
In Search of Small Molecule Inhibitors Targeting the Flexible CK2 Subunit Interface



 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure and Mode of Binding of CK2β on CK2α
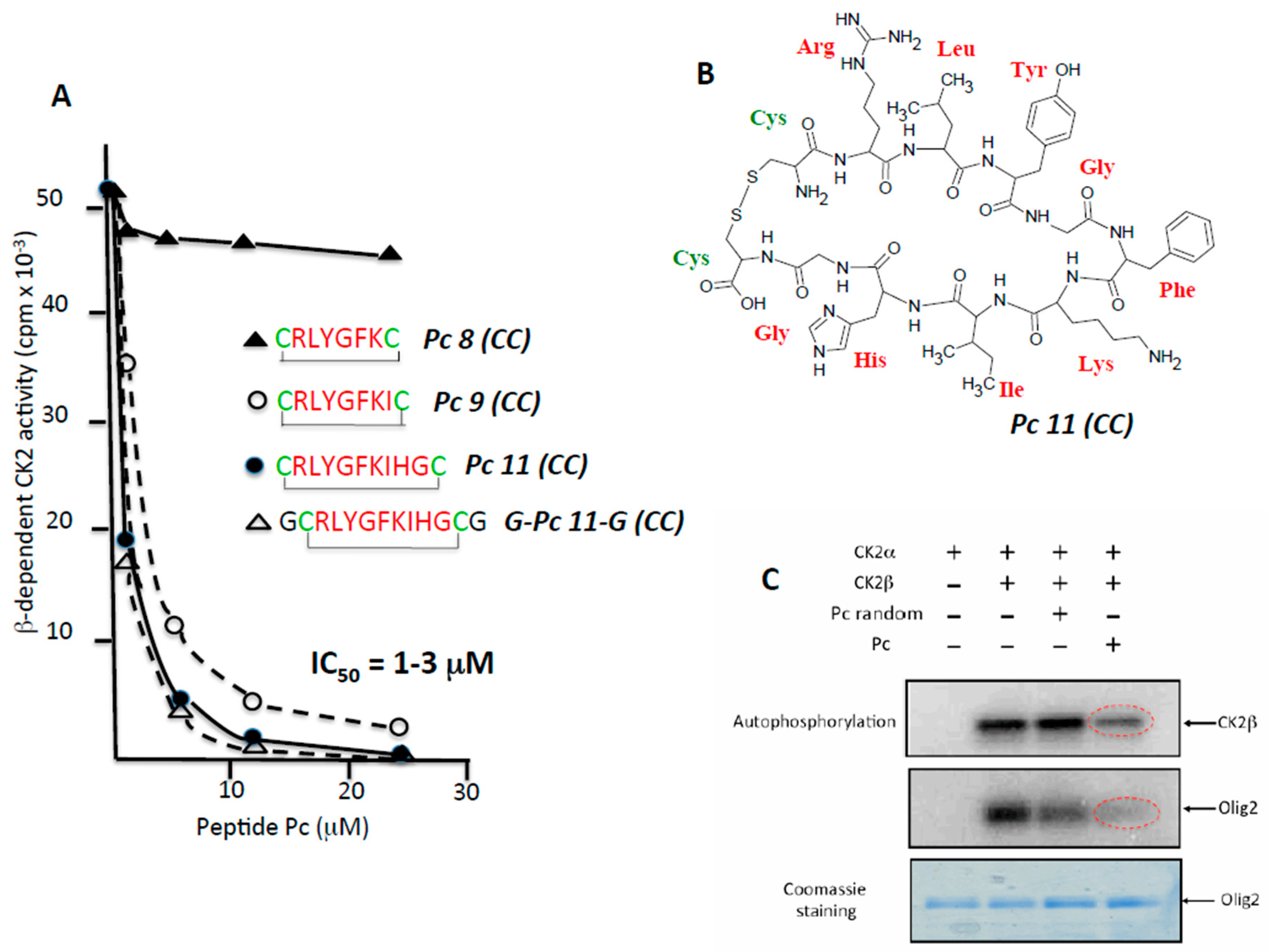
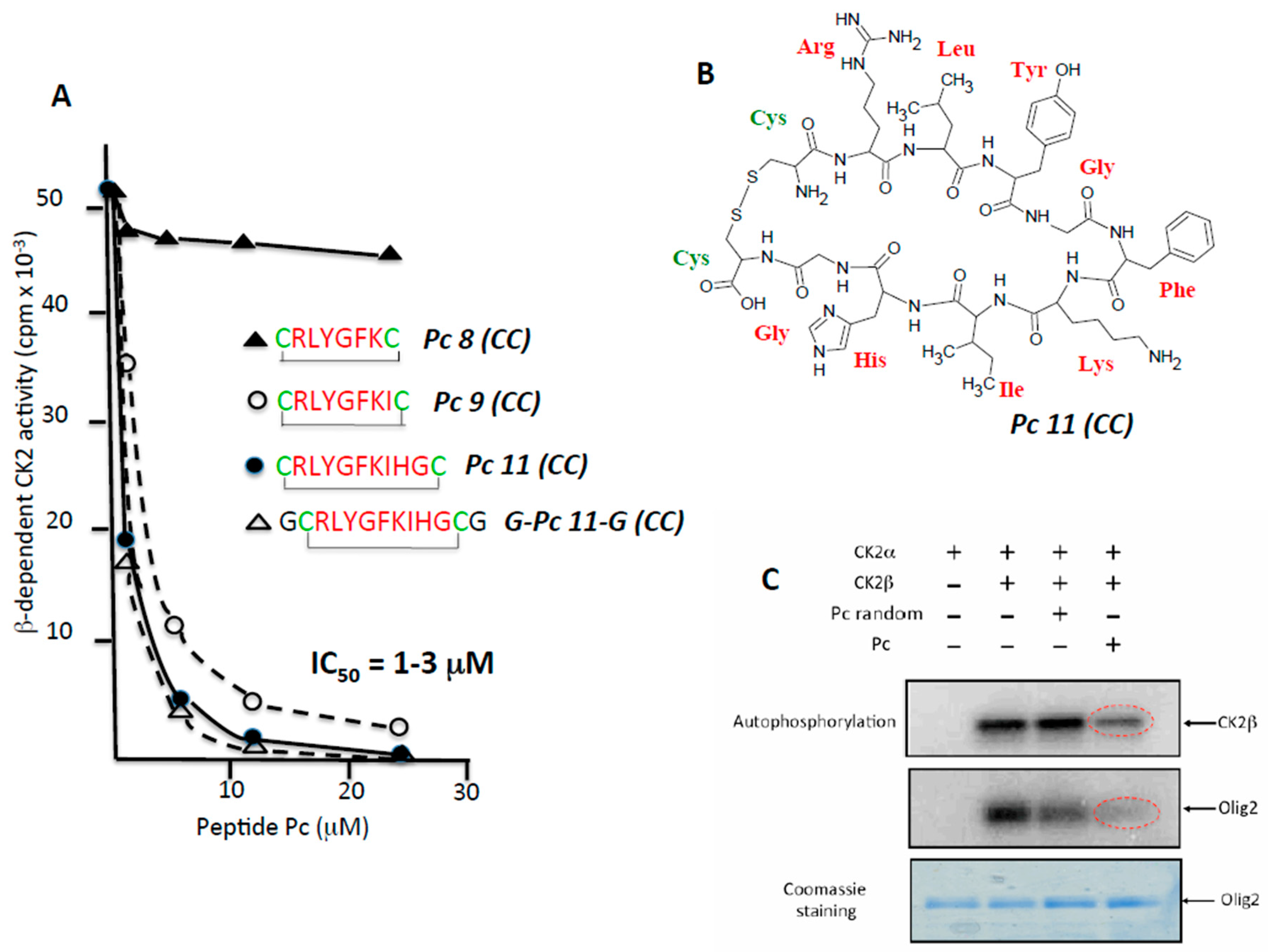
2.2. Rational Design of CK2β-Derived Cyclic Peptides
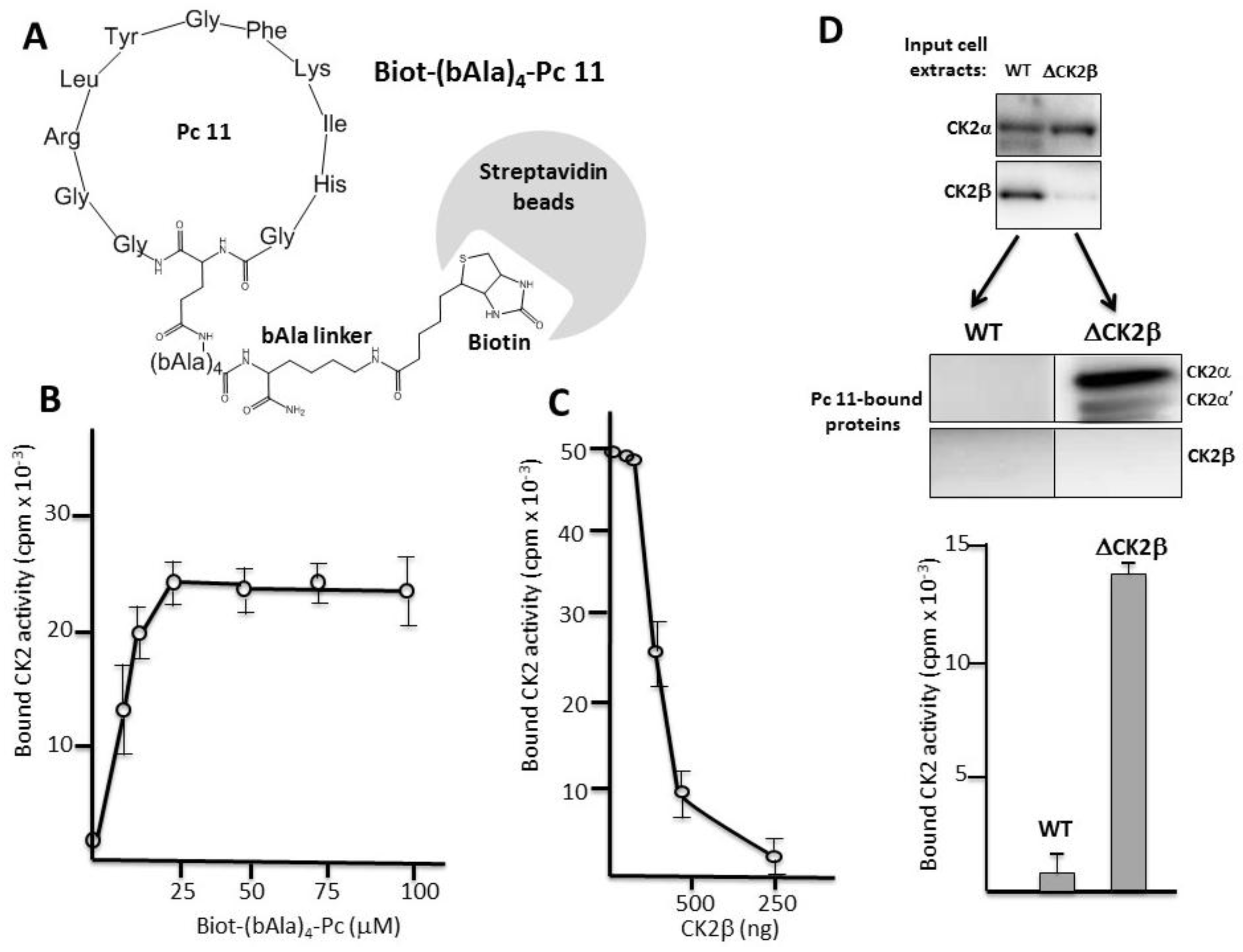
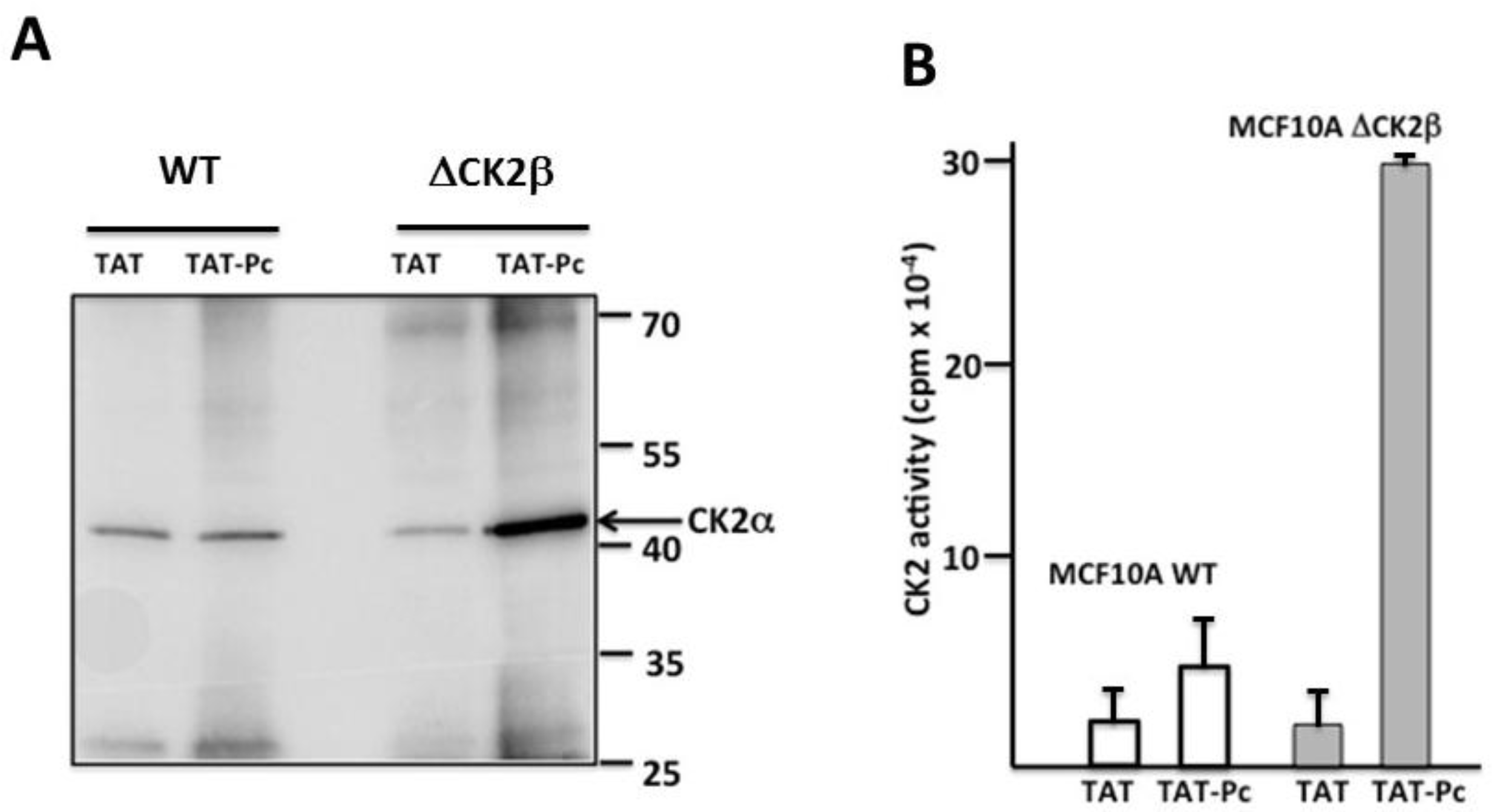
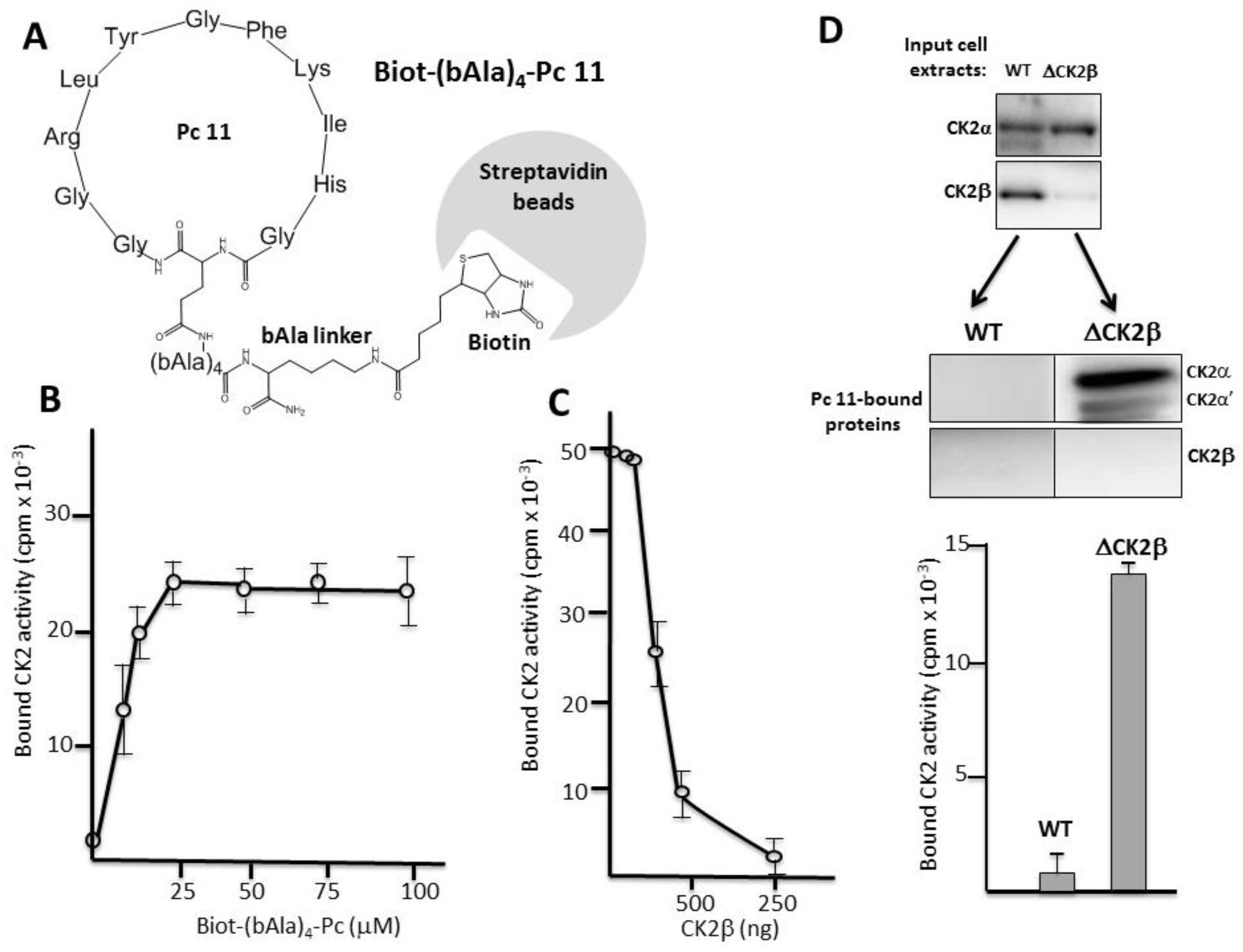
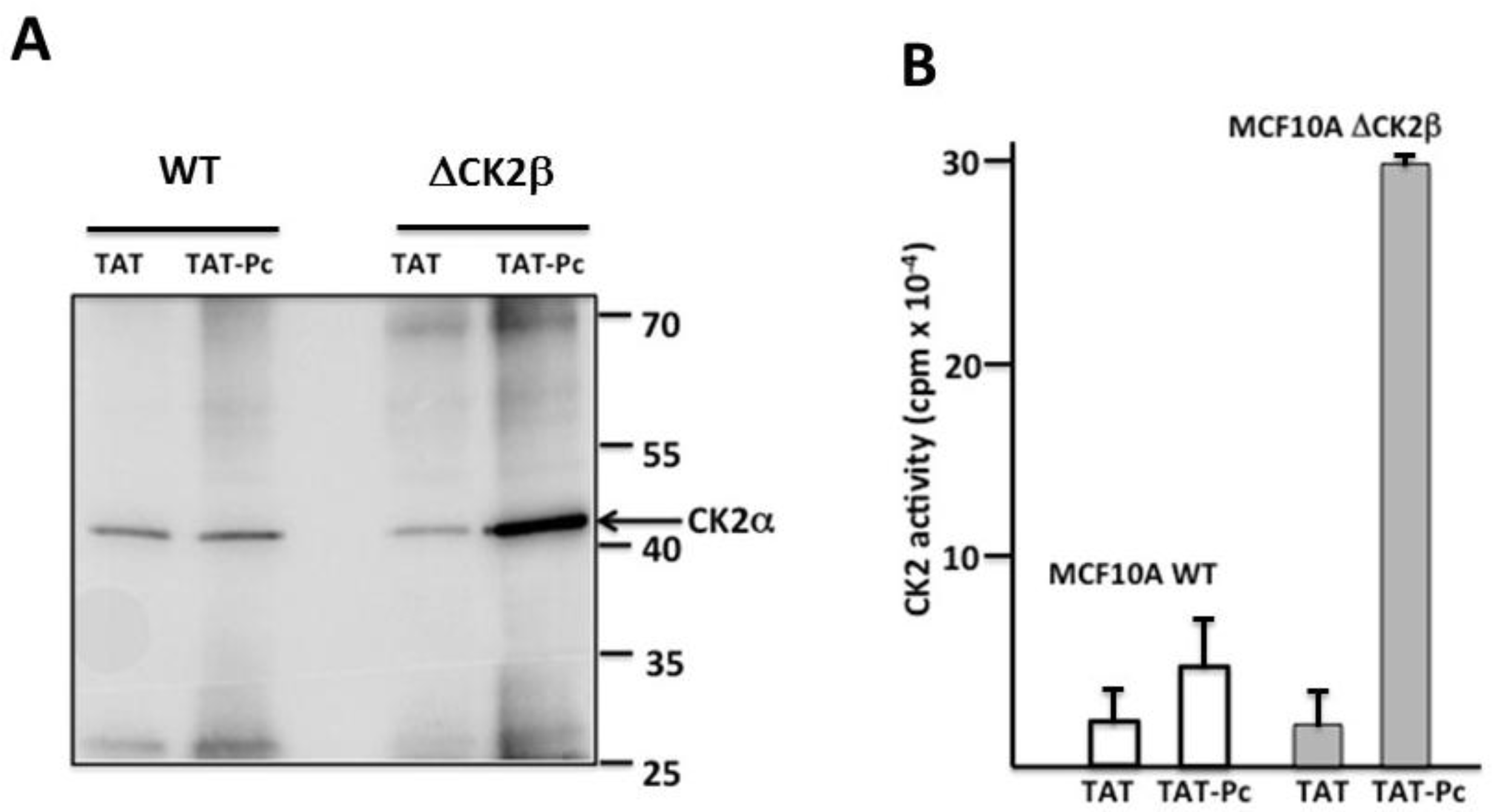
2.3. Pc Binds to CK2β in CK2β-Deficient Cell Extracts
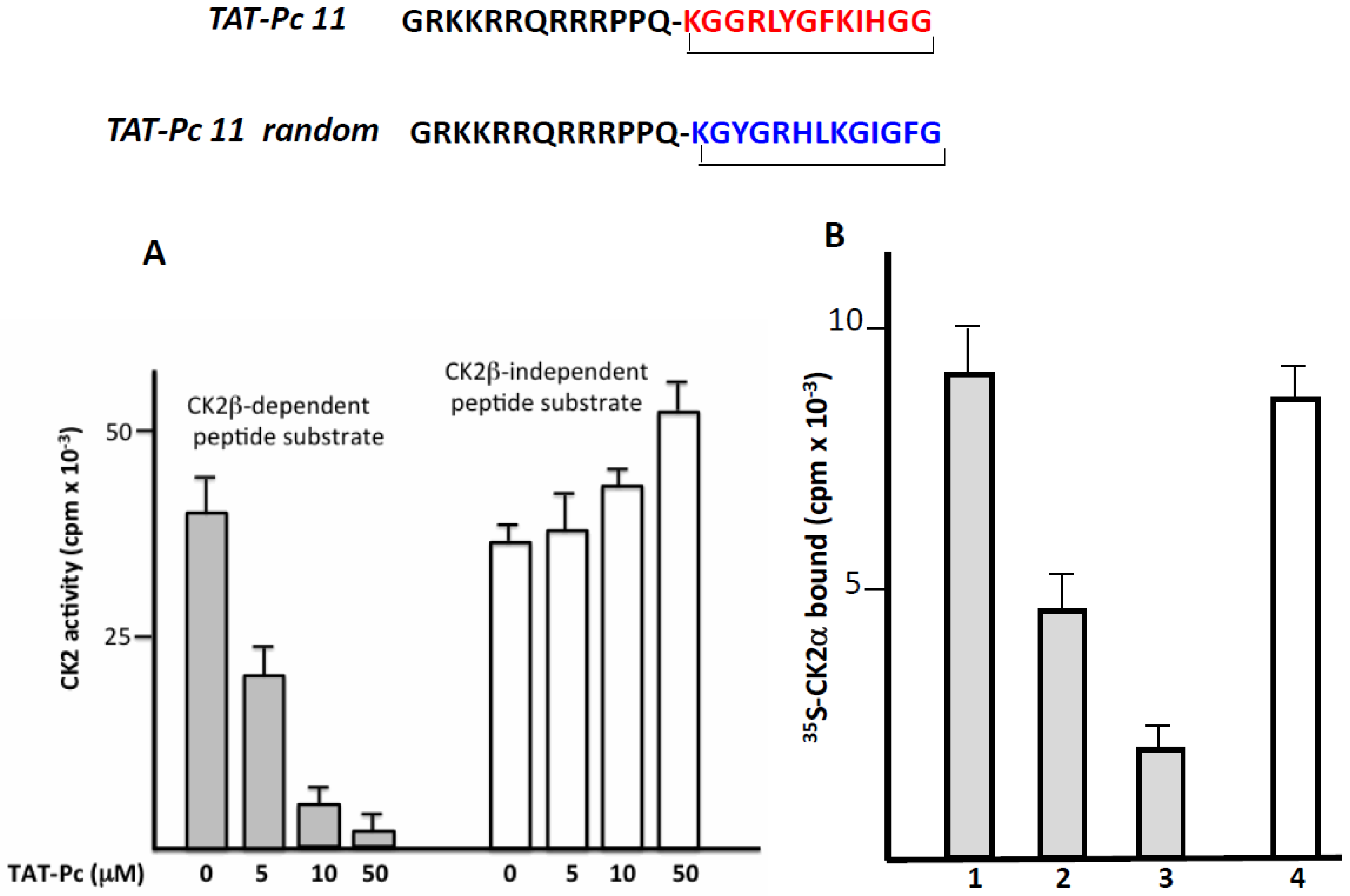
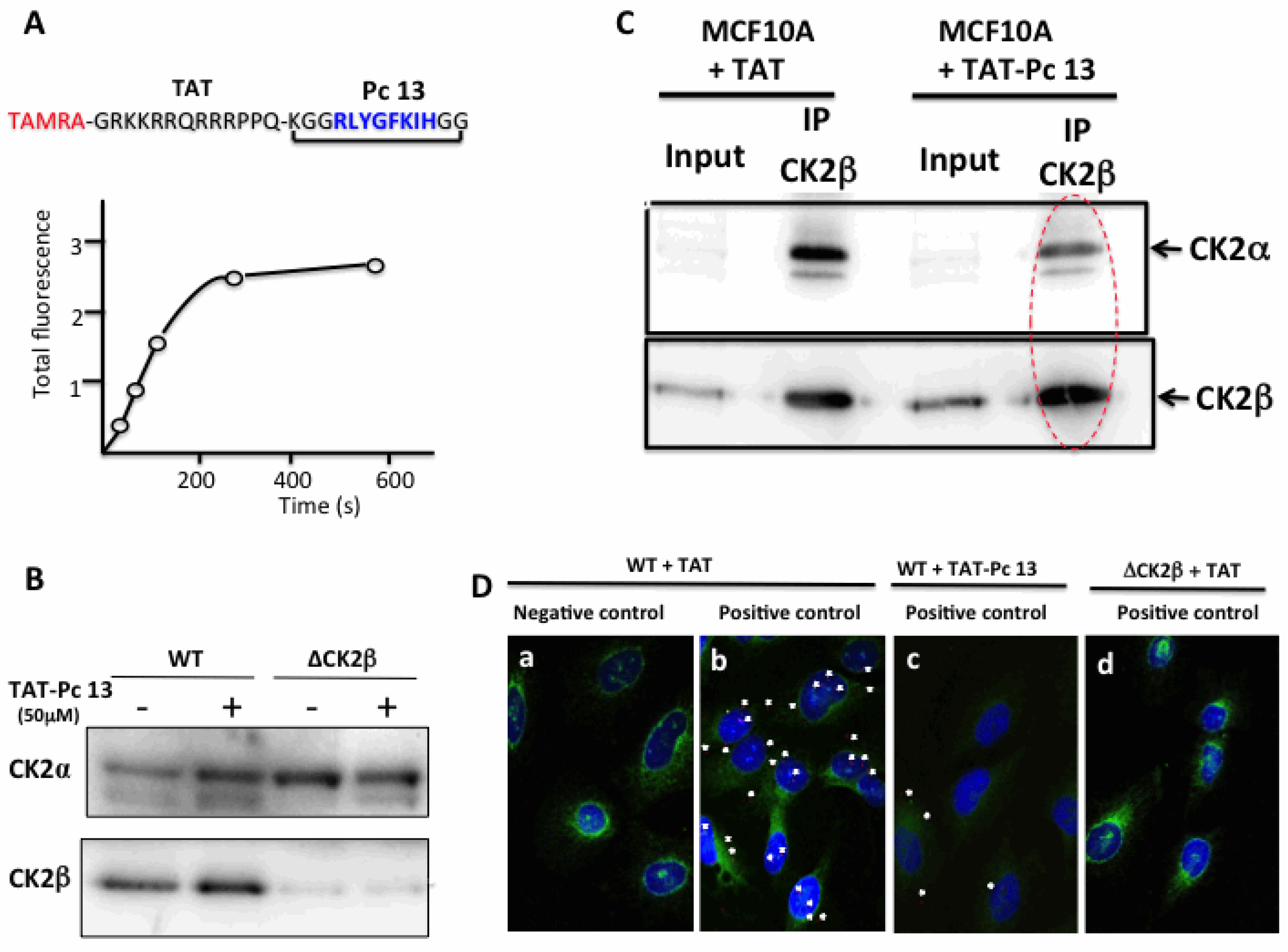
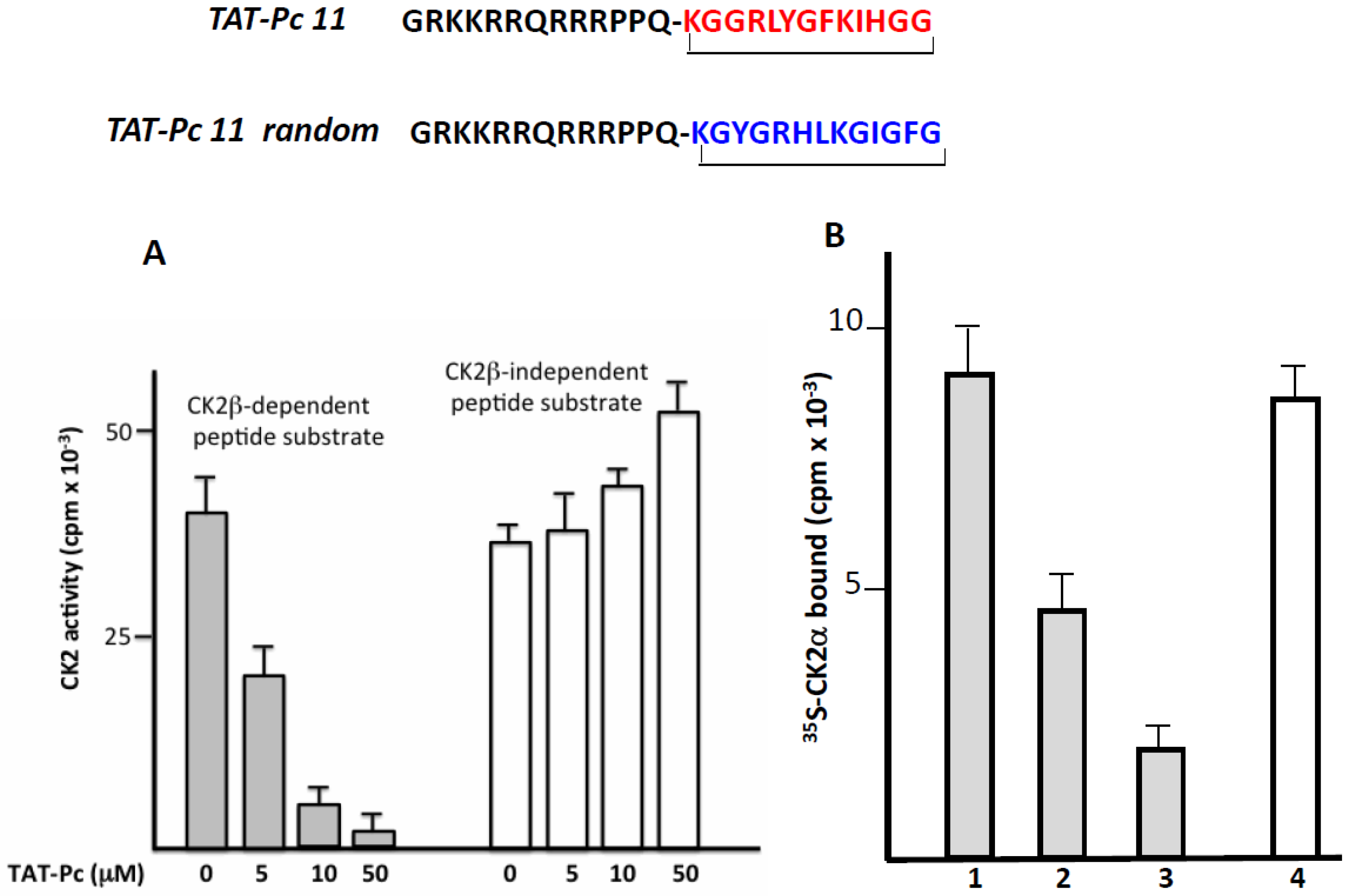
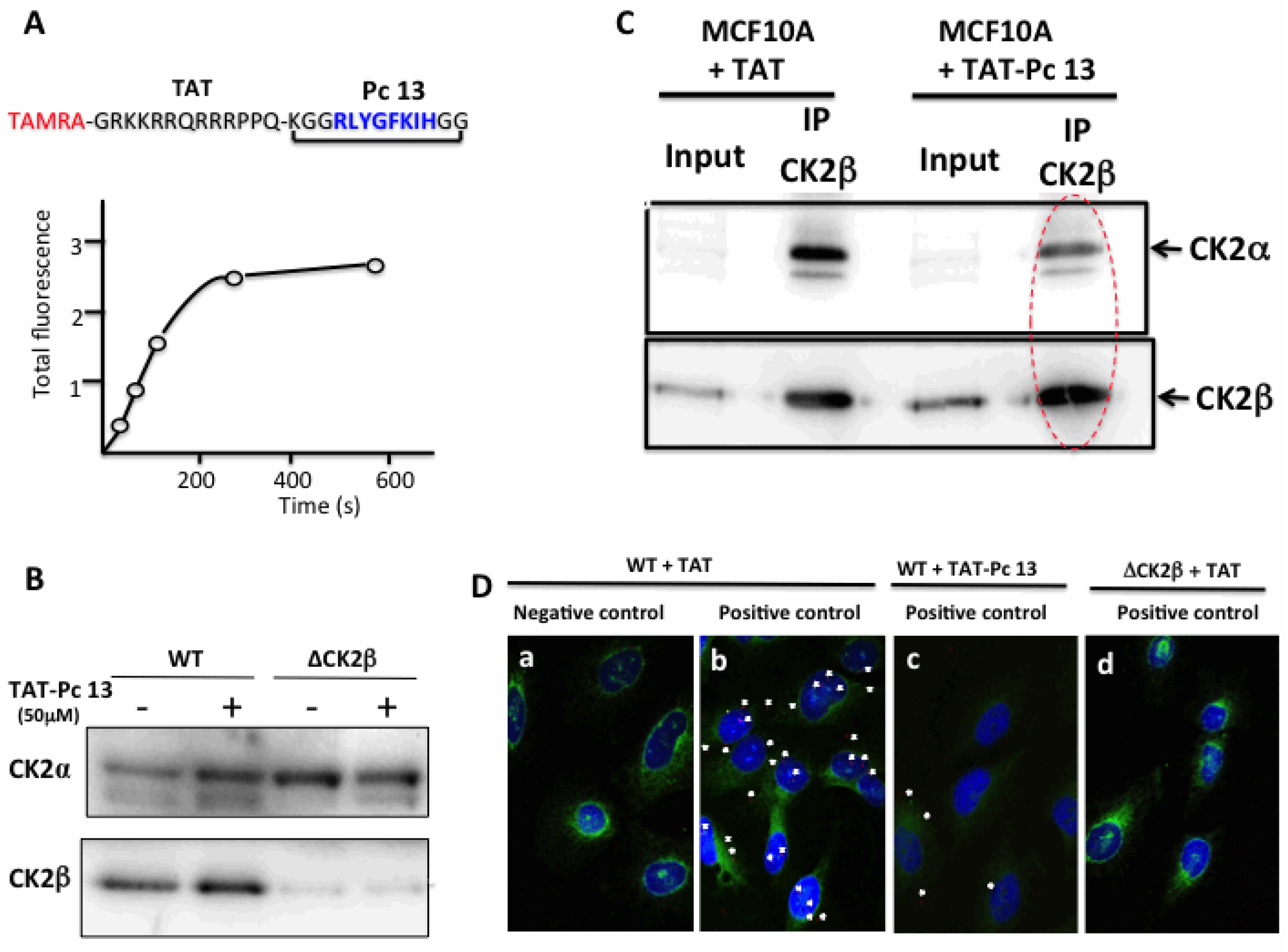
2.4. Design and Characterization of a TAT-Conjugated Pc Analogue
2.5. Bioactivity of TAT-Pc 13 in Cell Extracts
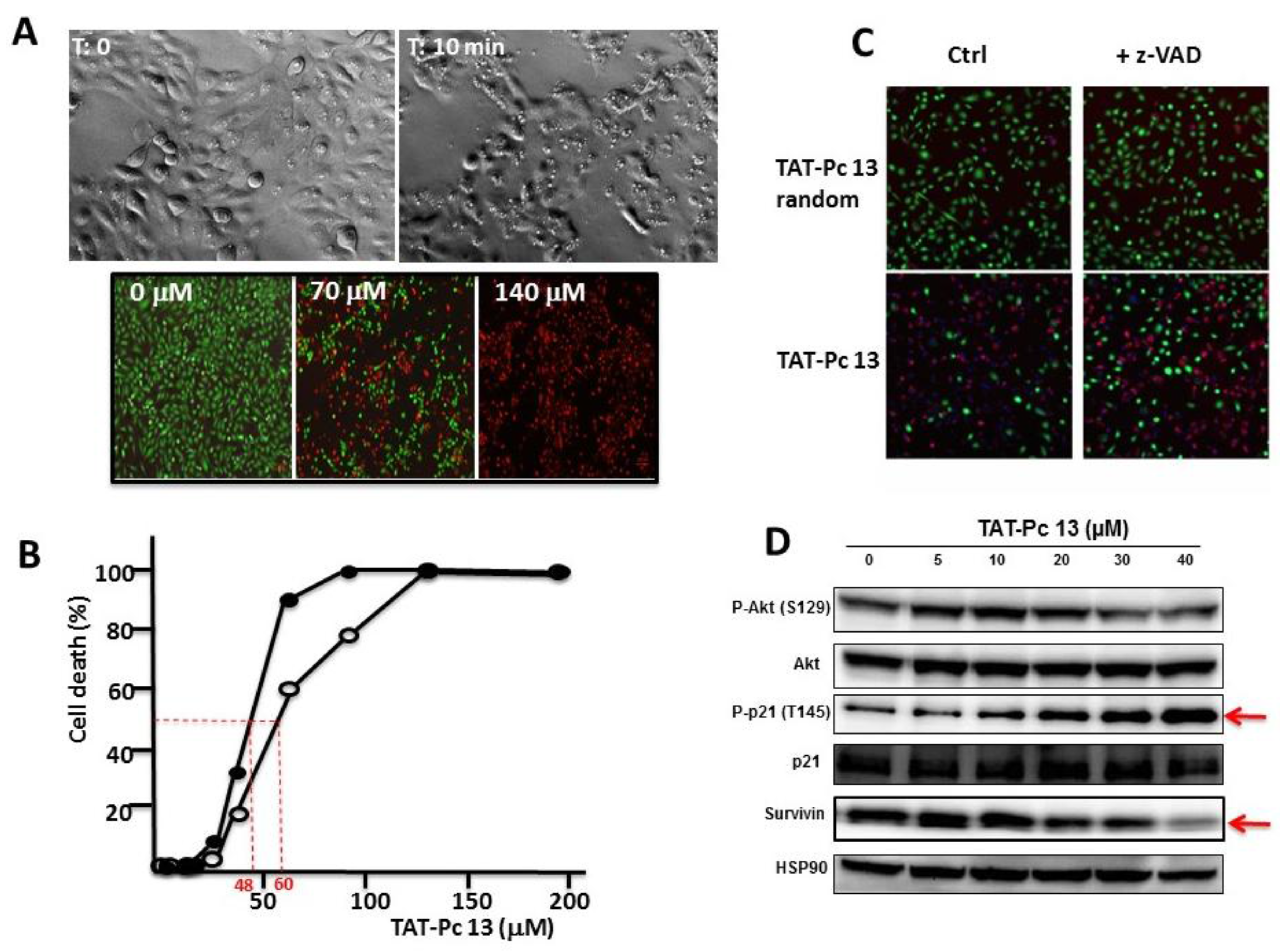
2.6. Uptake and Cellular Effects of TAT-Pc
2.7. Inhibition of Cell Viability by TAT-Pc 13
3. Materials and Methods
3.1. Materials
3.2. Peptide Synthesis
3.3. Proteins
3.4. In Vitro Kinase Assays
3.5. In Vitro Pull-Down Assay
3.6. In Vitro CK2α–CK2β Interaction Assay
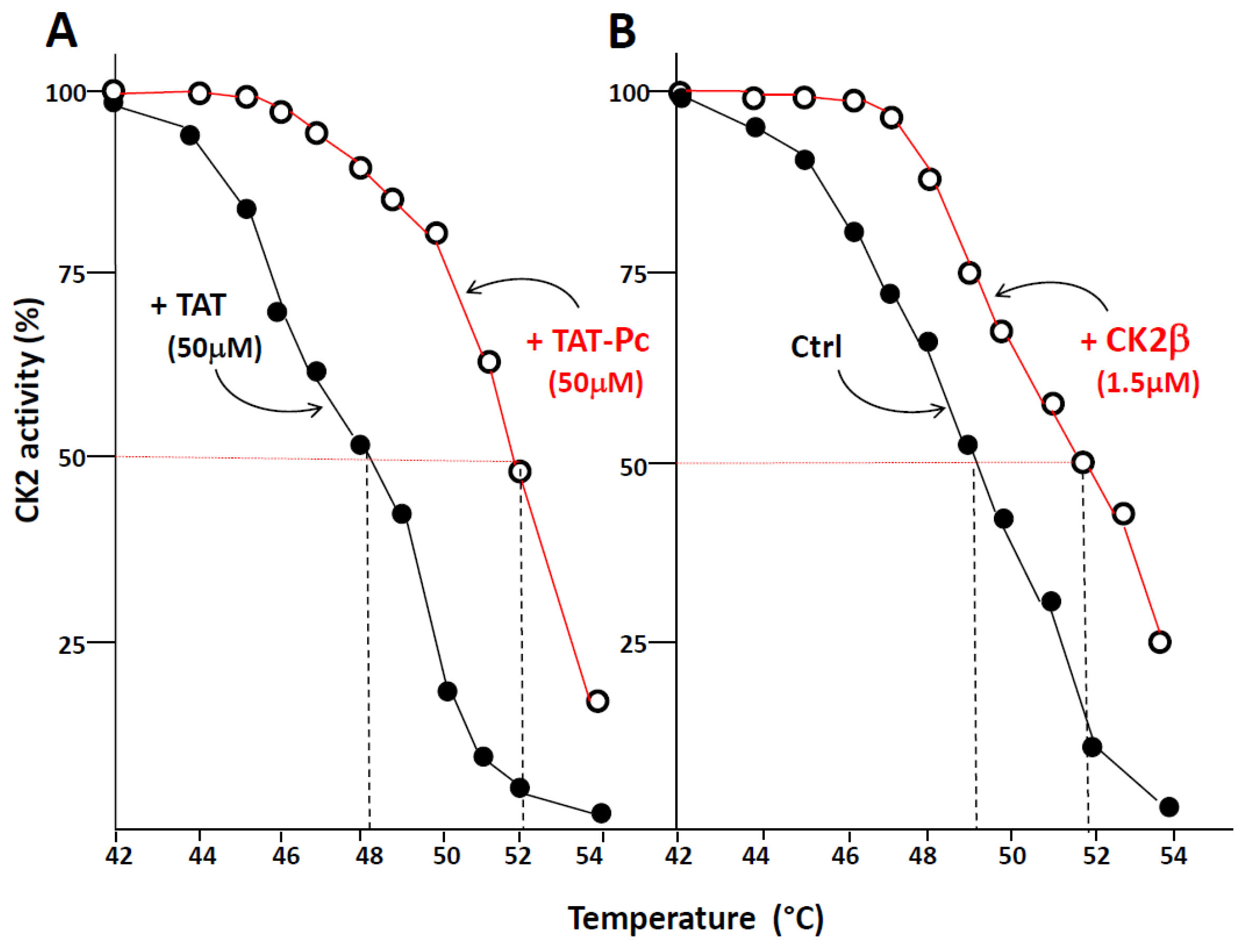
3.7. Thermal Shift Denaturation Assay
3.8. Proximity Ligation Assay
3.9. Cell Viability Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of interest
References
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Gerber, D.A.; Cochet, C. Joining the cell survival squad: An emerging role for protein kinase CK2. Trends Cell Biol. 2002, 12, 226–230. [Google Scholar] [CrossRef]
- Filhol, O.; Giacosa, S.; Wallez, Y.; Cochet, C. Protein kinase CK2 in breast cancer: The CK2beta regulatory subunit takes center stage in epithelial plasticity. Cell. Mol. Life Sci. 2015, 72, 3305–3322. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: A patent review. Expert Opin. Ther. Patents 2012, 22, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase CK2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 inhibition: An update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and sar of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine- 8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2010, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Marschke, R.F.; Borad, M.J.; McFarland, R.W.; Alvarez, R.H.; Lim, J.K.; Padgett, C.S.; Von Hoff, D.D.; O’Brien, S.E.; NorthfeltFront, D.W. Findings from the phase I clinical trials of CX-4945, an orally available inhibitor of CK2. J. Clin. Onc. 2011, 29, 3087. [Google Scholar]
- Prudent, R.; Cochet, C. New protein kinase CK2 inhibitors: Jumping out of the catalytic box. Chem. Biol. 2009, 16, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O.G. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. EMBO J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [PubMed]
- Filhol, O.; Nueda, A.; Martel, V.; Gerber-Scokaert, D.; Benitez, M.J.; Souchier, C.; Saoudi, Y.; Cochet, C. Live-cell fluorescence imaging reveals the dynamics of protein kinase CK2 individual subunits. Mol. Cell. Biol. 2003, 23, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Deshiere, A.; Duchemin-Pelletier, E.; Spreux, E.; Ciais, D.; Combes, F.; Vandenbrouck, Y.; Coute, Y.; Mikaelian, I.; Giusiano, S.; Charpin, C.; et al. Unbalanced expression of CK2 kinase subunits is sufficient to drive epithelial-to-mesenchymal transition by snail1 induction. Oncogene 2013, 32, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Raaf, J.; Brunstein, E.; Issinger, O.G.; Niefind, K. The CK2 alpha/CK2 beta interface of human protein kinase CK2 harbors a binding pocket for small molecules. Chem. Biol. 2008, 15, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 2007, 408, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raaf, J.; Guerra, B.; Neundorf, I.; Bopp, B.; Issinger, O.G.; Jose, J.; Pietsch, M.; Niefind, K. First structure of protein kinase CK2 catalytic subunit with an effective CK2beta-competitive ligand. ACS Chem. Biol. 2013, 8, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, N.; Chen, W.; Zhao, L.; Zhong, R. Underlying mechanisms of cyclic peptide inhibitors interrupting the interaction of CK2alpha/CK2beta: Comparative molecular dynamics simulation studies. Phys. Chem. Chem. Phys. 2016, 18, 9202–9210. [Google Scholar] [CrossRef] [PubMed]
- Poletto, G.; Vilardell, J.; Marin, O.; Pagano, M.A.; Cozza, G.; Sarno, S.; Falques, A.; Itarte, E.; Pinna, L.A.; Meggio, F. The regulatory beta subunit of protein kinase CK2 contributes to the recognition of the substrate consensus sequence. A study with an eIF2beta-derived peptide. Biochemistry 2008, 47, 8317–8325. [Google Scholar] [CrossRef] [PubMed]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.G.; Boldyreff, B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol. Cell. Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, A.; Olsen, B.B.; Issinger, O.G.; Niefind, K. The protein kinase CK2 (andante) holoenzyme structure supports proposed models of autoregulation and trans-autophosphorylation. J. Mol. Biol. 2014, 426, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Weibrecht, I.; Leuchowius, K.J.; Clausson, C.M.; Conze, T.; Jarvius, M.; Howell, W.M.; Kamali-Moghaddam, M.; Soderberg, O. Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Rev. Proteom. 2010, 7, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Heriche, J.K.; Lebrin, F.; Rabilloud, T.; Leroy, D.; Chambaz, E.M.; Goldberg, Y. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science 1997, 276, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Leroy, D.; Alghisi, G.C.; Roberts, E.; Filhol-Cochet, O.; Gasser, S.M. Mutations in the C-terminal domain of topoisomerase II affect meiotic function and interaction with the casein kinase 2 beta subunit. Mol. Cell. Biochem. 1999, 191, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, L.; Leroy, D.; Filhol, O.; Nueda, A.; Benitez, M.J.; Chambaz, E.M.; Cochet, C.; Dideberg, O. Crystal structure of the human protein kinase CK2 regulatory subunit reveals its zinc finger-mediated dimerization. EMBO J. 1999, 18, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Lu, K.P.; Kwon, Y.T.; Tsai, L.H.; Filhol, O.; Cochet, C.; Brickey, D.A.; Soderling, T.R.; Bartleson, C.; Graves, D.J.; et al. A structural basis for substrate specificities of protein ser/thr kinases: Primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and ERK1. Mol. Cell. Biol. 1996, 16, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Allalou, A.; Wahlby, C. Blobfinder, a tool for fluorescence microscopy image cytometry. Comput. Methods Programs Biomed. 2009, 94, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Siemer, S.; Boldyreff, B.; Issinger, O.G. Protein kinase CK2: Evidence for a protein kinase CK2beta subunit fraction, devoid of the catalytic CK2alpha subunit, in mouse brain and testicles. FEBS Lett. 1999, 462, 353–357. [Google Scholar] [CrossRef]
- Filhol, O.; Martiel, J.L.; Cochet, C. Protein kinase CK2: A new view of an old molecular complex. EMBO Rep. 2004, 5, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bhattacharya, N.; Mixter, P.F.; Wei, W.; Sedivy, J.; Magnuson, N.S. Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by PIM-1 kinase. Biochim. Biophys. Acta 2002, 1593, 45–55. [Google Scholar] [CrossRef]
- Burch, L.R.; Scott, M.; Pohler, E.; Meek, D.; Hupp, T. Phage-peptide display identifies the interferon-responsive, death-activated protein kinase family as a novel modifier of MDM2 and p21WAF1. J. Mol. Biol. 2004, 337, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Hupp, T.R. Chemical genetics approach to identify peptide ligands that selectively stimulate DAPK-1 kinase activity. Biochemistry 2007, 46, 2655–2673. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Inbal, B.; Kissil, J.L.; Raveh, T.; Berissi, H.; Spivak-Kroizaman, T.; Feinstein, E.; Kimchi, A. DAP-kinase participates in TNF-alpha- and FAS-induced apoptosis and its function requires the death domain. J. Cell Biol. 1999, 146, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wazir, U.; Sanders, A.J.; Wazir, A.M.; Ye, L.; Jiang, W.G.; Ster, I.C.; Sharma, A.K.; Mokbel, K. Effects of the knockdown of death-associated protein 3 expression on cell adhesion, growth and migration in breast cancer cells. Oncol. Rep. 2015, 33, 2575–2582. [Google Scholar] [PubMed]
- Engemann, H.; Heinzel, V.; Page, G.; Preuss, U.; Scheidtmann, K.H. DAP-like kinase interacts with the rat homolog of Schizosaccharomyces pombe CDC5 protein, a factor involved in pre-mRNA splicing and required for G2/M phase transition. Nucleic Acids Res. 2002, 30, 1408–1417. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bestgen, B.; Belaid-Choucair, Z.; Lomberget, T.; Le Borgne, M.; Filhol, O.; Cochet, C. In Search of Small Molecule Inhibitors Targeting the Flexible CK2 Subunit Interface. Pharmaceuticals 2017, 10, 16. https://doi.org/10.3390/ph10010016
Bestgen B, Belaid-Choucair Z, Lomberget T, Le Borgne M, Filhol O, Cochet C. In Search of Small Molecule Inhibitors Targeting the Flexible CK2 Subunit Interface. Pharmaceuticals. 2017; 10(1):16. https://doi.org/10.3390/ph10010016
Chicago/Turabian StyleBestgen, Benoît, Zakia Belaid-Choucair, Thierry Lomberget, Marc Le Borgne, Odile Filhol, and Claude Cochet. 2017. "In Search of Small Molecule Inhibitors Targeting the Flexible CK2 Subunit Interface" Pharmaceuticals 10, no. 1: 16. https://doi.org/10.3390/ph10010016
APA StyleBestgen, B., Belaid-Choucair, Z., Lomberget, T., Le Borgne, M., Filhol, O., & Cochet, C. (2017). In Search of Small Molecule Inhibitors Targeting the Flexible CK2 Subunit Interface. Pharmaceuticals, 10(1), 16. https://doi.org/10.3390/ph10010016