electrodes for adsorptive stripping analysis of nucleic acids. Electroanalysis 1996, 8, 753–758. [Google Scholar]
- Steenken, S.; Jovanovic, S. V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar]
- Palecek, E; Jelen, F. Electrochemistry of nucleic acids, in Electrochemistry of nucleic acids and proteins. Towards electrochemical sensors for genomics and proteomics; Palecek, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, 2005; pp. 74–174. [Google Scholar]
- Meric, B.; Kerman, K.; Ozkan, D.; Kara, P.; Ozsoz, M. Indicator-free electrochemical DNA biosensor based on adenine and guanine signals. Electroanalysis 2002, 14, 1245–50. [Google Scholar]
- Kerman, K.; Ozkan, D.; Kara, P.; Erdem, A.; Meric, B.; Nielsen, P. E.; Ozsoz, M. Label-free bioelectronic detection of point mutation by using peptide nucleic acid probes. Electroanalysis 2003, 15, 667–70. [Google Scholar]
- Mascini, M.; Bagni, G.; DiPietro, M. L.; Ravera, M.; Baracco, S.; Osella, D. Electrochemical biosensor evaluation of the interaction between DNA and metallo-drugs. Biometals 2006, 19, 409–418. [Google Scholar]
- Kerman, K.; Morita, Y.; Takamura, Y.; Tamiya, E. Escherichia coil Single-strand binding protein-DNA interactions on carbon nanotube-modified electrodes from a label-free electrochemical hybridization sensor. Anal. Bioanal. Chem. 2005, 381, 1114–1121. [Google Scholar]
- Erdem, A.; Kosmider, B.; Osiecka, R.; Zyner, E.; Ochocki, J.; Ozsoz, M. Electrochemical genosensing of the interaction between the potential chemotherapeutic agent, cis-bis(3-aminoflavone)dichloroplatinum(II) and DNA in comparison with cis-DDP. J. Pharm. Biomed. Anal. 2005, 38, 645–652. [Google Scholar]
- Xie, H.; Yang, D.; Heller, A.; Gao, Z. Electrocatalytic oxidation of guanine, guanosine, and guanosine monophosphate. Biohys. J. 2007, 92, L70–L72. [Google Scholar]
- So, H-M.; Won, K.; Hwan Kim, Y. H.; Kim, B-K.; Ryu, B. H.; Na, P. S.; Kim, H.; Lee, J-O. Single-walled carbon nanotube biosensors using aptamers as molecular recognition elements. J. Am. Chem. Soc. 2005, 127, 11906–11907. [Google Scholar]
- Rodriquez, M. C.; Kawde, A.-N.; Wang, J. Aptamer biosensor for label-free impedance spectroscopy detection of proteins based on recognition-induced switching of the surface charge. Chem. Commun. 2005, 4267–4269. [Google Scholar]
- Kawde, A.-N.; Rodriquez, M. C.; Lee, T. M. H.; Wang, J. Label-free bioelectronic detection of aptamer-protein interactions. Electrochem. Commun. 2005, 7, 537–540. [Google Scholar]
- Xu, D.; Xu, D.; Yu, X.; Liu, Z.; He, W.; Ma, Z. Label-free electrochemical detection of aptamer-based array electrodes. Anal. Chem. 2005, 77, 5107–5113. [Google Scholar]
- Cai, H.; Ming-Hung, T.; Hsing, I-M. Label-free protein recognition using an aptamer-based impedance measurement assay. Sens. Actuat. B Chem. 2006, 114, 433–437. [Google Scholar]
- Vestergaard, M.; Kerman, K.; Tamiya, E. The study of Alzheimer's disease biomarkers: current role and future prospects of nanosensor technology. NanoBiotechnol. 2006, 2, 5–16. [Google Scholar]
- Pumera, M.; Sánchez, S.; Ichinose, I.; Tang, J. Electrochemical nanobiosensors. Sens. Actuat. B Chem. 2007, 123, 1195–1205. [Google Scholar]
- Endo, T.; Kerman, K.; Nagatani, N.; Hiepa, H. M.; Kim, D-K.; Yonezawa, Y.; Nakano, K.; Tamiya, E. Multiple label-free detection of antigen-antibody reaction using localised surface plasmon resonance-based core-shell structured layer nanochip. Anal. Chem. 2006, 78, 6465–6475. [Google Scholar]
- Vestergaard, M.; Kerman, K.; Kim, D-K.; Hiep, H. M.; Tamiya, E. Detection of Alzheimer's tau protein using localised surface plasmon resonance-based immunochip. Talanta 2007. Published online. [Google Scholar]
- Haes, A. J.; Hal, P. W.; Chang, L.; Klein, W. L.; Van Duyne, R. P. A localised surface plasmon resonance biosensor: first steps toward an assay for Alzheimer's disease. Nano Lett. 2004, 4, 1029–1034. [Google Scholar]
- Haes, A.J.; Chang, L.; Klein, W.L.; Van Duyne, R. P. Detection of a biomarker for Alzheimer's disease from synthetic and clinical samples using a nanoscale optical biosensor. J. Am. Chem. Soc. 2005, 127, 2264–2271. [Google Scholar]
- Jensen, T.; Kelly, L.; Lazarides, A.; Schatz, G.C. Electrodynamics of noble metal nanoparticles and nanoparticle clusters. J. Clust. Sci. 1999, 10, 295–317. [Google Scholar]
- Haynes, C. L.; Van Duyne, R. P. Nanosphere Lithography: A versatile nanofabrication tool for studies of size-dependent nanoparticle optics. J. Phys. Chem. B 2001, 105, 5599–5611. [Google Scholar]
- Kerman, K.; Chikae, M.; Yamamura, S.; Tamiya, E. Gold nanoparticle-based electrochemical detection of protein phosphorylation. Anal. Chim. Acta 2007, 588, 26–33. [Google Scholar]
- Chen, Z-P.; Peng, Z-F.; Zhang, P.; Jin, X-F.; Jiang, J-H.; Zhang, X-B.; Shen, G-L.; Yu, R-Q. A sensitive immunosensor using colloidal gold as electrochemical label. Talanta 2007, 72, 1800–1804. [Google Scholar]
- Ambrosi, A.; Castaneda, M. T.; Killard, A. J.; Smyth, M. R.; Alegret, S.; Merkoci, A. Double-codified gold nanolabels for enhanced immunoanalysis. Anal. Chem. 2007, 79, 5232–5240. [Google Scholar]
- Pumera, M.; Castaneda, M. T.; Pividor, M. I.; Eritja, R.; Merkoci, A.; Alegret, S. Magnetically trigged direct electrochemical detection of DNA hybridization using Au67 quantum dot as electrical tracer. Langmuir 2005, 21, 9625–9629. [Google Scholar]
- Pumera, M.; Aldavert, M.; Mills, C.; Merkoci, A.; Alegret, S. Direct voltammetric determination of gold nanoparticles using graphite-epoxy composite electrode. Electrochim. Acta 2005, 50, 3702–3707. [Google Scholar]
- Gonzales-Garcia, M. B.; Costa-Garcia, A. Adsorptive stripping voltammetric behaviour of colloidal gold and immunogold on carbon paste electrode. Bioelectrochem. Bioenerg. 1995, 38, 389–395. [Google Scholar]
- Sánchez, S.; Pumera, M.; Cabruja, E.; Fàbregas, E. Carbon nanotube/polysulfone composite screen-printed electrochemical enzyme biosensors. Analyst. 2007, 132, 142. [Google Scholar]
- An, C. D.; Yoshiki, T.; Lee, G.; Okada, Y. Evaluation of a rapid qualitative prostate specific antigen assay, the One Step PSATM test. Cancer Lett. 2001, 162, 135–139. [Google Scholar]
- Tanaka, R.; Teruko, Y.; Nagatani, N.; Endo, T.; et al. A novel enhancement assay for immunochromatographic test strips using gold nanoparticles. Anal. Bioanal. Chem. 2006, 385, 1414–1420. [Google Scholar]
- Yuhi, T.; Nagatani, N.; Endo, T.; Kerman, K.; Takata, M.; Konaka, H.; Namiki, M.; Takamura, Y.; Tamiya, E. Resin-based micropipette tip for immunochromatographic assays in urine samples. J. Immunol. Meth. 2006, 312, 54–60. [Google Scholar]
- Ciesiolka, T.; Gabius, H.-J. An 8- to 10-fold enhancement in sensitivity for quantification of proteins by modified application of colloidal gold. Anal. Biochem. 1988, 168, 280–283. [Google Scholar]
- Anderson, N. L.; Anderson, N. G. The human plasma proteome. Mol. Cell Proteomics 2002, 1, 845–867. [Google Scholar]
- Fountoulakis, M.; Juranville, J.-F.; Jiang, L.; Avila, D.; Roder, D.; Jakob, P.; Berrndt, P.; Evers, S.; Langen, H. Depletion of high-abundance plasma proteins. Amino Acids. 2004, 27, 249–259. [Google Scholar]
- Greenough, C.; Jenkins, R. E.; Kitteringham, N.R.; Pirmohamed, M.; Park, K.; Pennington, S.R. A method for the repaid depletion of albumin and immunoglobulin from human plasma. Proteomics 2004, 4, 3107–3111. [Google Scholar]
- Vestergaard, M.; Tamiya, E. A rapid sample pretreatment protocol: improved sensitivity in the detection of a low-abundant serum biomarker for prostate cancer. Anal. Sci. 2007, 23, 1–4. [Google Scholar]
Figure 1.
Electrochemical responses of 80 μM Aβ-42 before (A) and after (B) aggregation following an incubation for 120 min in TBS at 37±1°C. Square wave voltammetric measurements were taken using a glassy carbon electrode as the working electrode in a three-electrode system including a reference electrode (Ag/AgCl) and a Pt wire as the counter electrode; (C) Plot for the dependence of the fluorescence responses of Thioflavin-T on incubation period in the presence of 80 μM Aβ-42 in TBS at 37±1°C.
Figure 1.
Electrochemical responses of 80 μM Aβ-42 before (A) and after (B) aggregation following an incubation for 120 min in TBS at 37±1°C. Square wave voltammetric measurements were taken using a glassy carbon electrode as the working electrode in a three-electrode system including a reference electrode (Ag/AgCl) and a Pt wire as the counter electrode; (C) Plot for the dependence of the fluorescence responses of Thioflavin-T on incubation period in the presence of 80 μM Aβ-42 in TBS at 37±1°C.
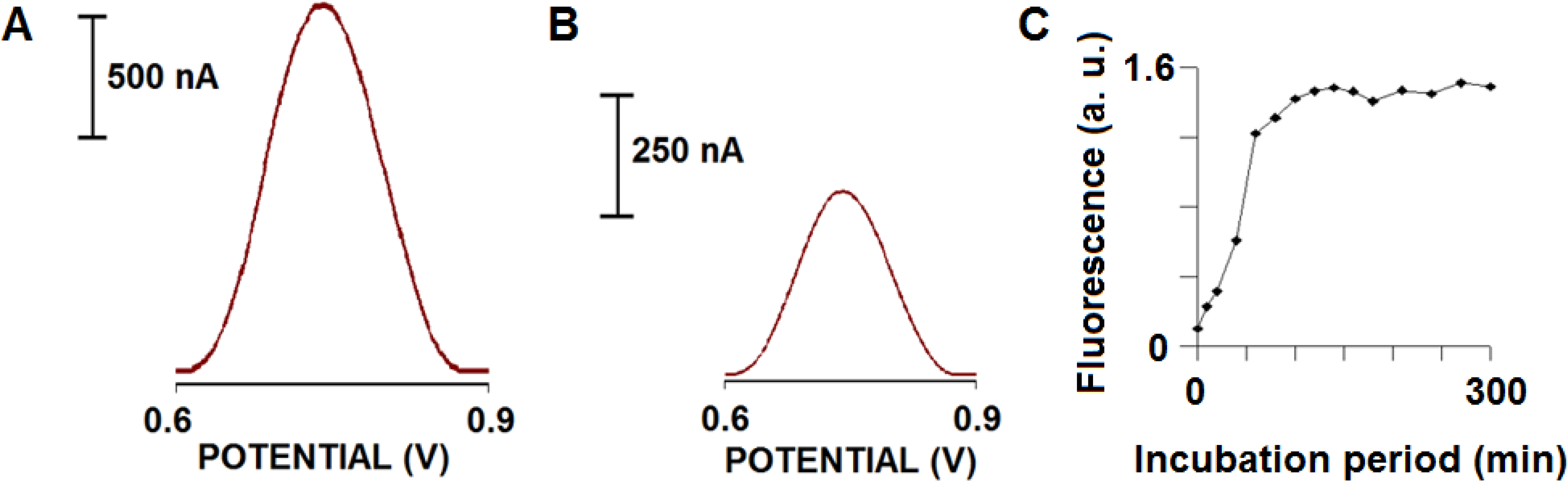
Figure 2.
Schematic illustration for the label-free detection of tyrosine-kinase catalysed peptide phosphorylation. The peptides that are conjugated with a magnetic bead (MB) contain a single phosphorylation site such as tyrosine (Tyr). Since Tyr has intrinsic electro-activity, the current response from its voltammetric oxidation is monitored. Under optimized conditions, Tyr residue is phosphorylated in the presence of a tyrosine kinase and ATP. During phosphorylation, the phosphate group at the γ-position of ATP is transferred to the hydroxyl group of Tyr. The intrinsic electro-activity of Tyr is lost upon phosphorylation and the current response decays with the increasing concentration of the tyrosine kinase.
Figure 2.
Schematic illustration for the label-free detection of tyrosine-kinase catalysed peptide phosphorylation. The peptides that are conjugated with a magnetic bead (MB) contain a single phosphorylation site such as tyrosine (Tyr). Since Tyr has intrinsic electro-activity, the current response from its voltammetric oxidation is monitored. Under optimized conditions, Tyr residue is phosphorylated in the presence of a tyrosine kinase and ATP. During phosphorylation, the phosphate group at the γ-position of ATP is transferred to the hydroxyl group of Tyr. The intrinsic electro-activity of Tyr is lost upon phosphorylation and the current response decays with the increasing concentration of the tyrosine kinase.
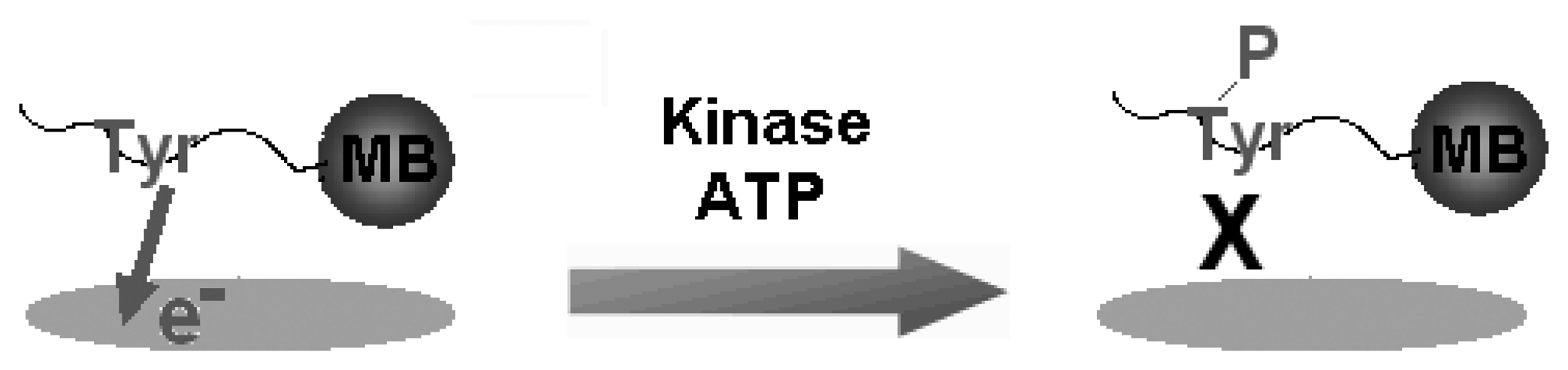
Figure 3.
Schematic illustration for the general immunosensor formats. (A) Sandwich-type immunoassay with a labeled-secondary antibody. After the binding of the target antigen to the primary antibody-modified surface, the secondary antibody with a label is introduced to the form the sandwich-type complex. Various molecules such an enzyme or a nanoparticle can act as the source of electrochemical responses. (B) In a competitive-type immunosensor, the target molecule competes with its labeled form to attach on the primary antibodies on the surface. The electrochemical responses obtained from the labeled antigens are inversely proportional to the concentration of the target antigen. High sensitivity levels can be reached using competitive immunosensing strategies.
Figure 3.
Schematic illustration for the general immunosensor formats. (A) Sandwich-type immunoassay with a labeled-secondary antibody. After the binding of the target antigen to the primary antibody-modified surface, the secondary antibody with a label is introduced to the form the sandwich-type complex. Various molecules such an enzyme or a nanoparticle can act as the source of electrochemical responses. (B) In a competitive-type immunosensor, the target molecule competes with its labeled form to attach on the primary antibodies on the surface. The electrochemical responses obtained from the labeled antigens are inversely proportional to the concentration of the target antigen. High sensitivity levels can be reached using competitive immunosensing strategies.
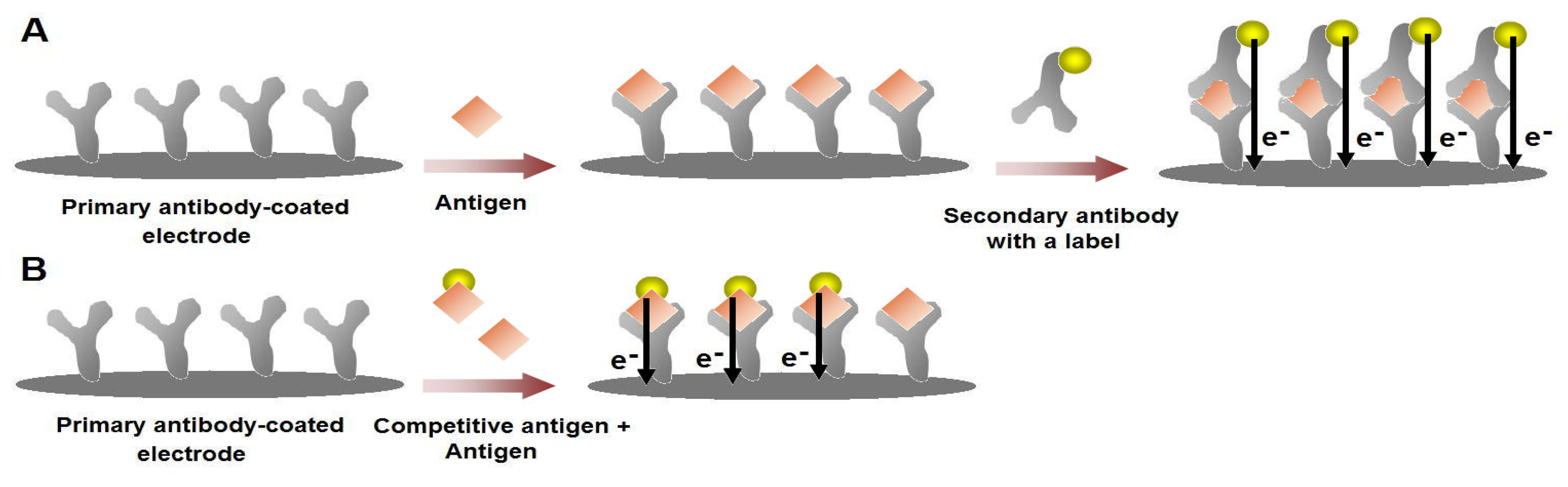
Figure 4.
Schematic illustration for the label-free voltammetric immunosensors. The primary antibody is coated on the electrode surface and the electro-active residues in the antibody structure give a specific current response. Upon the binding of the target antigen with the antibody, the specific current response of the antibody layer changes. This change can either be an increase or a decrease depending on the structure of the antigen and/or the conformational changes that would occur upon the formation of the antigen-antibody complex. Then, the current responses would be further altered with the binding of the secondary antibody, which would also contribute to the current response with its intrinsic electro-activity.
Figure 4.
Schematic illustration for the label-free voltammetric immunosensors. The primary antibody is coated on the electrode surface and the electro-active residues in the antibody structure give a specific current response. Upon the binding of the target antigen with the antibody, the specific current response of the antibody layer changes. This change can either be an increase or a decrease depending on the structure of the antigen and/or the conformational changes that would occur upon the formation of the antigen-antibody complex. Then, the current responses would be further altered with the binding of the secondary antibody, which would also contribute to the current response with its intrinsic electro-activity.
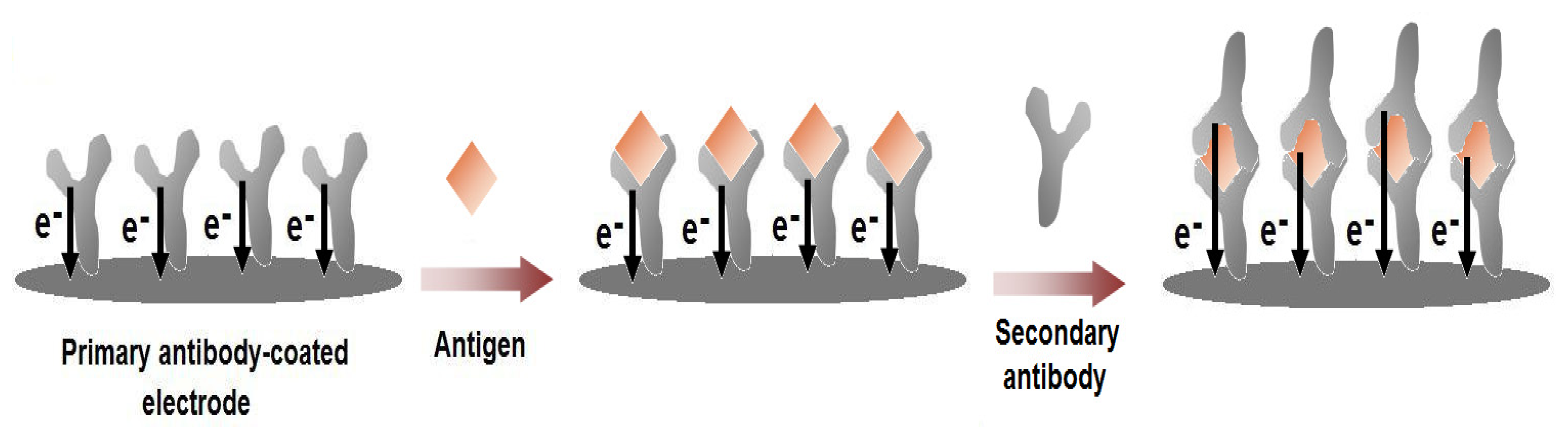
Figure 5.
Schematic illustration of the label-free immunosensors using single-walled carbon nanotubes (SWCNTs). SWCNTs were grown directly on the platinum electrode surface. Upon the covalent attachment of the antibodies on the SWCNTs, a current response is recorded from the intrinsic electro-activity of the antibodies. As the antigens are bound with the antibodies, the electrochemical responses change. The current height at the peak oxidation potential (∼0.5 V vs. Ag/AgCl) increases or decreases depending on the concentration of the bound antigens.
Figure 5.
Schematic illustration of the label-free immunosensors using single-walled carbon nanotubes (SWCNTs). SWCNTs were grown directly on the platinum electrode surface. Upon the covalent attachment of the antibodies on the SWCNTs, a current response is recorded from the intrinsic electro-activity of the antibodies. As the antigens are bound with the antibodies, the electrochemical responses change. The current height at the peak oxidation potential (∼0.5 V vs. Ag/AgCl) increases or decreases depending on the concentration of the bound antigens.
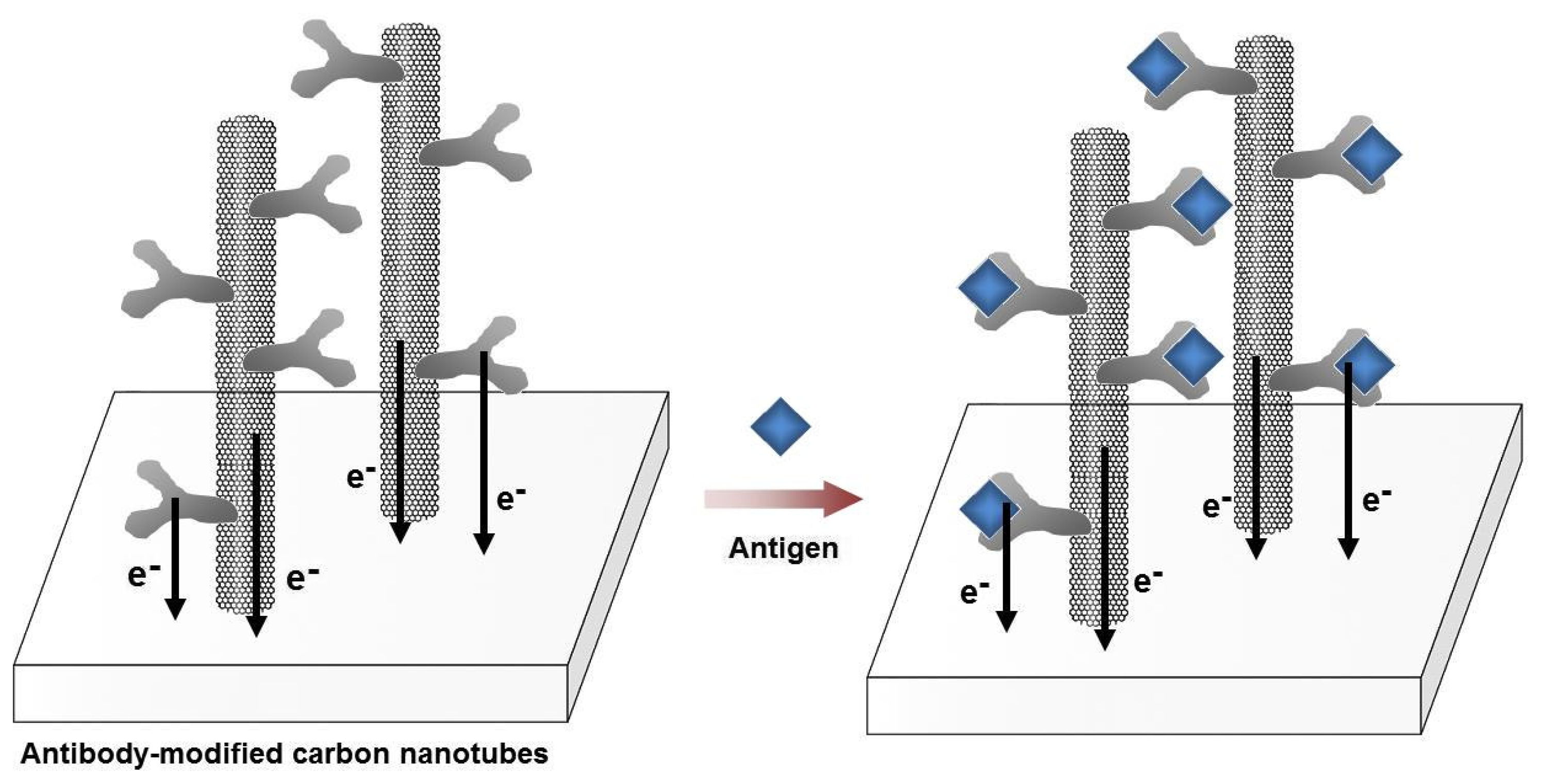
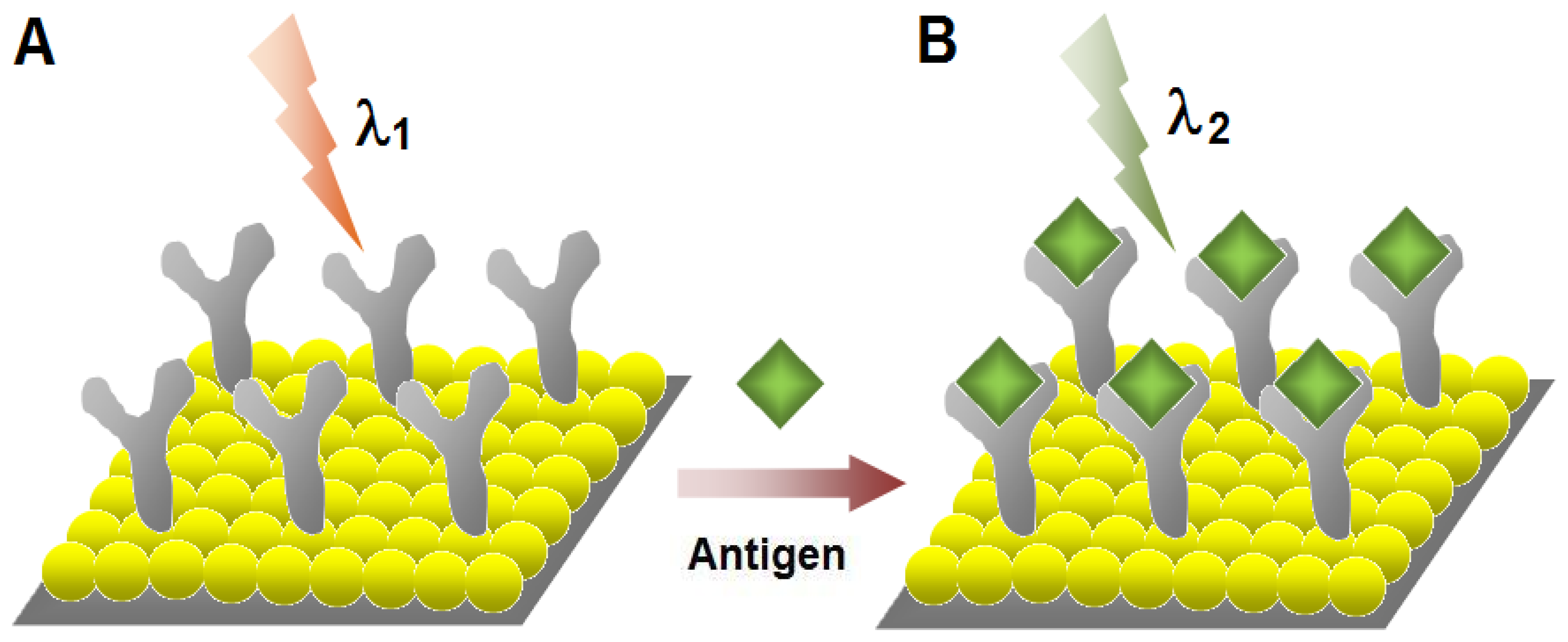
Figure 6.
Schematic illustration for the label-free optical detection of target biomolecules using localized surface plasmon resonance (LSPR) of metal nanoparticles (NPs). (A) LSPR absorbance of the metal NPs (λ1) change as the layer of antibodies forms on the surface. (B) The binding of the antigens with the antibodies increases the layers on the surface and the absorbance intensity of the metal NPs increase (λ2) leading to sensitive detection of biorecognition events.
Figure 6.
Schematic illustration for the label-free optical detection of target biomolecules using localized surface plasmon resonance (LSPR) of metal nanoparticles (NPs). (A) LSPR absorbance of the metal NPs (λ1) change as the layer of antibodies forms on the surface. (B) The binding of the antigens with the antibodies increases the layers on the surface and the absorbance intensity of the metal NPs increase (λ2) leading to sensitive detection of biorecognition events.

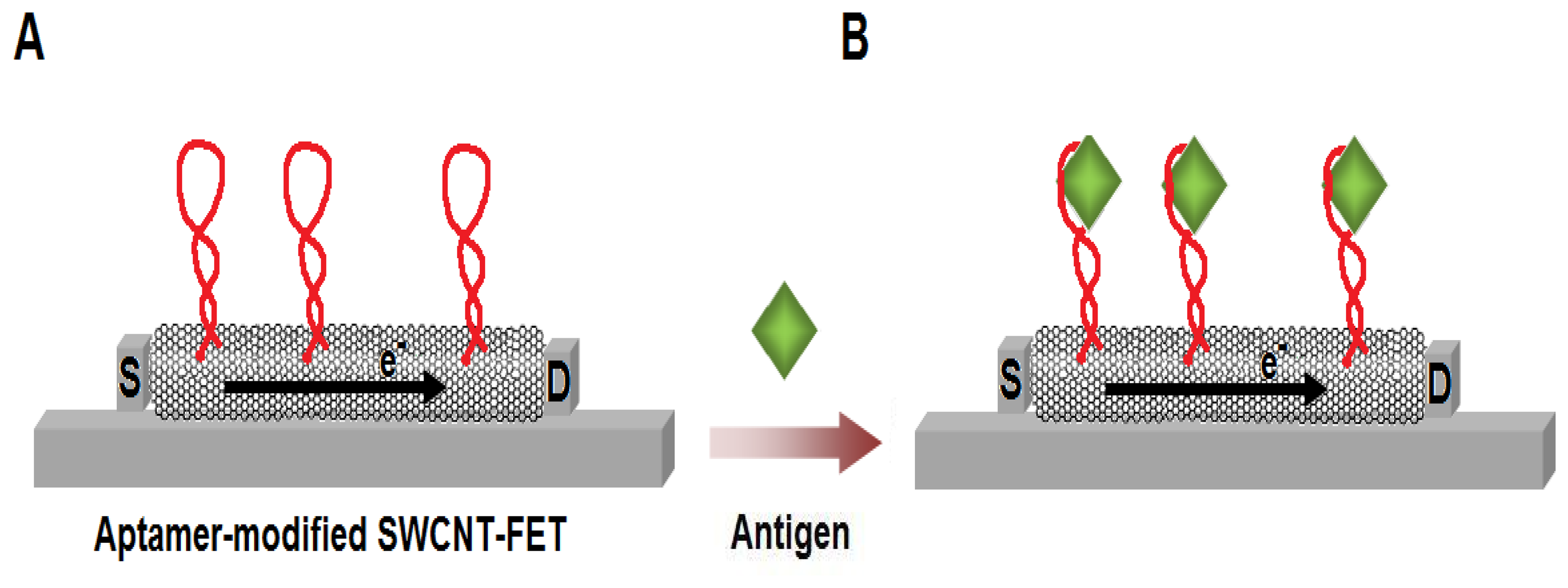
Figure 7.
Schematic illustration of the SWCNT-based field-effect transistor (SWCNT-FET) for the label-free aptamer-based sensing. (A) As the aptamers are attached on the SWCNT, the electrical flow from the source (S) to the drain (D) of the system changes. (B) The current flow is further altered upon the binding of the antigens to the aptamers.
Figure 7.
Schematic illustration of the SWCNT-based field-effect transistor (SWCNT-FET) for the label-free aptamer-based sensing. (A) As the aptamers are attached on the SWCNT, the electrical flow from the source (S) to the drain (D) of the system changes. (B) The current flow is further altered upon the binding of the antigens to the aptamers.
© 2007 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Vestergaard, M.; Kerman, K.; Tamiya, E. An Overview of Label-free Electrochemical Protein Sensors. Sensors 2007, 7, 3442-3458. https://doi.org/10.3390/s7123442
AMA Style
Vestergaard M, Kerman K, Tamiya E. An Overview of Label-free Electrochemical Protein Sensors. Sensors. 2007; 7(12):3442-3458. https://doi.org/10.3390/s7123442
Chicago/Turabian StyleVestergaard, Mun\'delanji, Kagan Kerman, and Eiichi Tamiya. 2007. "An Overview of Label-free Electrochemical Protein Sensors" Sensors 7, no. 12: 3442-3458. https://doi.org/10.3390/s7123442