Modifications of Poly(o-phenylenediamine) Permselective Layer on Pt-Ir for Biosensor Application in Neurochemical Monitoring

Abstract
:1. Introduction
2. Experimental Section
2.1. Instrumentation and Software
2.2. Reagents and Solutions
2.3. Electrode Preparation and Polymer Modification
2.4. Electrode Characterization and Data Analysis
3. Results and Discussion
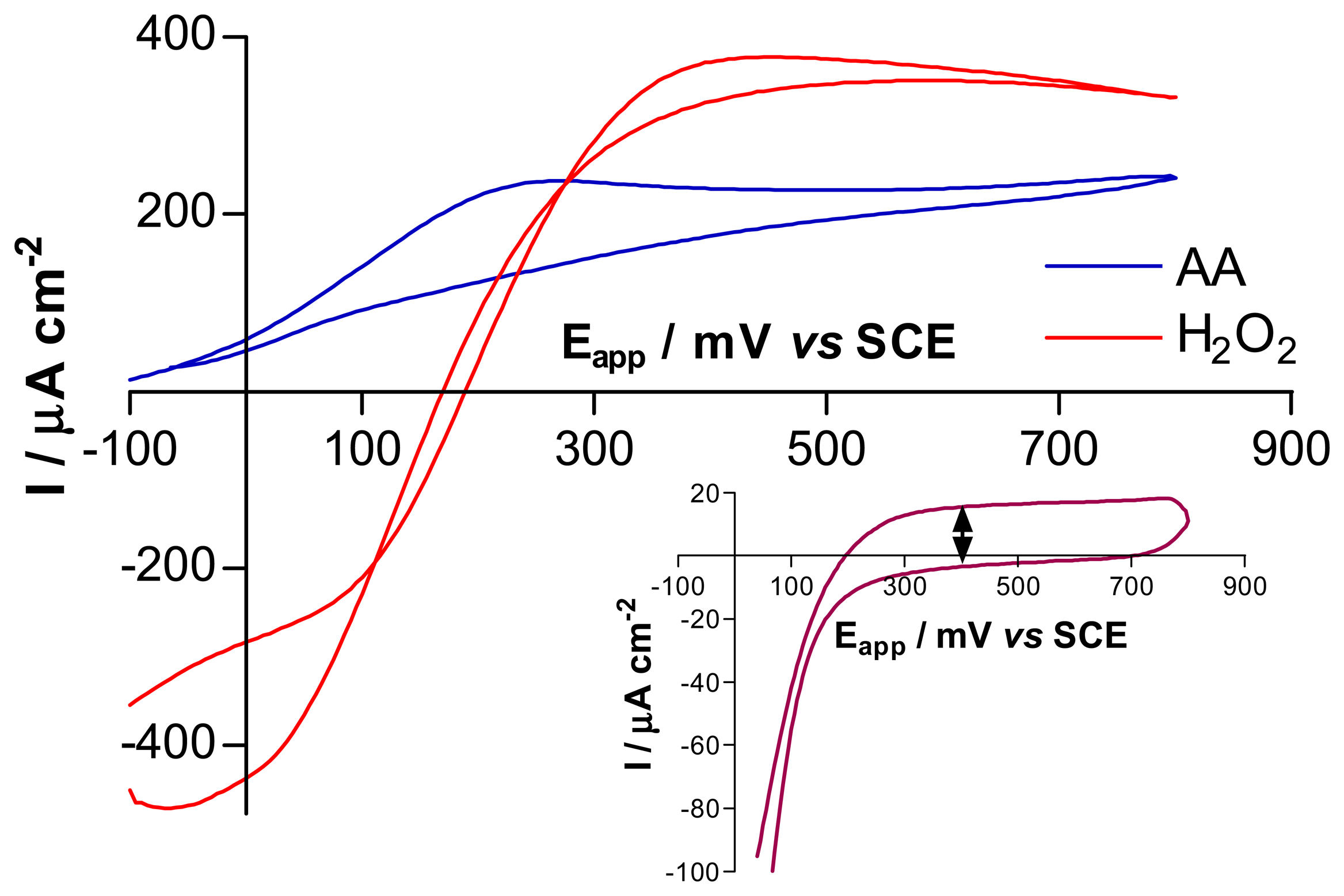
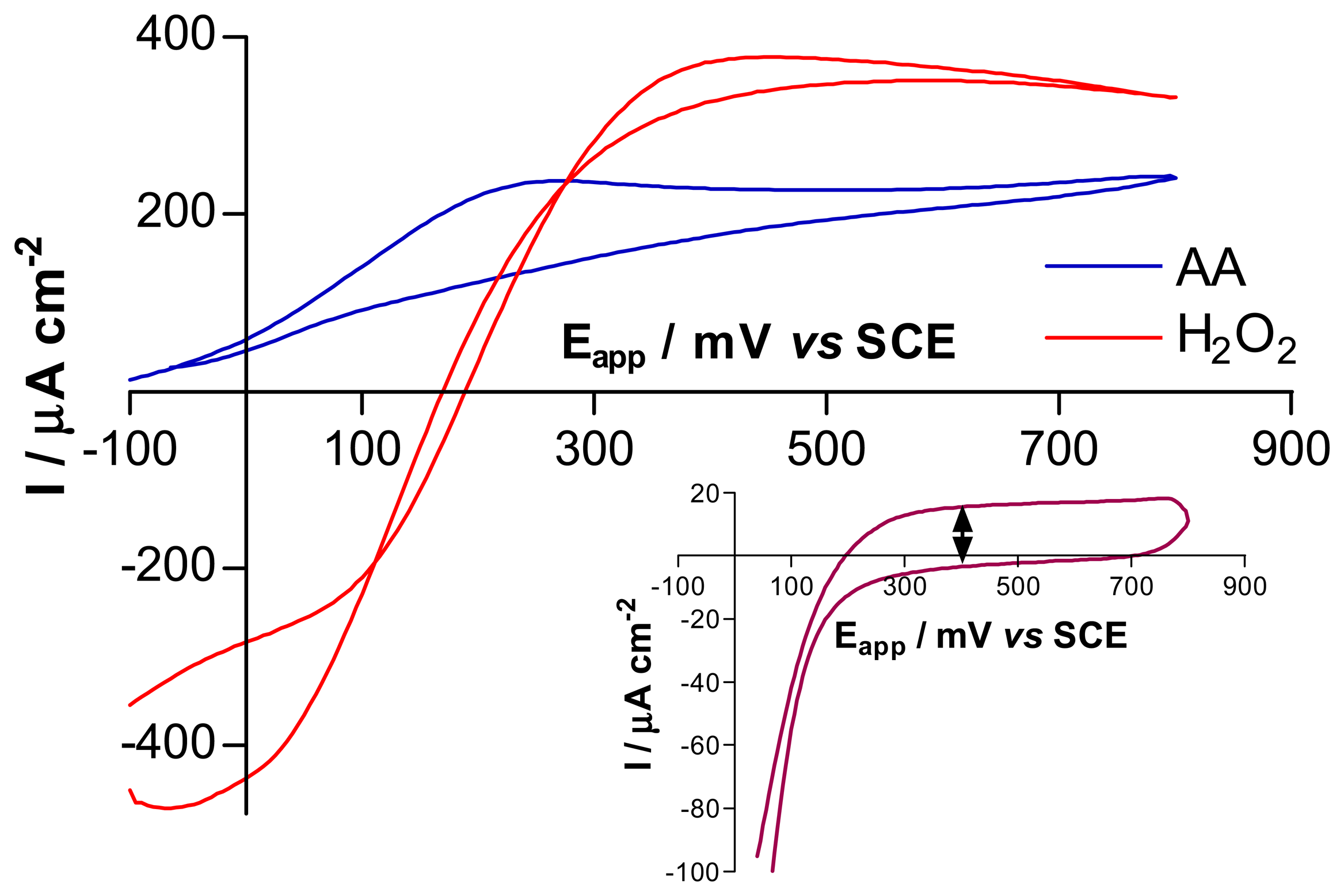
3.1. Comparison of Pt and Pt-Ir, using cyclic voltammetry and amperometry
3.2. Comparison of H2O2 sensitivity at bare and PPD-modified electrodes
3.3. Comparison of oPD and oPD-Me as monomers
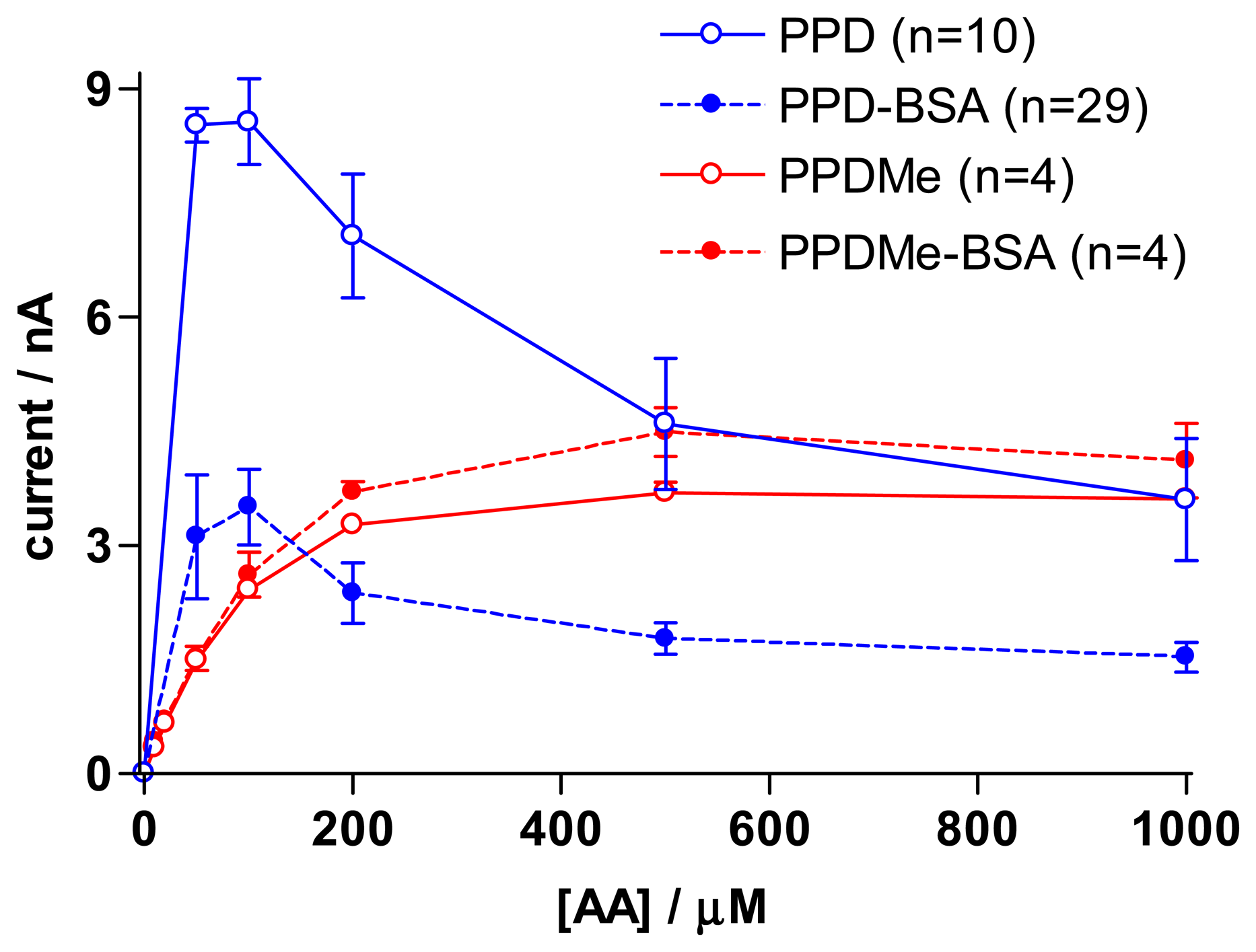
3.4. Effect of albumins in the monomer solution on resulting polymers
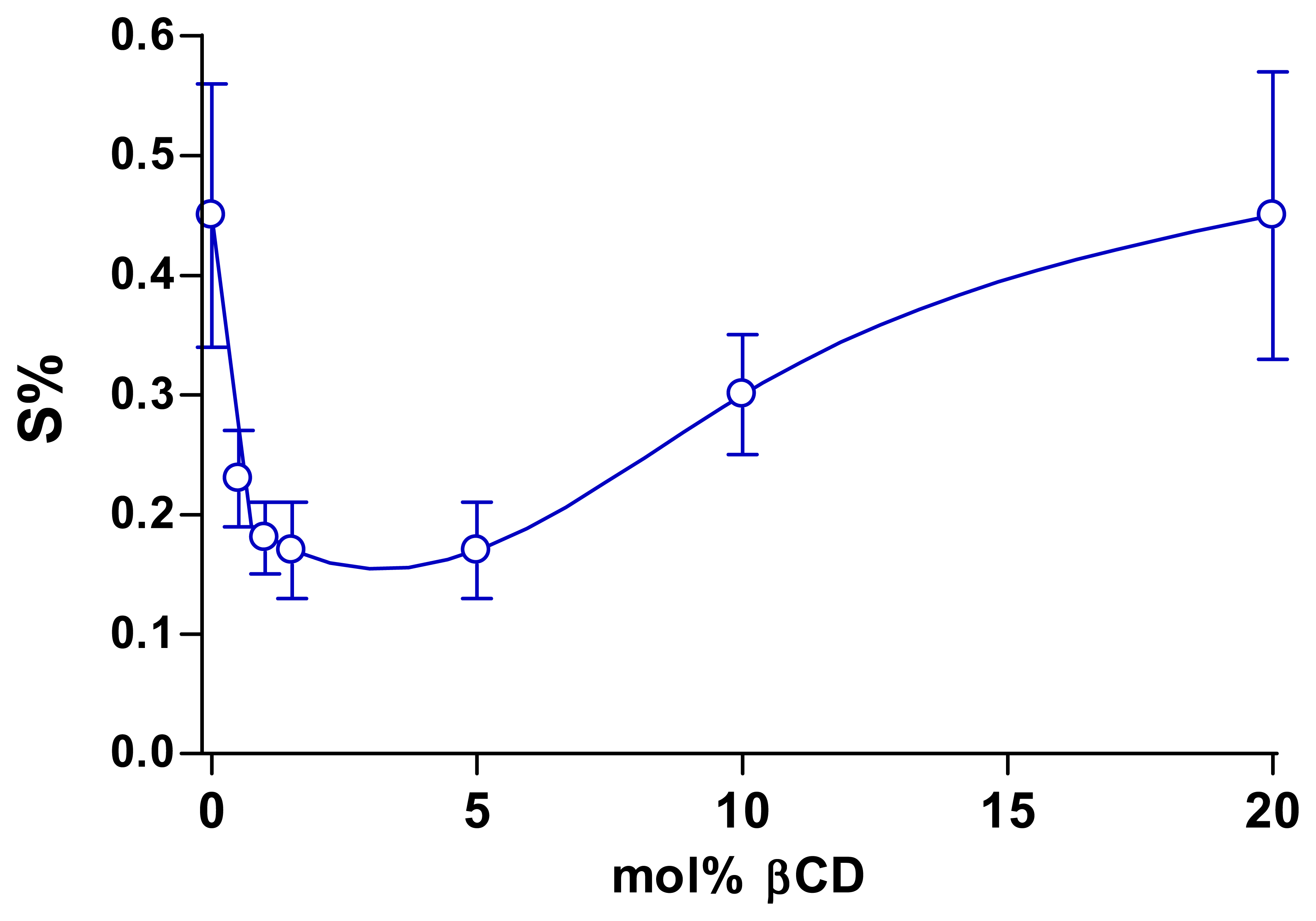
3.5. Effect of other modifiers in the monomer solution on resulting polymers
4. Conclusions
Acknowledgments
References and Notes
- Wilson, G.S.; Gifford, R. Biosensors for real-time in vivo measurements. Biosens. Bioelectron. 2005, 20, 2388–2403. [Google Scholar]
- Gorton, L. Carbon paste electrodes modified with enzymes, tissues, and cells. Electroanalysis 1995, 7, 23–45. [Google Scholar]
- Shear, J.B.; Fishman, H.A.; Allbritton, N.L.; Garigan, D.; Zare, R.N.; Scheller, R.H. Single cells as biosensors for chemical separations. Science 1994, 267, 74–77. [Google Scholar]
- Raoof, J.B.; Ojani, R.; Kiani, A. Apple-modified carbon paste electrode: A biosensor for selective determination of dopamine in pharmaceutical formulations. Bull. Electrochem. 2005, 21, 223–228. [Google Scholar]
- Wang, J.; Wu, L.H.; Martinez, S.; Sanchez, J. Tissue bioelectrode for eliminating protein interferences. Anal. Chem. 1991, 63, 398–400. [Google Scholar]
- Bernhardt, P.V. Enzyme Electrochemistry - Biocatalysis on an electrode. Aust. J. Chem. 2006, 59, 233–256. [Google Scholar]
- Zhang, M.N.; Mao, L.Q. Enzyme-based amperometric biosensors for continuous and on-line monitoring of cerebral extracellular microdialysate. Front. Biosci. 2005, 10, 345–352. [Google Scholar]
- Wilson, G.S.; Hu, Y.B. Enzyme based biosensors for in vivo measurements. Chem. Rev. 2000, 100, 2693–2704. [Google Scholar]
- Ikeda, T. Enzyme-modified electrodes with bioelectrocatalytic function (review). Bunseki Kagaku 1995, 44, 333–354. [Google Scholar]
- O'Neill, R.D.; Lowry, J.P.; Mas, M. Monitoring brain chemistry in vivo: voltammetric techniques, sensors and behavioral applications. Crit. Rev. Neurobiol. 1998, 12, 69–127. [Google Scholar]
- Kusakabe, H.; Midorikawa, Y.; Fujishima, T.; Kuninaka, A.; Yoshino, H. Purification and properties of a new enzyme, L-glutamate oxidase, from Streptomyces sp. X-119-6 grown on wheat bran. Agric. Biol. Chem. 1983, 47, 1323–1328. [Google Scholar]
- McMahon, C.P.; Rocchitta, G.; Kirwan, S.M.; Killoran, S.J.; Serra, P.A.; Lowry, J.P.; O'Neill, R.D. Oxygen tolerance of an implantable polymer/enzyme composite glutamate biosensor displaying polycation-enhanced substrate sensitivity. Biosens. Bioelectron. 2007, 22, 1466–1473. [Google Scholar]
- Hamdi, N.; Wang, J.J.; Walker, E.; Maidment, N.T.; Monbouquette, H.G. An electroenzymatic L-glutamate microbiosensor selective against dopamine. J. Electroanal. Chem. 2006, 591, 33–40. [Google Scholar]
- McMahon, C.P.; Rocchitta, G.; Serra, P.A.; Kirwan, S.M.; Lowry, J.P.; O'Neill, R.D. Control of the oxygen dependence of an implantable polymer/enzyme composite biosensor for glutamate. Anal. Chem. 2006, 78, 2352–2359. [Google Scholar]
- Oldenziel, W.H.; Dijkstra, G.; Cremers, T.I.F.H.; Westerink, B.H.C. Evaluation of hydrogel-coated glutamate microsensors. Anal. Chem. 2006, 78, 3366–3378. [Google Scholar]
- Kulagina, N.V.; Shankar, L.; Michael, A.C. Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal. Chem. 1999, 71, 5093–5100. [Google Scholar]
- Day, B.K.; Pomerleau, F.; Burmeister, J.J.; Huettl, P.; Gerhardt, G.A. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J. Neurochem. 2006, 96, 1626–1635. [Google Scholar]
- Lowry, J.P.; O'Neill, R.D. Neuroanalytical chemistry in vivo using electrochemical sensors. In Encyclopedia of Sensors; Grimes, C.A., Dickey, E.C., Pishko, M.V., Eds.; American Scientific Publishers: California, 2006; pp. 501–524. [Google Scholar]
- Dale, N.; Hatz, S.; Tian, F.M.; Llaudet, E. Listening to the brain: microelectrode biosensors for neurochemicals. Trends Biotechnol. 2005, 23, 420–428. [Google Scholar]
- Pantano, P.; Kuhr, W.G. Enzyme-modified microelectrodes for in vivo neurochemical measurements. Electroanalysis 1995, 7, 405–416. [Google Scholar]
- Wang, J. Electroanalysis and biosensors. Anal. Chem. 1995, 67, R487–R492. [Google Scholar]
- Ao, X.P.; Stenken, J.A. Microdialysis sampling of cytokines. Methods 2006, 38, 331–341. [Google Scholar]
- Watson, C.J.; Venton, B.J.; Kennedy, R.T. In vivo measurements of neurotransmitters by microdialysis sampling. Anal. Chem. 2006, 78, 1391–1399. [Google Scholar]
- Fillenz, M. In vivo neurochemical monitoring and the study of behaviour. Neurosci. Biobehav. Rev. 2005, 29, 949–962. [Google Scholar]
- Duff, A.; O'Neill, R.D. Effect of probe size on the concentration of brain extracellular uric acid monitored with carbon paste electrodes. J. Neurochem. 1994, 62, 1496–1502. [Google Scholar]
- O'Neill, R.D.; Gonzalez-Mora, J.L.; Boutelle, M.G.; Ormonde, D.E.; Lowry, J.P.; Duff, A.; Fumero, B.; Fillenz, M.; Mas, M. Anomalously high concentrations of brain extracellular uric acid detected with chronically implanted probes: implications for in vivo sampling techniques. J. Neurochem. 1991, 57, 22–29. [Google Scholar]
- Calvo, E.J.; Etchenique, R.; Danilowicz, C.; Diaz, L. Electrical communication between electrodes and enzymes mediated by redox hydrogels. Anal. Chem. 1996, 68, 4186–4193. [Google Scholar]
- El Atrash, S.S.; O'Neill, R.D. Characterisation in vitro of a naphthoquinone-mediated glucose oxidase-modified carbon paste electrode designed for neurochemical analysis in vivo. Electrochim. Acta 1995, 40, 2791–2797. [Google Scholar]
- Kulys, J.; Wang, L.; Hansen, H.E.; Buchrasmussen, T.; Wang, J.; Ozsoz, M. Methylene-green-mediated carbon paste glucose biosensor. Electroanalysis 1995, 7, 92–94. [Google Scholar]
- Nagy, G.; Rice, M.E.; Adams, R.N. A new type of enzyme electrode: the ascorbic acid eliminator electrode. Life Sci. 1982, 31, 2611–2616. [Google Scholar]
- Sternberg, R.; Bindra, D.S.; Wilson, G.S.; Thevenot, D.R. Covalent enzyme coupling on cellulose acetate membranes for glucose sensor development. Anal. Chem. 1988, 60, 2781–2786. [Google Scholar]
- Fan, Z.H.; Harrison, D.J. Permeability of glucose and other neutral species through recast perfluorosulfonated ionomer films. Anal. Chem. 1992, 64, 1304–1311. [Google Scholar]
- Malitesta, C.; Palmisano, F.; Torsi, L.; Zambonin, P.G. Glucose fast-response amperometric sensor based on glucose oxidase immobilized in an electropolymerized poly(o-phenylenediamine) film. Anal. Chem. 1990, 62, 2735–2740. [Google Scholar]
- Ohnuki, Y.; Matsuda, H.; Ohsaka, T.; Oyama, N. Permselectivity of films prepared by electrochemical oxidation of phenol and amino-aromatic compounds. J. Electroanal. Chem. 1983, 158, 55–67. [Google Scholar]
- Craig, J.D.; O'Neill, R.D. Electrosynthesis and permselective characterisation of phenol-based polymers for biosensor applications. Anal. Chim. Acta 2003, 495, 33–43. [Google Scholar]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar]
- Wightman, R.M. Probing cellular chemistry in biological systems with microelectrodes. Science 2006, 311, 1570–1574. [Google Scholar]
- Bolger, F.B.; Lowry, J.P. Brain tissue oxygen: in vivo monitoring with carbon paste electrodes. Sensors 2005, 5, 473–487. [Google Scholar]
- O'Neill, R.D. Long-term monitoring of brain dopamine metabolism in vivo with carbon paste electrodes. Sensors 2005, 5, 317–342. [Google Scholar]
- Macca, C.; Wang, J. Experimental procedures for the determination of amperometric selectivity coefficients. Anal. Chim. Acta 1995, 303, 265–274. [Google Scholar]
- Wang, J. Selectivity coefficients for amperometric sensors. Talanta 1994, 41, 857–863. [Google Scholar]
- McMahon, C.P.; Killoran, S.J.; Kirwan, S.M.; O'Neill, R.D. The selectivity of electrosynthesised polymer membranes depends on the electrode dimensions: implications for biosensor applications. J. Chem. Soc. Chem. Commun. 2004, 2128–2130. [Google Scholar]
- Schuvailo, O.M.; Soldatkin, O.O.; Lefebvre, A.; Cespuglio, R.; Soldatkin, A.P. Highly selective microbiosensors for in vivo measurement of glucose, lactate and glutamate. Anal. Chim. Acta 2006, 573, 110–116. [Google Scholar]
- Dixon, B.M.; Lowry, J.P.; O'Neill, R.D. Characterization in vitro and in vivo of the oxygen dependence of an enzyme/polymer biosensor for monitoring brain glucose. J. Neurosci. Meth. 2002, 119, 135–142. [Google Scholar]
- Lowry, J.P.; Miele, M.; O'Neill, R.D.; Boutelle, M.G.; Fillenz, M. An amperometric glucose-oxidase/poly(o-phenylenediamine) biosensor for monitoring brain extracellular glucose: in vivo characterisation in the striatum of freely-moving rats. J. Neurosci. Meth. 1998, 79, 65–74. [Google Scholar]
- Lowry, J.P.; O'Neill, R.D.; Boutelle, M.G.; Fillenz, M. Continuous monitoring of extracellular glucose concentrations in the striatum of freely moving rats with an implanted glucose biosensor. J. Neurochem. 1998, 70, 391–396. [Google Scholar]
- McNay, E.C.; McCarty, R.C.; Gold, P.E. Fluctuations in brain glucose concentration during behavioral testing: Dissociations between brain areas and between brain and blood. Neurobiol. Learn. Memory 2001, 75, 325–337. [Google Scholar]
- Miele, M.; Fillenz, M. In vivo determination of extracellular brain ascorbate. J. Neurosci. Meth. 1996, 70, 15–19. [Google Scholar]
- Oldenziel, W.H.; Dijkstra, G.; Cremers, T.I.F.H.; Westerink, B.H.C. In vivo monitoring of extracellular glutamate in the brain with a microsensor. Brain Res. 2006, 1118, 34–42. [Google Scholar]
- Baker, D.A.; Xi, Z.X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar]
- Miele, M.; Berners, M.; Boutelle, M.G.; Kusakabe, H.; Fillenz, M. The determination of the extracellular concentration of brain glutamate using quantitative microdialysis. Brain Res. 1996, 707, 131–133. [Google Scholar]
- De Giglio, E.; Losito, I.; Torsi, L.; Sabbatini, L.; Zambonin, P.G. Electroanalytical and spectroscopic characterization of poly(o-phenylenediamine) grown on highly oriented pyrolytic graphite. Annali Chim. 2003, 93, 209–221. [Google Scholar]
- Lowry, J.P.; O'Neill, R.D. Partial characterization in vitro of glucose oxidase-modified poly(phenylenediamine)-coated electrodes for neurochemical analysis in vivo. Electroanalysis 1994, 6, 369–379. [Google Scholar]
- Ryan, M.R.; Lowry, J.P.; O'Neill, R.D. Biosensor for neurotransmitter L-glutamic acid designed for efficient use of L-glutamate oxidase and effective rejection of interference. Analyst 1997, 122, 1419–1424. [Google Scholar]
- Craig, J.D.; O'Neill, R.D. Comparison of simple aromatic amines for electrosynthesis of permselective polymers in biosensor fabrication. Analyst 2003, 128, 905–911. [Google Scholar]
- O'Neill, R.D.; Chang, S.C.; Lowry, J.P.; McNeil, C.J. Comparisons of platinum, gold, palladium and glassy carbon as electrode materials in the design of biosensors for glutamate. Biosens. Bioelectron. 2004, 19, 1521–1528. [Google Scholar]
- McAteer, K.; O'Neill, R.D. Strategies for decreasing ascorbate interference at glucose oxidase-modified poly(o-phenylenediamine)-coated electrodes. Analyst 1996, 121, 773–777. [Google Scholar]
- Centonze, D.; Malitesta, C.; Palmisano, F.; Zambonin, P.G. Permeation of solutes through an electropolymerized ultrathin poly-o-phenylenediamine film used as an enzyme-entrapping membrane. Electroanalysis 1994, 6, 423–429. [Google Scholar]
- Lowry, J.P.; McAteer, K.; El Atrash, S.S.; Duff, A.; O'Neill, R.D. Characterization of glucose oxidase-modified poly(phenylenediamine)-coated electrodes in vitro and in vivo: homogeneous interference by ascorbic acid in hydrogen peroxide detection. Anal. Chem. 1994, 66, 1754–1761. [Google Scholar]
- Oldham, K.B. The steepness of voltammetric waves. J. Electroanal. Chem. 1985, 184, 257–267. [Google Scholar]
- Lyne, P.D.; O'Neill, R.D. Stearate-modified carbon paste electrodes for detecting dopamine in vivo: decrease in selectivity caused by lipids and other surface-active agents. Anal. Chem. 1990, 62, 2347–2351. [Google Scholar]
- O'Neill, R.D. Microvoltammetric techniques and sensors for monitoring neurochemical dynamics in vivo - a review. Analyst 1994, 119, 767–779. [Google Scholar]
- O'Neill, R.D.; Grunewald, R.A.; Fillenz, M.; Albery, W.J. The effect of unilateral cortical lesions on the circadian changes in rat striatal ascorbate and homovanillic acid levels measured in vivo using voltammetry. Neurosci. Lett. 1983, 42, 105–110. [Google Scholar]
- Zhang, Y.; Wilson, G.S. Electrochemical oxidation of H2O2 on Pt and Pt + Ir electrodes in physiological buffer and its applicability to H2O2-based biosensors. J. Electroanal. Chem. 1993, 345, 253–271. [Google Scholar]
- Casella, I.G. Electrooxidation of ascorbic acid on the dispersed platinum glassy carbon electrode and its amperometric determination in flow injection analysis. Electroanalysis 1996, 8, 128–134. [Google Scholar]
- Deakin, M.R.; Kovach, P.M.; Stutts, K.J.; Wightman, R.M. Heterogeneous mechanisms of the oxidation of catechols and ascorbic acid at carbon electrodes. Anal. Chem. 1986, 58, 1474–1480. [Google Scholar]
- Hu, I.F.; Kuwana, T. Oxidative mechanism of ascorbic acid at glassy carbon electrodes. Anal. Chem. 1986, 58, 3235–3239. [Google Scholar]
- Evans, J.F.; Kuwana, T.; Henne, M.T.; Royer, G.P. Electrocatalysis of solution species using modified electrodes. J. Electroanal. Chem. 1977, 80, 409–416. [Google Scholar]
- Ormonde, D.E.; O'Neill, R.D. Altered response of carbon paste electrodes after contact with brain tissue. Implications for modified electrode use in vivo. J. Electroanal. Chem. 1989, 261, 463–469. [Google Scholar]
- Hall, S.B.; Khudaish, E.A.; Hart, A.L. Electrochemical oxidation of hydrogen peroxide at platinum electrodes. Part 1. An adsorption-controlled mechanism. Electrochim. Acta 1998, 43, 579–588. [Google Scholar]
- Aoki, K.; Ishida, M.; Tokuda, K. Electrode kinetics of the oxidation of hydrogen peroxide at pretreated glassy carbon and carbon fibre electrodes. J. Electroanal. Chem. 1988, 251, 63–71. [Google Scholar]
- Wang, J.; Rivas, G.; Chicharro, M. Iridium-dispersed carbon paste enzyme electrodes. Electroanalysis 1996, 8, 434–437. [Google Scholar]
- Yang, Y.F.; Denuault, G. Scanning electrochemical microscopy (SECM): study of the formation and reduction of oxides on platinum electrode surfaces in Na2SO4 solution (pH=7). J. Electroanal. Chem. 1998, 443, 273–282. [Google Scholar]
- Burke, L.D.; Buckley, D.T. Formation and reduction of hydrous oxide films on platinum in aqueous solution at 273 K. J. Appl. Electrochem. 1995, 25, 913–922. [Google Scholar]
- Lingane, J.J.; Lingane, P.J. Chronopotentiometry of hydrogen peroxide with a platinum wire electrode. J. Electroanal. Chem. 1963, 5, 411–419. [Google Scholar]
- Mikkelsen, S.R.; Lennox, R.B. Rotating disc electrode characterization of immobilized glucose oxidase. Anal. Biochem. 1991, 195, 358–363. [Google Scholar]
- Ormonde, D.E.; O'Neill, R.D. The oxidation of ascorbic acid at carbon paste electrodes. Modified response following contact with surfactant, lipid and brain tissue. J. Electroanal. Chem. 1990, 279, 109–121. [Google Scholar]
- Sohn, T.W.; Stoecker, P.W.; Carp, W.; Yacynych, A.M. Microarray electrodes as biosensors. Electroanalysis 1991, 3, 763–766. [Google Scholar]
- Myler, S.; Eaton, S.; Higson, S.P.J. Poly(o-phenylenediamine) ultra-thin polymer-film composite membranes for enzyme electrodes. Anal. Chim. Acta 1997, 357, 55–61. [Google Scholar]
- Harrar, J.E. Controlled-potential coulometric determination of hydrogen peroxide. Anal. Chem. 1963, 35, 893–896. [Google Scholar]
- Jones, D.A.; Parkin, M.C.; Langemann, H.; Landolt, H.; Hopwood, S.E.; Strong, A.J.; Boutelle, M.G. On-line monitoring in neurointensive care - Enzyme-based electrochemical assay for simultaneous, continuous monitoring of glucose and lactate from critical care patients. J. Electroanal. Chem. 2002, 538, 243–252. [Google Scholar]
- Collinge, J.; Sidle, K.C.L.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996, 383, 685–690. [Google Scholar]
- Losito, I.; Palmisano, F.; Zambonin, P.G. o-Phenylenediamine electropolymerization by cyclic voltammetry combined with electrospray ionization-ion trap mass spectrometry. Anal. Chem. 2003, 75, 4988–4995. [Google Scholar]
- Cooper, J.M.; Foreman, P.L.; Glidle, A.; Ling, T.W.; Pritchard, D.J. Glutamate oxidase enzyme electrodes: microsensors for neurotransmitter determination using electrochemically polymerized permselective films. J. Electroanal. Chem. 1995, 388, 143–149. [Google Scholar]
- Murphy, L.J. Reduction of interference response at a hydrogen peroxide detecting electrode using electropolymerized films of substituted naphthalenes. Anal. Chem. 1998, 70, 2928–2935. [Google Scholar]
- Schofield, W.C.E.; McGettrick, J.D.; Badyal, J.P.S. A substrate-independent approach for cyclodextrin functionalized surfaces. J. Phys. Chem. B 2006, 110, 17161–17166. [Google Scholar]
- Duo, J.; Fletcher, H.; Stenken, J.A. Natural and synthetic affinity agents as microdialysis sampling mass transport enhancers: Current progress and future perspectives. Biosens. Bioelectron. 2006, 22, 449–457. [Google Scholar]
- Ferancova, A.; Labuda, J.; Barek, J.; Zima, J. Cyclodextrins as supramolecular complex agents in electroanalytical chemistry: Review 1995-2001. Chem. Listy 2002, 96, 856–862. [Google Scholar]
- Ferancova, A.; Korgova, E.; Labuda, J.; Zima, J.; Barek, J. Cyclodextrin modified carbon paste based electrodes as sensors for the determination of carcinogenic polycyclic aromatic amines. Electroanalysis 2002, 14, 1668–1673. [Google Scholar]
- Ganjali, M.R.; Norouzi, P.; Rezapour, M.; Faridbod, F.; Pourjavid, M.R. Supramolecular based membrane sensors. Sensors 2006, 6, 1018–1086. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Pt | Pt-Ir | p-value |
|---|---|---|---|
| Background scans | |||
| Current at the CV reversal potential (μA cm-2); n = 4 × 10 | 17.4 ± 1.1 | 16.6 ± 0.9 | 0.62 |
| Double-layer capacitance, Cdl (Eqn. 1; mF cm-2); n = 4 × 10 | 0.48 ± 0.02 | 0.45 ± 0.01 | 0.15 |
| H2O2 scans | |||
| Potential of maximum slope (Es,max, mV); n = 2 × 20 | 200 ± 5 | 195 ± 5 | 0.55 |
| Normalized maximum slope (Smax,N, mV-1) ; n = 2 × 20 | 0.45 ± 0.01 | 0.43 ± 0.01 | 0.30 |
| AA scans | |||
| Potential of maximum slope (Es,max, mV); n = 2 × 20 | 110 ± 5 | 115 ± 5 | 0.55 |
| Normalized maximum slope (Smax,N, mV-1) ; n = 2 × 20 | 0.59 ± 0.01 | 0.51 ± 0.07 | 0.38 |
| Albumin | iL(AA) nA | imax(AA) nA | H2O2 slope nA mM-1 | S% (Eqn. 2) |
|---|---|---|---|---|
| BSA | 1.5 ± 0.2 (29) | 3.5 ± 0.3 (29) | 661 ± 92 (6) | 0.23 ± 0.04 |
| CEA | 1.6 ± 0.2 (12) | 3.1 ± 1.2 (12) | 620 ± 10 (8) | 0.27 ± 0.03 |
| p-value | p > 0.74 | p > 0.69 | p > 0.61 | p > 0.43 |
| Modifier | iL(AA) nA | H2O2 slope nA mM-1 | S% (Eqn. 2) | p-value |
|---|---|---|---|---|
| Gelatin | 1.1 ± 0.1 (6) | 117 ± 17 (6) | 0.90 ± 0.15 | p < 0.001 |
| Casein | 0.7 ± 0.1 (4) | 182 ± 54 (4) | 0.37 ± 0.12 | p > 0.30 |
| βCD | 0.9 ± 0.2 (10) | 533 ± 60 (8) | 0.17 ± 0.04 | p = 0.06 |
© 2007 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Kirwan, S.M.; Rocchitta, G.; McMahon, C.P.; Craig, J.D.; Killoran, S.J.; O’Brien, K.B.; Serra, P.A.; Lowry, J.P.; O’Neill, R.D. Modifications of Poly(o-phenylenediamine) Permselective Layer on Pt-Ir for Biosensor Application in Neurochemical Monitoring. Sensors 2007, 7, 420-437. https://doi.org/10.3390/s7040420
Kirwan SM, Rocchitta G, McMahon CP, Craig JD, Killoran SJ, O’Brien KB, Serra PA, Lowry JP, O’Neill RD. Modifications of Poly(o-phenylenediamine) Permselective Layer on Pt-Ir for Biosensor Application in Neurochemical Monitoring. Sensors. 2007; 7(4):420-437. https://doi.org/10.3390/s7040420
Chicago/Turabian StyleKirwan, Sarah M., Gaia Rocchitta, Colm P. McMahon, Jennifer D. Craig, Sarah J. Killoran, Kylie B. O’Brien, Pier A. Serra, John P. Lowry, and Robert D. O’Neill. 2007. "Modifications of Poly(o-phenylenediamine) Permselective Layer on Pt-Ir for Biosensor Application in Neurochemical Monitoring" Sensors 7, no. 4: 420-437. https://doi.org/10.3390/s7040420