Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine

 ,
,
Abstract
:1. Introduction
- -
- Class I (structural mimetics): show a structural analogy with the native substrate in a well- defined spatial orientation, they carry all the functionalities responsible for the interaction with a receptor or an enzyme.
- -
- Class II (functional mimetics): the analogy of this type is based on the interaction with the target enzyme or receptor regardless of their apparent structural similarity with the native compound.
- -
- Class III (functional-structural mimetics): all the functional groups required for biological activity are mounted in a scaffoldwith a structure different from that of the starting compound.
2. Materials and Methods
2.1. Peptide Design, Synthesis and Purification
2.2. Antibiotics
2.3. Bacterial Strains
2.4. Bacterial Susceptibility Assays
2.4.1. MIC and MBC Values of UP-5
2.4.2. MICs and MBC Determination of Individual Antibiotics
2.4.3. MIC and MBC Determination for Peptide and Antibiotics in Combinations
2.4.4. FIC Determination
2.5. Antibiofilm Assay
2.6. MTT Cell Proliferation Assay
2.7. Erythrocyte Hemolytic Assay
2.8. Stability Assays
2.9. Statistical Analysis
3. Results
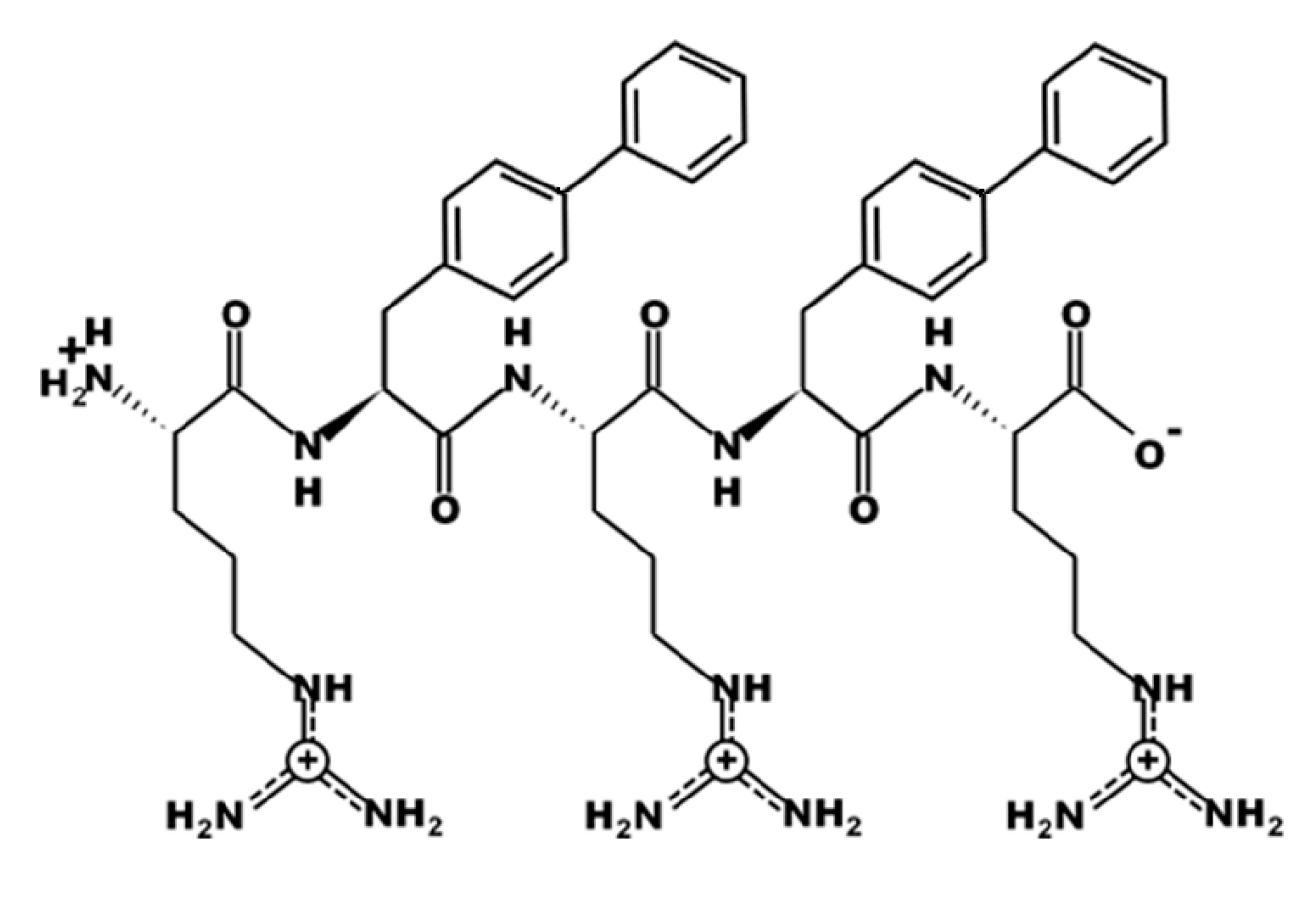
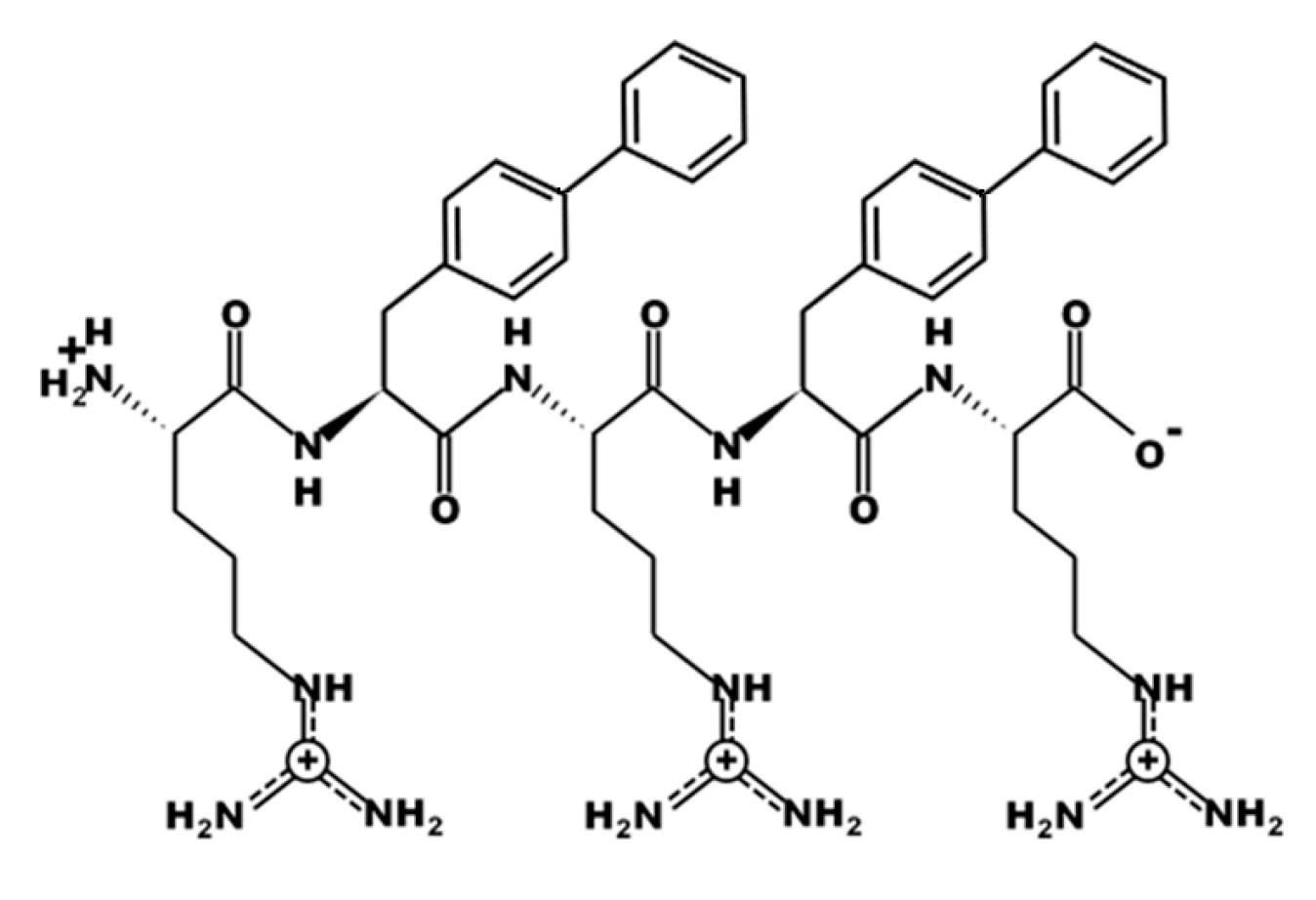
3.1. Design of UP-5
3.2. In Vitro Antimicrobial Activity of UP-5
3.3. Antimicrobial Synergistic Assays
3.3.1. Determination of the MIC and MBC of the Individual Antibiotics
3.3.2. Checkerboard Assay Results
3.3.3. Determination of MBCs of UP-5 and Antibiotics in Combinations
3.4. Antibiofilm Activity of UP-5
3.5. Hemolytic Assay
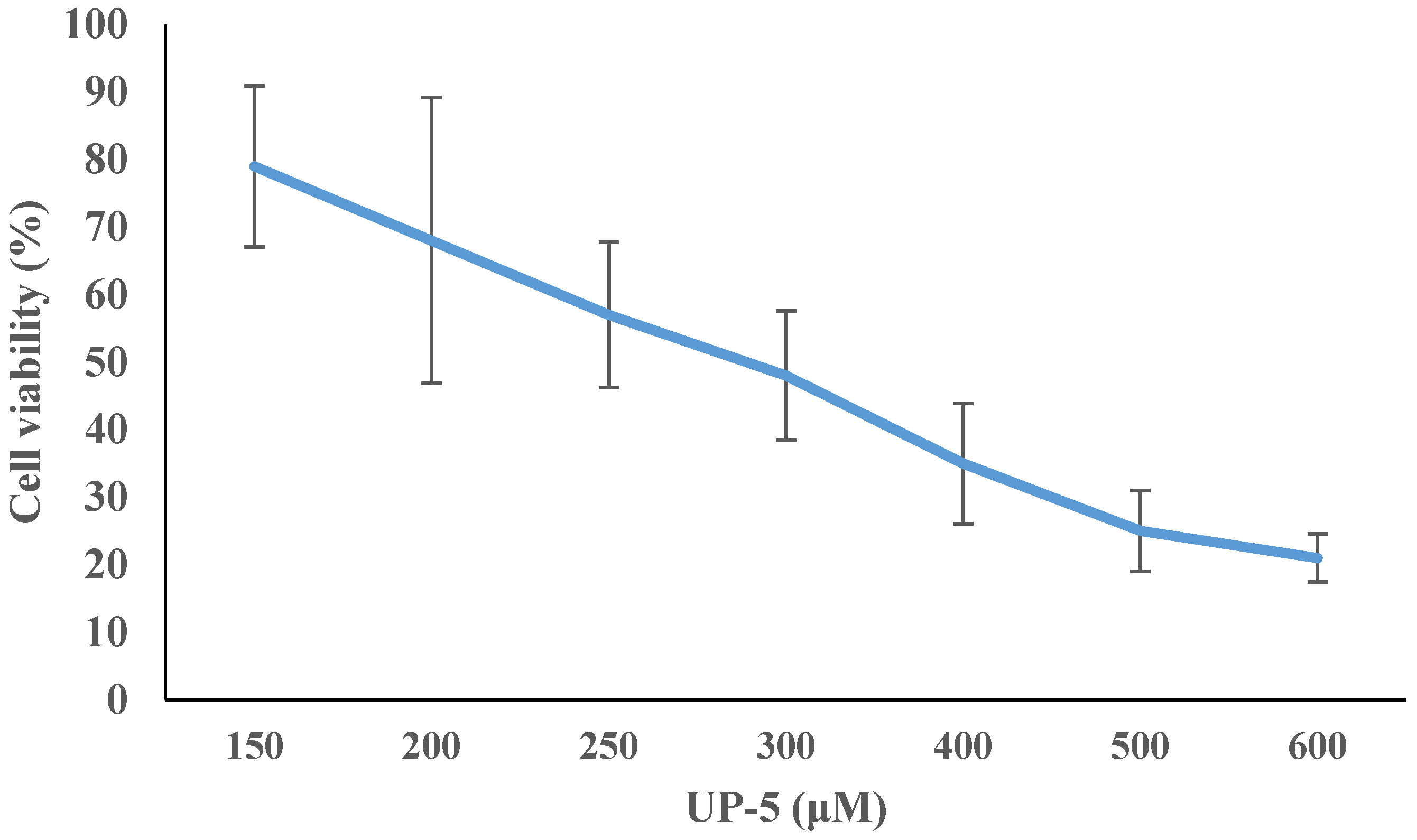
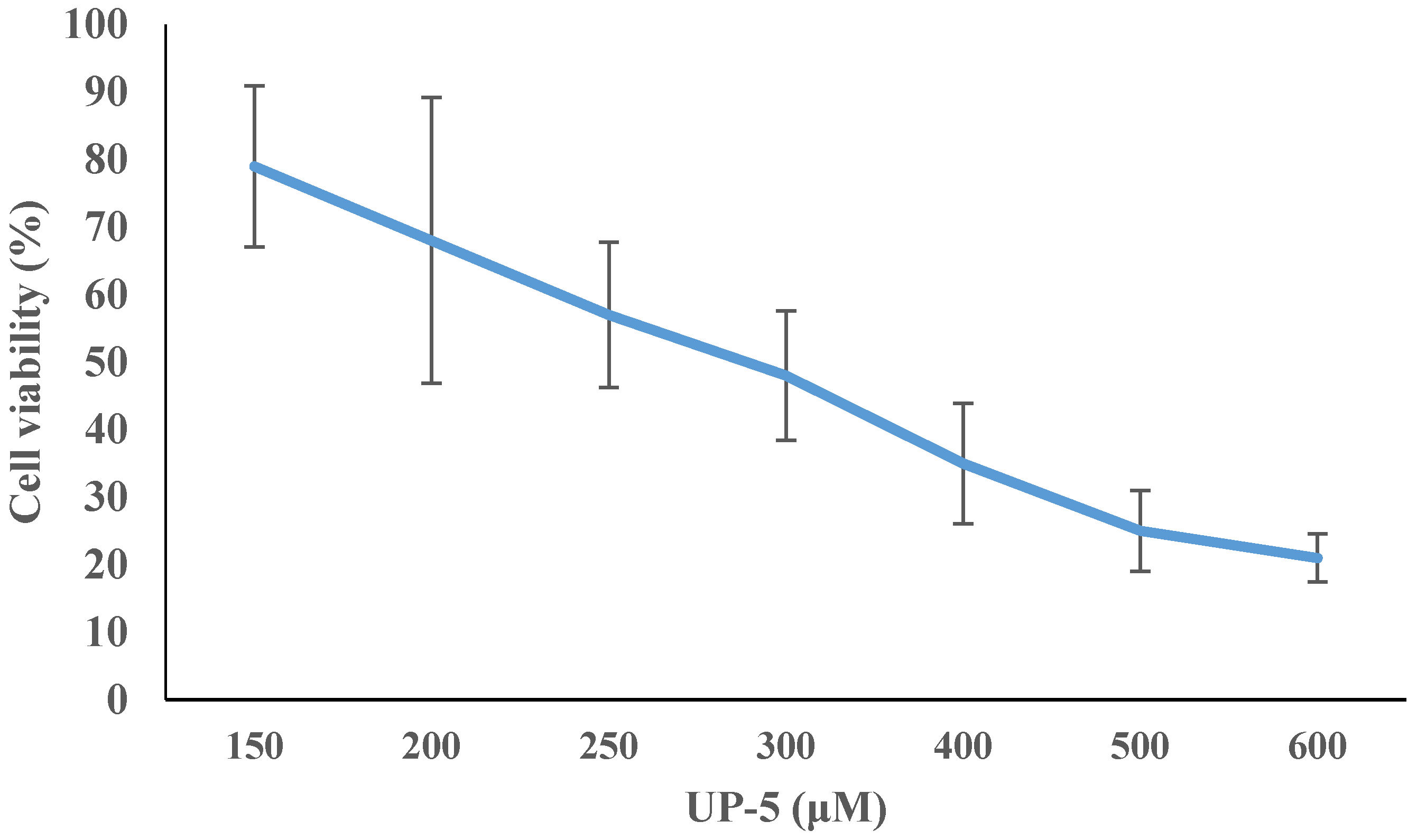
3.6. Cell Cytotoxicity
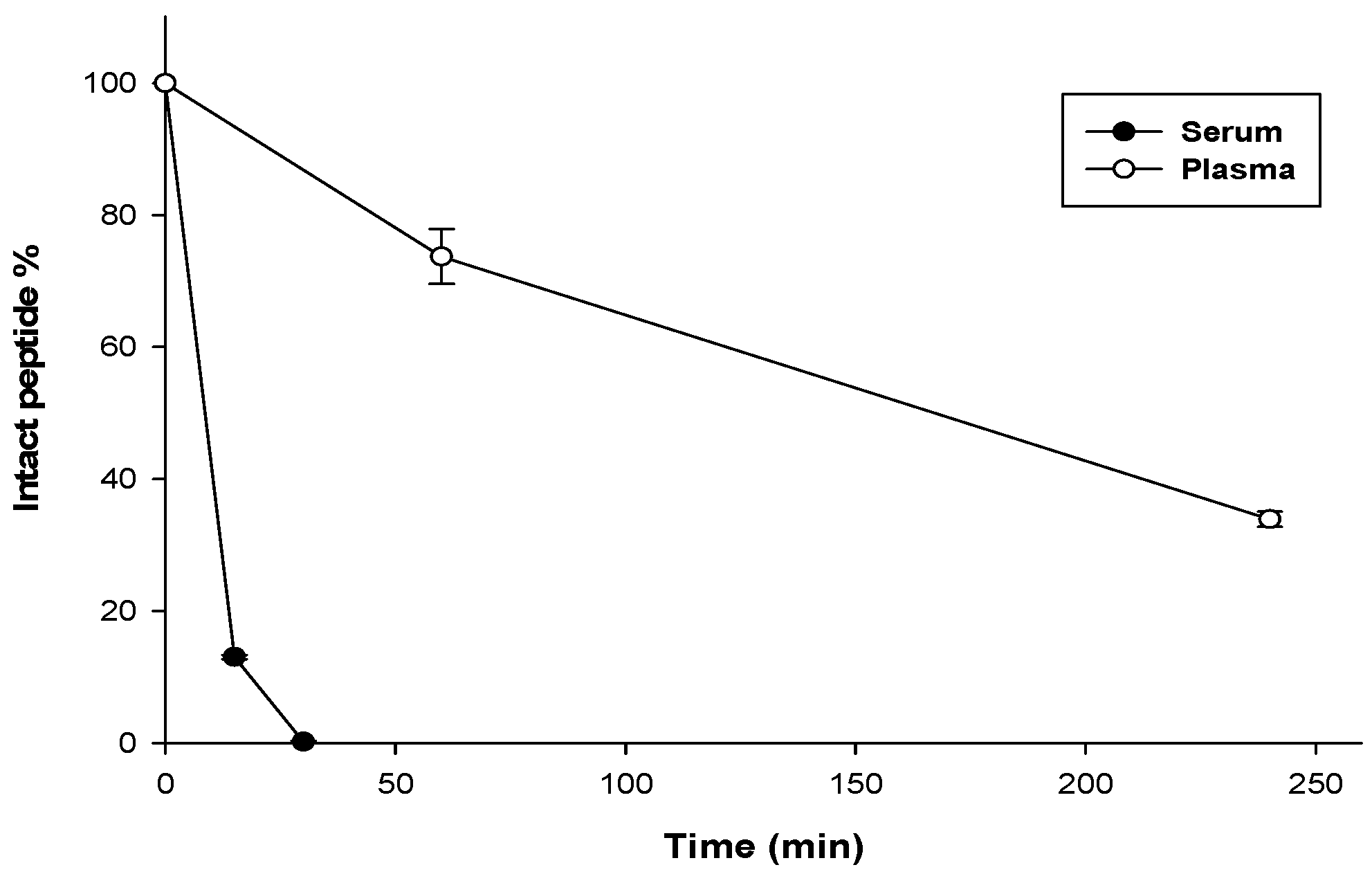
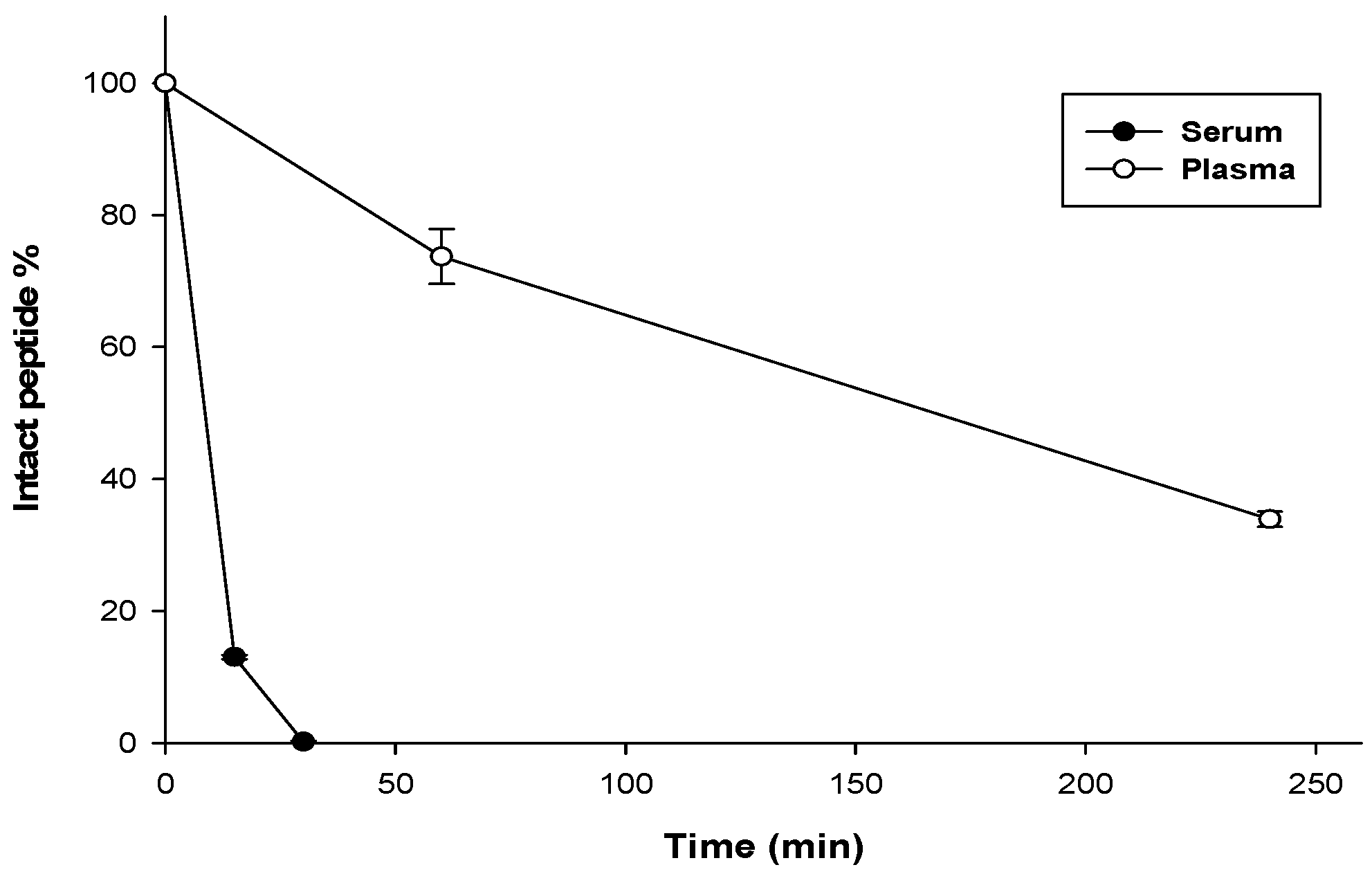
3.7. Stability Assay
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Antimicrobial Resistance. Available online: http://www.who.int/mediacentre/factsheets/fs194/en/ (accessed on 13 November 2017).
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.; Wertheim, H.F.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Otero-González, A.J.; Magalhães, B.S.; Garcia-Villarino, M.; López-Abarrategui, C.; Sousa, D.A.; Dias, S.C.; Franco, O.L. Antimicrobial peptides from marine invertebrates as a new frontier for microbial infection control. FASEB J. 2010, 24, 1320–1334. [Google Scholar]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.; Gellatly, S.L.; Hancock, R.E. Multifunctional cationic host defence peptides and their clinical applications. Cell. Mol. Life Sci. 2011, 68, 2161. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Gao, L.; Fang, W. Theoretical insight into the relationship between the structures of antimicrobial peptides and their actions on bacterial membranes. J. Phys. Chem. B 2014, 119, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; Hristova, K. Antimicrobial Peptides: Successes, Challenges and Unanswered Questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Porto, W.F.; Silva, O.N.; Franco, O.L. Prediction and rational design of antimicrobial peptides. InProtein Struct. 2012. [Google Scholar] [CrossRef]
- Albada, H.B.; Prochnow, P.; Bobersky, S.; Langklotz, S.; Bandow, J.E.; Metzler-Nolte, N. Short Antibacterial Peptides with Significantly Reduced Hemolytic Activity can be Identified by a Systematic l-to-d Exchange Scan of their Amino Acid Residues. ACS Comb. Sci. 2013, 15, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Deliv. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The host antimicrobial peptide Bac7 1-35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem. Biol. 2014, 21, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiang, Q.; Zhang, Q.; Huang, Y.; Su, Z. Overview on the recent study of antimicrobial peptides: Origins, functions, relative mechanisms and application. Peptides 2012, 37, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Petersen, A.P.; Lau, C.K.; Jing, W.; Storey, D.G.; Vogel, H.J. Mechanism of action of puroindoline derived tryptophan-rich antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Balhara, V.; Schmidt, R.; Gorr, S.U.; DeWolf, C. Membrane selectivity and biophysical studies of the antimicrobial peptide GL13K. Biochim. Biophys. Acta (BBA)-Biomembr. 2013, 1828, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.N.; Hall, K.; Aguilar, M.I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Trabocchi, A.; Guarna, A. The basics of peptidomimetics. In Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs; John Wiley & Sons: New York, NY, USA, 2014; pp. 1–7. [Google Scholar]
- Rekha, R.S. Role of Antimicrobial Peptides in Tuberculosis and Respiratory Tract Infections: Clinical and Mechanistic Studies. Ph.D. Thesis, Inst för laboratoriemedicin/Dept of Laboratory Medicine, Karolinska Institute, Stockholm, Sweden, 14 October 2015. [Google Scholar]
- Liskamp, R.M. Conformationally restricted amino acids and dipeptides, (non) peptidomimetics and secondary structure mimetics. Recl. Trav. Chim. Pays-Bas 1994, 113, 1–9. [Google Scholar] [CrossRef]
- Olson, G.L.; Bolin, D.R.; Bonner, M.P.; Bos, M.; Cook, C.M.; Fry, D.C.; Graves, B.J.; Hatada, M.; Hill, D.E. Concepts and progress in the development of peptide mimetics. J. Med. Chem. 1993, 36, 3039–3049. [Google Scholar] [CrossRef] [PubMed]
- Ripka, A.S.; Rich, D.H. Peptidomimetic design. Curr. Opin. Chem. Biol. 1998, 2, 441–452. [Google Scholar] [CrossRef]
- Daily, A.E.; Greathouse, D.V.; van der Wel, P.C.; Koeppe, R.E., 2nd. Helical Distortion in Tryptophan- and Lysine-Anchored Membrane-Spanning α-Helices as a Function of Hydrophobic Mismatch: A Solid-State Deuterium NMR Investigation Using the Geometric Analysis of Labeled Alanines Method. Biophys. J. 2008, 94, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.L.; Yu, H.Y.; Yip, B.S.; Chih, Y.H.; Liang, C.W.; Cheng, H.T.; Cheng, J.W. Boosting salt resistance of short antimicrobial peptides. Antimicrob. Agents Chemother. 2013, 57, 4050–4052. [Google Scholar] [CrossRef] [PubMed]
- Lau, Q.Y.; Ng, F.M.; Cheong, J.W.D.; Yap, Y.Y.A.; Tan, Y.Y.F.; Jureen, R.; Hill, J.; Chia, C.S.B. Discovery of an ultra-short linear antibacterial tetrapeptide with anti-MRSA activity from a structure-activity relationship study. Eur. J. Med. Chem. 2015, 105, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Arnusch, C.J.; Ulm, H.; Josten, M.; Shadkchan, Y.; Osherov, N.; Sahl, H.G.; Shai, Y. Ultrashort peptide bioconjugates are exclusively antifungal agents and synergize with cyclodextrin and amphotericin B. Antimicrob. Agents Chemother. 2012, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bisht, G.S.; Rawat, D.S.; Kumar, A.; Kumar, R.; Pasha, S. Antimicrobial activity of rationally designed amino terminal modified peptides. Bioorg. Med. Chem. Lett. 2007, 17, 4343–4346. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Seo, M.D.; Won, H.S.; Kim, J.H.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial peptides for therapeutic applications: A review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.N.; Zhanel, G.G.; Schweizer, F. Antibacterial activity of ultrashort cationic lipo-β-peptides. Antimicrob. Agents Chemother. 2009, 53, 2215–2217. [Google Scholar]
- Chen, X.; Zhang, M.; Zhou, C.; Kallenbach, N.R.; Ren, D. Control of bacterial persister cells by Trp/Arg-containing antimicrobial peptides. Appl. Environ. Microbiol. 2011, 77, 4878–4885. [Google Scholar] [CrossRef] [PubMed]
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Alst, T.; Havelkova, M.; Strøm, M.B. Antimicrobial Activity of Small β-Peptidomimetics Based on the Pharmacophore Model of Short Cationic Antimicrobial Peptides. J. Med. Chem. 2010, 53, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochi. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Zarena, D.; Mishra, B.; Lushnikova, T.; Wang, F.; Wang, G. The π Configuration of the WWW Motif of a Short Trp-Rich Peptide Is Critical for Targeting Bacterial Membranes, Disrupting Preformed Biofilms, and Killing Methicillin-Resistant Staphylococcus aureus. Biochemistry 2017, 56, 4039–4043. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hanke, M.L.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Chittezham Thomas, V.; Bayles, K.W.; Kielian, T. Transformation of human cathelicidin LL-37 into selective, stable, and potent antimicrobial compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Deepankumar, K.; Prabhu, N.S.; Kim, J.H.; Yun, H. Protein engineering for covalent immobilization and enhanced stability through incorporation of multiple noncanonical amino acids. Biotechnol. Bioprocess Eng. 2017, 22, 248–255. [Google Scholar] [CrossRef]
- Schnaider, L.; Brahmachari, S.; Schmidt, N.W.; Mensa, B.; Shaham-Niv, S.; Bychenko, D.; Adler-Abramovich, L.; Shimon, L.J.W.; Kolusheva, S.; DeGrado, W.F.; et al. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 2017, 8, 1365. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R. Synthesis of Modified Amino Acids and Insertion in Peptides and Mimetics. Structural Aspects and Impact on Biological Activity. ph.D. Thesis, University of Bologna, Bologna, Italy, 2012. [Google Scholar]
- Brown, D.F.J.; Edwards, D.I.; Hawkey, P.M.; Morrison, D.; Ridgway, G.L.; Towner, K.J.; Wren, M.W. Guidelines for the laboratory diagnosis and susceptibility testing of methicillin-resistant Staphylococcus aureus (MRSA). J. Antimicrob. Chemother. 2005, 56, 1000–1018. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.M.; Gomes, R.T.; Freire, N.R.; Aguiar, E.G.; Brandão, M.D.; Santos, V.R. In vitro antimicrobial activity of Brazilian medicinal plant extracts against pathogenic microorganisms of interest to dentistry. Planta Medica 2011, 77, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Sueke, H.; Kaye, S.B.; Neal, T.; Hall, A.; Tuft, S.; Parry, C.M. An in vitro investigation of synergy or antagonism between antimicrobial combinations against isolates from bacterial keratitis. Invest. Ophthalmol. Vis. Sci. 2010, 51, 4151–4155. [Google Scholar] [CrossRef] [PubMed]
- Schwering, M.; Song, J.; Louie, M.; Turner, R.J.; Ceri, H. Multi-species biofilms defined from drinking water microorganisms provide increased protection against chlorine disinfection. Biofouling 2013, 29, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Zhou, M.; Wang, L.; Chen, T.; Walker, B.; Shaw, C. Antimicrobial/cytolytic peptides from the venom of the North African scorpion, Androctonus amoreuxi: Biochemical and functional characterization of natural peptides and a single site-substituted analog. Peptides 2012, 35, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Knappe, D.; Henklein, P.; Hoffmann, R.; Hilpert, K. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob. Agents Chemother. 2010, 54, 4003–4005. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Canton, R.; Cornaglia, G. Emergence of antibiotic resistance: Need for a new paradigm. Clin. Microbiol. Infect. 2012, 18, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Mataraci, E.; Dosler, S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob. Agents Chemother. 2012, 56, 6366–6371. [Google Scholar] [CrossRef] [PubMed]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of Short Cationic Antibacterial Peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar]
- Wang, G. Determination of solution structure and lipid micelle location of an engineered membrane peptide by using one NMR experiment and one sample. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 3271–3281. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, A.M.; Balaji, K.; Girilal, M.; Yadav, R.; Kalaichelvan, P.T.; Venketesan, R. Biogenic synthesis of silver nanoparticles and their synergistic effect with antibiotics: A study against gram-positive and gram-negative bacteria. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ruden, S.; Hilpert, K.; Berditsch, M.; Wadhwani, P.; Ulrich, A.S. Synergistic interaction between silver nanoparticles and membrane-permeabilizing antimicrobial peptides. Antimicrob. Agents Chemother. 2009, 53, 3538–3540. [Google Scholar] [CrossRef] [PubMed]
- Maróti, G.; Kereszt, A.; Kondorosi, E.; Mergaert, P. Natural roles of antimicrobial peptides in microbes, plants and animals. Res. Microbiol. 2011, 162, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Ramamoorthy, A.; Chen, Z. Membrane orientation of MSI-78 measured by sum frequency generation vibrational spectroscopy. Langmuir 2011, 27, 7760–7767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Sun, Y.; Liu, Q.; Wang, X.; Li, Z.; Hao, J. In vitro synergistic activities of antimicrobial peptide brevinin-2CE with five kinds of antibiotics against multidrug-resistant clinical isolates. Curr. Microbial. 2014, 68, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hirt, H.; Gorr, S.-U. Antimicrobial peptide GL13K is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Individual and combined effects of engineered peptides and antibiotics on Pseudomonas aeruginosa biofilms. Pharmaceuticals 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Laverty, G.; McLaughlin, M.; Shaw, C.; Gorman, S.P.; Gilmore, B.F. Antimicrobial Activity of Short, Synthetic Cationic Lipopeptides. Chem. Biol. Drug Des. 2010, 75, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Corzo, G.; Escoubas, P.; Villegas, E.; Barnham, K.J.; He, W.; Norton, R.S.; Nakajima, T. Characterization of unique amphipathic antimicrobial peptides from venom of the scorpion Pandinus imperator. Biochem. J. 2001, 359 Pt 1, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Kang, J.H.; Lee, M.K.; Kim, S.Y.; Kim, Y.; Hahm, K.S. Cecropin A - magainin 2 hybrid peptides having potent antimicrobial activity with low hemolytic effect. Biochem. Mol. Biol. Int. 1998, 44, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Tarazi, S.; Alsheyab, F.; Al-Balas, Q.; Mukattash, T. Antimicrobial and Antibiofilm Activity of Mauriporin, a Multifunctional Scorpion Venom Peptide. Int. J. Pept. Res. Ther. 2014, 20, 397–408. [Google Scholar] [CrossRef]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.D.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; De Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan-and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | ATCC # | MIC Value (μM) | MBC Value (μM) |
|---|---|---|---|
| Staphylococcus aureus | 29231 | 10 | 10 |
| Staphylococcus aureus | 43300 | 15 | 15 |
| Staphylococcus aureus | BAA-41 | 15 | 15 |
| Staphylococcus aureus | 33591 | 10 | 10 |
| Enterococcus faecalis | BAA-2316 | 70 | 70 |
| Escherichia coli | 25922 | 55 | 55 |
| Escherichia coli | 35218 | 60 | 60 |
| Pseudomonas aeruginosa | 27853 | 65 | 65 |
| Pseudomonas aeruginosa | BAA-2114 | 55 | 55 |
| Bacterial Species | Antibiotics | UP-5 | FIC Index | |||
|---|---|---|---|---|---|---|
| MIC/MBC Alone (μM) | MIC in Combination (μM) | Reduction in MIC (%) | MIC in Combination (μM) | |||
| S. aureus ATCC 29213 | Levofloxacin | 0.5/0.5 | 0.05 | 90 | 0.125 | 0.11 |
| Chloramphenicol | 20/30 | 10 | <50 | 2.5 | 0.75 | |
| Rifampicin | 0.025/0.025 | 0.025 | <50 | 10 | 2 | |
| Ampicillin | 2.5/2.5 | 2.5 | <50 | 0.25 | 1.025 | |
| Erythromycin | 0.5/1.5 | 0.5 | <50 | 10 | 2 | |
| S. aureus ATCC 33591 | Levofloxacin | 10/10 | 1.25 | 87.5 | 0.125 | 0.13 |
| Chloramphenicol | 130/150 | 65 | 50 | 5 | 1 | |
| Rifampicin | 0.04/0.04 | 0.005 | 87.5 | 0.125 | 0.13 | |
| Ampicillin | 85/85 | 12.5 | 85.3 | 1.25 | 0.27 | |
| Erythromycin | 8/10 | 8 | <50 | 0.125 | 1 | |
| S. aureus ATCC 43300 | Levofloxacin | 20/20 | 5 | <50 | 0.025 | 0.25 |
| Chloramphenicol | 15/20 | 7.5 | <50 | 7.5 | 1 | |
| Rifampicin | 1/1 | 0.125 | 87.5 | 5 | 0.45 | |
| Ampicillin | 20/20 | 2.5 | <50 | 7.5 | 0.62 | |
| Erythromycin | 100/150 | 100 | <50 | 7.5 | 1.5 | |
| S. aureus ATCC BAA-41 | Levofloxacin | 10/10 | 10 | <50 | 15 | 2 |
| Chloramphenicol | 25/30 | 7.5 | 70 | 7.5 | 0.8 | |
| Rifampicin | 0.005/0.005 | 0.002 | 60 | 15 | 1.4 | |
| Ampicillin | 40/40 | 20 | <50 | 7.5 | 1 | |
| Erythromycin | 350/400 | 125 | 64.3 | 15 | 1.35 | |
| P. auroginosa ATCC BAA-2114 | Levofloxacin | 12/12 | 5 | 58.3 | 20 | 0.78 |
| Chloramphenicol | 200/325 | 12.5 | 93.7 | 10 | 0.24 | |
| Rifampicin | 50/50 | 0.25 | 99.5 | 25 | 0.46 | |
| Ampicillin | >500/>500 | 250 | >50 | 20 | 0.86 | |
| Erythromycin | 125/150 | 50 | 60 | 27.5 | 0.9 | |
| Peptide Concentration (μM) | Viable Biofilm Cells (%) | ±SD |
|---|---|---|
| 100 | 0 | 0.0002 |
| 80 | 0 | 0.0005 |
| 60 | 0 | 0.0006 |
| 40 | 0 | 0.002 |
| 30 | 0.054 | 0.005 |
| 20 | 0.108 | 0.03 |
| 10 | 0.35 | 0.09 |
| MBEC value | 20 | |
| MBCb | 20 |
| Hemolysis (%) | Concentration (μM) | ±SD |
|---|---|---|
| 0.09 | 40 | 0.0005 |
| 1.01 | 60 | 0.004 |
| 1.04 | 80 | 0.001 |
| 1.04 | 100 | 0.0005 |
| 2.06 | 125 | 0.002 |
| 3.0 | 150 | 0.001 |
| 3.04 | 175 | 0.001 |
| 3.09 | 200 | 0.01 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almaaytah, A.; Qaoud, M.T.; Khalil Mohammed, G.; Abualhaijaa, A.; Knappe, D.; Hoffmann, R.; Al-Balas, Q. Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine. Pharmaceuticals 2018, 11, 3. https://doi.org/10.3390/ph11010003
Almaaytah A, Qaoud MT, Khalil Mohammed G, Abualhaijaa A, Knappe D, Hoffmann R, Al-Balas Q. Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine. Pharmaceuticals. 2018; 11(1):3. https://doi.org/10.3390/ph11010003
Chicago/Turabian StyleAlmaaytah, Ammar, Mohammed T. Qaoud, Gubran Khalil Mohammed, Ahmad Abualhaijaa, Daniel Knappe, Ralf Hoffmann, and Qosay Al-Balas. 2018. "Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine" Pharmaceuticals 11, no. 1: 3. https://doi.org/10.3390/ph11010003