A Role for Adenosine A1 Receptors in GABA and NMDA-Receptor Mediated Modulation of Dopamine Release: Studies Using Fast Cyclic Voltammetry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
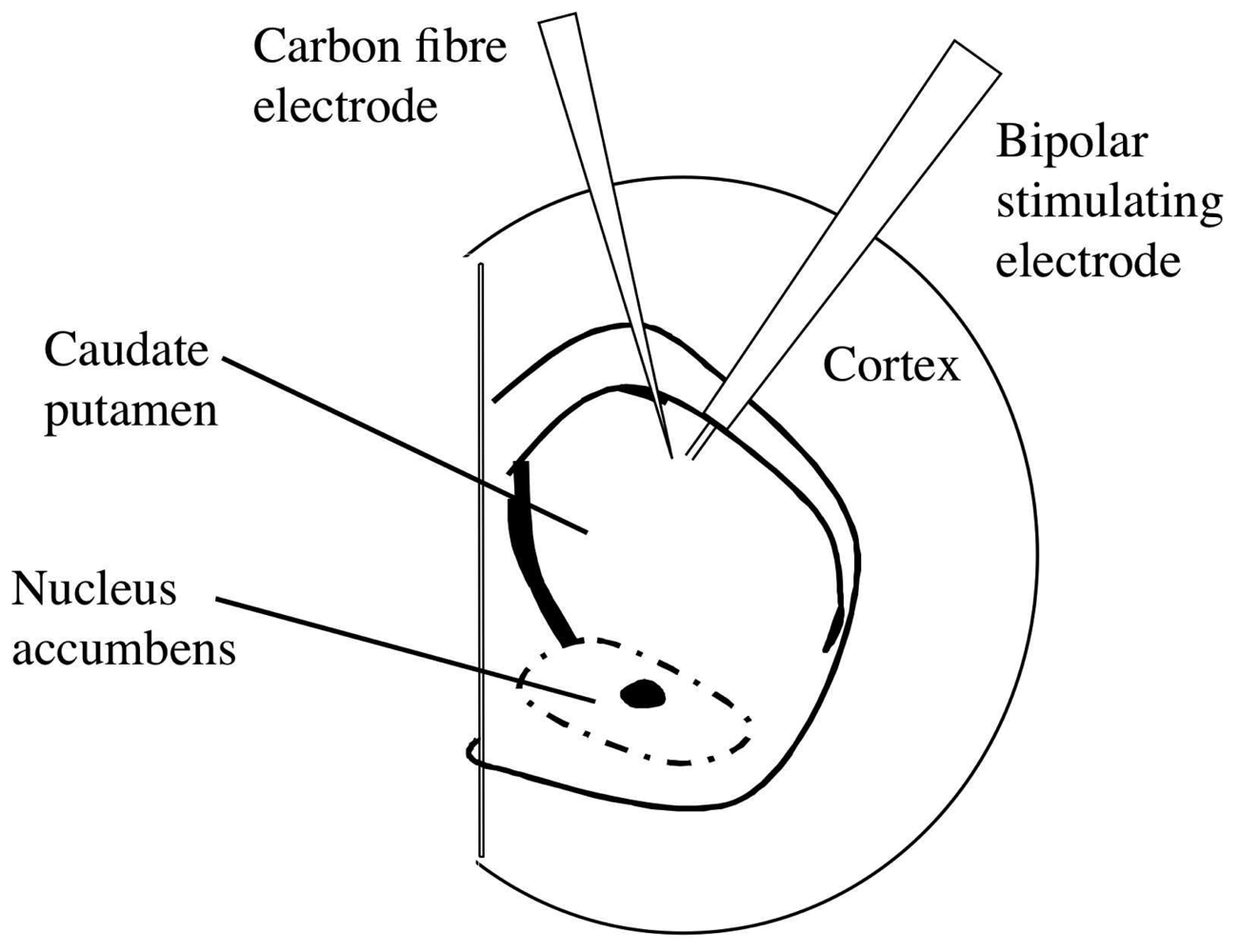
2.1 Brain slices
2.2 Measurement of endogenous dopamine release
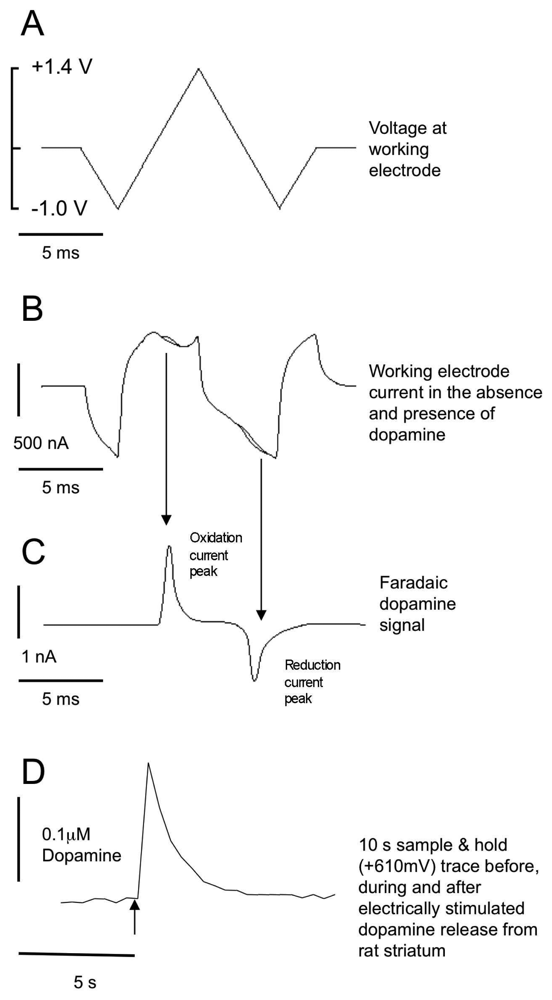
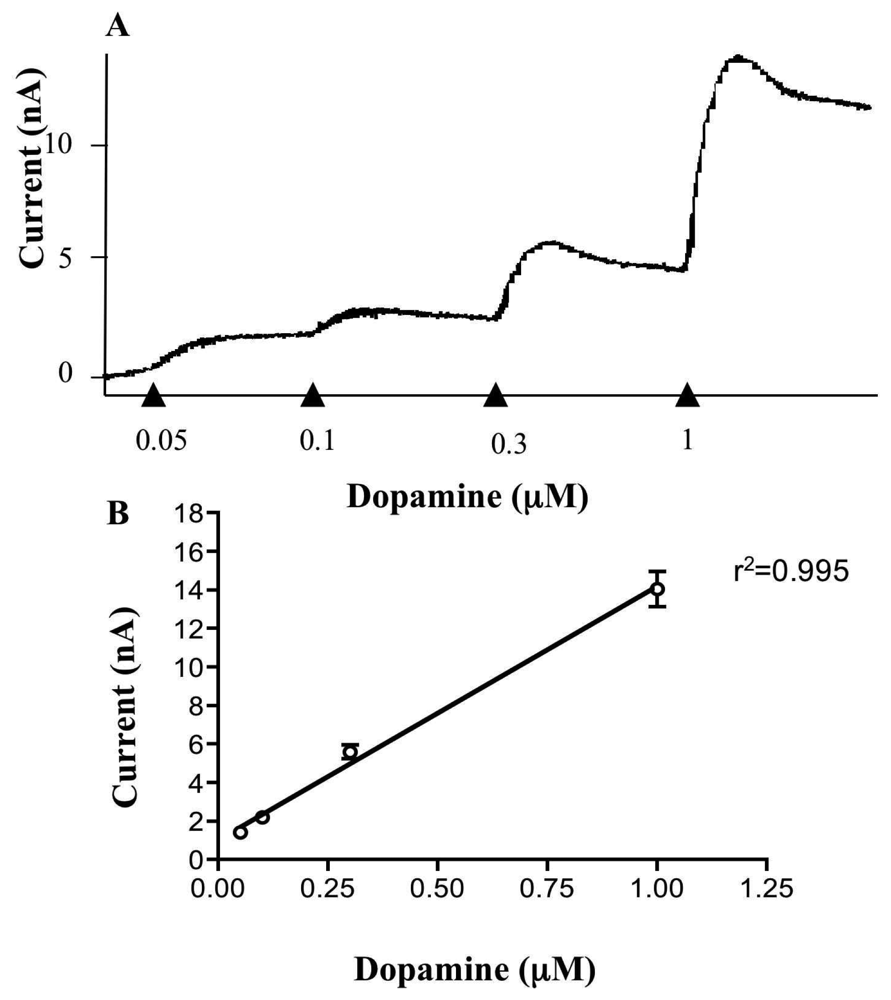
2.3 Electrode calibration
2.4 Buffers and Drugs
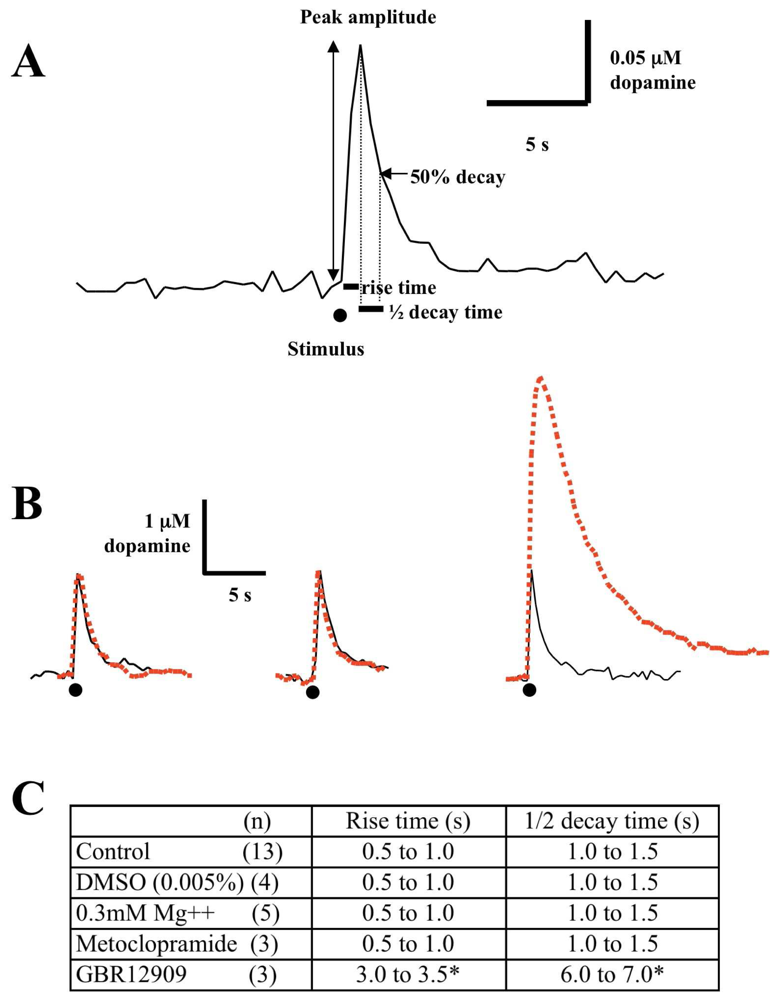
2.5 Data analysis
3. Results
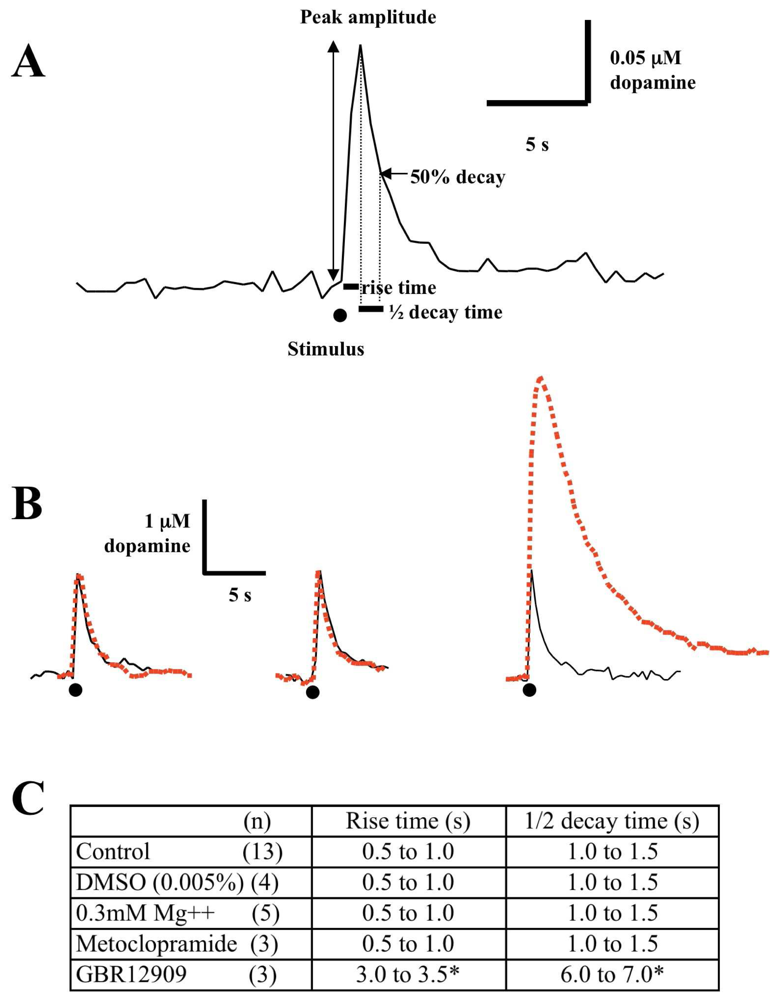
3.1 Effect of low magnesium on single pulse evoked dopamine release
3.2 Role of A1 receptors in the NMDA modulation of dopamine release
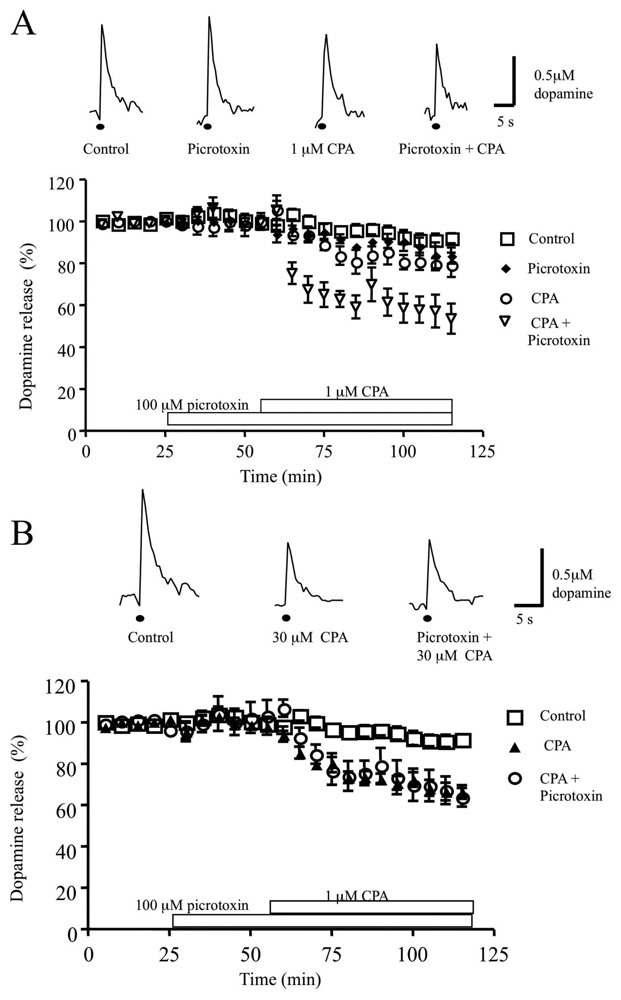
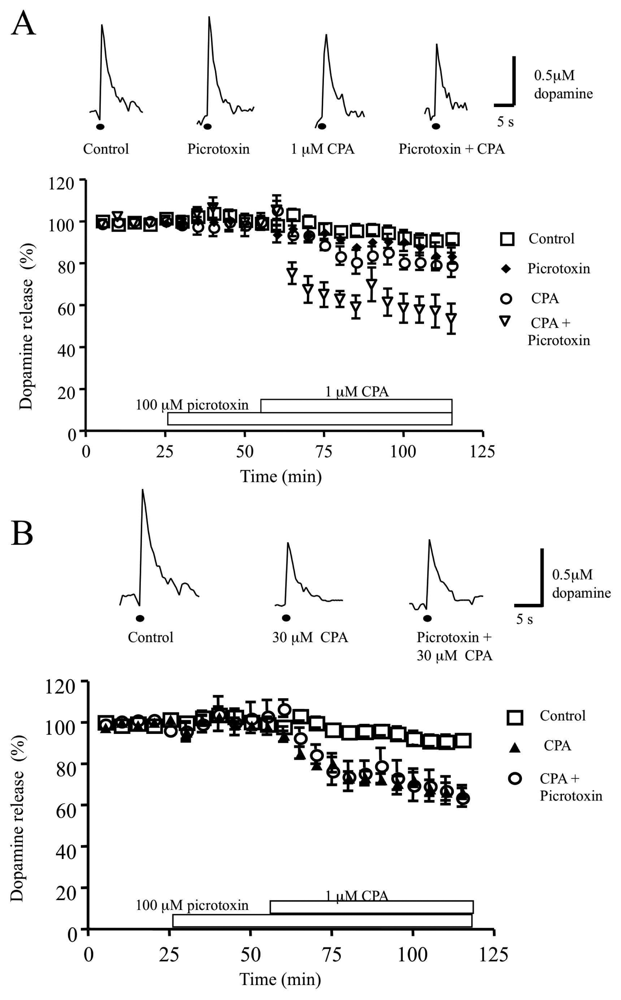
3.3 Role of the GABAA receptor antagonist picrotoxin, in the CPA-induced inhibition of dopamine release
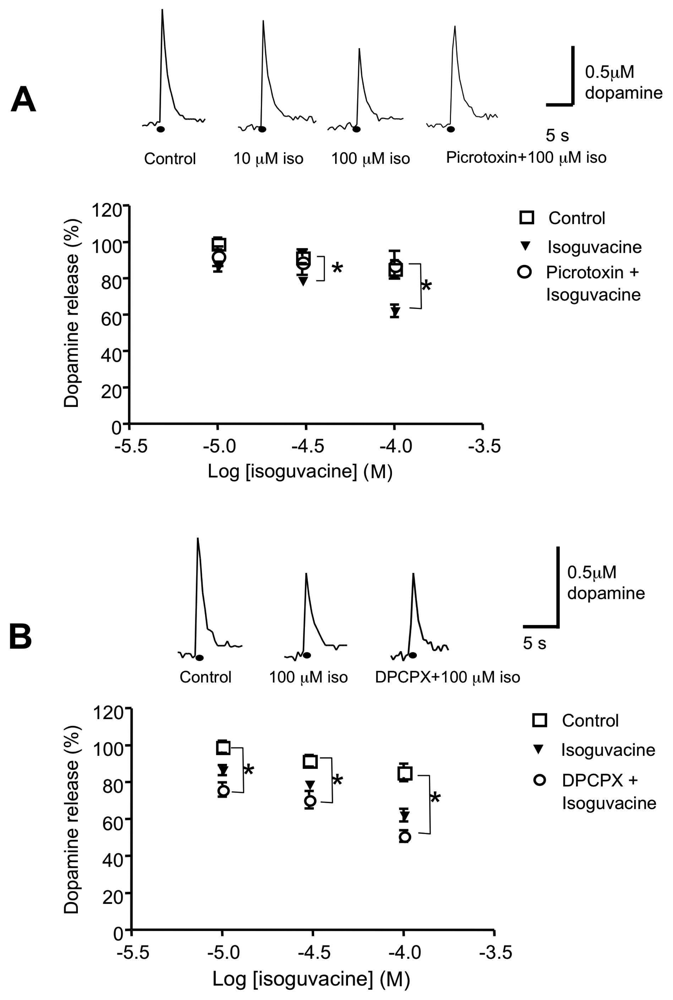
3.4 Role of the GABAA receptor agonist, isoguvacine and the A1 receptor antagonist DPCPX in dopamine release
4. Discussion
4.1 Glutamate transmission
4.2 GABA transmission
Conclusion
Acknowledgments
References
- Centonze, D.; Grande, C.; Usiello, A.; Gubellini, P.; Erbs, E.; Martin, A.B.; Pisani, A.; Tognazzi, N.; Bernardi, G.; Moratalla, R.; Borrelli, E.; Calabresi, P. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J. Neurosci. 2003, 23(15), 6245–6254. [Google Scholar]
- Calabresi, P.; Centonze, D.; Gubellini, P.; Marfia, G.A.; Pisani, A.; Sancesario, G.; Bernardi, G. Synaptic transmission in the striatum: from plasticity to neurodegeneration. Prog. Neurobiol. 2000, 61(3), 231–265. [Google Scholar]
- Ferre, S.; Popoli, P.; Gimenez-Llort, L.; Finnman, U.B.; Martinez, E.; Scotti de Carolis, A.; Fuxe, K. Postsynaptic antagonistic interaction between adenosine A1 and dopamine D1 receptors. Neuroreport 1994, 6(1), 73–76. [Google Scholar]
- Franco, R.; Ferre, S.; Agnati, L.; Torvinen, M.; Gines, S.; Hillion, J.; Casado, V.; Lledo, P.; Zoli, M.; Lluis, C.; Fuxe, K. Evidence for adenosine/dopamine receptor interactions: indications for heteromerization. Neuropsychopharmacology 2000, 23 (4 Suppl), S50–59. [Google Scholar]
- Torvinen, M.; Gines, S.; Hillion, J.; Latini, S.; Canals, M.; Ciruela, F.; Bordoni, F.; Staines, W.; Pedata, F.; Agnati, L. F.; Lluis, C.; Franco, R.; Ferre, S.; Fuxe, K. Interactions among adenosine deaminase, adenosine A(1) receptors and dopamine D(1) receptors in stably cotransfected fibroblast cells and neurons. Neuroscience 2002, 113(3), 709–719. [Google Scholar]
- Fuxe, K.; Marcellino, D.; Genedani, S.; Agnati, L. Adenosine A (2A) receptors, dopamine D(2) receptors and their interactions in Parkinson's disease. Mov. Disord. 2007, 22(14), 1990–2017. [Google Scholar]
- Fuxe, K.; Canals, M.; Torvinen, M.; Marcellino, D.; Terasmaa, A.; Genedani, S.; Leo, G.; Guidolin, D.; Diaz-Cabiale, Z.; Rivera, A.; Lundstrom, L.; Langel, U.; Narvaez, J.; Tanganelli, S.; Lluis, C.; Ferre, S.; Woods, A.; Franco, R.; Agnati, L.F. Intramembrane receptor-receptor interactions: a novel principle in molecular medicine. J. Neural. Transm. 2007, 114(1), 49–75. [Google Scholar]
- Franco, R.; Lluis, C.; Canela, E.I.; Mallol, J.; Agnati, L.; Casado, V.; Ciruela, F.; Ferre, S.; Fuxe, K. Receptor-receptor interactions involving adenosine A1 or dopamine D1 receptors and accessory proteins. J. Neural. Transm. 2007, 114(1), 93–104. [Google Scholar]
- Wood, P.L.; Kim, H.S.; Boyar, W.C.; Hutchison, A. Inhibition of nigrostriatal release of dopamine in the rat by adenosine receptor agonists: A1 receptor mediation. Neuropharmacology 1989, 28(1), 21–25. [Google Scholar]
- Ballarin, M.; Reiriz, J.; Ambrosio, S.; Mahy, N. Effect of locally infused 2-chloroadenosine, an A1 receptor agonist, on spontaneous and evoked dopamine release in rat neostriatum. Neurosci. Lett. 1995, 185(1), 29–32. [Google Scholar]
- Quarta, D.; Ferre, S.; Solinas, M.; You, Z.B.; Hockemeyer, J.; Popoli, P.; Goldberg, S.R. Opposite modulatory roles for adenosine A1 and A2A receptors on glutamate and dopamine release in the shell of the nucleus accumbens. Effects of chronic caffeine exposure. J. Neurochem. 2004, 88(5), 1151–1158. [Google Scholar]
- Quarta, D.; Borycz, J.; Solinas, M.; Patkar, K.; Hockemeyer, J.; Ciruela, F.; Lluis, C.; Franco, R.; Woods, A.S.; Goldberg, S.R.; Ferre, S. Adenosine receptor-mediated modulation of dopamine release in the nucleus accumbens depends on glutamate neurotransmission and N-methyl-D-aspartate receptor stimulation. J. Neurochem. 2004, 91(4), 873–880. [Google Scholar]
- Jin, S.; Johansson, B.; Fredholm, B.B. Effects of adenosine A1 and A2 receptor activation on electrically evoked dopamine and acetylcholine release from rat striatal slices. J. Pharmacol. Exp. Ther. 1993, 267(2), 801–808. [Google Scholar]
- Okada, M.; Mizuno, K.; Kaneko, S. Adenosine A1 and A2 receptors modulate extracellular dopamine levels in rat striatum. Neurosci. Lett. 1996, 212(1), 53–56. [Google Scholar]
- Golembiowska, K.; Zylewska, A. Agonists of A1 and A2A adenosine receptors attenuate methamphetamine-induced overflow of dopamine in rat striatum. Brain Res. 1998, 806(2), 202–209. [Google Scholar]
- Yabuuchi, K.; Kuroiwa, M.; Shuto, T.; Sotogaku, N.; Snyder, G. L.; Higashi, H.; Tanaka, M.; Greengard, P.; Nishi, A. Role of adenosine A1 receptors in the modulation of dopamine D1 and adenosine A2A receptor signaling in the neostriatum. Neuroscience 2006, 141(1), 19–25. [Google Scholar]
- O′Neill, C.; Nolan, B.J.; Macari, A.; O′Boyle, K.M.; O′Connor, J.J. Adenosine A1 receptor-mediated inhibition of dopamine release from rat striatal slices is modulated by D1 dopamine receptors. Eur. J. Neurosci. 2007, 26(12), 3421–3428. [Google Scholar]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: from structure to function. Physiol. Rev. 1998, 78(1), 189–225. [Google Scholar]
- Schoffelmeer, A.N.; Vanderschuren, L.J.; De Vries, T.J.; Hogenboom, F.; Wardeh, G.; Mulder, A. H. Synergistically interacting dopamine D1 and NMDA receptors mediate nonvesicular transporter-dependent GABA release from rat striatal medium spiny neurons. J. Neurosci. 2000, 20(9), 3496–3503. [Google Scholar]
- Tarazi, F.I.; Campbell, A.; Yeghiayan, S.K.; Baldessarini, R.J. Localization of ionotropic glutamate receptors in caudate-putamen and nucleus accumbens septi of rat brain: comparison of NMDA, AMPA, and kainate receptors. Synapse 1998, 30(2), 227–235. [Google Scholar]
- Ochi, M.; Inoue, H.; Koizumi, S.; Shibata, S.; Watanabe, S. Long-term enhancement of dopamine release by high frequency tetanic stimulation via a N-methyl-D-aspartate-receptor-mediated pathway in rat striatum. Neuroscience 1995, 66(1), 29–36. [Google Scholar]
- David, H.N.; Ansseau, M.; Abraini, J.H. Dopamine-glutamate reciprocal modulation of release and motor responses in the rat caudate-putamen and nucleus accumbens of “intact” animals. Brain Res. Brain Res. Rev. 2005, 50(2), 336–360. [Google Scholar]
- Iravani, M.M.; Kruk, Z.L. Real-time effects of N-methyl-D-aspartic acid on dopamine release in slices of rat caudate putamen: a study using fast cyclic voltammetry. J. Neurochem. 1996, 66(3), 1076–1085. [Google Scholar]
- Wu, Y.; Pearl, S.M.; Zigmond, M.J.; Michael, A.C. Inhibitory glutamatergic regulation of evoked dopamine release in striatum. Neuroscience 2000, 96(1), 65–72. [Google Scholar]
- McGinty, J.F. Regulation of neurotransmitter interactions in the ventral striatum. Ann. N.Y. Acad. Sci. 1999, 877, 129–139. [Google Scholar]
- Bamford, N.S.; Zhang, H.; Schmitz, Y.; Wu, N.P.; Cepeda, C.; Levine, M.S.; Schmauss, C.; Zakharenko, S.S.; Zablow, L.; Sulzer, D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004, 42(4), 653–663. [Google Scholar]
- Ambrosio, A.F.; Malva, J.O.; Carvalho, A.P.; Carvalho, C.M. Modulation of Ca2+ channels by activation of adenosine A1 receptors in rat striatal glutamatergic nerve terminals. Neurosci. Lett. 1996, 220(3), 163–166. [Google Scholar]
- Huang, Q.; Zhou, D.; Chase, K.; Gusella, J.F.; Aronin, N.; DiFiglia, M. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc. Natl. Acad. Sci. U.S.A. 1992, 89(24), 11988–11992. [Google Scholar]
- Le Moine, C.; Bloch, B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J. Comp. Neurol. 1995, 355(3), 418–426. [Google Scholar]
- Limberger, N.; Spath, L.; Starke, K. A search for receptors modulating the release of gamma-[3H]aminobutyric acid in rabbit caudate nucleus slices. J. Neurochem. 1986, 46(4), 1109–1117. [Google Scholar]
- Floran, B.; Aceves, J.; Sierra, A.; Martinez-Fong, D. Activation of D1 dopamine receptors stimulates the release of GABA in the basal ganglia of the rat. Neurosci. Lett. 1990, 116(1-2), 136–140. [Google Scholar]
- Floran, B.; Gonzalez, B.; Floran, L.; Erlij, D.; Aceves, J. Interactions between adenosine A(2a) and dopamine D2 receptors in the control of [(3)H]GABA release in the globus pallidus of the rat. Eur. J. Pharmacol. 2005, 520(1-3), 43–50. [Google Scholar]
- Harsing, L. G., Jr; Zigmond, M. J. Influence of dopamine on GABA release in striatum: evidence for D1-D2 interactions and non-synaptic influences. Neuroscience 1997, 77(2), 419–429. [Google Scholar]
- Avshalumov, M.V.; Chen, B.T.; Marshall, S.P.; Pena, D.M.; Rice, M.E. Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J. Neurosci. 2003, 23(7), 2744–2750. [Google Scholar]
- Starr, M.S. GABA potentiates potassium-stimulated 3H-dopamine release from slices of rat substantia nigra and corpus striatum. Eur. J. Pharmacol. 1978, 48(3), 325–328. [Google Scholar]
- Krebs, M.O.; Kemel, M.L.; Gauchy, C.; Desban, M.; Glowinski, J. Local GABAergic regulation of the N-methyl-D-aspartate-evoked release of dopamine is more prominent in striosomes than in matrix of the rat striatum. Neuroscience 1993, 57(2), 249–260. [Google Scholar]
- Ronken, E.; Mulder, A.H.; Schoffelmeer, A.N. Interacting presynaptic kappa-opioid and GABAA receptors modulate dopamine release from rat striatal synaptosomes. J. Neurochem. 1993, 61(5), 1634–1639. [Google Scholar]
- Blanchet, F.; Kemel, M.L.; Gauchy, C.; Desban, M.; Perez, S.; Glowinski, J. N-methyl-D-aspartate-evoked release of [3H]acetylcholine in striatal compartments of the rat: regulatory roles of dopamine and GABA. Neuroscience 1997, 81(1), 113–127. [Google Scholar]
- Cheramy, A.; Nieoullon, A.; Glowinski, J. Blockade of the picrotoxin-induced in vivo release of dopamine in the cat caudate nucleus by diazepam. Life Sci. 1977, 20(5), 811–816. [Google Scholar]
- Mayfield, R.D.; Jones, B.A.; Miller, H.A.; Simosky, J.K.; Larson, G.A.; Zahniser, N.R. Modulation of endogenous GABA release by an antagonistic adenosine A1/dopamineD1 receptor interaction in rat brain limbic regions but not basal ganglia. Synapse 1999, 33(4), 274–281. [Google Scholar]
- Armstrong-James, M.; Millar, J. Carbon fibre microelectrodes. J. Neurosci. Methods 1979, 1(3), 279–287. [Google Scholar]
- Palij, P.; Bull, D. R.; Sheehan, M. J.; Millar, J.; Stamford, J.; Kruk, Z. L.; Humphrey, P. P. Presynaptic regulation of dopamine release in corpus striatum monitored in vitro in real time by fast cyclic voltammetry. Brain Res. 1990, 509(1), 172–174. [Google Scholar]
- Millar, J.; Barnett, T.G. Basic instrumentation for fast cyclic voltammetry. J. Neurosci. Methods 1988, 25(2), 91–95. [Google Scholar]
- Kruk, Z.L.; O'Connor, J.J. Fast electrochemical studies in isolated tissues. Trends Pharmacol. Sci. 1995, 16(5), 145–149. [Google Scholar]
- Sidlo, Z.; Reggio, P.H.; Rice, M.E. Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochem. Int. 2008, 52(1-2), 80–88. [Google Scholar]
- Limberger, N.; Trout, S.J.; Kruk, Z.L.; Starke, K. “Real time” measurement of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn. Schmiedebergs Arch. Pharmacol. 1991, 344(6), 623–629. [Google Scholar]
- Pazzagli, M.; Pedata, F.; Pepeu, G. Effect of K+ depolarization, tetrodotoxin, and NMDA receptor inhibition on extracellular adenosine levels in rat striatum. Eur. J. Pharmacol. 1993, 234(1), 61–65. [Google Scholar]
- Chieng, B.; Bekkers, J.M. Inhibition of calcium channels by opioid- and adenosine-receptor agonists in neurons of the nucleus accumbens. Br. J. Pharmacol. 2001, 133(3), 337–344. [Google Scholar]
- Moore, K.A.; Nicoll, R.A.; Schmitz, D. Adenosine gates synaptic plasticity at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2003, 100(24), 14397–14402. [Google Scholar]
- Sebastiao, A.M.; de Mendonca, A.; Moreira, T.; Ribeiro, J.A. Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. J. Neurosci. 2001, 21(21), 8564–8571. [Google Scholar]
- Zhang, H.; Sulzer, D. Glutamate spillover in the striatum depresses dopaminergic transmission by activating group I metabotropic glutamate receptors. J. Neurosci. 2003, 23(33), 10585–10592. [Google Scholar]
- DeBoer, P.; Westerink, B. H. GABAergic modulation of striatal cholinergic interneurons: an in vivo microdialysis study. J Neurochem 1994, 62(1), 70–75. [Google Scholar]
- Ikarashi, Y.; Yuzurihara, M.; Takahashi, A.; Hirohisa, I.; Shiobara, T.; Maruyama, Y. Modulation of acetylcholine release via GABAA and GABAB receptors in rat striatum. Brain Res. 1999, 816(1), 238–240. [Google Scholar]
- Shen, K.Z.; Johnson, S.W. Potentiation of GABA(A) receptor agonists by GABA uptake inhibitors in the rat ventral midbrain. Eur. J. Pharmacol. 2001, 428(1), 1–7. [Google Scholar]
- Fujiyama, F.; Fritschy, J. M.; Stephenson, F. A.; Bolam, J. P. Synaptic localization of GABA(A) receptor subunits in the striatum of the rat. J. Comp. Neurol. 2000, 416(2), 158–172. [Google Scholar]
- Zhang, W.; Yamada, M.; Gomeza, J.; Basile, A.S.; Wess, J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J. Neurosci. 2002, 22(15), 6347–6352. [Google Scholar]
- Girault, J.A.; Spampinato, U.; Savaki, H.E.; Glowinski, J.; Besson, M.J. In vivo release of [3H]gamma-aminobutyric acid in the rat neostriatum--I. Characterization and topographical heterogeneity of the effects of dopaminergic and cholinergic agents. Neuroscience 1986, 19(4), 1101–1108. [Google Scholar]
- Bracci, E.; Centonze, D.; Bernardi, G.; Calabresi, P. Dopamine excites fast-spiking interneurons in the striatum. J. Neurophysiol. 2002, 87(4), 2190–2194. [Google Scholar]





© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O’Connor, J.J.; O’Neill, C. A Role for Adenosine A1 Receptors in GABA and NMDA-Receptor Mediated Modulation of Dopamine Release: Studies Using Fast Cyclic Voltammetry. Sensors 2008, 8, 5516-5534. https://doi.org/10.3390/s8095516
O’Connor JJ, O’Neill C. A Role for Adenosine A1 Receptors in GABA and NMDA-Receptor Mediated Modulation of Dopamine Release: Studies Using Fast Cyclic Voltammetry. Sensors. 2008; 8(9):5516-5534. https://doi.org/10.3390/s8095516
Chicago/Turabian StyleO’Connor, John J., and Carmel O’Neill. 2008. "A Role for Adenosine A1 Receptors in GABA and NMDA-Receptor Mediated Modulation of Dopamine Release: Studies Using Fast Cyclic Voltammetry" Sensors 8, no. 9: 5516-5534. https://doi.org/10.3390/s8095516