Microsensors for in vivo Measurement of Glutamate in Brain Tissue

Abstract
:
1. Introduction
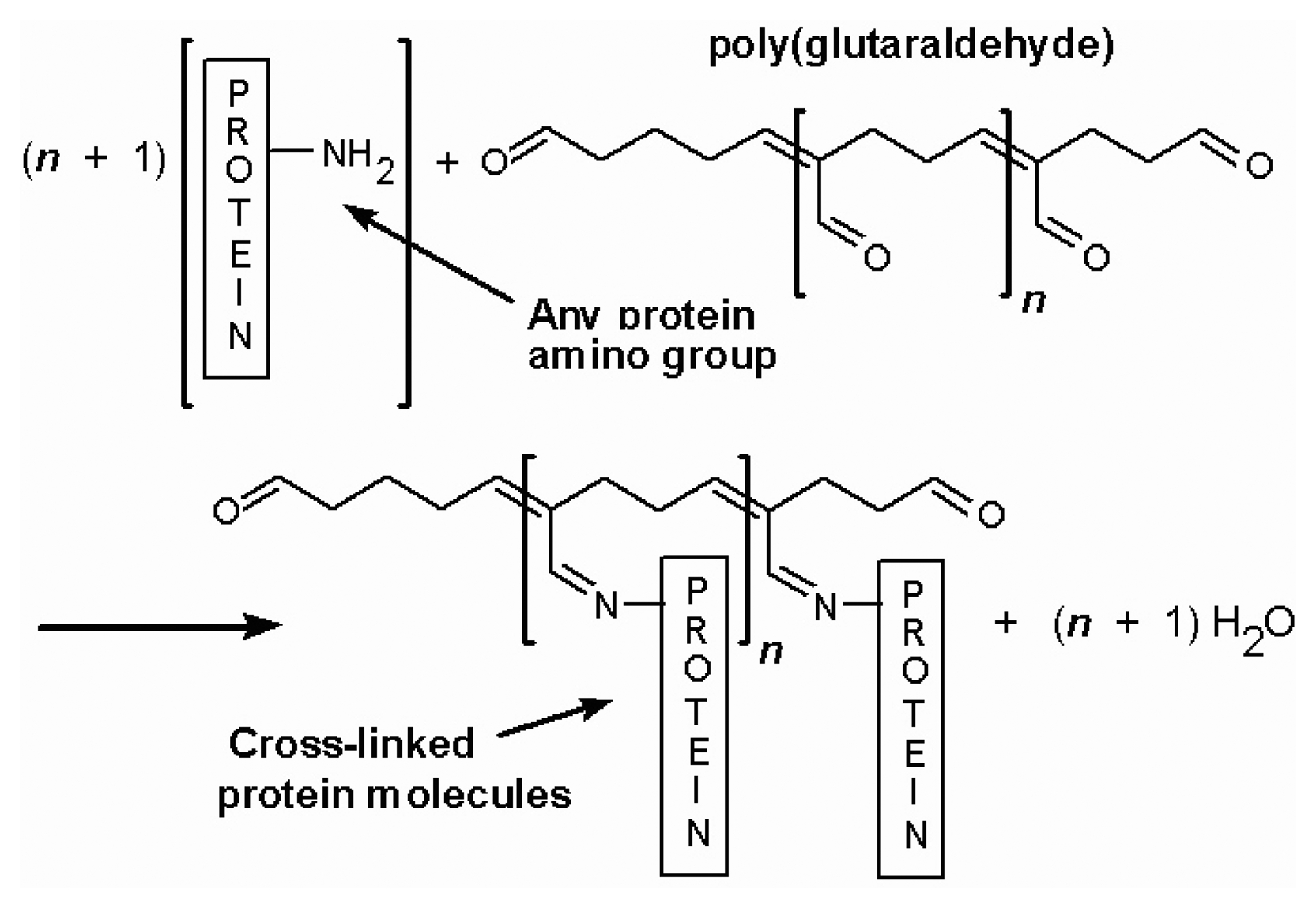
2. Structure, material, binding system of sensors and working mechanisms
- I.
- II.
- III.
- 2Os3+ + 2e- → 2Os2+
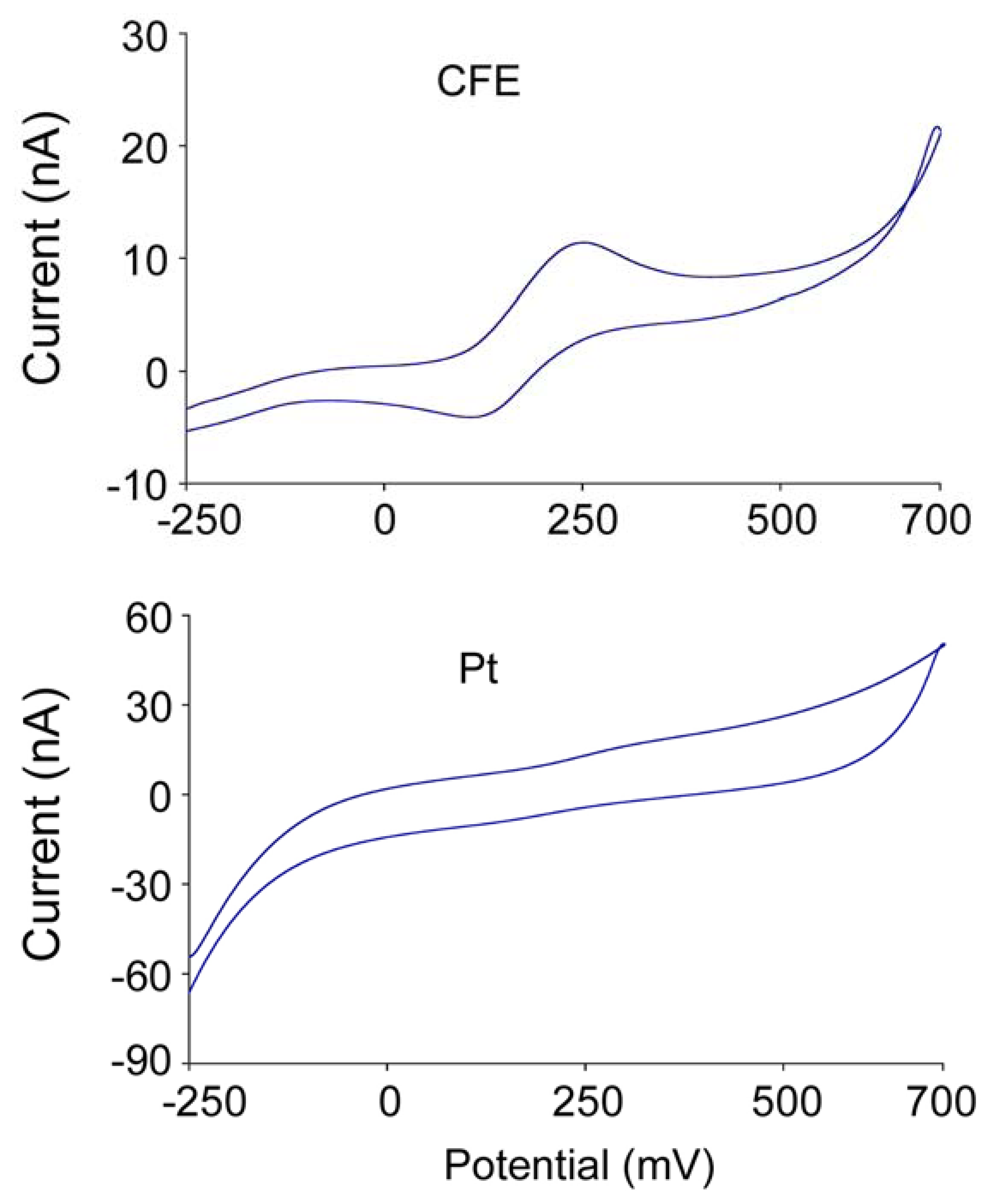
3. Characteristics of sensors in vitro
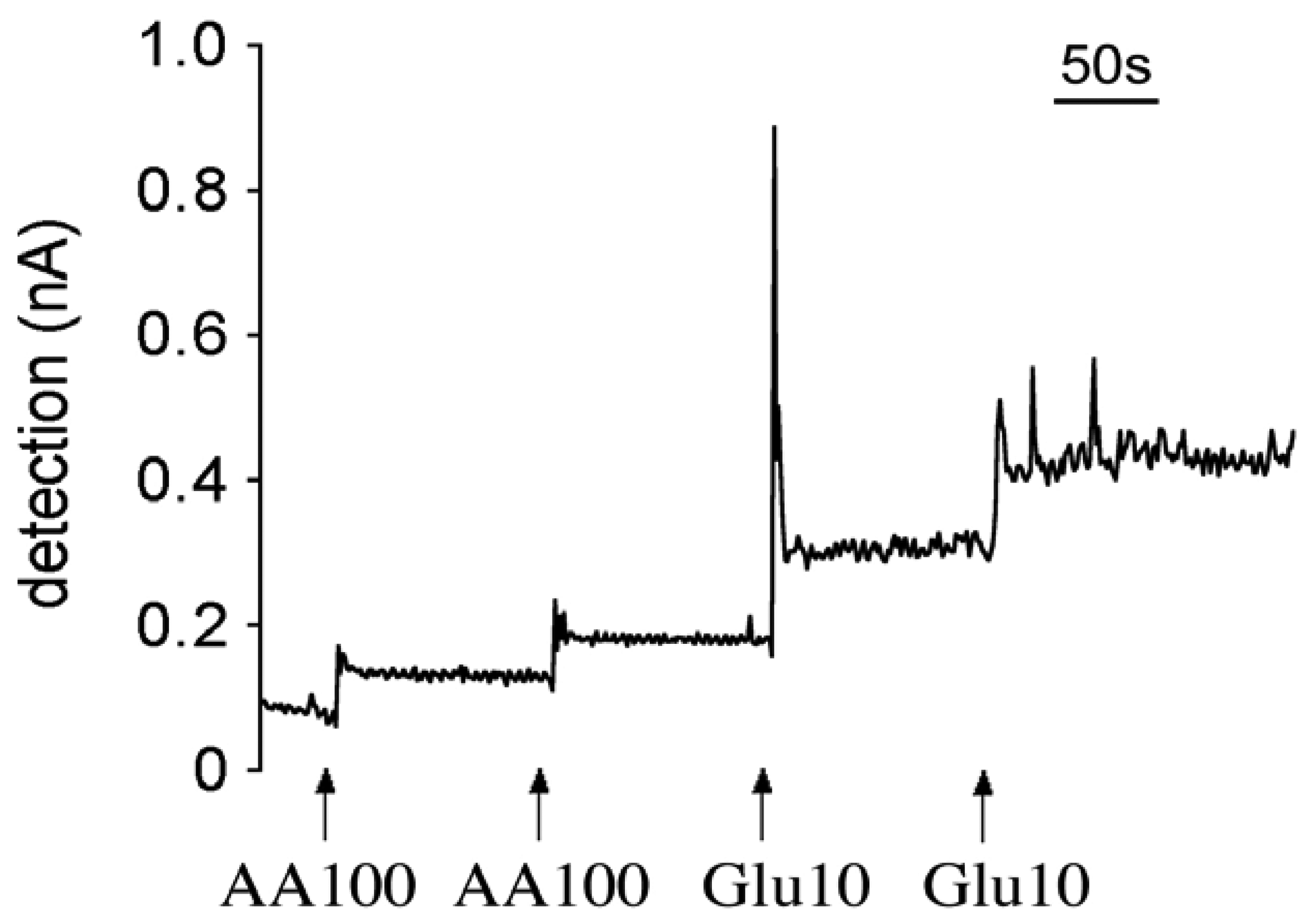
3.1 Response time
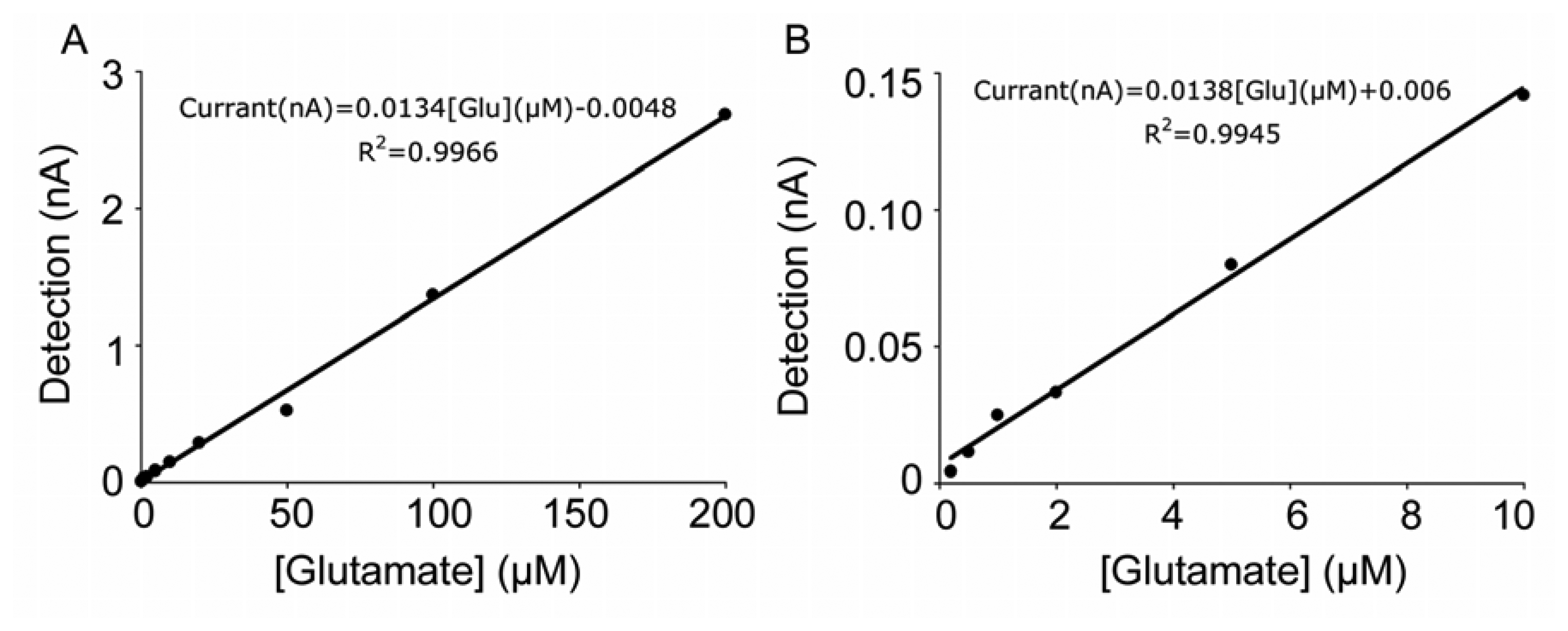
3.2 Sensitivity and linearity of the glutamate response
3.3 Specificity
4. Interference and solutions
5. In vivo detection of glutamate
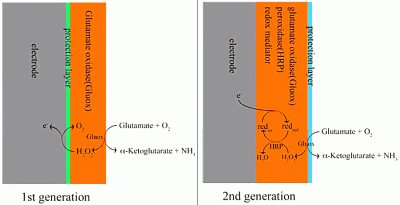
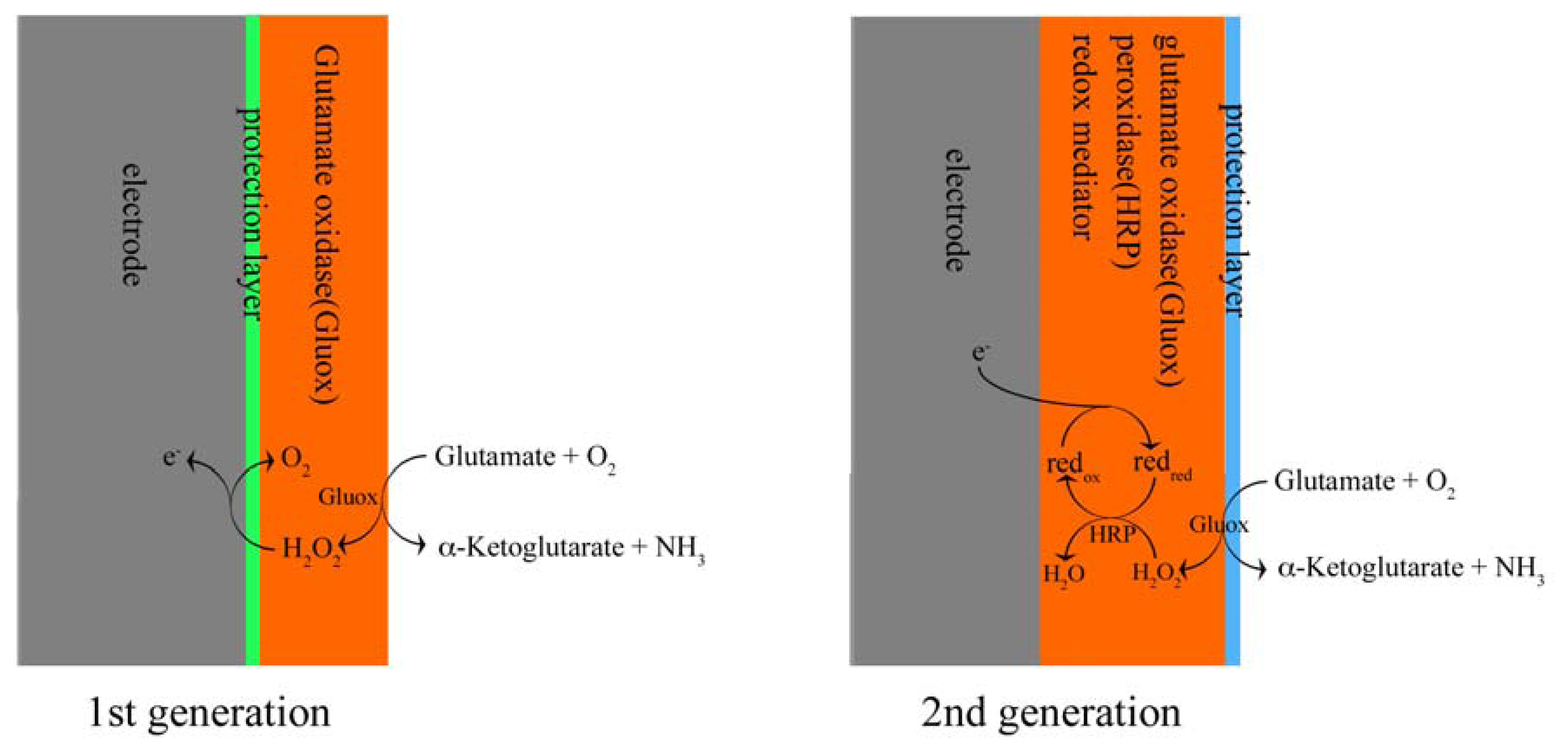
5.1 First generation sensors
5.2 Second generation sensors
5.3. The extracellular concentration of glutamate
6. Discussion
6.1. Sensitivity of the sensors
6.2. Speed and selectivity of the sensors
6.3. Possible synaptic origin of recorded glutamate
References
- Goff, D.C.; Coyle, J.T. The Emerging Role of Glutamate in the Pathophysiology and Treatment of Schizophrenia. Am. J. Psychiatry 2001, 158, 1367–1377. [Google Scholar]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, S14–23. [Google Scholar]
- Javitt, D.C. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry 2004, 9, 984–997. [Google Scholar]
- Amiel, J.; Mathew, S. Glutamate and anxiety disorders. Curr. Psychiatry Rep. 2007, 9, 278–283. [Google Scholar]
- Hascup, K.N.; Hascup, E.R.; Pomerleau, F.; Huettl, P.; Gerhardt, G.A. Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J. Pharmacol Exp. Ther. 2008, 324, 725–31. [Google Scholar]
- Rutherford, E.C.; Pomerleau, F.; Huettl, P.; Strömberg, I.; Gerhardt, G.A. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J. Neurochem. 2007, 102, 712–22. [Google Scholar]
- Day, B.K.; Pomerleau, F.; Burmeister, J.J.; Huettl, P.; Gerhardt, G.A. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J. Neurochem. 2006, 96, 1626–35. [Google Scholar]
- Kulagina, N.V.; Shankar, L.; Michael, A.C. Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal. Chem. 1999, 71, 5093–100. [Google Scholar]
- Rahman, M.A.; Kwon, N.; Won, M.; Choe, E.S.; Shim, Y. Functionalized conducting polymer as an enzyme-immobilizing substrate: an amperometric glutamate microbiosensor for in vivo measurements. Anal. Chem. 2005, 77, 4854–60. [Google Scholar]
- Schuvailo, O.M.; Soldatkin, O.O.; Lefebvre, A.; Cespuglio, R.; Soldatkin, A.P. Highly selective microbiosensors for in vivo measurement of glucose, lactate and glutamate. Anal. Chim. Acta. 2006, 573-574, 110–6. [Google Scholar]
- Oldenziel, W.; Dijkstra, G.; Cremers, T.; Westerink, B. In vivo monitoring of extracellular glutamate in the brain with a microsensor. Brain Res. 2006, 1118, 34–42. [Google Scholar]
- Hu, Y.; Mitchell, K.M.; Albahadily, F.N.; Michaelis, E.K.; Wilson, G.S. Direct measurement of glutamate release in the brain using a dual enzyme-based electrochemical sensor. Brain Res. 1994, 659, 117–25. [Google Scholar]
- Clark, L.C.; Lyons, C. Electrode Systems for Continuous Monitoring in Cardiovascular Surgery. Ann. N.Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar]
- Guilbault, G.G.; Lubrano, G.J. An enzyme electrode for the amperometric determination of glucose. Anal. Chim. Acta 1973, 64, 439–455. [Google Scholar]
- Updike, S.J.; Hicks, G.P. The Enzyme Electrode. Nature 1967, 214, 986–988. [Google Scholar]
- Cass, A.E.; Davis, G.; Francis, G.D.; Hill, H.A.; Aston, W.J.; Higgins, I.J.; Plotkin, E.V.; Scott, L.D.L.; Turner, A.P.F. Ferrocene-mediated enzyme electrode for amperometric determination of glucose. Anal. Chem. 1984, 56, 667–71. [Google Scholar]
- Pandey, P.C.; Glazier, S.; Weetall, H.H. An Amperometric Flow-Injection Analysis Biosensor for Glucose Based on Graphite Paste Modified with Tetracyanoquinodimethane. Anal. Biochem. 1993, 214, 233–237. [Google Scholar]
- Oldenziel, W.; Dijkstra, G.; Cremers, T.; Westerink, B. Evaluation of Hydrogel-Coated Glutamate Microsensors. Anal. Chem. 2006, 78, 3366–3378. [Google Scholar]
- McMahon, C.P.; Rocchitta, G.; Kirwan, S.M.; Killoran, S.J.; Serra, P.A.; Lowry, J.P.; O'Neill, R.D. Oxygen tolerance of an implantable polymer/enzyme composite glutamate biosensor displaying polycation-enhanced substrate sensitivity. Biosens. Bioelectron. 2007, 22, 1466–1473. [Google Scholar]
- Maalouf, R.; Chebib, H.; Saïkali, Y.; Vittori, O.; Sigaud, M.; Jaffrezic-Renault, N. Amperometric and impedimetric characterization of a glutamate biosensor based on Nafion and a methyl viologen modified glassy carbon electrode. Biosens. Bioelectron. 2007, 22, 2682–8. [Google Scholar]
- O'Neill, R.D.; Chang, S.; Lowry, J.P.; McNeil, C.J. Comparisons of platinum, gold, palladium and glassy carbon as electrode materials in the design of biosensors for glutamate. Biosens. Bioelectron. 2004, 19, 1521–8. [Google Scholar]
- Ricci, F.; Amine, A.; Moscone, D.; Palleschi, G. A probe for NADH and H2O2 amperometric detection at low applied potential for oxidase and dehydrogenase based biosensor applications. Biosens. Bioelectron. 2007, 22, 854–62. [Google Scholar]
- Chaubey, A.; Malhotra, B.D. Mediated biosensors. Biosens. Bioelectron. 2002, 17, 441–456. [Google Scholar]
- Oldenziel, W.H.; Westerink, B.H.C. Improving glutamate microsensors by optimizing the composition of the redox hydrogel. Anal. Chem. 2005, 77, 5520–8. [Google Scholar]
- Oldenziel, W.H.; Beukema, W.; Westerink, B.H.C. Improving the reproducibility of hydrogel-coated glutamate microsensors by using an automated dipcoater. J. Neurosci. Methods 2004, 140, 117–26. [Google Scholar]
- Bravo, R.; Hsueh, C.C.; Jaramillo, A.; Brajter-Toth, A. Possibilities and limitations in miniaturized sensor design for uric acid. Analyst 1998, 123, 1625–30. [Google Scholar]
- Borsook, H.; Keighley, G. Oxidation-Reduction Potential of Ascorbic Acid (Vitamin C). Proc. Natl. Acad. Sci. U. S. A. 1933, 19, 875–8. [Google Scholar]
- Bohmer, A.; Muller, A.; Passarge, M.; Liebs, P.; Honeck, H.; Muller, H. A Novel L-Glutamate Oxidase from Streptomyces Endus. Purification and Properties. Eur. J. Biochem. 1989, 182, 327–332. [Google Scholar]
- Kusakabe, H.; Midorikawa, Y.; Fujishima, T.; Kuninaka, A.; Yoshino, H. Purification and Properties of a New Enzyme, L-Glutamate Oxidase, from Streptomyces sp. X-119-6 Grown on Wheat Bran. Agric. Biol. Chem. 1983, 47, 1323–1328. [Google Scholar]
- Sternberg, R.; Bindra, D.S.; Wilson, G.S.; Thévenot, D.R. Covalent enzyme coupling on cellulose acetate membranes for glucose sensor development. Anal. Chem. 1988, 60, 2781–6. [Google Scholar]
- Mu, S.; Xue, H. Bioelectrochemical characteristics of glucose oxidase immobilized in a polyaniline film. Sensors & Actuators: B. Chemical 1996, 31, 155–160. [Google Scholar]
- Rubinstein, I.; Bard, A.J. Polymer films on electrodes. 4. Nafion-coated electrodes and electrogenerated chemiluminescence of surface-attached tris(2,2′-bipyridine)ruthenium(2+). J. Am. Chem. Soc. 1980, 102, 6641–6642. [Google Scholar]
- Gerhardt, G.A.; Ok, e A.F.; Nagy, G.; Moghaddam, B.; Adams, R.N. Nafion-coated electrodes with high selectivity for CNS electrochemistry. Brain. Res. 1984, 290, 390–5. [Google Scholar]
- Fan, Z.; Jed Harrison, D. Permeability of glucose and other neutral species through recast perfluorosulfonated ionomer films. Anal. Chem. (Washington, DC, U. S.) 1992, 64, 1304–1311. [Google Scholar]
- Moore, R.B., III; Martin, C.R. Chemical and morphological properties of solution-cast perfluorosulfonate ionomers. Macromolecules 1988, 21, 1334–1339. [Google Scholar]
- Zook, L.A.; Leddy, J. Density and Solubility of Nafion: Recast, Annealed, and Commercial Films. Anal. Chem. 1996, 68, 3793–3796. [Google Scholar]
- Hsueh, C.C.; Brajter-Toth, A. Electrochemical Preparation and Analytical Applications of Ultrathin Overoxidized Polypyrrole Films. Anal. Chem. 1994, 66, 2458–2464. [Google Scholar]
- Wang, J.; Chen, S.; Lin, M. Use of different electropolymerization conditions for controlling the size-exclusion selectivity at polyaniline, polypyrrole and polyphenol films. J. electroanal. chem. interfacial electrochem. 1989, 273, 231–242. [Google Scholar]
- Cooper, J.M.; Foreman, P.L.; Glidle, A.; Ling, T.W.; Pritchard, D.J. Glutamate oxidase enzyme electrodes: microsensors for neurotransmitter determination using electrochemically polymerized permselective films. J. Electroanal. Chem. 1995, 388, 143–149. [Google Scholar]
- Shin, M.C.; Kim, H.S. Electrochemical characterization of polypyrrole/glucose oxidase biosensor: Part II. Optimal preparation conditions for the biosensor. Biosens. Bioelectron. 1996, 11, 171–178. [Google Scholar]
- Umana, M.; Waller, J. Protein-modified electrodes. The glucose oxidase/polypyrrole system. Anal. Chem. 1986, 58, 2979–2983. [Google Scholar]
- Malitesta, C.; Palmisano, F.; Torsi, L.; Zambonin, P.G. Glucose fast-response amperometric sensor based on glucose oxidase immobilized in an electropolymerized poly (o-phenylenediamine) film. Anal. Chem. 1990, 62, 2735–2740. [Google Scholar]
- Moussy, F.; Harrison, D.J.; O'Brienm, D.W.; Rajotte, R.V. Performance of subcutaneously implanted needle-type glucose sensors employing a novel trilayer coating. Anal. Chem. 1993, 65, 2072–2077. [Google Scholar]
- Yacynych, A.M.; Mark, J. The Spectroelectrochemical Study of the Oxidation of 1,2-Diaminobenzene: Alone and in the Presence of Ni(II). J. Electrochem. Soc. 1976, 123, 1346–1351. [Google Scholar]
- Elliott, J.M.; Cabuché, L.M.; Bartlett, P.N. Electrochemical characterization of a templated insulating polymer-modified electrode. Anal. Chem. 2001, 73, 2855–61. [Google Scholar]
- Berners, M.O.; Boutelle, M.G.; Fillenz, M. On-line measurement of brain glutamate with an enzyme/polymer-coated tubular electrode. Anal. Chem. 1994, 66, 2017–21. [Google Scholar]
- Hendry, S.; Cardosi, M.; Turner, A.; Neuse, E. Polyferrocenes as mediators in amperometric biosensors for glucose: Biosensors. Anal. Chim. Acta 1993, 281, 453–459. [Google Scholar]
- Dicks, J.M.; Hattori, S.; Karube, I.; Turnerm, A.P.; Yokozawa, T. Ferrocene modified polypyrrole with immobilised glucose oxidase and its application in amperometric glucose microbiosensors. Ann. Biol. Clin.(Paris) 1989, 47, 607–19. [Google Scholar]
- Bartlett, P.N.; Ali, Z.; Eastwick-Field, V. Electrochemical immobilisation of enzymes. Part 4.-Co-immobilisation of glucose oxidase and ferro/ferricyanide in poly(N-methylpyrrole) films. Faraday Trans. 1992, 88, 2677–2683. [Google Scholar]
- Kajiya, Y.; Sugai, H.; Iwakura, C.; Yoneyama, H. Glucose sensitivity of polypyrrole films containing immobilized glucose oxidase and hydroquinonesulfonate ions. Anal. Chem. 1991, 63, 49–54. [Google Scholar]
- Karyakin, A.A.; Karyakina, E.E.; Gorton, L. Amperometric biosensor for glutamate using Prussian blue-based Òartificial peroxidaseÓ as a transducer for hydrogen peroxide. Anal. Chem 2000, 72, 1720–1723. [Google Scholar]
- Gregg, B.A.; Heller, A. Cross-linked redox gels containing glucose oxidase for amperometric biosensor applications. Anal. Chem. 1990, 62, 258–263. [Google Scholar]
- Ballarin, B.; Cassani, M.C.; Mazzoni, R.; Scavetta, E.; Tonelli, D. Enzyme electrodes based on sono-gel containing ferrocenyl compounds. Biosens. Bioelectron. 2007, 22, 1317–1322. [Google Scholar]
- Mulchandani, A.; Pan, S. Ferrocene-Conjugatedm-Phenylenediamine Conducting Polymer-Incorporated Peroxidase Biosensors. Anal. Biochem. 1999, 267, 141–147. [Google Scholar]
- Gülce, A.; Gülce, H. Polyvinylferrocenium modified Pt electrode for anaerobic glucose monitoring. J. Biochem. Biophys. Methods 2005, 62, 81–92. [Google Scholar]
- Linke, B.; Kerner, W.; Kiwit, M.; Pishko, M.; Heller, A. Amperometric biosensor for in vivo glucose sensing based on glucose oxidase immobilized in a redox hydrogel. Biosens. Bioelectron. 1994, 9, 151–8. [Google Scholar]
- Oldenziel, W.; deJong, L.; Dijkstra, G.; Cremers, T.; Westerink, B. Improving the Performance of Glutamate Microsensors by Purification of Ascorbate Oxidase. Anal. Chem. 2006, 78, 2456–2460. [Google Scholar]
- Rahman, M.A.; Lee, K.; Park, D.; Won, M.; Shim, Y. An amperometric bilirubin biosensor based on a conductive poly-terthiophene-Mn(II) complex. Biosens. Bioelectron. 2008, 23, 857–864. [Google Scholar]
- Morita, H.; Abe, C.; Awazu, C.; Tanaka, K. Long-term hypergravity induces plastic alterations in vestibulo-cardiovascular reflex in conscious rats. Neurosci. Lett. 2007, 412, 201–205. [Google Scholar]
- Cairns, B.E.; Dong, X.; Mann, M.K.; Svensson, P.; Sessle, B.J.; Arendt-Nielsen, L.; McErlane, K. Systemic administration of monosodium glutamate elevates intramuscular glutamate levels and sensitizes rat masseter muscle afferent fibers. Pain 2007, 132, 33–41. [Google Scholar]
- Lee, K.H.; Blaha, C.D. Apparatus and method for modulating neurochemical levels in the brain. US Patent WO/2006/041871; IPC: A61N 1/36 (2006.01), 2006. [Google Scholar]
- Burmeister, J.J.; Gerhardt, G.A. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal. Chem. 2001, 73, 1037–1042. [Google Scholar]
- Burmeister, J.J.; Moxon, K.; Gerhardt, G.A. Ceramic-based multisite microelectrodes for electrochemical recordings. Anal. Chem. 2000, 72, 187–192. [Google Scholar]
- Burmeister, J.J.; Pomerleau, F.; Palmer, M.; Day, B.K.; Huettl, P.; Gerhardt, G.A. Improved ceramic-based multisite microelectrode for rapid measurements of l-glutamate in the CNS. J. Neurosci. Methods 2002, 119, 163–171. [Google Scholar]
- Nickell, J.; Pomerleau, F.; Allen, J.; Gerhardt, G.A. Age-related changes in the dynamics of potassium-evoked L-glutamate release in the striatum of Fischer 344 rats. J Neural Transm 2005, 112, 87–96. [Google Scholar]
- Nickell, J.; Salvatore, M.F.; Pomerleau, F.; Apparsundaram, S.; Gerhardt, G.A. Reduced plasma membrane surface expression of GLAST mediates decreased glutamate regulation in the aged striatum. Neurobiol. Aging 2007, 28, 1737–1748. [Google Scholar]
- Quintero, J.E.; Day, B.K.; Zhang, Z.; Grondin, R.; Stephens, M.L.; Huettl, P.; Pomerleau, F.; Gash, D.M.; Gerhardt, G.A. Amperometric measures of age-related changes in glutamate regulation in the cortex of rhesus monkeys. Exp. Neurol. 2007, 208, 238–46. [Google Scholar]
- Thomas, T.C.; Grandy, D.K.; Gerhardt, G.A.; Glaser, P.E.A. Decreased Dopamine D4 Receptor Expression Increases Extracellular Glutamate and Alters Its Regulation in Mouse Striatum. Neuropsychopharmacology advance online publication. 2008. [Google Scholar] [CrossRef]
- Borland, L.M.; Shi, G.; Yang, H.; Michael, A.C. Voltammetric study of extracellular dopamine near microdialysis probes acutely implanted in the striatum of the anesthetized rat. J. Neurosci. Methods 2005, 146, 149–58. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research group | Characteristics of sensorin vitro | Generation | Reference | |||||
|---|---|---|---|---|---|---|---|---|
| Electrode | Surface (mm2) | Potential (mV) vs. Ag/AgCl | Response time (s) | Sensitivity (nA/μM) | glutamate detection limit(μM) | |||
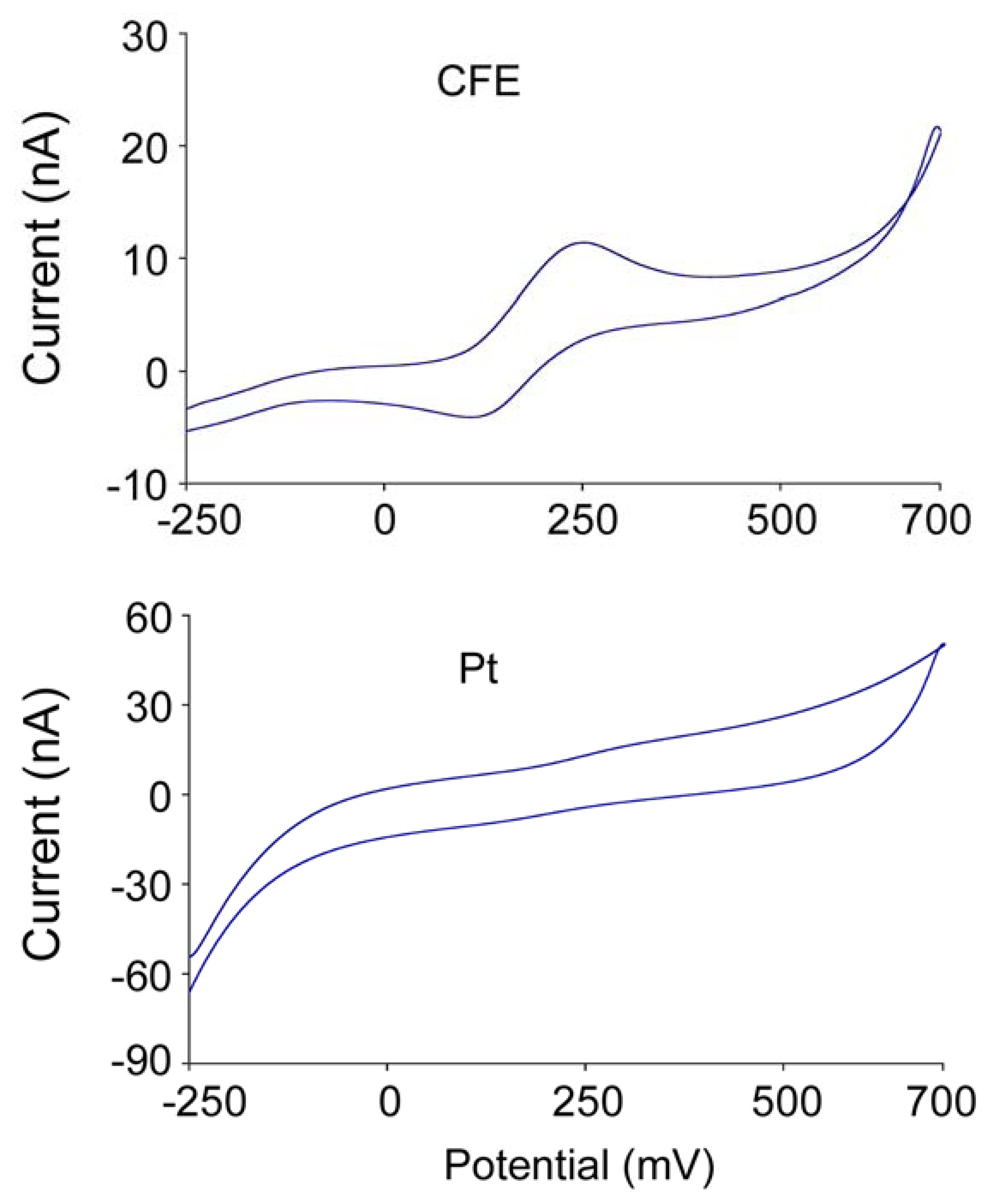
| Gerhardt | Pt site | 0.005 | +700 | ∼ 1 | 0.016 ± 0.001 | 1.82 ±0.17 | 1st | [7] |
| Michael | CFE | 0.0095 ∼0.0126 | -100 | 20 ∼ 40 | 0.0034 ± 0.001 | 1∼3 | 2nd | [8] |
| Shim | CP coated Pt cylinder | 0.024 | +450 | ∼ 10 | 14.0 ± 0.2 | 0.1± 0.03 | 1st | [9] |
| Soldatkin | Ru coated CFE | 0.048 | +400 | - | 0.029 | 2.5 | 1st | [10] |
| Westerink | CFE | 0.0095 ∼0.016 | -150 | ∼ 8 | 0.0055 ± 0.00007 | 5 | 2nd | [11] |
| Wilson | Pt/Ir | 0.183 | +600 | ∼ 1 | 0.1 | 2 | 1st | [12] |
| AA 200 μM | UA 50 μM | dopamine 5 μM | cysteine 5 μM | ||
|---|---|---|---|---|---|
| 1st generation | + Glu 100μM | 122±4 (n=4) | 101±5 (n=4) | 116±4 (n=4) | 117±9 (n=4) |
| - | 17±2 (n=4) | 4±0 (n=4) | 23±4 (n=4) | 3±0 (n=4) | |
| 2nd generation | + Glu 100μM | 78±5 (n=19) | 72±27 (n=14) | 98±6 (n=9) | 108±11 (n=9) |
| - | 3±3 (n=19) | 0±0 (n=14) | 12±7 (n=9) | 18±10 (n=9) |
| Enzymatic biosensor | Electrode | Mediator | Redox potential | Reference |
|---|---|---|---|---|
| Glucose oxidase | sono-gel carbon composite (SCC) electrode | ferrocene | 0.30 V vs. SCE | [53] |
| Glucose oxidase | Glass carbon electrode | Poly(m-aminoanilino methylferrocene) | -0.05 V vs. Ag/AgCl | [54] |
| Glucose oxidase | Pt | polyvinylferrocenium | 0.30 V vs. SCE | [55] |
| Glutamate oxidase | Pt | Prussian Blue | 0.0 V vs. Ag/AgCl | [51] |
| Glucose oxidase | Carbon rotating disk electrodes | [Os(bpy)2Cl]+1/+2 | 0.40 V vs. Ag/AgCl | [56] |
| Glutamate oxidase | CFE | [Os(bpy)2Cl]+2/+3 | -0.15 V vs. Ag/AgCl | [18] |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qin, S.; Van der Zeyden, M.; Oldenziel, W.H.; Cremers, T.I.F.H.; Westerink, B.H.C. Microsensors for in vivo Measurement of Glutamate in Brain Tissue. Sensors 2008, 8, 6860-6884. https://doi.org/10.3390/s8116860
Qin S, Van der Zeyden M, Oldenziel WH, Cremers TIFH, Westerink BHC. Microsensors for in vivo Measurement of Glutamate in Brain Tissue. Sensors. 2008; 8(11):6860-6884. https://doi.org/10.3390/s8116860
Chicago/Turabian StyleQin, Si, Miranda Van der Zeyden, Weite H. Oldenziel, Thomas I.F.H. Cremers, and Ben H.C. Westerink. 2008. "Microsensors for in vivo Measurement of Glutamate in Brain Tissue" Sensors 8, no. 11: 6860-6884. https://doi.org/10.3390/s8116860