Cross-Reactive Sensor Array for Metal Ion Sensing Based on Fluorescent SAMs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
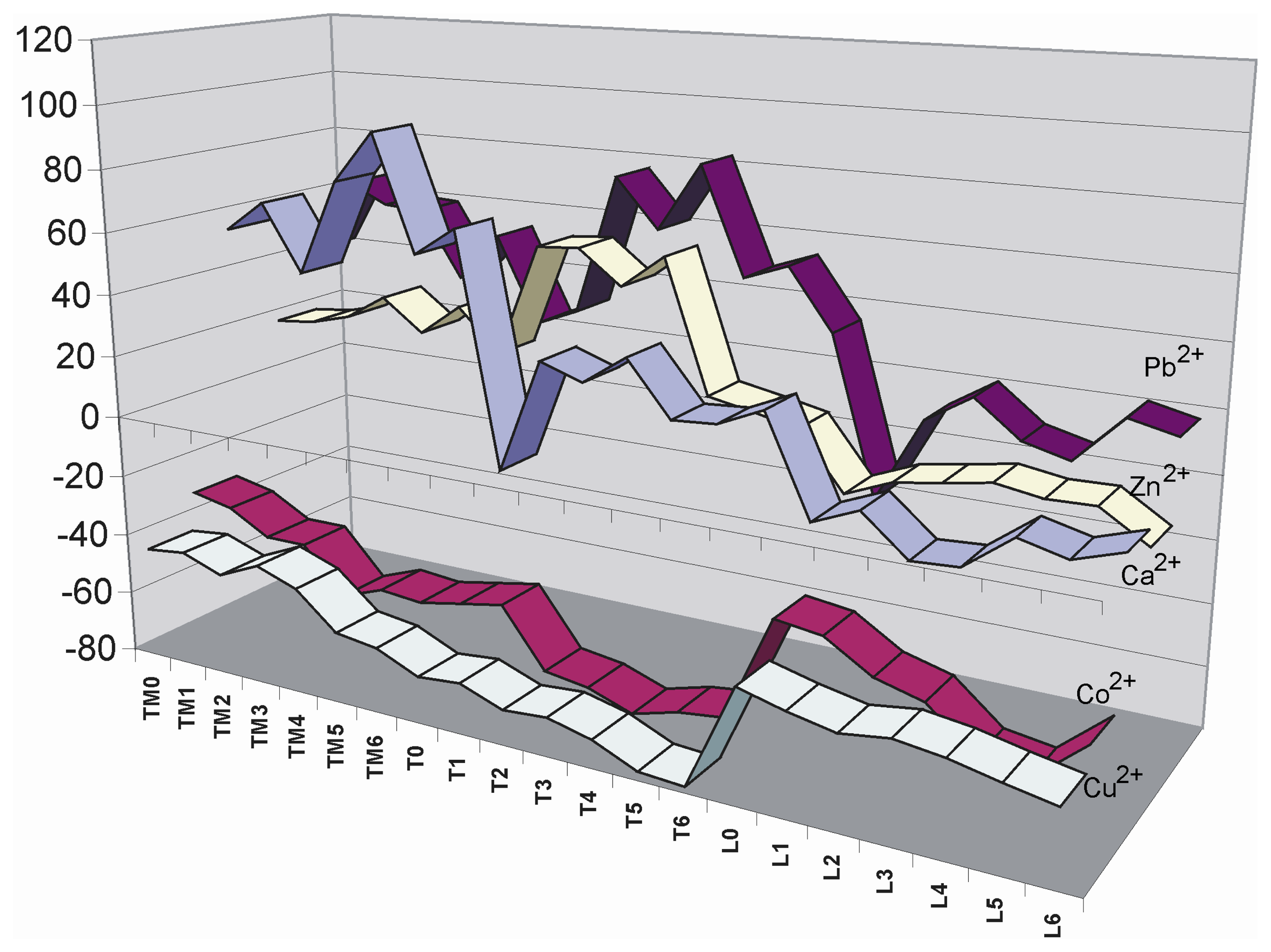
2. Results and Discussion
3. Experimental Section
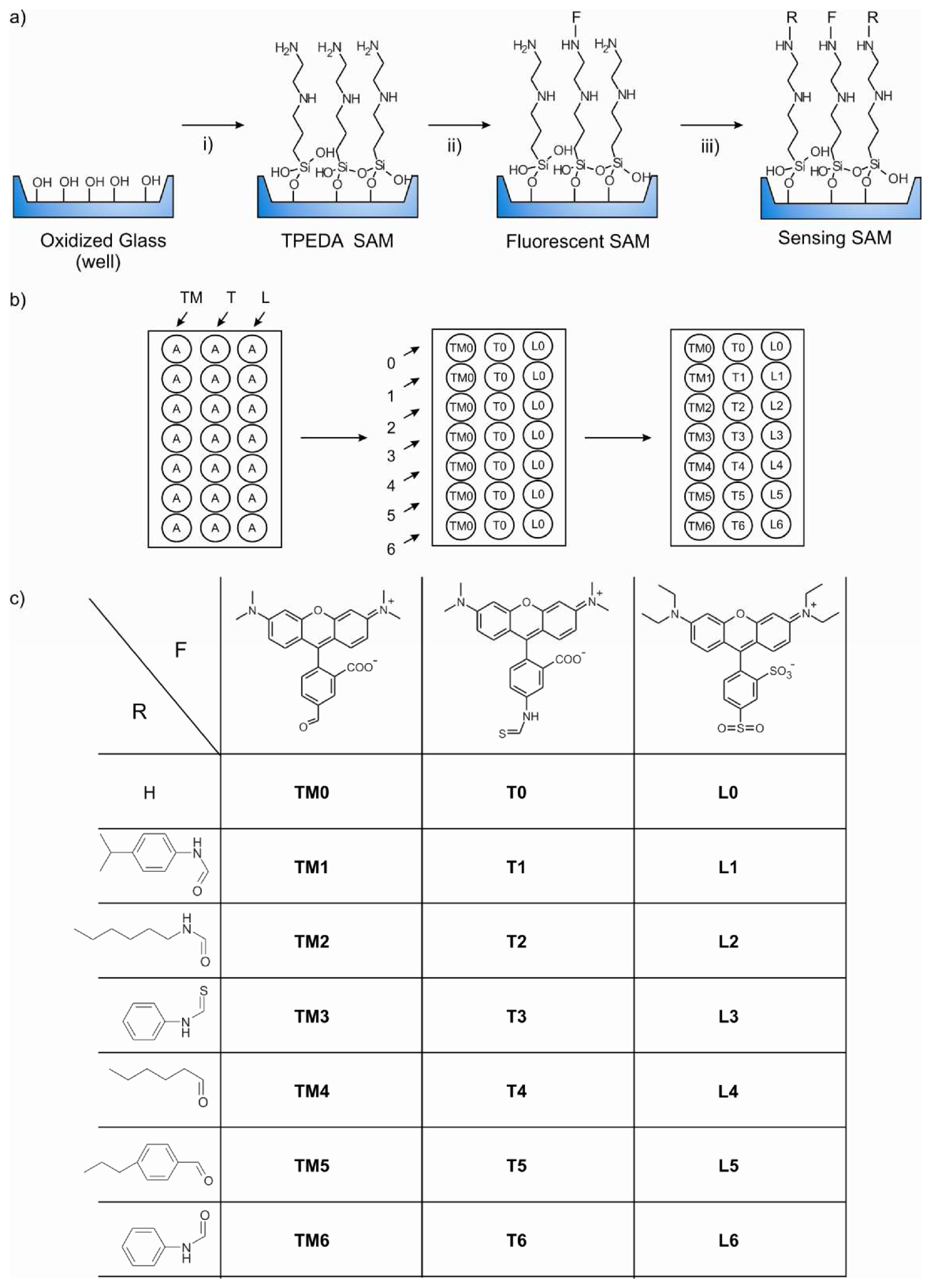
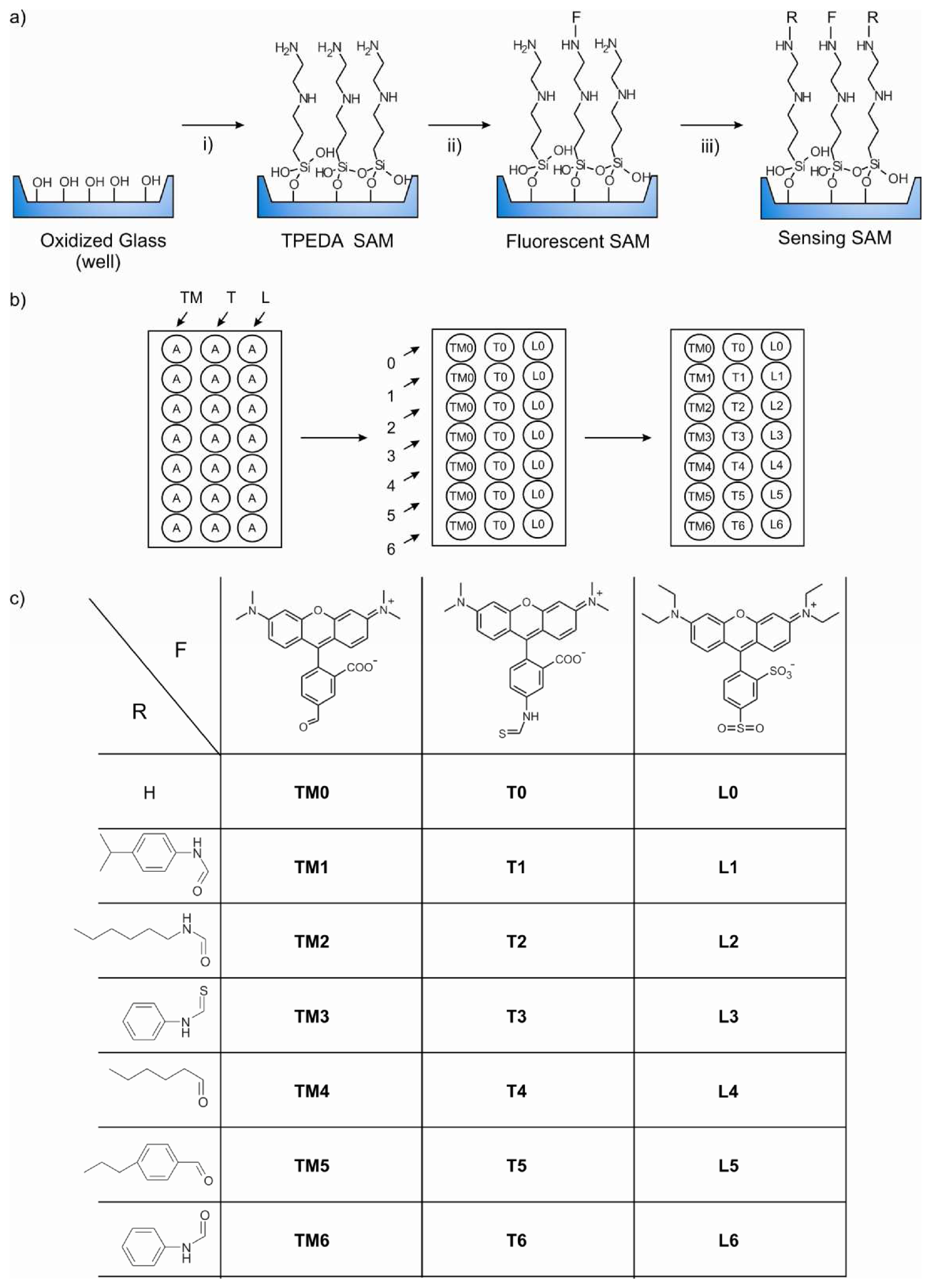
Monolayer microarray fabrication
Synthesis of TPEDA monolayers in the MTP wells
Immobilization of the fluorophores: Synthesis of the TM0, L0, and T0 SAMs
Immobilization of the ligands: Synthesis of the layers L1-L6, T1-T6, and TM1-TM6
Metal ion sensing with the microtiter plate
Imaging Instrumentation
Laser scanning confocal microscopy
Laser confocal fluorescence scanner
Acknowledgments
References and Notes
- Czarnik, A. W. Desperatel Seeking Sensors. Chemistry & Biology 1995, 2, 423–428. [Google Scholar]
- Behr, J.P. The Lock and Key Principle, The State of the Art - 100 Years On; Wiley: New York, 1994; Vol. 1. [Google Scholar]
- Jurs, P. C.; Bakken, G. A.; Mcclelland, H. E. Computational Methods for the Analysis of Chemical Sensor Array Data From Volatile Analytes. Chem. Rev. 2000, 100, 2649–2678. [Google Scholar]
- Doty, R. L. Handbook of Olfaction and Gustation, Neurological Disease and Therapy., 2nd ed.; Marcel Dekker: New York, 2003; Vol. 57. [Google Scholar]
- Lavigne, J. J.; Anslyn, E. V. Sensing a Paradigm Shift in the Field of Molecular Recognition: From Selective to Differential Receptors. Angew. Chem., Int. Ed. 2001, 40, 3119–3130. [Google Scholar]
- Albert, K. J.; Lewis, N. S.; Schauer, C. L.; Sotzing, G. A.; Stitzel, S. E.; Vaid, T. P.; Walt, D. R. Cross-Reactive Chemical Sensor Arrays. Chem. Rev. 2000, 100, 2595–2626. [Google Scholar]
- Stopfer, M.; Jayaraman, V.; Laurent, G. Intensity Versus Identity Coding in an Olfactory System. Neuron 2003, 39, 991–1004. [Google Scholar]
- Lundstrom, I. Artificial Noses - Picture the Smell. Nature 2000, 406, 682–683. [Google Scholar]
- Shepherd, G. M. Discrimination of Molecular Signals by the Olfactory Receptor Neuron. Neuron 1994, 13, 771–790. [Google Scholar]
- Lundstrom, I.; Erlandsson, R.; Frykman, U.; Hedborg, E.; Spetz, A.; Sundgren, H.; Welin, S.; Winquist, F. Artificial Olfactory Images From a Chemical Sensor Using a Light-Pulse Technique. Nature 1991, 352, 47–50. [Google Scholar]
- Lancet, D. Olfaction - the Strong Scent of Success. Nature 1991, 351, 275–276. [Google Scholar]
- Bishop, C. M. Neural Networks for Pattern Recognition; Bishop, C. M., Ed.; Oxford University Press: Oxford, U.K., 1995. [Google Scholar]
- Lyons, W. B.; Lewis, E. Neural Networks and Pattern Recognition Techniques Applied to Optical Fibre Sensors. Trans. Inst. Measurm. Control 2000, 22, 385–404. [Google Scholar]
- Wright, A. T.; Anslyn, E. V. Differential Receptor Arrays and Assays for Solution-Based Molecular Recognition. Chem. Soc. Rev. 2006, 35, 14–28. [Google Scholar]
- Persaud, K.; Dodd, G. Analysis of Discrimination Mechanisms in the Mammalian Olfactory System Using a Model Nose. Nature 1982, 299, 352–355. [Google Scholar]
- Freund, M. S.; Lewis, N. S. A Chemically Diverse Conducting Polymer-Based Electronic Nose. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2652–2656. [Google Scholar]
- Lonergan, M. C.; Severin, E. J.; Doleman, B. J.; Beaber, S. A.; Grubb, R. H.; Lewis, N. S. Array-Based Vapor Sensing Using Chemically Sensitive, Carbon Black-Polymer Resistors. Chem. Mater. 1996, 8, 2298–2312. [Google Scholar]
- Heilig, A.; Barsan, N.; Weimar, U.; Schweizer-Berberich, M.; Gardner, J. W.; Gopel, W. Gas Identification by Modulating Temperatures of SnO2-Based Thick Film Sensors. Sens. Actuators, B 1997, 43, 45–51. [Google Scholar]
- Gardner, J. W.; Shurmer, H. V.; Tan, T. T. Application of an Electronic Nose to the Discrimination of Coffees. Sens. Actuators, B 1992, 6, 71–75. [Google Scholar]
- Crooks, R. M.; Ricco, A. J. New Organic Materials Suitable for Use in Chemical Sensor Arrays. Acc. Chem. Res. 1998, 31, 219–227. [Google Scholar]
- Grate, J. W.; Abraham, M. H. Solubility Interactions and the Design of Chemically Selective Sorbent Coatings for Chemical Sensors and Arrays. Sens. Actuators, B 1991, 3, 85–111. [Google Scholar]
- Brunink, J. A. J.; Dinatale, C.; Bungaro, F.; Davide, F. A. M.; Damico, A.; Paolesse, R.; Boschi, T.; Faccio, M.; Ferri, G. The Application of Metalloporphyrins as Coating Material for Quartz Microbalance-Based Chemical Sensors. Anal. Chim. Acta 1996, 325, 53–64. [Google Scholar]
- Walt, D. R. Fiber Optic Imaging Sensors. Acc. Chem. Res. 1998, 31, 267–278. [Google Scholar]
- Mayr, T.; Liebsch, G.; Klimant, I.; Wolfbeis, O. S. Multi-Ion Imaging Using Fluorescent Sensors in a Microtiterplate Array Format. Analyst 2002, 127, 201–203. [Google Scholar]
- Dickinson, T. A.; White, J.; Kauer, J. S.; Walt, D. R. A Chemical-Detecting System Based on a Cross-Reactive Optical Sensor Array. Nature 1996, 382, 697–700. [Google Scholar]
- Chojnacki, P.; Werner, T.; Wolfbeis, O. S. Combinatorial Approach Towards Materials for Optical Ion Sensors. Microchim. Acta 2004, 147, 87–92. [Google Scholar]
- Mayr, T.; Igel, C.; Liebsch, G.; Klimant, I.; Wolfbeis, O. S. Cross-Reactive Metal Ion Sensor Array in a Micro Titer Plate Format. Anal. Chem. 2003, 75, 4389–4396. [Google Scholar]
- Greene, N. T.; Morgan, S. L.; Shimizu, K. D. Molecularly Imprinted Polymer Sensor Arrays. Chem. Commun. 2004, 1172–1173. [Google Scholar]
- Suslick, K. S.; Rakow, N. A.; Sen, A. Colorimetric Sensor Arrays for Molecular Recognition. Tetrahedron 2004, 60, 11133–11138. [Google Scholar]
- Rakow, N. A.; Suslick, K. S. Novel Materials and Applications of Electronic Noses and Tongues (Vol 406, Pg 710, 2000). MRS Bulletin 2004, 29, 913. [Google Scholar]
- Rakow, N. A.; Suslick, K. S. A Colorimetric Sensor Array for Odour Visualization. Nature 2000, 406, 710–713. [Google Scholar]
- Goodey, A.; Lavigne, J. J.; Savoy, S. M.; Rodriguez, M. D.; Curey, T.; Tsao, A.; Simmons, G.; Wright, J.; Yoo, S. J.; Sohn, Y.; Anslyn, E. V.; Shear, J. B.; Neikirk, D. P.; Mcdevitt, J. T. Development of Multianalyte Sensor Arrays Composed of Chemically Derivatized Polymeric Microspheres Localized in Micromachined Cavities. J. Am. Chem. Soc. 2001, 123, 2559–2570. [Google Scholar]
- Wright, A. T.; Anslyn, E. V.; Mcdevitt, J. T. A Differential Array of Metalated Synthetic Receptors for the Analysis of Tripeptide Mixtures. J. Am. Chem. Soc. 2005, 127, 17405–17411. [Google Scholar]
- Ho, C. K.; Hughes, R. C. In-Situ Chemiresistor Sensor Package for Real-Time Detection of Volatile Organic Compounds in Soil and Groundwater. Sensors 2002, 2, 23–34. [Google Scholar]
- Zhang, C.; Suslick, K. S. A Colorimetric Sensor Array for Organics in Water . J. Am. Chem. Soc. 2005, 127, 11548–11549. [Google Scholar]
- Schauer, C. L.; Steemers, F. J.; Walt, D. R. A Cross-Reactive, Class-Selective Enzymatic Array Assay. J. Am. Chem. Soc. 2001, 123, 9443–9444. [Google Scholar]
- Lavigne, J. J.; Savoy, S.; Clevenger, M. B.; Ritchie, J. E.; Mcdoniel, B.; Yoo, S. J.; Anslyn, E. V.; Mcdevitt, J. T.; Shear, J. B.; Neikirk, D. Solution-Based Analysis of Multiple Analytes by a Sensor Array: Toward the Development of an “Electronic Tongue”. J. Am. Chem. Soc. 1998, 120, 6429–6430. [Google Scholar]
- Potyrailo, R. A.; Sivavec, T. M. Dual-Response Resonant Chemical Sensors for Multianalyte Analysis. Sens. Actuators, B 2005, 106, 249–252. [Google Scholar]
- Biran, I.; Rissin, D. M.; Ron, E. Z.; Walt, D. R. Optical Imaging Fiber-Based Live Bacterial Cell Array Biosensor. Anal. Biochem. 2003, 315, 106–113. [Google Scholar]
- Zimmerman, R. S.; Basabe-Desmonts, L.; Van der Baan, F.; Reinhoudt, D. N.; Crego-Calama, M. A Combinatorial Approach to Surface-Confined Cation Sensors in Water. J. Mater. Chem. 2005, 15, 2772–2277. [Google Scholar]
- Carrascosa, L. G.; Moreno, M.; Alvarez, M.; Lechuga, L. M. Nanomechanical Biosensors: a New Sensing Tool. Trac-Trends in Analytical Chemistry 2006, 25, 196–206. [Google Scholar]
- Fritz, J.; Baller, M. K.; Lang, H. P.; Strunz, T.; Meyer, E.; Guntherodt, H. J.; Delamarche, E.; Gerber, C.; Gimzewski, J. K. Stress at the Solid-Liquid Interface of Self-Assembled Monolayers on Gold Investigated With a Nanomechanical Sensor. Langmuir 2000, 16, 9694–9696. [Google Scholar]
- Balakirev, M. Y.; Porte, S.; Vernaz-Gris, M.; Berger, M.; Arie, J. P.; Fouque, B.; Chatelain, F. Photochemical Patterning of Biological Molecules Inside a Glass Capillary. Anal. Chem. 2005, 77, 5474–5479. [Google Scholar]
- Dyukova, V. I.; Shilova, N. V.; Galanina, O. E.; Rubina, A. Y.; Bovin, N. V. Design of Carbohydrate Multiarrays. Biochimica Et Biophysica Acta-General Subjects 2006, 1760, 603–609. [Google Scholar]
- Mack, N. H.; Dong, R.; Nuzzo, R. G. Quantitative Imaging of Protein Adsorption on Patterned Organic Thin-Film Arrays Using Secondary Electron Emission. J. Am. Chem. Soc. 2006, 128, 7871–7881. [Google Scholar]
- Ji, H. F.; Hansen, K. M.; Hu, Z.; Thundat, T. Detection of Ph Variation Using Modified Microcantilever Sensors. Sens. Actuators, B 2001, 72, 233–238. [Google Scholar]
- Birkert, O.; Tunnernann, R.; Jung, G.; Gauglitz, G. Label-Free Parallel Screening of Combinatorial Triazine Libraries Using Reflectometric Interference Spectroscopy. Anal. Chem. 2002, 74, 834–840. [Google Scholar]
- Crego-Calama, M.; Reinhoudt, D. N. New Materials for Metal Ion Sensing by Self-Assembled Monolayers on Glass. Adv. Mater. 2001, 13, 1171–1174. [Google Scholar]
- Basabe-Desmonts, L.; Beld, J.; Zimmerman, R. S.; Hernando, J.; Mela, P.; García-Parajó, M. F. G.; Van Hulst, N. F.; Van den Berg, A.; Reinhoudt, D. N.; Crego-Calama, M. A Simple Approach to Sensor Discovery and Fabrication on Self-Assembled Monolayers on Glass. J. Am. Chem. Soc. 2004, 126, 7293–7299. [Google Scholar]
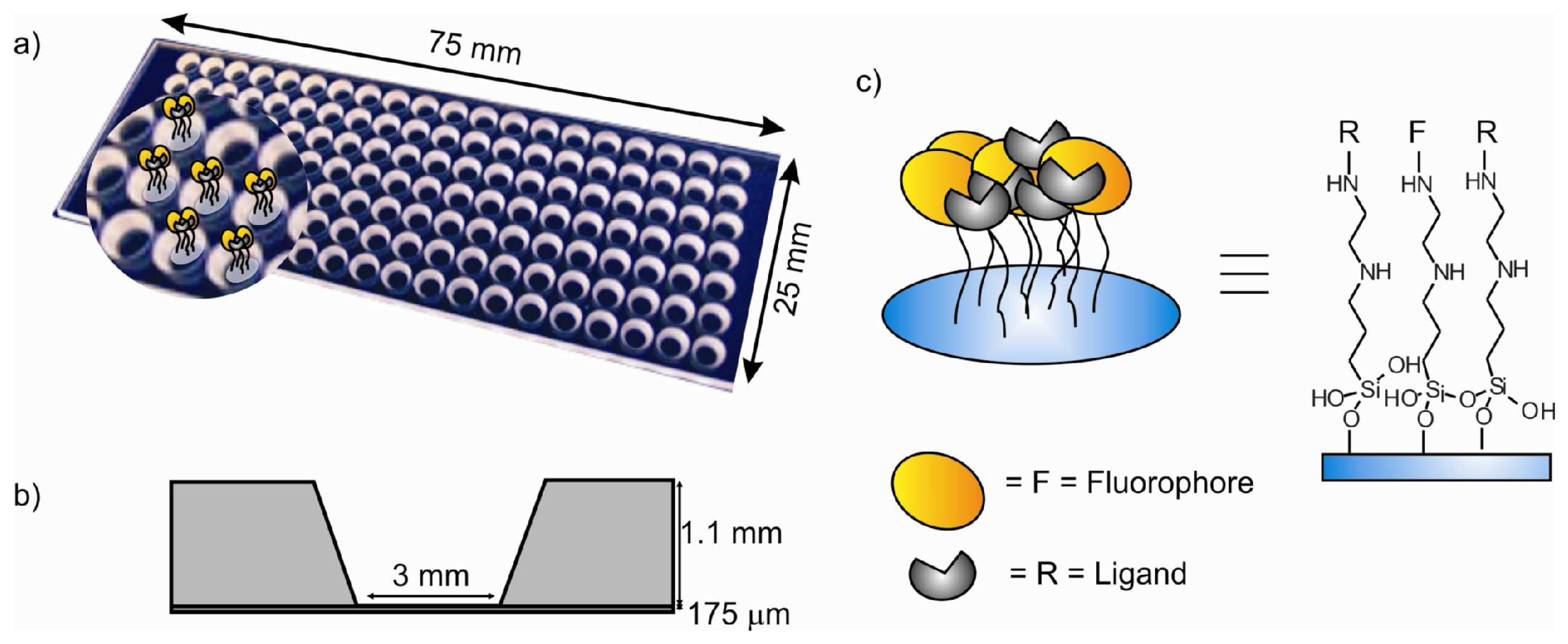
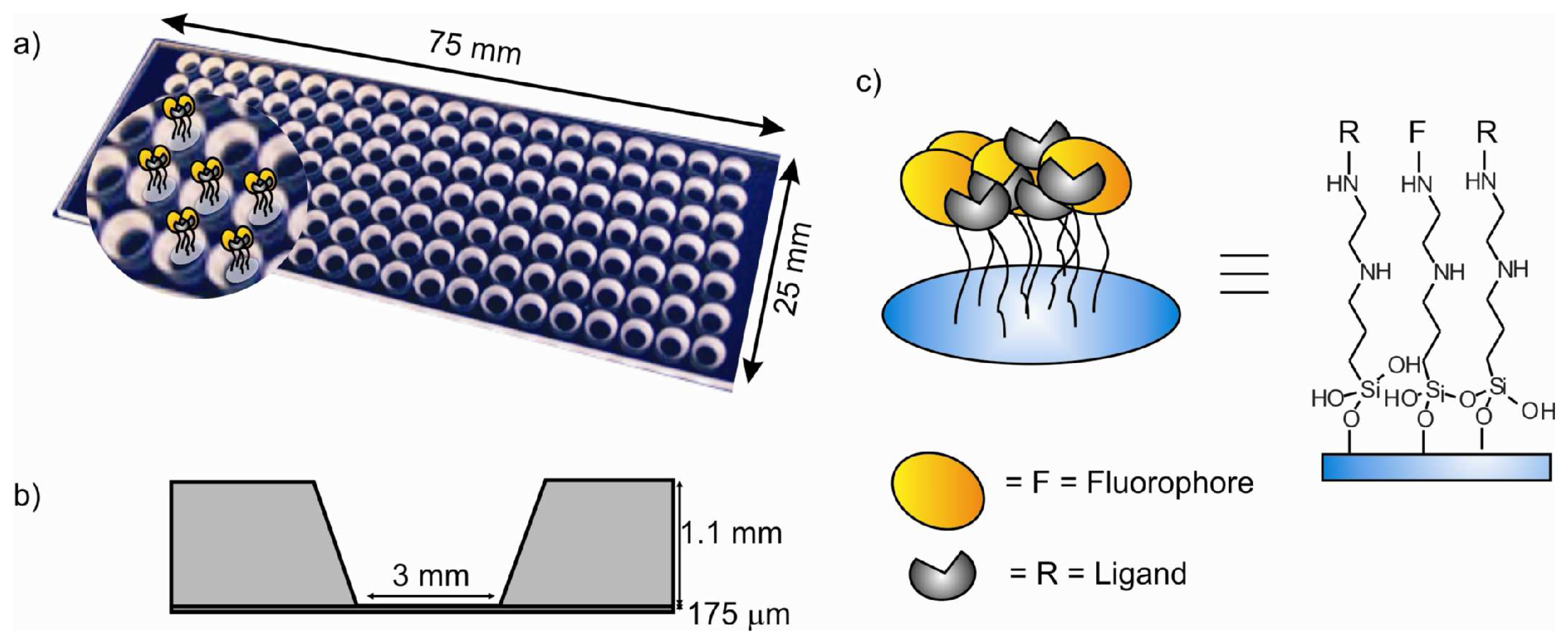
- The thickness of the glass bottom plate was 175 μm to allow fluorescent microscopy imaging of the bottom surface of the channel even with oil immersion objectives for acquisition (if needed) of higher resolution images.
- Due to steric hindrance, some amino groups remain unreacted after addition of the fluorophore molecules. Thus, these groups can be reacted with a small molecule to form the complexing functionalities yielding the final sensitive fluorescent self-assembled monolayer (SAMs).
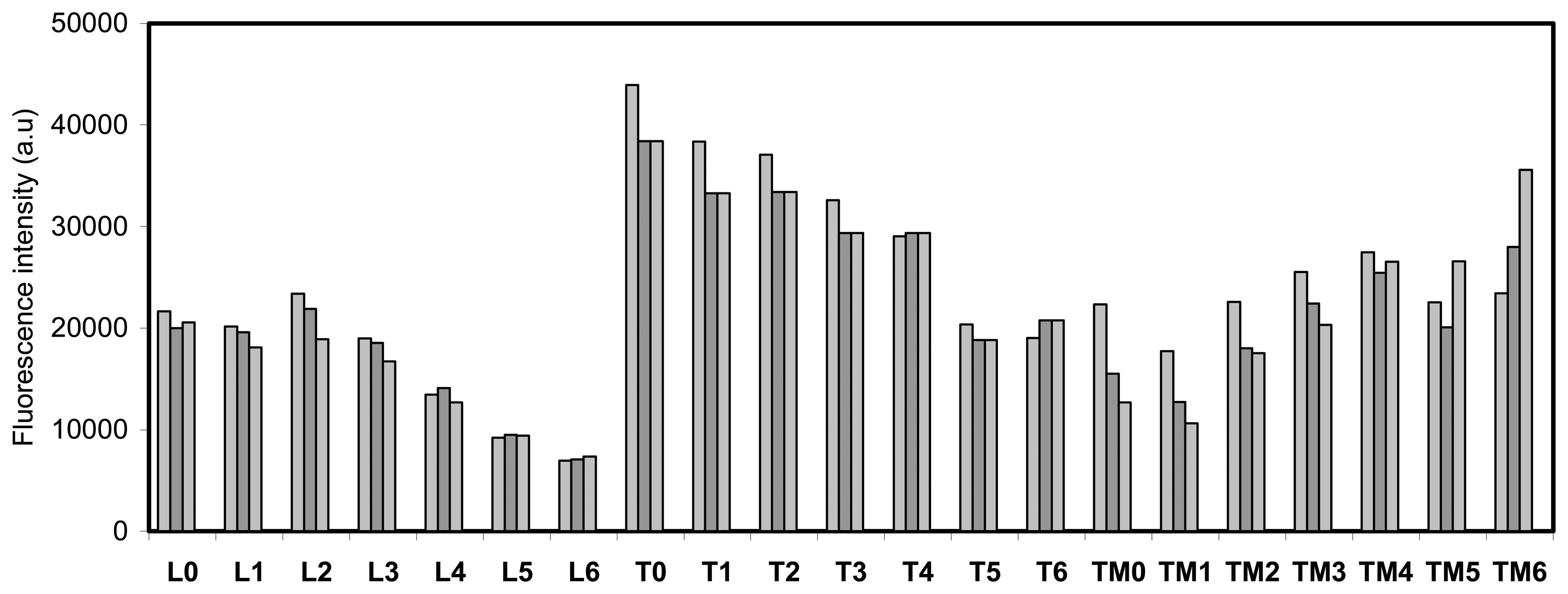
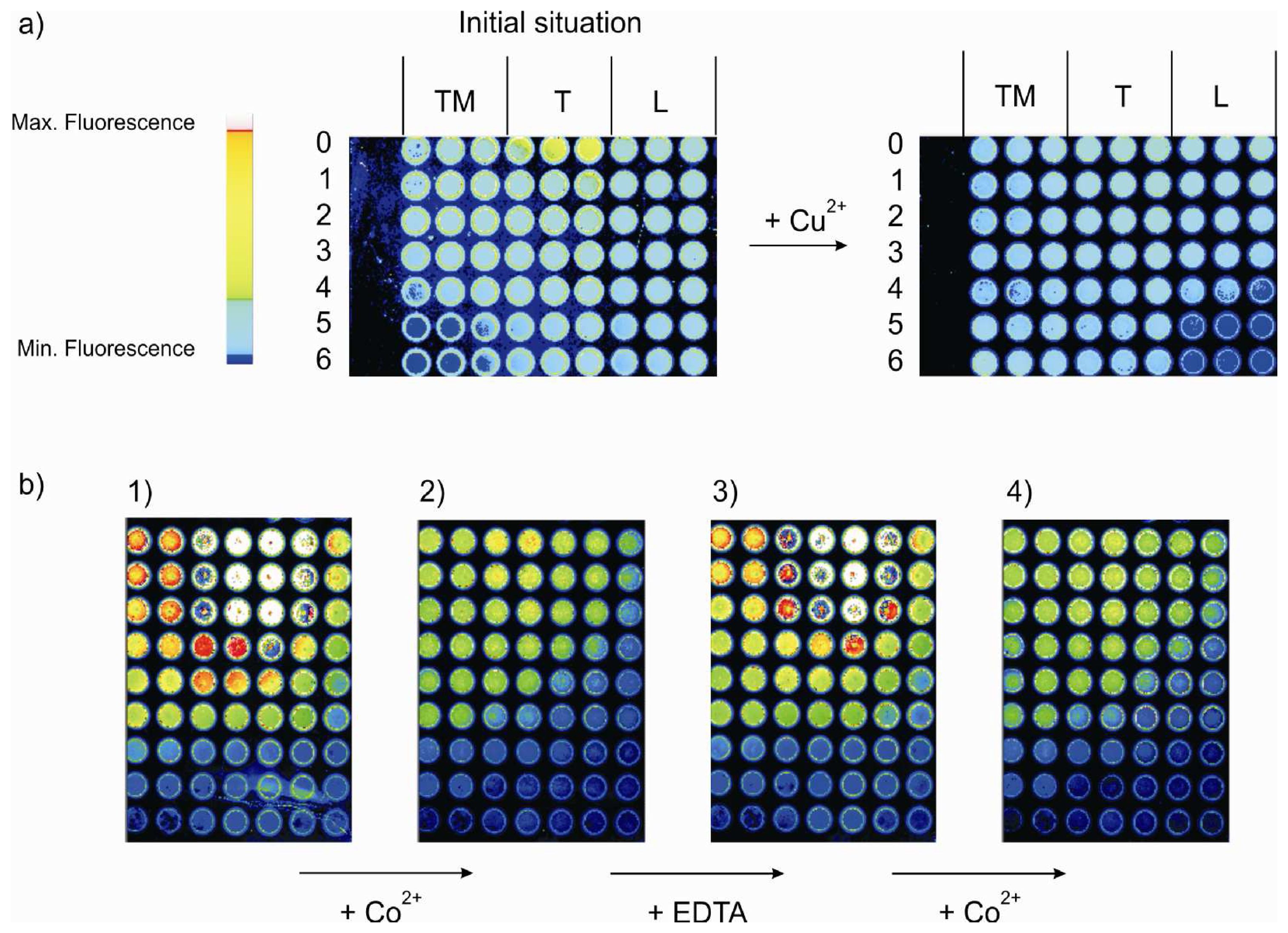
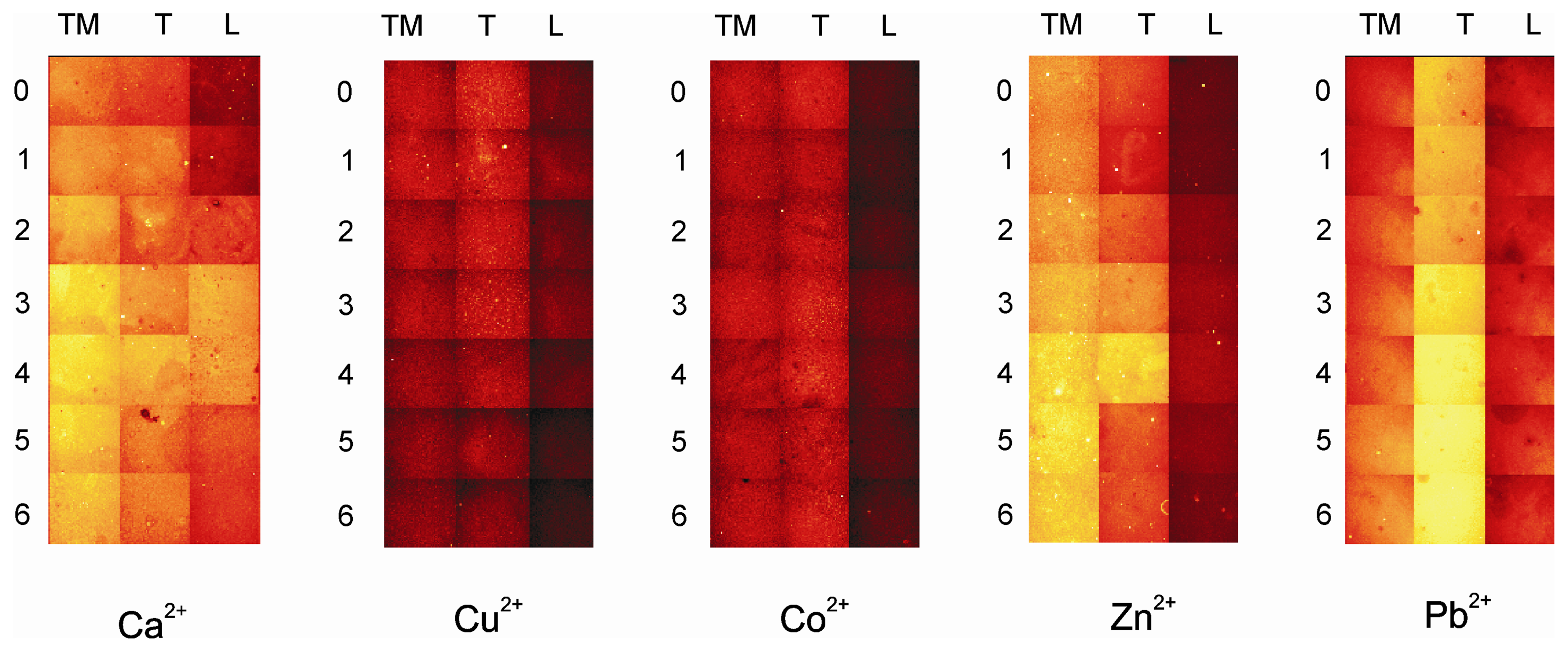
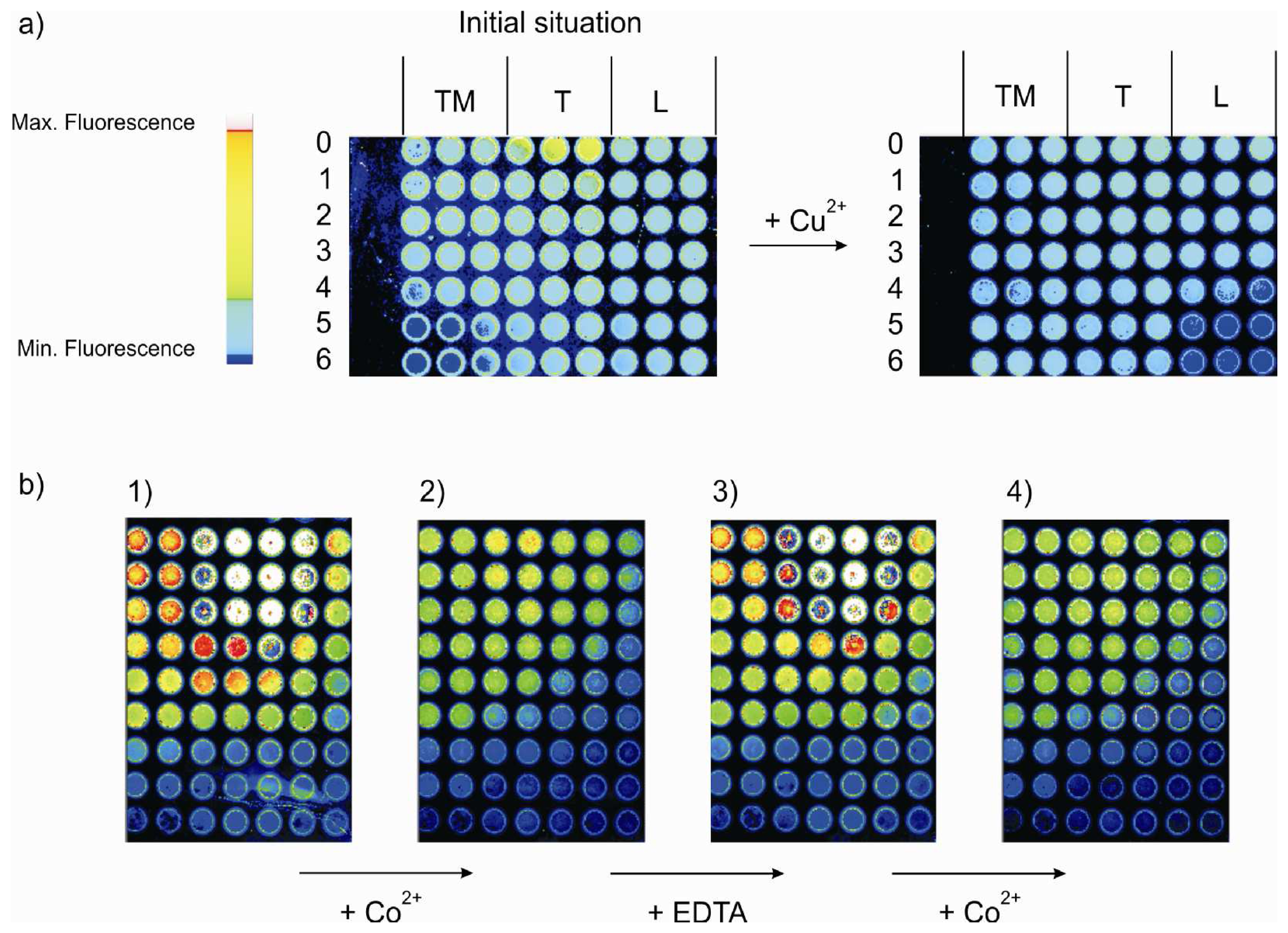
- All the shown fluorescence images are made in air. After incubation of the plate with the analyte, it is rinsed with dichloromethene, dried and then imaged.
- By setting threshold values luminescence intensity of the array can be scale up and down, in order to get high or low fluorescence intensity, into a visible pattern by eye. Different color within a gray scale is assigned to different fluorescence intensity values, and with a particular graphical software the gray scale pictures are transformed into colored photos with a red to yellow scale.
- The imaging of the array in absence and in contact with the analytes was done keeping constant all the parameters of the imaging set-up. The threshold for the fluorescence intensity of the reference library was set differently for layers TM0-TM6, T0-T6 and L0-L6 (see experimental).
- Every monolayer was made in triple in consecutive wells in the analysis library of the MTP. One image of each well is obtained after the incubation with the analytes. But only one image (out of three) for each of the 21 systems in the array is selected for the final picture.
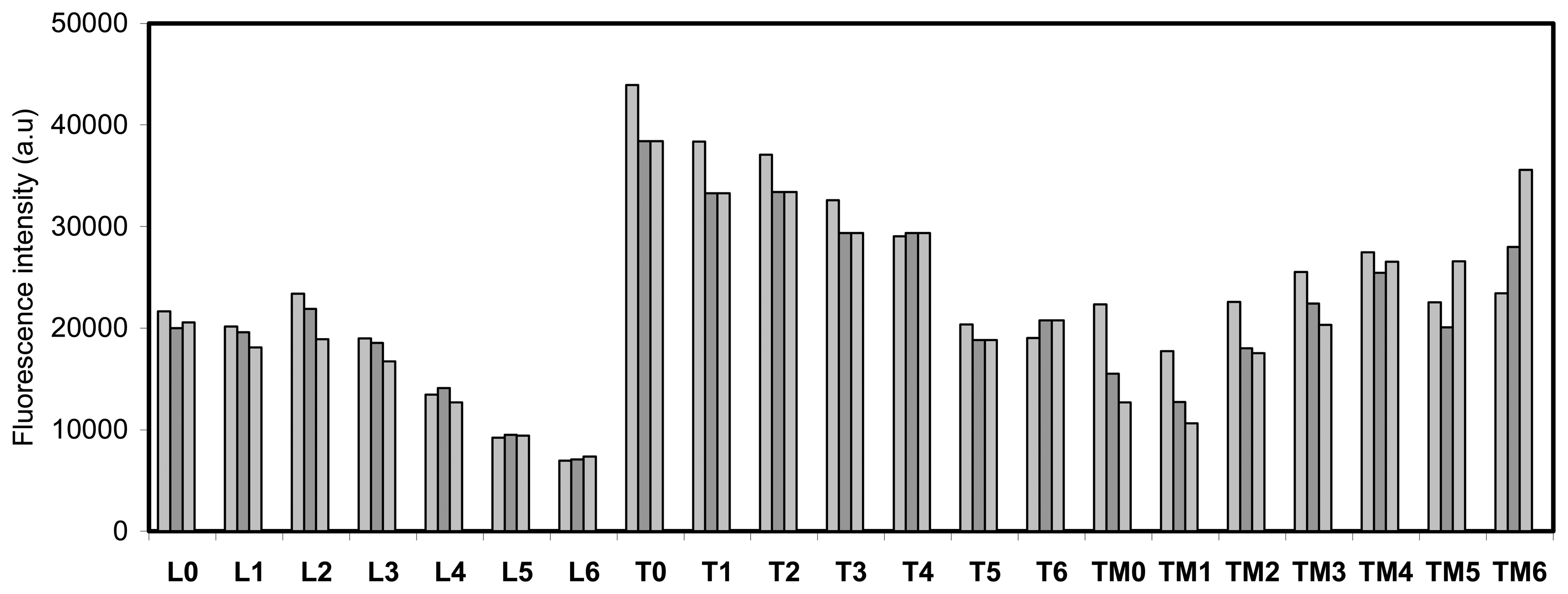
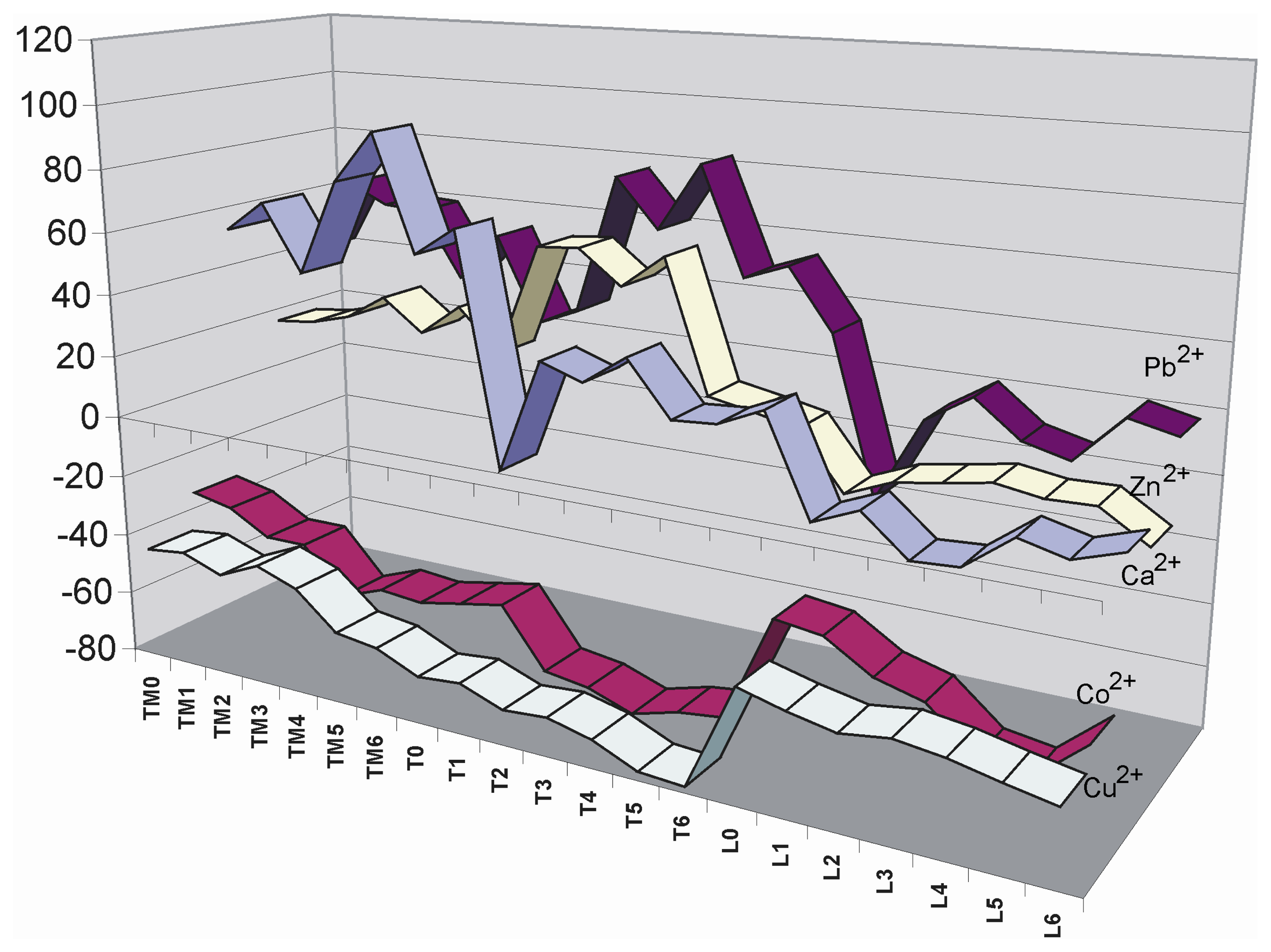
- Each point is the average of at least 3 measurements. The average deviation of these values is lower than 5 %.
- A deviation of the scanner laser focus calibration did not permit the scanning of the whole microtiter plate with high precision. Due to this reason only the half of the plate was imaged each time in order to get the maximum precision along the scanned area.

© 2007 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Basabe-Desmonts, L.; Van der Baan, F.; Zimmerman, R.S.; Reinhoudt, D.N.; Crego-Calama, M. Cross-Reactive Sensor Array for Metal Ion Sensing Based on Fluorescent SAMs. Sensors 2007, 7, 1731-1746. https://doi.org/10.3390/s7091731
Basabe-Desmonts L, Van der Baan F, Zimmerman RS, Reinhoudt DN, Crego-Calama M. Cross-Reactive Sensor Array for Metal Ion Sensing Based on Fluorescent SAMs. Sensors. 2007; 7(9):1731-1746. https://doi.org/10.3390/s7091731
Chicago/Turabian StyleBasabe-Desmonts, Lourdes, Frederieke Van der Baan, Rebecca S. Zimmerman, David N. Reinhoudt, and Mercedes Crego-Calama. 2007. "Cross-Reactive Sensor Array for Metal Ion Sensing Based on Fluorescent SAMs" Sensors 7, no. 9: 1731-1746. https://doi.org/10.3390/s7091731