The Biochemical Basis of Hydroxymethylglutaryl-CoA Reductase Inhibitors as Neuroprotective Agents in Aneurysmal Subarachnoid Hemorrhage

{kind=link}
{kind=link}
{kind=link}
Abstract
:Background
Aneurysmal subarachnoid hemorrhage and delayed cerebral ischemic deficits
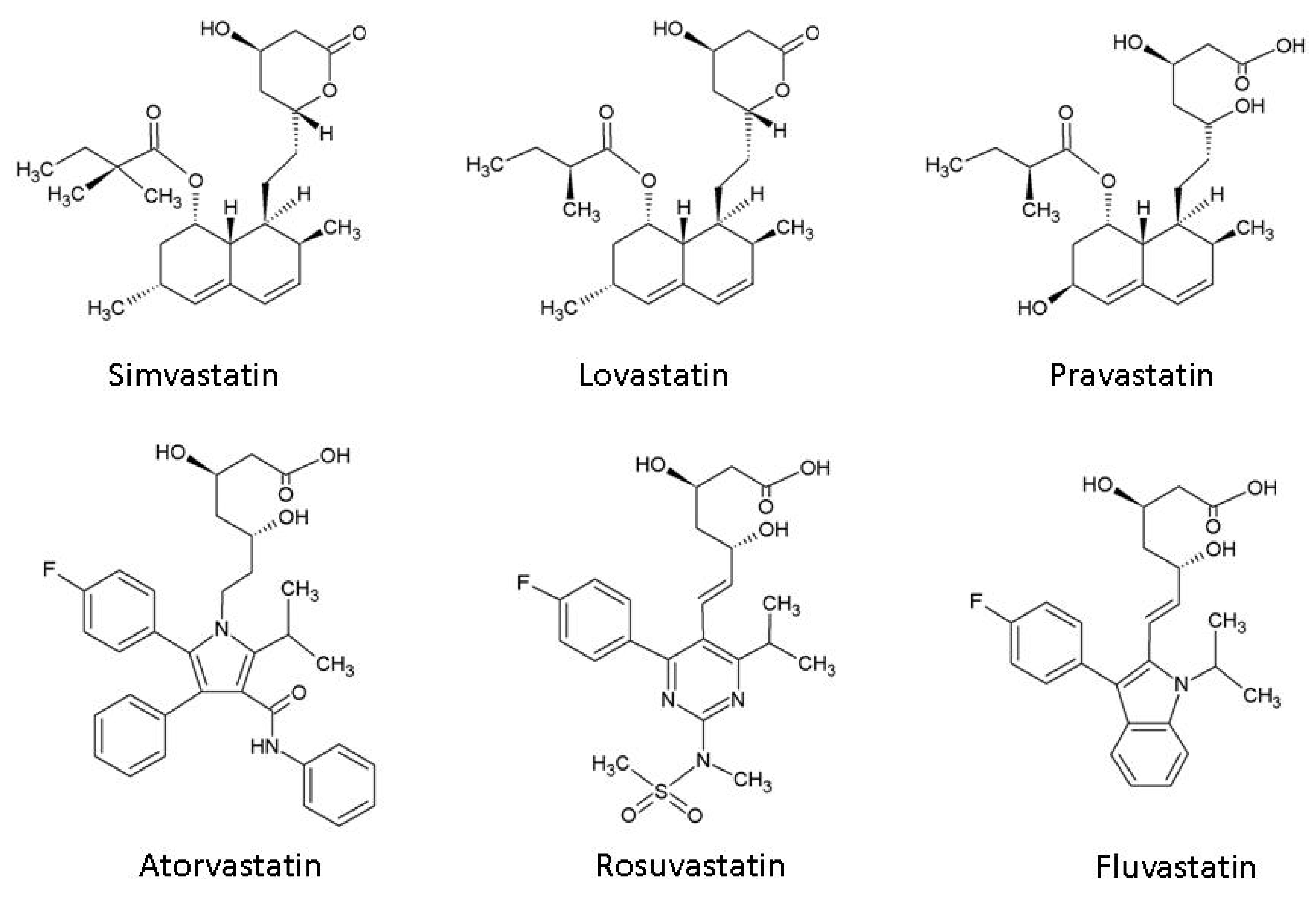
Statins (hydroxymethylglutaryl-CoA reductase inhibitors)


Ongoing multi-center clinical trials of the use of simvastatin for aneurysmal subarachnoid hemorrhage
Objective of this Review
Search Methodology
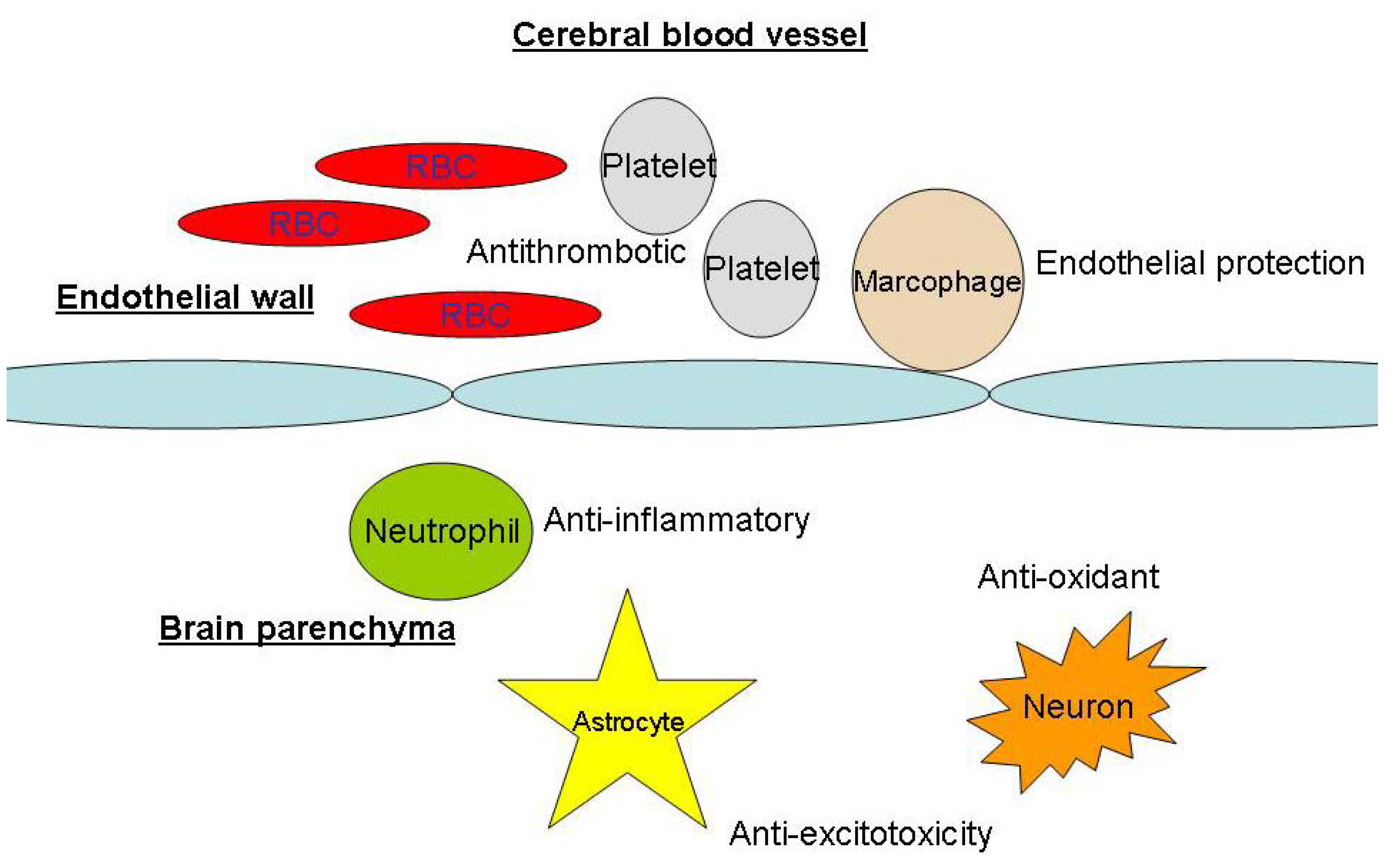
Biochemical Basis of Neuroprotection in Aneurysmal Subarachnoid Hemorrhage and Cerebral Vasospasm
Decrease in cerebral blood flow reduction and brain injury during cerebral artery occlusion
Anti-inflammatory effects
Anti-oxidant effects
Anti-platelet effects
Anti-excitotoxicity
Other neuroprotective mechanisms
Limitations of clinical translation of experimental data
Conclusions
Acknowledgements
References and Notes
- Macdonald, R.L.; Pluta, R.M.; Zhang, J.H. Cerebral vasospasm after subarachnoid hemorrhage: The emerging revolution. Nat. Clin. Pract. Neurol. 2007, 3, 256–263. [Google Scholar]
- Wong, G.K.; Ng, R.Y.; Poon, W.S. Aneurysmal subarachnoid hemorrhage. Surg. Pract. 2008, 12, 51–55. [Google Scholar] [CrossRef]
- Pluta, R.M.; Hansen-Schwartz, J.; Drier, J.; Vajkoczy, P.; Macdonald, R.L.; Nishizawa, S.; Kasuya, H.; Wellman, G.; Keller, E.; Zauner, A.; Dorsch, N.; Clark, J.; Ono, S.; Kiris, T.; LeRoux, P.; Zhang, J.H. Cerebral vasospasm following subarachnoid hemorrhage: Time for a new world of thought. Neurol. Res. 2009, 31, 151–158. [Google Scholar]
- Wong, G.K.; Wong, R.; Mok, V.C.; Fan, D.S.; Leung, G.; Wong, A.; Chan, A.S.; Zhu, C.X.; Poon, W.S. Clinical study on cognitive dysfunction after spontaneous subarachnoid haemorrhage: Patient profiles and relationship to cholinergic dysfunction. Acta Neurochir. (Wien) 2009, 151, 1601–1607. [Google Scholar]
- Weir, B.; Grace, M.; Hansen, J.; Rothberg, C. Time course of vasospasm in man. J. Neurosurg. 1978, 48, 173–178. [Google Scholar]
- Pickard, J.D.; Murray, G.D.; Illingworth, R.; Shaw, M.D.; Teasdale, G.M.; Foy, P.M.; Humphrey, P.R.; Lang, D.A.; Nelson, R.; Richard, P. Effect of oral nimodipine on cerebral infraction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. Brit. Med. J. 1989, 11, 636–642. [Google Scholar]
- Pluta, R. Delayed cerebral ischemia and nitric oxide: Review, new hypothesis, and proposed treatment. Pharmacol. Ther. 2005, 105, 23–56. [Google Scholar]
- Alabadi, J.A.; Salom, J.; Torregrosa, G.; Miranda, F.J.; Jover, T.; Alborch, E. Changes in the cerebrovascular effects of endothelin-1 and nicardipine after experimental subarachnoid hemorrhage. Neurosurgery 1993, 33, 707–714. [Google Scholar]
- Nishizawa, S.; Laher, I. Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc. Med. 2005, 15, 24–34. [Google Scholar]
- Ishiguro, M.; Wellman, T.L.; Honda, A.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Emergence of R-type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ. Res. 2005, 96, 419–426. [Google Scholar]
- Kusaka, G.; Ishikawa, M.; Nanda, A.; Granger, D.N.; Zhang, J.H. Signaling pathways for early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2004, 24, 916–925. [Google Scholar]
- Dreier, J.P.; Major, S.; Manning, A.; Woitzik, J.; Drenckhahn, C.; Steinbrink, J.; Tolias, C.; Oliveira-Ferreira, A.I.; Fabricius, M.; Hartings, J.A.; Vajkoczy, P.; Lauritzen, M.; Dirnagl, U.; Bohner, G.; Strong, A.J. COSBID study group. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009, 132, 1866–1881. [Google Scholar] [PubMed]
- van der Most, P.J.; Dolga, A.M.; Nijholt, I.M.; Luiten, P.G.; Eisel, U.L. Statins: Mechanisms of neuroprotection. Prog. Neurobiol. 2009, 88, 64–75. [Google Scholar]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar]
- Bjorkhem, I.; Lutjohann, D.; Diczfalusy, U.; Stahle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar]
- Saheki, A.; Terasaki, T.; Tamai, I.; Tsuji, A. In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm. Res. 1994, 11, 305–311. [Google Scholar]
- Suzuki, H.; Hasegawa, Y.; Kanamaru, K.; Zhang, J.H. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke 2010, 41, 1783–1790. [Google Scholar]
- Vergouwen, M.D.; de Haan, R.J.; Vermeulen, M.; Roos, Y.B. Effect of statin treatment on vasospasm, delayed cerebral ischemia, and functional outcome in patients with aneurysmal subarachnoid hemorrhage: a systemic review and meta-analysis update. Stroke 2010, 41, e47–e52. [Google Scholar]
- Kramer, A.H.; Fletcher, J.J. Statins in the management of patients with aneurysmal subarachnoid hemorrhage: A systemic review and meta-analysis. Neurocritical. Care 2010, 12, 285–296. [Google Scholar]
- Tseng, M.Y.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: A phase II randomised placebo-controlled trial. Stroke 2005, 36, 1627–1632. [Google Scholar]
- Tseng, M.Y.; Hutchison, P.J.; Czosnyka, M.; Richards, H.; Pickard, J.D.; Kirkpatrick, P.J. Effects of acute pravastatin on intensity of rescue therapy, length of inpatient stay and 6-month outcome in patients after subarachnoid haemorrhage. Stroke 2007, 38, 1545–1550. [Google Scholar]
- Endres, M.; Laufs, U.; Liao, J.K.; Moskowitz, M.D. Targeting eNOS for stroke protection. Trends Neurosci. 2004, 27, 283–289. [Google Scholar]
- Sironi, L.; Cimino, M.; Guerrini, U.; Calvio, A.M.; Lodetti, B.; Asdente, M.; Balduini, W.; Paoletti, R.; Tremoli, E. Treatment with statins after induction of focal ischemia in rats reduced the extent of brain damage. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 322–327. [Google Scholar]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, P.; Moskowitz, M.A.; Liao, J.K. Stroke protection by 3-hydroxyl-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar]
- Calabro, P.; Yeh, E.T. The pleotropic effects of statins. Curr. Opin. Cardiol. 2005, 20, 541–546. [Google Scholar]
- Rikitake, Y.; Liao, J.K. Rho GTPase, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar]
- Amarenco, P.; Moskowitz, M.A. The dynamics of statins: From event prevention to neuroprotection. Stroke 2006, 37, 294–296. [Google Scholar]
- Hernandez-Perera, O.; Perez-Sala, D.; Navarro-Antolin, J.; Sanchez-Pascuala, R.; Hernandez, G.; Diaz, C.; Lamas, S. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorstatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthetase in vascular endothelial cells. J. Clin. Invest. 1998, 101, 2711–2719. [Google Scholar]
- Morikawa, S.; Takabe, W.; Mataki, C.; Kanke, T.; Itoh, T.; Wada, Y.; Izumi, A.; Saito, Y.; Hamakubo, T.; Kodama, T. The effect of statins in mRNA levels of genes related to inflammation, coagulation, and vascular constriction in human umbilical vein endothelial cells. Atheroscler. Thromb. 2002, 9, 178–183. [Google Scholar] [CrossRef]
- Tanaka, N.; Katayama, Y.; Katsumata, T.; Otori, T.; Nishiyama, Y. Effects of long-term administration of HMG-CoA reductase inhibitor, atorvastatin, on stroke events and local cerebral blood flow in stroke-prone spontaneous hypertensive rats. Brain Res. 2007, 1169, 125–132. [Google Scholar]
- Gholami, M.R.; Abolhassani, F.; Pasbakhsh, P.; Akbari, M.; Sobhani, A.; Eshraghian, M.R.; Kamalian, N.; Amoli, F.A.; Dehpoor, A.R.; Sohrabi, D. The effects of simvastatin on ischemic-reperfusion injury of sciatic nerve in adult rats. Eur. J. Pharmacol. 2008, 590, 111–114. [Google Scholar]
- Urban, P.; Pavlikova, M.; Sivonova, M.; Kaplan, P.; Tatarkova, Z.; Kaminska, B.; Lehotsky, J. Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: Effect of neuroprotectant simvastatin. Cell Mol. Neurobiol. 2009, 29, 181–192. [Google Scholar]
- Huber, R.; Riepe, M.W. Improved posthypoxic recovery in vitro on treatment with drugs used for secondary stroke prevention. Neuropharmacology 2005, 48, 558–565. [Google Scholar]
- Kumagai, R.; Oki, C.; Muramatsu, Y.; Kurosaki, R.; Kato, H.; Araki, T. Pitavastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, reduces hippocampal damage after transient cerebral ischemia in gerbils. J. Neural. Transm. 2004, 111, 1103–1120. [Google Scholar] [PubMed]
- Lim, J.H.; Lee, J.C.; Lee, Y.H.; Choi, I.Y.; Oh, Y.K.; Kim, H.S.; Park, J.S.; Kim, W.K. Simvastatin prevents oxygen and glucose deprivation/reoxygenation-induced death of cortical neurons by reducing the production and toxicity of 4-hydroxy-2E-nonenal. J. Neurochem. 2006, 97, 140–150. [Google Scholar]
- Bassuk, S.S.; Rifai, N.; Ridker, P.M. High-sensitivity C-reactive protein: Clinical importance. Curr. Probl. Cardiol. 2004, 29, 439–493. [Google Scholar]
- Mora, S.; Ridker, P.M. Justification for the use of statins in primary prevention: An intervention trial evaluating rosuvastatin (JUPITER)- Can C-reactive protein be used to target statin therapy in primary prevention? Am. J. Cardiol. 2006, 97, 33A–41A. [Google Scholar]
- Jialal, I.; Stein, D.; Balis, D.; Grundy, S.M.; Adams-Huet, B.; Devaraj, S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation 2001, 103, 1933–1935. [Google Scholar]
- Plenge, J.K.; Hernandez, T.L.; Weil, K.M.; Poirer, P.; Grunwald, G.K.; Marcovina, S.M.; Eckel, R.H. Simvastatin lowers C-reactive protein within 14 days: An effect independent of low-density lipoprotein cholesterol reduction. Circulation 2002, 106, 1447–1452. [Google Scholar]
- Albert, M.A.; Danielson, E.; Rifai, N.; Ridker, P.M. PRINCE Investigators. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA 2001, 286, 64–70. [Google Scholar] [PubMed]
- van Wissen, S.; Trip, M.D.; Smilde, T.J.; de Graaf, J.; Stalenhoef, A.F.; Kastelein, J.J. Differential hs-CRP reduction in patients with familial hypercholesterolemia treated with aggressive or conventional statin therapy. Atherosclerosis 2002, 165, 361–366. [Google Scholar]
- Bonnet, J.; McPherson, R.; Tedgui, A.; Simoneau, D.; Nozza, A.; Martineau, P.; Davignon, J. CAP investigators. Comparative effects of 10-mg versus 80-mg Atorvastatin on high-sensitivity C-reactive protein in patients with stable coronary artery disease: results of the CAP (Comparative Atorvastatin Pleiotropic effect) study. Clin. Ther. 2008, 30, 2298–2313. [Google Scholar] [PubMed]
- Keidar, S.; Aviram, M.; Maor, I.; Oiknine, J.; Brook, G. Pravastatin inhibits cellular cholesterol synthesis and increases low density lipoprotein receptor activity in marcophages: In vitro and in vivo studies. Br. J. Clin. Pharmac. 1994, 38, 513–519. [Google Scholar]
- Weber, C.; Erl, W.; Weber, K.S.; Weber, P.C. HMG-CoA reductase inhibitors decrease CD11b expression and CD11b-dependent adhesion of monocytes to endothelium and reduce increased adhesiveness of monocytes isolated from patients with hypercholesterolemia. J. Am. Coll. Cardiol. 1997, 30, 1212–1217. [Google Scholar]
- Chopp, M.; Zhang, R.L.; Chen, H.; Li, Y.; Jiang, N.; Rusche, J.R. Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats. Stroke 1994, 25, 869–876. [Google Scholar]
- Kimura, M.; Kurose, I.; Russell, J.; Granger, D.N. Effects of fluvastatin on leukocyte-endothelial cell adhesion in hypercholesterolemic rats. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1521–1526. [Google Scholar]
- Lehr, H.A.; Seemuller, J.; Hubner, C.; Menger, M.D.; Messmer, K. Oxidized LDL-induced leukocyte/endothelial interaction in vivo involves the receptor for platelet activating factor. Arterioscler. Thromb. 1993, 13, 1013–1018. [Google Scholar]
- Pantoni, L.; Sarti, C.; Inzitari, D. Cytokines and cell adhesion molecules in cerebral ischemia: Experimental bases and therapeutic perspectives. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 503–513. [Google Scholar]
- Jung, K.H.; Chu, K.; Jeong, S.W.; Han, S.Y.; Lee, S.T.; Kim, J.Y.; Kim, M.; Roh, J.K. HMG-CoA reductase inhibitor, Atorvastatin, promotes sensorimotor recovery, suppressing acute inflammatory reaction after experimental intracerebral hemorrhage. Stroke 2004, 35, 1744–1749. [Google Scholar] [PubMed]
- Clarke, R.M.; Lyons, A.; O’Connell, F.; Deighan, B.F.; Barry, C.E.; Anyakoha, N.G.; Nicolaou, A.; Lynch, M.A. A pivotal role for interleukin-4 in Atorvastatin-associated neuroprotection in rat brain. J. Biol. Chem. 2008, 283, 1808–1817. [Google Scholar]
- Balduini, W.; Mazzoni, E.; Carloni, S.; de Simoni, M.G.; Perego, C.; Sironi, L.; Cimino, M. Prophylactic but not delayed administration of simvastatin protects against long-lasting cognitive and morphological consequences of neonatal hypoxic-ischemic brain injury, reduces interleukin-1β and tumor necrosis factor-α mRNA induction, and does not affect endothelial nitric oxide synthase expression. Stroke 2003, 34, 2007–2012. [Google Scholar] [PubMed]
- Chan, P.H. Role of oxidants in ischemic brain damage. Stroke 1996, 27, 1124–1129. [Google Scholar]
- Vaughan, C.; Delanty, N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke 1999, 30, 1969–1973. [Google Scholar]
- Hussein, O.; Schlezinger, S.A.; Rosenblat, M.; Keidar, S.; Aviram, M. Reduced susceptibility of low-density lipoprotein (LDL) to lipid peroxidation after fluvastatin therapy is associated with the hypocholesterolemic effects of the drug and its binding to LDL. Atherosclerosis 1997, 128, 11–18. [Google Scholar]
- Chen, L.; Haught, W.H.; Yang, B.; Saldeen, T.G.; Parthasarathy, S.; Mehta, J.L. Preservation of endogenous antioxidant activity and inhibition of lipid peroxidation as common mechanisms of antiatherosclerotic effects of vitamin E, lovastatin and amlodipine. J. Am. Coll. Cardiol. 1997, 30, 569–575. [Google Scholar]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis 1998, 138, 271–280. [Google Scholar] [PubMed]
- Humans, J.A.; Ubbink, J.B.; Jerling, J.J.; Delport, R.; Vermaak, W.J.; Vorster, H.H.; Lagendijk, J.; Potgieter, H.C. The effect of simvastatin on the plasma antioxidant concentrations in patients with hypercholesterolemia. Clin. Chem. Acta 1997, 263, 67–77. [Google Scholar]
- Giroux, L.M.; Davignon, J.; Naruszewicz, M. Simvastatin inhibits the oxidation of low-density lipoproteins by activated human monocyte-derived marcophages. Biochem. Biophys. Acta 1993, 1165, 335–338. [Google Scholar]
- Wassmann, S.; Laufs, U.; Baumer, A.T.; Muller, K.; Ahlbory, K.; Linz, W.; Rosen, R.; Bohm, M.; Nickenig, G. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001, 37, 1450–1457. [Google Scholar]
- Westermaier, T.; Stetter, C.; Vince, G.H.; Pham, M.; Tejon, J.P.; Eriskat, J.; Kunze, E.; Matthies, C.; Ernestus, R.I.; Solymosi, L.; Roosen, K. Prophylactic intravenous magnesium sulfate for treatment of aneurysmal subarachnoid hemorrhage: A randomized placebo-controlled, clinical study. Crit. Care. Med. 2010, 38, 1284–1290. [Google Scholar]
- Huhle, G.; Abletshauser, C.; Mayer, N.; Weidinger, G.; Harenberg, J.; Heene, D.L. Reduction of platelet activity markers in type II hypercholesterolemic patients by a HMG-CoA-reductase inhibitor. Thromb. Res. 1999, 95, 229–234. [Google Scholar]
- Hale, L.P.; Craver, K.T.; Berrier, A.M.; Sheffield, M.V.; Case, L.D.; Owen, J. Combination of fosinopirl and pravastatin decreases platelet response to thrombin receptor agonist in monkeys. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1643–1646. [Google Scholar]
- Laufs, U.; Gertz, K.; Huang, P.; Nickenig, G.; Bohm, M.; Dirnagl, U.; Endres, M. Atorvastatin upregulates type III nitric oxide synthease in thrombocytes, decreases, platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke 2000, 31, 2442–2449. [Google Scholar]
- Alfon, J.; Royo, T.; Garcia-Moll, X.; Badimon, L. Platelet deposition on eroded vessel walls at a stenotic shear rate is inhibited by lipid-lowering treatment with atorvastatin. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1812–1817. [Google Scholar]
- Zacco, A.; Togo, J.; Spence, K.; Ellis, A.; Lloyd, D.; Furlong, S.; Piser, T. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J. Neurosci. 2003, 23, 11104–11111. [Google Scholar]
- Bosel, J.; Gandor, F.; Harms, C.; Synowitz, U.; Djoufack, P.C.; Megow, D.; Dirnagi, U.; Hortnagl, H.; Fink, K.B.; Endres, M. Neuroprotective effects of atorvastatin against glutamate-induced excitotoxicity in primary cortical neurons. J. Neurochem. 2005, 92, 1386–1398. [Google Scholar]
- Wong, G.K.; Wong, R.; Mok, V.C.; Fan, D.S.; Leung, G.; Wong, A.; Chan, A.S.; Zhu, C.X.; Poon, W.S. Clinical study on cognitive dysfunction after spontaneous subarachnoid haemorrhage: Patient profiles and relationship to cholinergic dysfunction. Acta Neurochir. (Wien) 2009, 151, 1601–1607. [Google Scholar]
- Cramer, C.; Haan, M.N.; Galea, S.; Langa, K.M.; Kalbfleisch, J.D. Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology 2008, 71, 344–350. [Google Scholar]
- Lu, D.; Qu, C.; Goussev, A.; Jiang, H.; Lu, C.; Schallert, T.; Mahmood, A.; Chen, J.; Chopp, M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J. Neurotrauma. 2007, 24, 1132–1146. [Google Scholar]
- Gao, C.; Liu, W.; Sun, Z.D.; Zhao, S.G.; Liu, X.Z. Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits capase-dependent apoptosis pathway. BMC Neurosci. 2009, 10. [Google Scholar]
- Maples, K.R.; Green, A.R.; Floyd, R.A. Nitrone-related therapeutics: Potential of NXY-059 for the treatment of acute ischemic stroke. CNS Drugs. 2004, 18, 1071–1084. [Google Scholar]
- Marshall, J.W.; Cummings, R.M.; Bowes, L.J.; Ridley, R.M.; Green, A.R. Functional and histological evidence for the protective effect of NXY-059 in a primate model of stroke when given 4 hours after occlusion. Stroke 2003, 34, 2228–2233. [Google Scholar]
- Lees, K.R.; Zivin, J.A.; Ashwood, T.; Davalos, A.; Davis, S.M.; Diener, H.C.; Grotta, J.; Lyden, P.; Shuaib, A.; Hardemark, H.G.; Wasiewski, W.W. Stroke-Acute Iscehmic NXY Treatment (SAINT I) Trial Investigators. NXY-059 for acute ischemic stroke. N. Engl. J. Med. 2006, 354, 588–600. [Google Scholar] [PubMed]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. SAINT II Trial Investigators. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [PubMed]
- Diener, H.C.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Shuaib, A.; Ashwood, T.; Wasiewski, W.; Alderfer, V.; Hardemark, H.G.; Rodichok, L. SAINT I and II Investigators. NXY-059 for the treatment of acute stroke. Pooled analysis of the SAINT I and II trials. Stroke 2008, 39, 1751–1758. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wong, G.K.C.; Poon, W.S. The Biochemical Basis of Hydroxymethylglutaryl-CoA Reductase Inhibitors as Neuroprotective Agents in Aneurysmal Subarachnoid Hemorrhage. Pharmaceuticals 2010, 3, 3186-3199. https://doi.org/10.3390/ph3103186
Wong GKC, Poon WS. The Biochemical Basis of Hydroxymethylglutaryl-CoA Reductase Inhibitors as Neuroprotective Agents in Aneurysmal Subarachnoid Hemorrhage. Pharmaceuticals. 2010; 3(10):3186-3199. https://doi.org/10.3390/ph3103186
Chicago/Turabian StyleWong, George Kwok Chu, and Wai Sang Poon. 2010. "The Biochemical Basis of Hydroxymethylglutaryl-CoA Reductase Inhibitors as Neuroprotective Agents in Aneurysmal Subarachnoid Hemorrhage" Pharmaceuticals 3, no. 10: 3186-3199. https://doi.org/10.3390/ph3103186