Modernization of Enoxaparin Molecular Weight Determination Using Homogeneous Standards

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
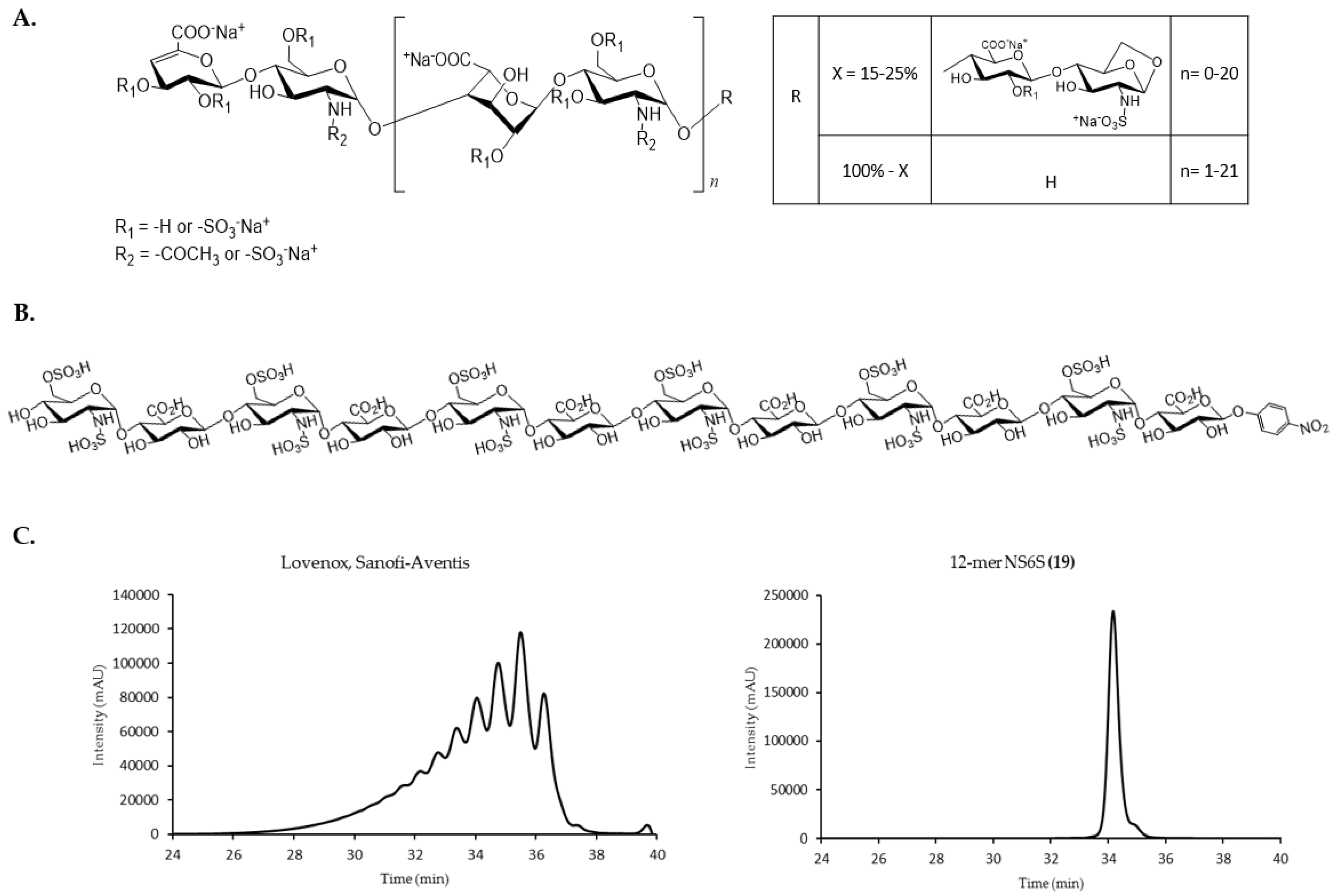
2.1. Chemoenzymatic Synthesis of Oligosaccharide Panel
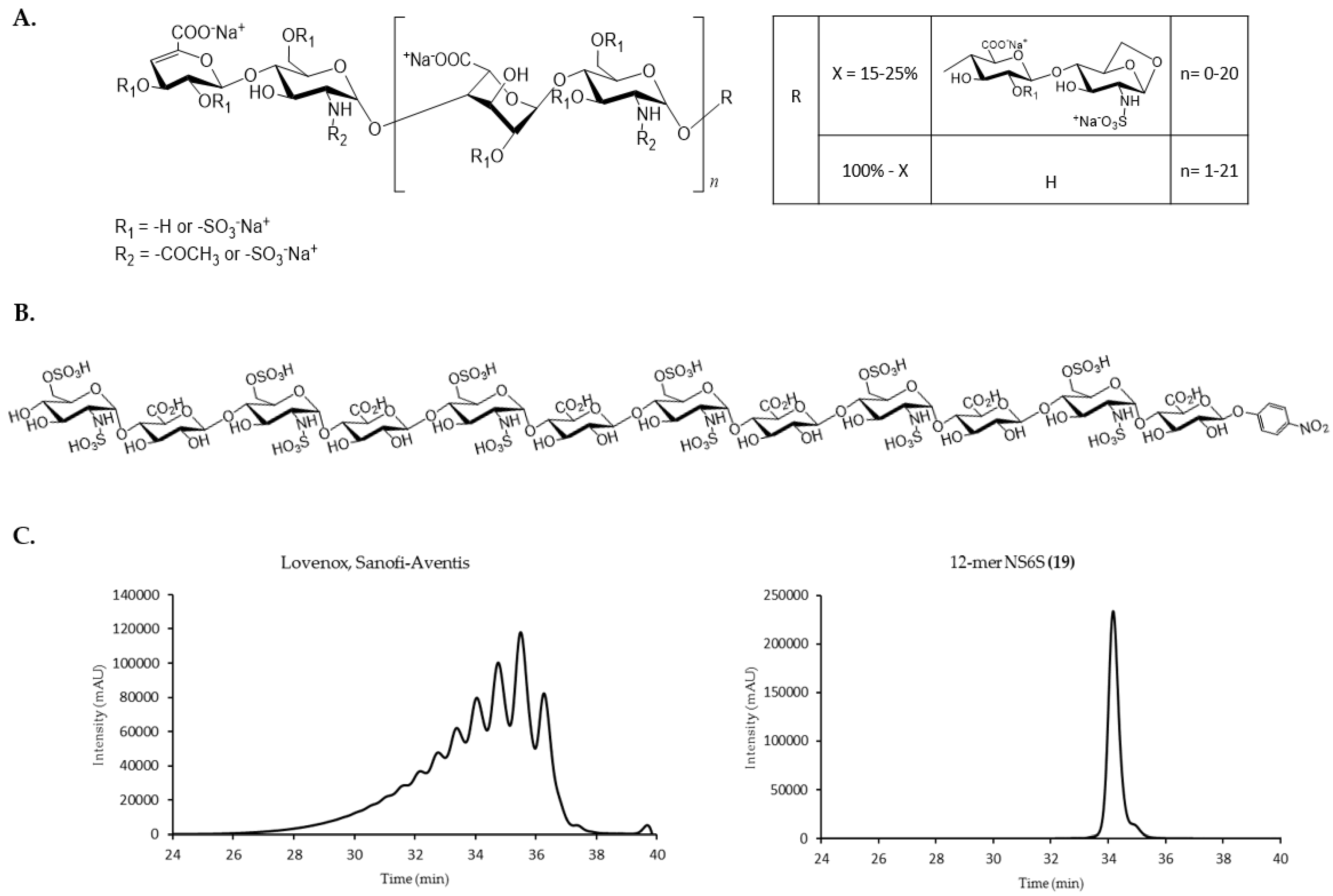
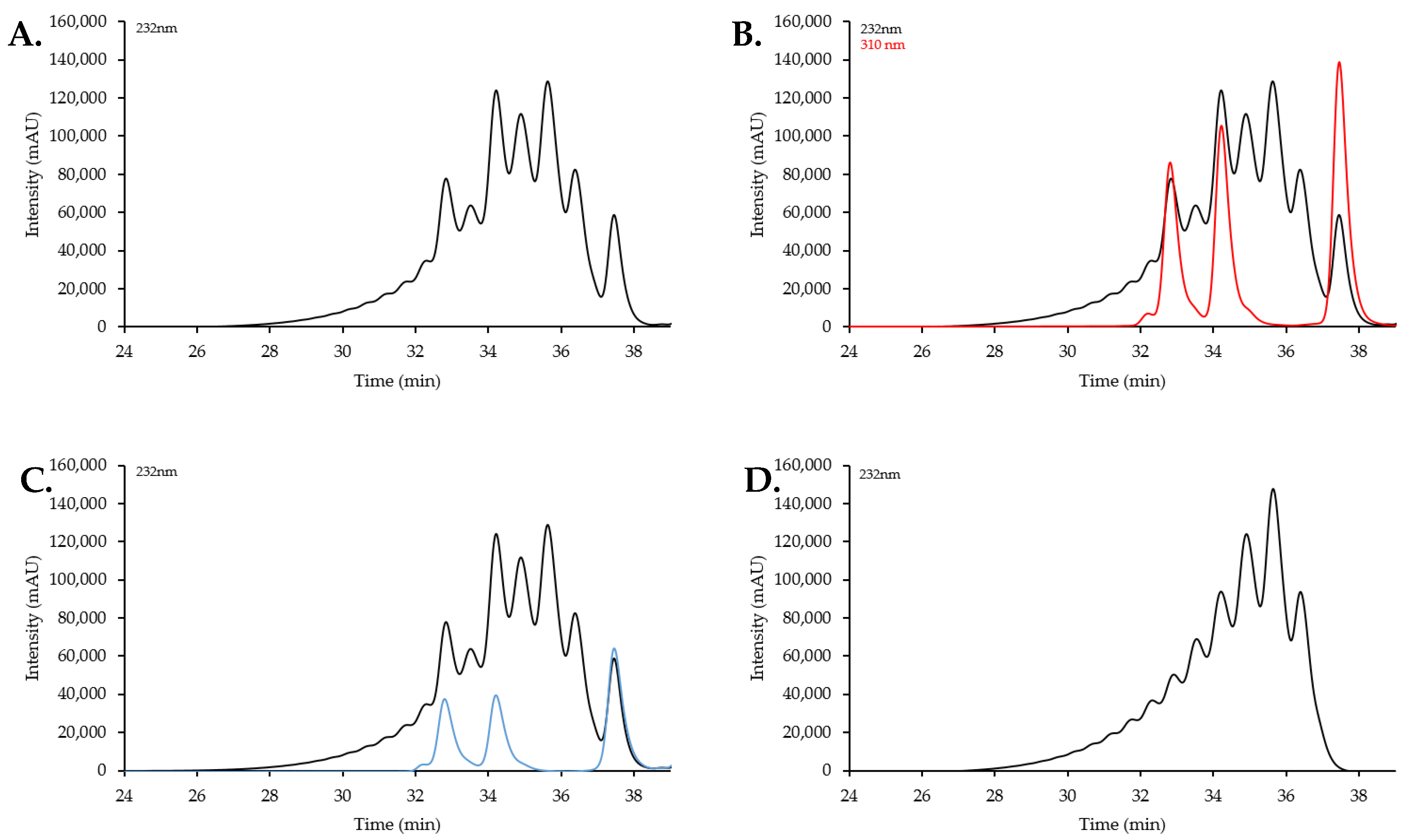
2.2. Size Exclusion Chromatography Profiles of Synthetic Oligosaccharides
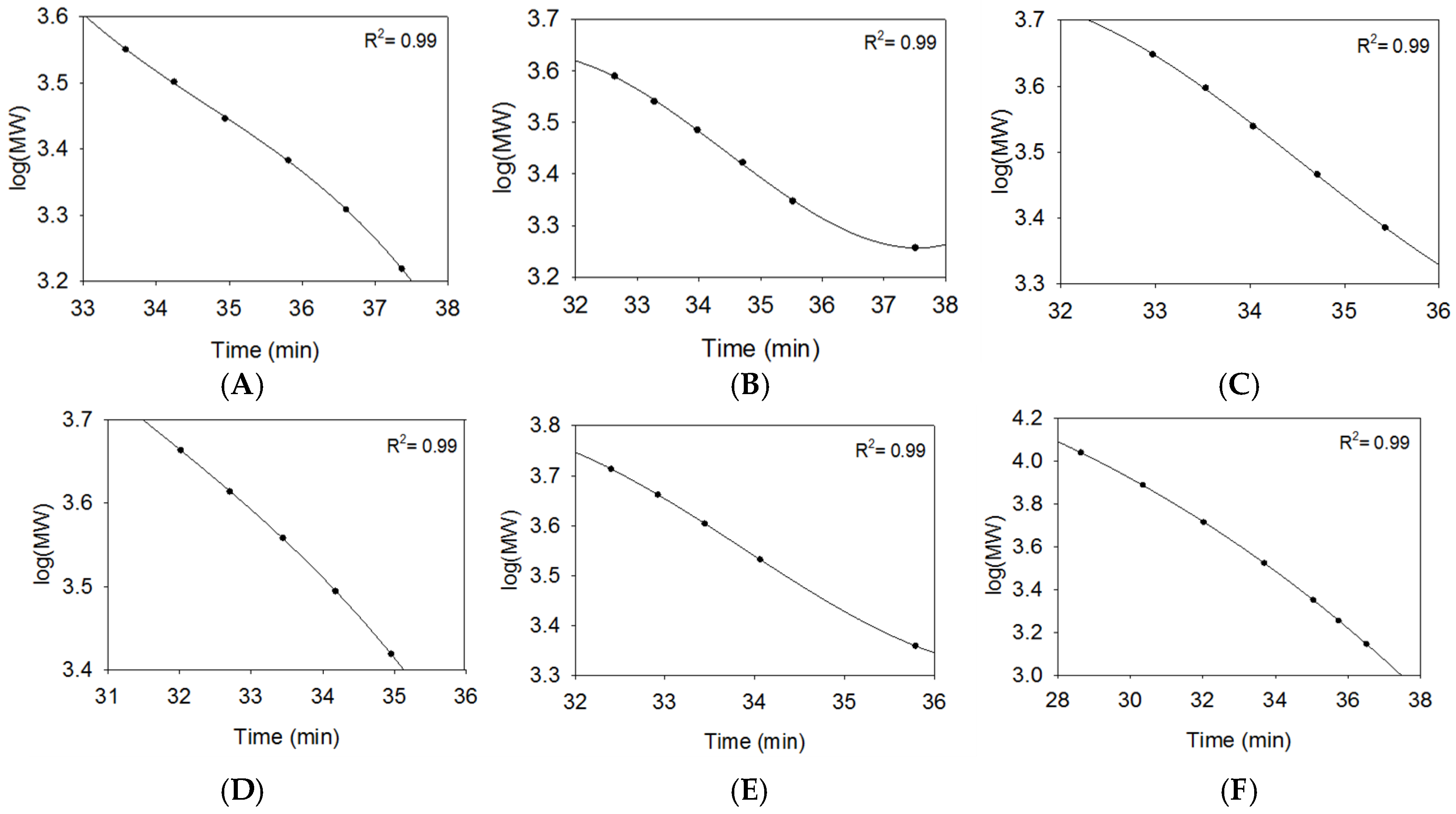
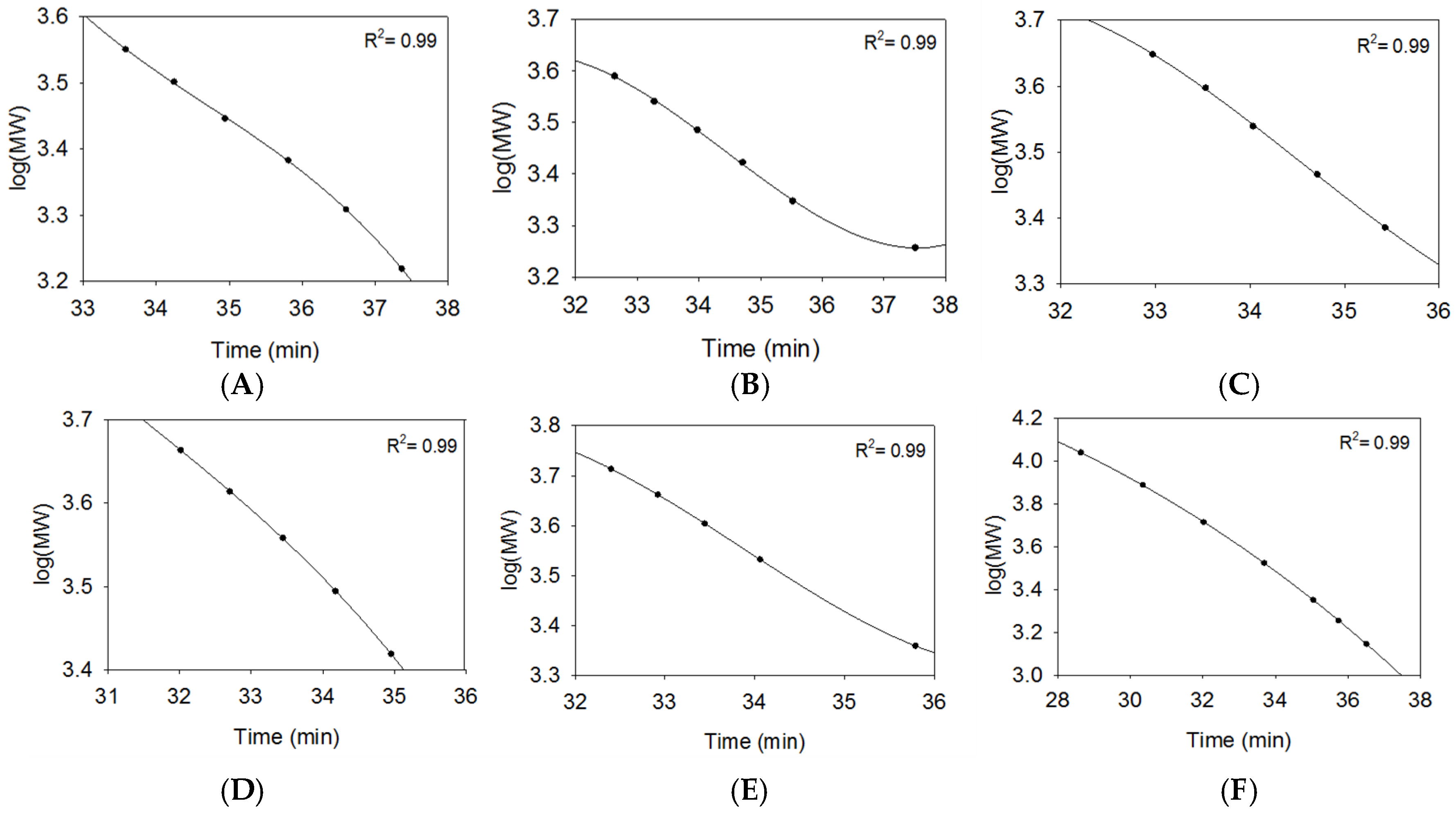
2.2.1. SEC Characterization of Synthetic Oligosaccharides
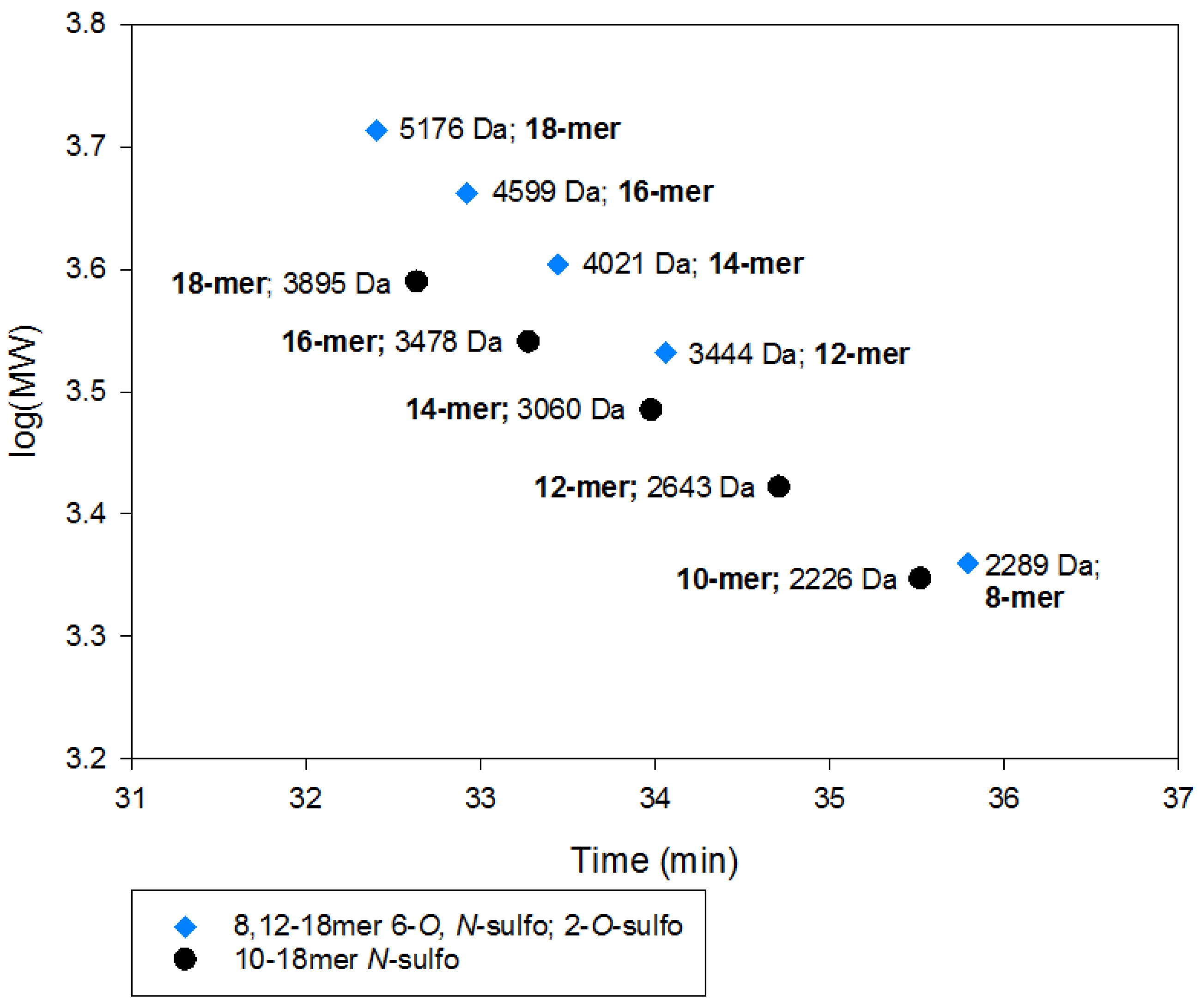
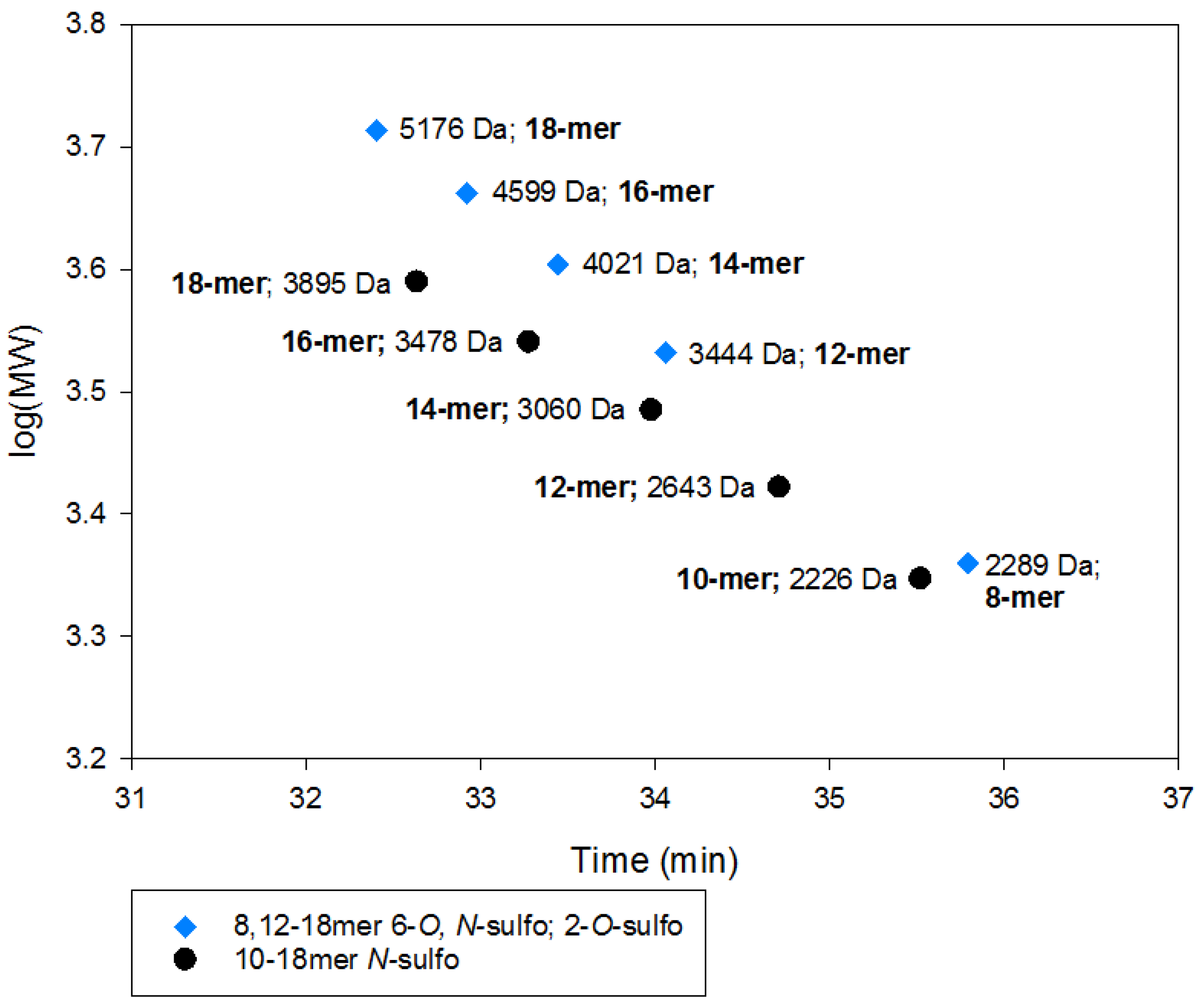
2.2.2. SEC RT Depends on Both Oligosaccharide Shape and Size (MW)
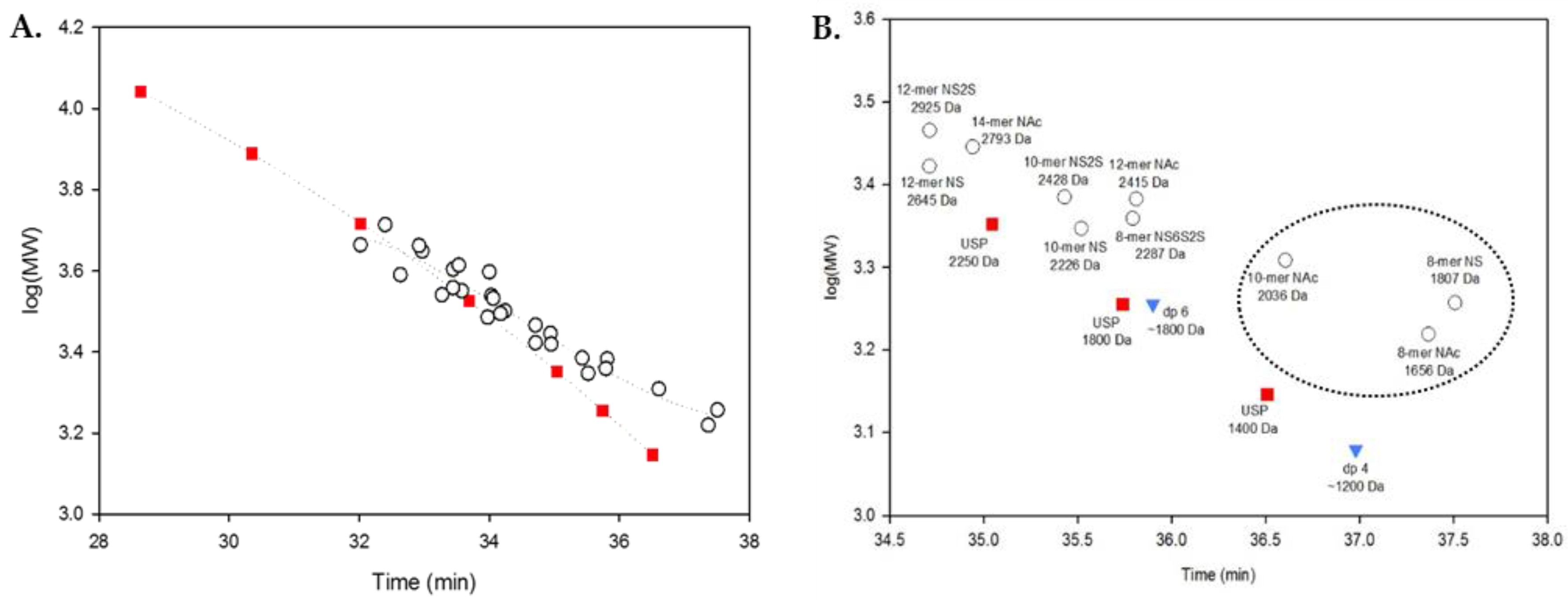
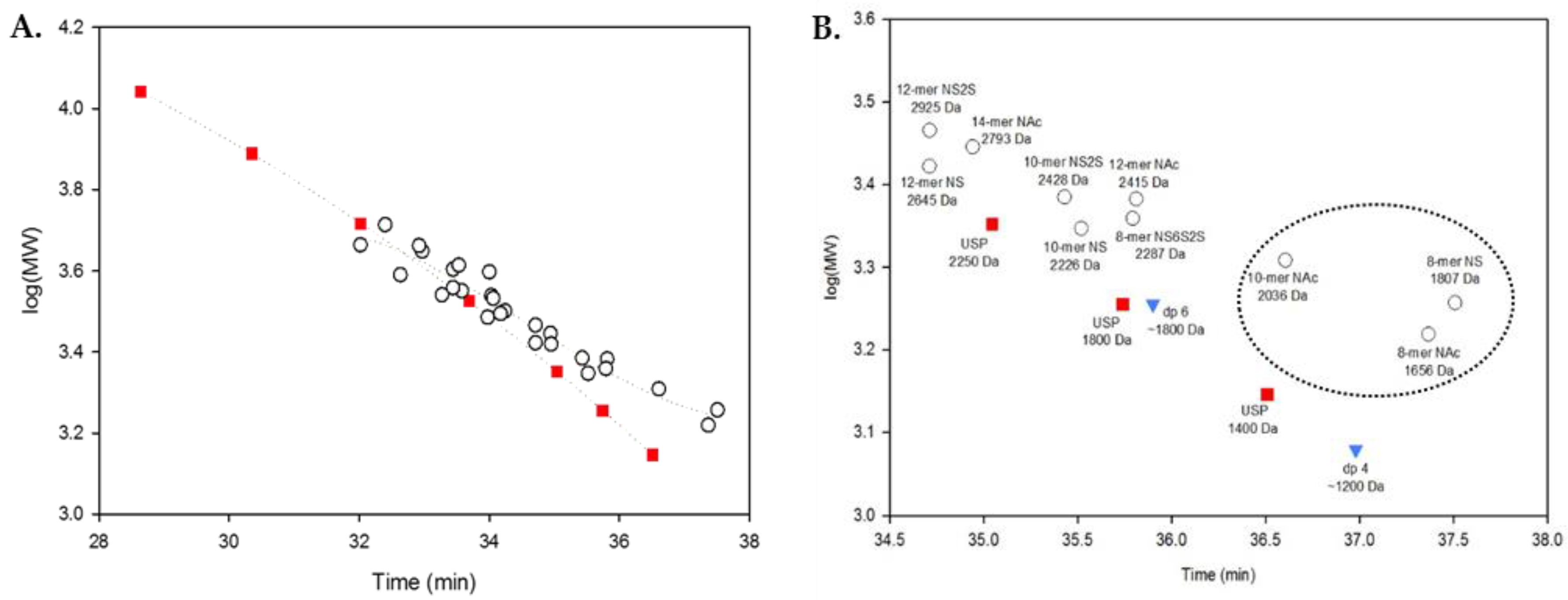
2.3. Using Calibration Curves to Determine Mw of USP Enoxaparin RS
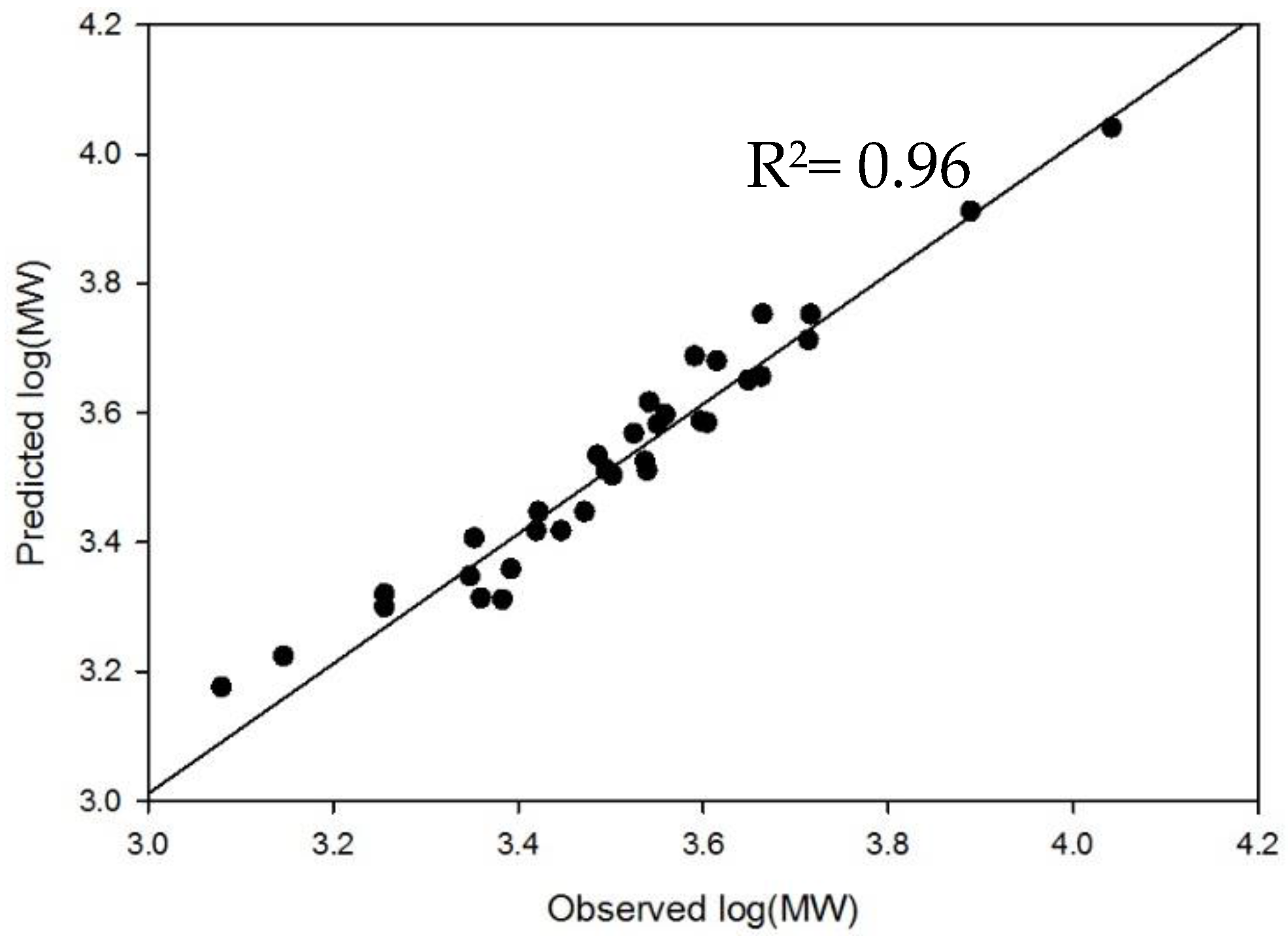
2.4. Method Modernization: SVM Modeling Allows for Synthetic Oligosaccharides to be Used in the Appropriate Proportions to Reflect the Various Quantities of Components in Enoxaparin
2.5. System Suitability: Applying the SVM Model in a Compendial Technique
2.6. Analysis of Commercially Available Enoxaparin Sodium Using Oligosaccharide SVM Model
3. Discussion
4. Materials and Methods
4.1. Chemoenzymatic Synthesis
4.2. MW Distribution and Mw
- The determination of USP Enoxaparin RS within 150 Da of labeled Mw value.
- The determination of an enoxaparin test sample with Mw ~4500 Da, the range being between 3800 and 5000 Da.
- The determination of an enoxaparin test sample MW distribution:
- ○
- ~16.0% below 2000 Da, the range being between 12.0 and 20.0%;
- ○
- ~74.0% between 2000 and 8000 Da, the range being between 68.0 and 82.0%;
- ○
- Not more than 18.0% higher than 8,000 Da.
4.3. Development and External Validation of SVM Model
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alquwaizani, M.; Buckley, L.; Adams, C.; Fanikos, J. Anticoagulants: A Review of the Pharmacology, Dosing, and Complications. Curr. Emerg. Hosp. Med. Rep. 2013, 1, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Jeske, W.P.; Walenga, J.M.; Hoppensteadt, D.A.; Vandenberg, C.; Brubaker, A.; Adiguzel, C.; Bakhos, M.; Fareed, J. Differentiating low-molecular-weight heparins based on chemical, biological, and pharmacologic properties: Implications for the development of generic versions of low-molecular-weight heparins. Semin. Thromb. Hemost. 2008, 34, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Raw, A.; Yu, L.; Lionberger, R.; Ya, N.; Verthelyi, D.; Rosenberg, A.; Kozlowski, S.; Webber, K.; Woodcock, J. Scientific considerations in the review and approval of generic enoxaparin in the United States. Nat. Biotechnol. 2013, 31, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Szajek, A.Y.; Chess, E.; Johansen, K.; Gratzl, G.; Gray, E.; Keire, D.; Linhardt, R.J.; Liu, J.; Morris, T.; Mulloy, B.; et al. The US regulatory and pharmacopeia response to the global heparin contamination crisis. Nat. Biotechnol. 2016, 34, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Bisio, A.; Mantegazza, A.; Vecchietti, D.; Bensi, D.; Coppa, A.; Torri, G.; Bertini, S. Determination of the molecular weight of low-molecular-weight heparins by using high-pressure size exclusion chromatography on line with a triple detector array and conventional methods. Molecules 2015, 20, 5085–5098. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Heath, A.; Shriver, Z.; Jameison, F.; Al Hakim, A.; Morris, T.S.; Szajek, A.Y. USP compendial methods for analysis of heparin: Chromatographic determination of molecular weight distributions for heparin sodium. Anal. Bioanal. Chem. 2014, 406, 4815–4823. [Google Scholar] [CrossRef] [PubMed]
- USP. Enoxaparin Sodium United States Pharmacopeia; USP: Rockville, MD, USA, 2016; Volume 39, pp. 3695–3697. [Google Scholar]
- Mulloy, B.; Hogwood, J. Chromatographic molecular weight measurements for heparin, its fragments and fractions, and other glycosaminoglycans. Methods Mol. Biol. 2015, 1229, 105–118. [Google Scholar] [PubMed]
- Sommers, C.D.; Ye, H.; Kolinski, R.E.; Nasr, M.; Buhse, L.F.; Al-Hakim, A.; Keire, D.A. Characterization of currently marketed heparin products: Analysis of molecular weight and heparinase-I digest patterns. Anal. Bioanal. Chem. 2011, 401, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cai, C.; Chandarajoti, K.; Hsieh, P.H.; Li, L.; Pham, T.Q.; Sparkenbaugh, E.M.; Sheng, J.; Key, N.S.; Pawlinski, R.; et al. Homogeneous low-molecular-weight heparins with reversible anticoagulant activity. Nat. Chem. Biol. 2014, 10, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Masuko, S.; Takieddin, M.; Xu, H.; Liu, R.; Jing, J.; Mousa, S.A.; Linhardt, R.J.; Liu, J. Chemoenzymatic Synthesis of Homogeneous Ultralow Molecular Weight Heparins. Science 2011, 334, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, V.; Zachary, S.; Malikarjun, S.; Yi-Wei, Q.I.; Ram, S. Analysis of Sulfated Polysaccharides. U.S. Patent 7575886, 18 August 2009. [Google Scholar]
- Ivanciuc, O. Applications of Support Vector Machines in Chemistry. In Reviews in Computational Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 23, pp. 291–400. [Google Scholar]
- Mulloy, B.; Hogwood, J.; Gray, E. Assays and reference materials for current and future applications of heparins. Biologicals 2010, 38, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Shastri, M.D.; Johns, C.; Hutchinson, J.P.; Khandagale, M.; Patel, R.P. Ion exchange chromatographic separation and isolation of oligosaccharides of intact low-molecular-weight heparin for the determination of their anticoagulant and anti-inflammatory properties. Anal. Bioanal. Chem. 2013, 405, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Steppich, J.; Wang, Z.; Sun, Y.; Xue, C.; Linhardt, R.J.; Li, L. Bottom-Up Low Molecular Weight Heparin Analysis Using Liquid Chromatography-Fourier Transform Mass Spectrometry for Extensive Characterization. Anal. Chem. 2014, 86, 6626–6632. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Li, G.; Yang, B.; Onishi, A.; Li, L.; Sun, P.; Zhang, F.; Linhardt, R.J. Structural Characterization of Pharmaceutical Heparins Prepared from Different Animal Tissues. J. Pharm. Sci. 2013, 102, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, B.; Ly, M.; Solakyildirim, K.; Xiao, Z.; Wang, Z.; Beaudet, J.M.; Torelli, A.Y.; Dordick, J.S.; Linhardt, R.J. Structural characterization of heparins from different commercial sources. Anal. Bioanal. Chem. 2011, 401, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Compound Name | Repeating Unit Structure 1 | MW (Da) |
|---|---|---|---|
| A. | 8-mer NAc (1) | GlcNAc-(GlcA-GlcNAc)3-GlcA-pNP | 1656 |
| 10-mer NAc (2) | GlcNAc-(GlcA-GlcNAc)4-GlcA-pNP | 2036 | |
| 12-mer NAc (3) | GlcNAc-(GlcA-GlcNAc)5-GlcA-pNP | 2415 | |
| 14-mer NAc (4) | GlcNAc-(GlcA-GlcNAc)6-GlcA-pNP | 2794 | |
| 16-mer NAc (5) | GlcNAc-(GlcA-GlcNAc)7-GlcA-pNP | 3174 | |
| 18-mer NAc (6) | GlcNAc-(GlcA-GlcNAc)8-GlcA-pNP | 3553 | |
| B. | 8-mer NS (7) | GlcNS-(GlcA-GlcNS)3-GlcA-pNP | 1808 |
| 10-mer NS (8) | GlcNS-(GlcA-GlcNS)4-GlcA-pNP | 2226 | |
| 12-mer NS (9) | GlcNS-(GlcA-GlcNS)5-GlcA-pNP | 2643 | |
| 14-mer NS (10) | GlcNS-(GlcA-GlcNS)6-GlcA-pNP | 3060 | |
| 16-mer NS (11) | GlcNS-(GlcA-GlcNS)7-GlcA-pNP | 3478 | |
| 18-mer NS (12) | GlcNS-(GlcA-GlcNS)8-GlcA-pNP | 3895 | |
| C. | 10-mer NS2S (13) | GlcNS-GlcA-(GlcNS-IdoA2S)3-GlcNS-GlcA-pNP | 2466 |
| 12-mer NS2S (14) | GlcNS-GlcA-(GlcNS-IdoA2S)4-GlcNS-GlcA-pNP | 2963 | |
| 14-mer NS2S (15) | GlcNS-GlcA-(GlcNS-IdoA2S)5-GlcNS-GlcA-pNP | 3461 | |
| 16-mer NS2S (16) | GlcNS-GlcA-(GlcNS-IdoA2S)6-GlcNS-GlcA-pNP | 3958 | |
| 18-mer NS2S (17) | GlcNS-GlcA-(GlcNS-IdoA2S)7-GlcNS-GlcA-pNP | 4456 | |
| D. | 10-mer NS6S (18) | GlcNS6S-(GlcA-GlcNS6S)4-GlcA-pNP | 2626 |
| 12-mer NS6S (19) | GlcNS6S-(GlcA-GlcNS6S)5-GlcA-pNP | 3124 | |
| 14-mer NS6S (20) | GlcNS6S-(GlcA-GlcNS6S)6-GlcA-pNP | 3621 | |
| 16-mer NS6S (21) | GlcNS6S-(GlcA-GlcNS6S)7-GlcA-pNP | 4118 | |
| 18-mer NS6S (22) | GlcNS6S-(GlcA-GlcNS6S)8-GlcA-pNP | 4616 | |
| E. | 8-mer NS6S2S (23) | GlcNS6S-GlcA-(GlcNS6S-IdoA2S)2-GlcNS6S-GlcA-pNP | 2289 |
| 12-mer NS6S2S (24) | GlcNS6S-GlcA-(GlcNS6S-IdoA2S)4-GlcNS6S-GlcA-pNP | 3444 | |
| 14-mer NS6S2S (25) | GlcNS6S-GlcA-(GlcNS6S-IdoA2S)5-GlcNS6S-GlcA-pNP | 4021 | |
| 16-mer NS6S2S (26) | GlcNS6S-GlcA-(GlcNS6S-IdoA2S)6-GlcNS6S-GlcA-pNP | 4599 | |
| 18-mer NS6S2S (27) | GlcNS6S-GlcA-(GlcNS6S-IdoA2S)7-GlcNS6S-GlcA-pNP | 5176 | |
| F. | dp4 (28) | ∆HexA,2S-GlcNS6S-IdoA2S-GlcNS6S | ~1200 |
| dp6 (29) | ∆HexA,2S-GlcNS6S-(IdoA2S-GlcNS6S)2 | ~1800 | |
| G. | USP A.1 | 11000 | |
| USP A.2 | 5200 | ||
| USP A.3 | 2250 | ||
| USP A.4 | 1400 | ||
| USP B.1 | 7750 | ||
| USP B.2 | 3350 | ||
| USP B.3 | 1800 |
| Series | USP Enoxaparin RS Mw (4370 ± 150 Da) | Deviation from Acceptability Range (100 ± 3.4%) |
|---|---|---|
| USP Calibrants | 4300 | −1.6% |
| NAc (group A) | 22,300 | +410.3% |
| NS (group B) | 3100 | −29.1% |
| NS2S (group C) | 3500 | −19.9% |
| NS6S (group D) | 3350 | −23.3 % |
| NS6S2S (group E) | 3700 | −15.3% |
| All oligosaccharides | 3250 | −25.6% |
| Disaccharide Structure | Molar % |
|---|---|
| ∆UA2S-GlcNS6S | ~70% |
| ∆UA-GlcNS6S | ~10% |
| ∆UA2S-GlcNS | ~6% |
| ∆UA-GlcNS | ~2% |
| ∆UA-GlcNAc | ~1% |
| USP Calibrants | Oligosaccharide SVM Model | |
|---|---|---|
| USP Enoxaparin RS (Mw = 4370 ± 150 Da) | 4300 Da | 4450 Da |
| Sample | Mw | Distribution |
|---|---|---|
| Lovenox | Mw = 4650 ± 35 Da | M2000: 14.0 ± 0.5% |
| M2000–8000: 82.0 ± 0.5% | ||
| M8000: 4.0 ± 0.5% | ||
| Enoxaparin Sodium; Sandoz | Mw = 4350 ± 0 Da | M2000: 16.5 ± 1.0% |
| M2000–8000: 81.0 ± 1.0% | ||
| M8000: 2.5 ± 0.0% | ||
| Enoxaparin Sodium; Amphastar | Mw = 4350 ± 27 Da | M2000: 13.5 ± 1.0% |
| M2000–8000: 83.5 ± 1.0% | ||
| M8000: 3.5 ± 1.0% |
| USP Method | Proposed Method | |
|---|---|---|
| Required materials | USP Enoxaparin MW Calibrant A USP Enoxaparin MW Calibrant B USP Enoxaparin RS Enoxaparin sample | Oligosaccharide Standard Solution Enoxaparin sample |
| Required HPLC time 1 | ~5.3 h | ~1.3 h |
| Analysis |
|
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, K.M.; Capuzzi, S.J.; Xu, Y.; Muratov, E.N.; Carrick, K.; Szajek, A.Y.; Tropsha, A.; Liu, J. Modernization of Enoxaparin Molecular Weight Determination Using Homogeneous Standards. Pharmaceuticals 2017, 10, 66. https://doi.org/10.3390/ph10030066
Arnold KM, Capuzzi SJ, Xu Y, Muratov EN, Carrick K, Szajek AY, Tropsha A, Liu J. Modernization of Enoxaparin Molecular Weight Determination Using Homogeneous Standards. Pharmaceuticals. 2017; 10(3):66. https://doi.org/10.3390/ph10030066
Chicago/Turabian StyleArnold, Katelyn M., Stephen J. Capuzzi, Yongmei Xu, Eugene N. Muratov, Kevin Carrick, Anita Y. Szajek, Alexander Tropsha, and Jian Liu. 2017. "Modernization of Enoxaparin Molecular Weight Determination Using Homogeneous Standards" Pharmaceuticals 10, no. 3: 66. https://doi.org/10.3390/ph10030066