Beta-Adrenergic Agonists

1
Preventive and Occupational Medicine Unit, Respiratory Pathophysiology Laboratory, University Hospital San Martino, Largo R. Benzi, 10 - 16132 Genoa, Italy
2
Respiratory Pathophysiology Unit, Department of Internal Medicine, University of Genoa, Viale Benedetto XV - 16132 Genoa, Italy
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2010, 3(4), 1016-1044; https://doi.org/10.3390/ph3041016
Submission received: 2 February 2010
/
Revised: 15 March 2010
/
Accepted: 26 March 2010
/
Published: 30 March 2010
(This article belongs to the Special Issue Antiasthmatic Drugs)
{kind=link}
{kind=link}
Abstract
:Inhaled β2-adrenoceptor (β2-AR) agonists are considered essential bronchodilator drugs in the treatment of bronchial asthma, both as symptoms-relievers and, in combination with inhaled corticosteroids, as disease-controllers. In this article, we first review the basic mechanisms by which the β2-adrenergic system contributes to the control of airway smooth muscle tone. Then, we go on describing the structural characteristics of β2-AR and the molecular basis of G-protein-coupled receptor signaling and mechanisms of its desensitization/ dysfunction. In particular, phosphorylation mediated by protein kinase A and β-adrenergic receptor kinase are examined in detail. Finally, we discuss the pivotal role of inhaled β2-AR agonists in the treatment of asthma and the concerns about their safety that have been recently raised.
1. Introduction
In the past, β-adrenoceptor (β-AR) agonists have been utilized in many clinical settings. Nowadays, they are considered as first-line medications in the treatment of airway narrowing, the hallmark feature of bronchial asthma and chronic obstructive pulmonary disease (COPD). Epinephrine was the first bronchodilator used at the beginning of the past century. Ephedrine was introduced into Western medicine in 1924, although it had been used in China for thousands of years [1]. The next major advance in this field was the development in the 1940s of isoproterenol [2], a β-AR selective agonist void of α-adrenergic activity. More recently, the development of selective β2-AR agonists has made available drugs with reduced cardiovascular effects [3]. Therefore, β2-AR agonists represent today a mainstay of the management of COPD [4] and asthma. In the latter condition, they are used both as symptoms-relievers and, in combination with inhaled corticosteroids, as disease-controllers [5,6].
The aim of this article is to review the molecular mechanisms by which β2-AR contribute to control the tone of airway smooth muscle (ASM) and their implications for practical use of β2-AR agonists in the treatment of asthma.
2. Adrenergic Control of ASM Tone
In humans, unlike in other species, there is no evidence that adrenergic nerves directly supply the ASM [7], the only inhibitory nervous system being the non-adrenergic, non-cholinergic one [8,9,10]. The sympathetic system affects ASM tone via circulating cathecolamines [7,8,9] acting on the β2-AR present on ASM cell membrane and on parasympathetic nerve endings [11,12].
2.1. Circulating Catecholamines
Epinephrine, nor-epinephrine, and dopamine are natural circulating compounds, but only the first has significant physiological effects. Following secretion from adrenal medulla and at plasma concentrations ranging from 0.2 to 0.4 nM/L, epinephrine provides a low-level stimulation of specific receptors [13], while spillover of nor-epinephrine from adjacent structures may also exert some minor effects. The action of epinephrine is rapidly (within about 2 min) terminated in various tissues as a consequence of oxidative deamination and methylation processes catalyzed by monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT), respectively [14].
In asthmatic but not healthy subjects, β-AR antagonists (β-blockers) often elicit bronchoconstriction, which may be interpreted as suggesting a protective effect of β-AR stimulation against excessive airway narrowing. However, no evidence of plasmatic epinephrine was provided during acute exacerbations of asthma or airway narrowing induced by a variety of challenges, including intravenously administered propranolol. Therefore, it does not seem that severe bronchoconstriction and activation of ASM elicit a protective epinephrine release [15].
2.2. Neural Interactions
Complex interactions between various components of the autonomic nervous system have long been recognized in various species. This means that changes in the function of one neural pathway may affects other pathways. In humans, ASM tone is mainly maintained by acetylcholine (ACh) released from the parasympathetic nervous system [16]. An early study, based on isometric measurement of tension in bronchial rings, suggested that stimulation of β2-AR could inhibit cholinergic neurotransmission [17]. As tyramine-induced release of nor-epinephrine from sympathetic nerves had no effect, this modulation was probably due to circulating epinephrine [17]. Therefore, apart from the relaxing effect on ASM, β2-AR may have a modulatory effect on cholinergic neurotransmission [17,18] (see below). It has been proposed that an impairment of modulation of ACh release in asthmatic airways may contribute to bronchoconstriction induced by β-blockers [19].
3. Adrenergic Receptors
Ahlquist [2] proposed in 1948 that adrenergic receptors, stimulating a variety of physiological responses in various organs, could be classified into two primary types, α and β. Stimulation of the former causes ASM contraction with the following rank of potency: epinephrine > nor-epinephrine > isoproterenol. By contrast, stimulation of β-AR, by the same agonists, relaxes ASM with a different rank of potency, i.e., isoproterenol > epinephrine > nor-epinephrine.
As a result of studies conducted by Lands et al. [20], β-AR have been further classified into β1- and β2-subtypes. The former show an almost equal affinity for epinephrine and nor-epinephrine, the latter are considered to be more sensitive to epinephrine than nor-epinephrine. Autoradiographic mapping has demonstrated that β-AR are widely distributed in the lung and are present in several cell types, including ASM from trachea down to the terminal bronchioles [21]. The presence of β1-AR in different species depends on the density of ASM adrenergic supply and the level of bronchial tree [22]. Consistent with the absence of sympathetic innervation to ASM in humans is the autoradiographic evidence of β2-AR only in ASM at any airway generation [21]. The amount of β2-messenger ribonucleic acid (mRNA) in ASM is high relative to the low receptor density, which may indicate a rapid turnover of β2-AR and may account for the relative resistance to the development of tolerance [23].
Apart from ASM relaxation, other effects of β2-AR activation have been reported, including increase in ciliary beat-frequency, changes in vascular permeability, decrease in ACh release, and modulation of immune cells function [21,23]. Whether these responses may contribute to the therapeutic efficacy of β2-AR agonists in asthma treatment remains unclear. More recently, a third subtype (namely, β3-AR) has been demonstrated in isolated canine [24] but not in humans [25] ASM. Its main action seems to be the enhancement of lipolysis in adipose tissue and thermogenesis (“brown” fat) in skeletal muscle [26].
Functional studies have shown that ASM relaxation, at the level of both central and peripheral human airways, is mediated solely by β2-AR [27,28]. In asthmatics, selective stimulation of β1-AR by prenalterol has no bronchodilator action [29]. Most importantly, β2-AR agonists act as functional antagonists and inhibit or reverse contractile responses, irrespective of constrictor stimuli [30,31]. This is a property that is of particular interest in asthma, where several physical or chemical spasmogens are likely to be involved. However, in COPD anticholinergics may produce equivalent or even greater bronchodilation than β2-AR agonists because vagal tone is the major reversible element in such patients [32].
4. G-Protein-Coupled Receptor Signaling
4.1. ASM Relaxation
The β2-AR belongs to the ubiquitously expressed 7-transmembrane receptors superfamily, which classically signals through heterotrimeric G-proteins [33,34]. They are commonly referred to as G-protein-coupled receptors because accomplish signal transduction to the interior of the cell via interactions with guanine nucleotide regulatory binding proteins [35]. The receptor-coupled G-proteins function as “molecular switches” alternating from an inactive guanosine-diphosphate to an active guanosine-triphosphate (GTP) state, which proceeds to regulate downstream cell processes [35]. Signaling via many hormones and neurotransmitters, as well as photons and odors, follows the same basic scheme, i.e., by binding of an extracellular ligand to the receptor, which then interacts with the membrane-bound G-protein. This complex, often referred to as the ternary complex, then acts through the activated G-protein to modulate an effector, such as adenylyl cyclase, phospholipase C, or ion channels [35]. For more details on this argument, we refer the reader to the review by Dohlman [36].
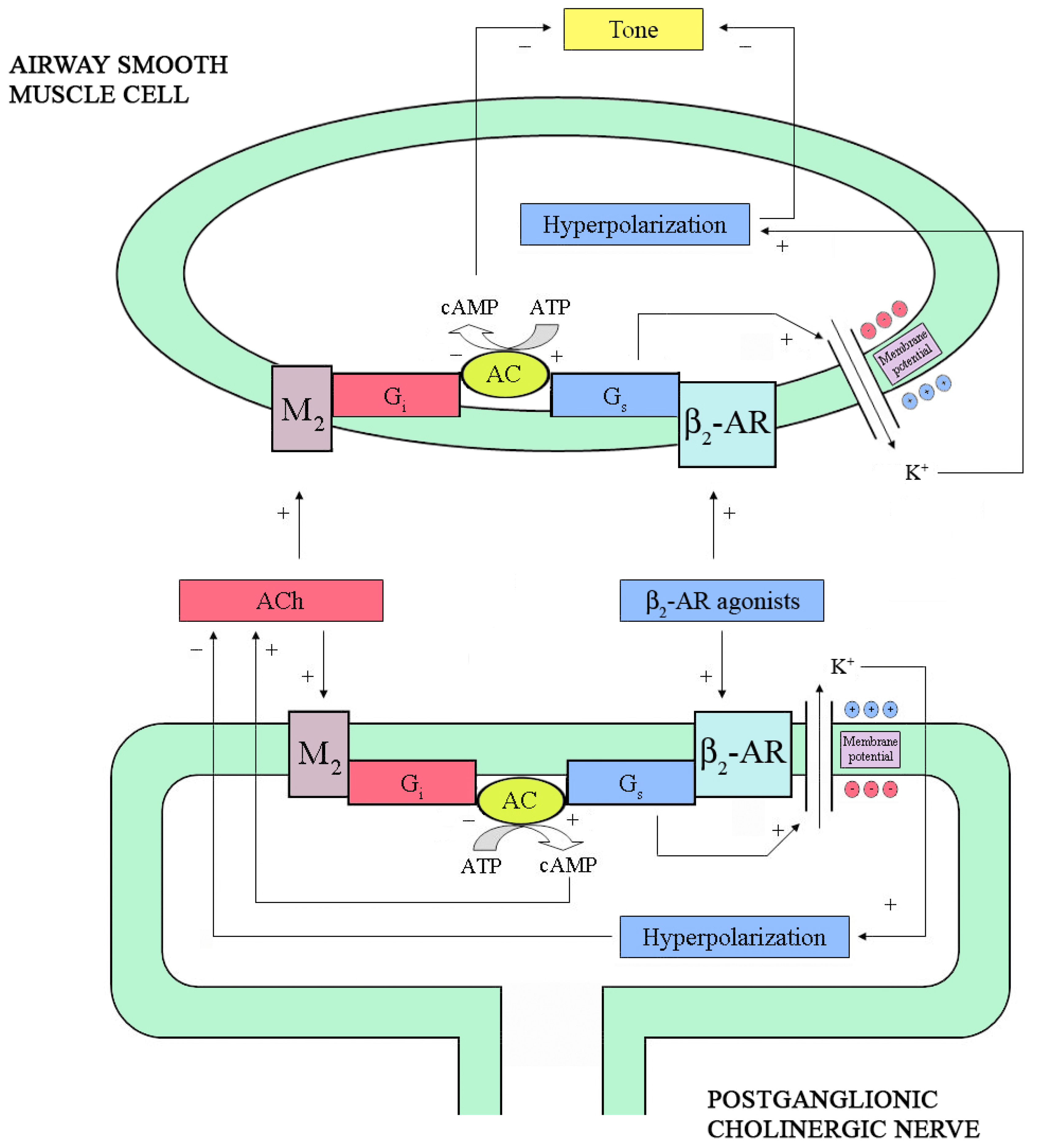
Activated β2-AR promotes the binding of a heterotrimeric stimulatory G-protein termed Gs (consisting of α, β, and γ subunits) to the receptors [37]. Upon binding of GTP, the Gs-protein subunits disassociate into Gα and Gβγ. Traditionally, Gα was considered to be the subunit that stimulates adenylyl cyclase (AC), but Gβγ can also activate certain pathways [37]. The resulting increase in intracellular cyclic 3',5'-adenosine monophosphate (cAMP) concentrations activates protein kinase (PKA), which phosphorylates several proteins causing relaxation [38]. The intrinsic GTPase activity of Gα subsequently terminates the process, with re-association of heterotrimeric protein [37]. In bronchial smooth muscle, PKA inhibits both myosin-light-chain kinase [39] and phosphoinositide hydrolysis [40]. Moreover, it promotes Ca2+/Na+ exchange [41], thus resulting in a decrease of intracellular [Ca2+], and stimulates Na+/K+ ATPase [42]. However, these effects are only observed at relatively high concentrations of β2-AR agonists, when maximal relaxation responses have been exceeded. In the 1990’s, Jones et al. [43,44] showed that two potent inhibitors of the opening of the large-conductance Ca2+-activated K+ (BKCa) channels, i.e., charybdotoxin and iberiotoxin, may inhibit the bronchodilator responses to β2-AR agonists and to other agents that can increase cAMP. These effects have been subsequently confirmed by other studies [45,46] and are observed at low concentrations of β2-AR agonists in isolated human airways [47]. This suggest that β2-AR may activate BKCa channels in ASM directly via the α-subunit of Gs [48] (Figure 1).
Studies, based on direct measurement of ACh release from guinea pig [49,50] and horse [51] trachealis, have shown that stimulation of β2-AR enhances cholinergic neurotransmission in the absence, but not in the presence, of epithelium [52]. Consistent with these results, direct activation of the β-AR-coupled Gs subunit by cholera toxin, which increases the activity of AC [33], caused an increase of ACh release in epithelium-denuded guinea pig trachealis [49].
Figure 1.
Pre- and post-junctional intracellular mechanisms modulating cholinergic neurotransmission and airway smooth muscle (ASM) cell tone. At pre-junctional level, stimulation of β2-adrenoceptor (β2-AR) by agonists opens Ca2+-activated K+ (BKCa) channels leading to cell membrane hyperpolarization and reduction of acetylcholine (ACh) release. By contrast, direct activation of adenylyl cyclase (AC) enhances ACh release. In the ASM cell, stimulation of β2-AR as well as direct stimulation of AC, opens BKCa channels determining cell membrane hyperpolarization and relaxation. The ACh released by postganglionic cholinergic nerves binds M2-muscarinic receptors expressed both at pre- and post-junctional level, thus inhibiting ACh release and increasing ASM cell tone. cAMP: cyclic 3',5'-adenosine monophosphate; ATP: adenosine trisphosphate; Gs and Gi: stimulatory and inhibitory subunits of the receptor-coupled G-protein, respectively.
Figure 1.
Pre- and post-junctional intracellular mechanisms modulating cholinergic neurotransmission and airway smooth muscle (ASM) cell tone. At pre-junctional level, stimulation of β2-adrenoceptor (β2-AR) by agonists opens Ca2+-activated K+ (BKCa) channels leading to cell membrane hyperpolarization and reduction of acetylcholine (ACh) release. By contrast, direct activation of adenylyl cyclase (AC) enhances ACh release. In the ASM cell, stimulation of β2-AR as well as direct stimulation of AC, opens BKCa channels determining cell membrane hyperpolarization and relaxation. The ACh released by postganglionic cholinergic nerves binds M2-muscarinic receptors expressed both at pre- and post-junctional level, thus inhibiting ACh release and increasing ASM cell tone. cAMP: cyclic 3',5'-adenosine monophosphate; ATP: adenosine trisphosphate; Gs and Gi: stimulatory and inhibitory subunits of the receptor-coupled G-protein, respectively.

Moreover, direct stimulation of AC by forskolin [53] or incubation with a cAMP analog, increased ACh release [49]. In ASM cells, however, stimulation of Gs protein directly opens BKCa channels [48], which has been found to decrease ACh release in guinea pig trachealis [54]. By simultaneously measuring electrically induced ACh release and force development, Brichetto et al. [55] from our laboratory have recently shown that β2-AR agonists attenuates cholinergic neurotransmission in bovine trachealis by a mechanism involving BKCa channels. Therefore, it can be hypothesized that β2-AR agonists have the potential to increase or decrease ACh release depending on whether they mainly act through AC or BKCa channels.
Previous studies have shown that β2-AR can activate PKA-independent mechanisms to elicit functional responses [45,46]. Freund-Michel et al. [56], have recently found that the anti-tussive properties of terbutaline were abolished by an inhibitor of protein kinase G (PKG) present in vagal sensory fibres but were unaffected by a PKA inhibitor. As the level of cyclic 3’,5’-guanosine monophosphate was not elevated in the vagus nerve, these data suggest that a cAMP-dependent cross-activation of PKG and, subsequently, the opening of the BKCa channels may account for the anti-tussive properties of β2-AR agonists. If bronchodilator and anti-tussive activity of β2-AR agonists is mediated by different signaling pathways, then some discrepancies, reported in the clinical literature [57,58,59,60] regarding their anti-tussive and bronchodilator efficacies, could be explained.
4.2. Anti-Inflammatory Effects
It has been proposed that actions other than ASM relaxation may play a role in the protective effect of long-acting β2-AR agonists against allergen-induced late asthmatic reactions [61]. In diseases such as asthma, ASM cells play a synthetic role by secreting inflammatory mediators [62]. In other words, ASM may produce multiple inflammatory mediators (prostanoids, cyto- and chemokines), thus being able to perpetuate and amplify the inflammatory process within the airway wall. Evidence is rapidly accumulating that the asthmatic ASM is abnormal, in that it proliferates faster [63,64], produces more chemokines and cytokines as well as a different profile of extracellular matrix proteins than in its non-asthmatic counterpart [62]. These abnormalities may stem from altered calcium homeostasis leading to increased mitochondrial biogenesis and/or decreased levels of key transcription factors, such as CCAAT enhancer binding protein-α [65]. Recent data indicate that both short- and long-acting β2-AR agonists are able to reduce the expression of surface intercellular adhesion molecule-1 and the release of granulocyte-macrophage colony-stimulating factor evoked by interleukin-1β in ASM [66]. Interestingly, the repression of these inflammatory indexes was prevented by adenoviral over-expression of PKI-α, a highly selective PKA inhibitor. These data indicate a PKA-dependent mechanism of repression and suggest that agents that elevate intracellular cAMP, and thereby activate PKA, may have an anti-inflammatory effect in ASM cells [66].
5. β2-AR Dysfunction
When a cell or tissue is first exposed to a β2-AR agonist, there is a brisk initial response in the production of cAMP, followed by a decline to nearly basal level despite the continued presence of the drug. The cell becomes “desensitized” or “tolerant” to further stimulation. Desensitization depends on both the concentration of β2-AR agonist and time of exposure. In any cell type, the process of desensitization occurs through an acute and a chronic phase [67].
Over the years, there has been some confusion regarding the terminology used to describe receptors dysfunction. More precisely, desensitization is defined as a warning of a functional response despite the continuous presence of a stimulus of constant intensity [37]. Typically, a decrease in receptor function over time during exposure of cells or tissues to agonist is one form of desensitization. For β2-AR, this would entail measurements of cAMP or AC activities. In intact animal models and in clinical setting, agonist-promoted desensitization is referred to as “tachyphylaxis” or “refractoriness” [37].
There is both in vivo and in vitro evidence that β2-AR are less responsive in asthmatic than healthy individuals [68,69]. It is not completely clear, however, whether this decreased responsiveness is the cause of asthma [70] or a byproduct of therapy with β2-AR agonists [68]. Trian et al. [71] have recently shown that epithelial infection with rhinovirus reduces β2-AR function in ASM. This may explain the clinical observation of β2-AR dysfunction during virus-induced asthma exacerbations. Some authors reported that the inhibitory effects of interleukin-1β on the isoproterenol-induced bronchodilation are abolished by COX-2 inhibitors [72]. Thus, it appears more likely that β2-AR hyporesponsiveness is a consequence of the disease process rather than its cause.
5.1. Desensitization by Phosphorylation
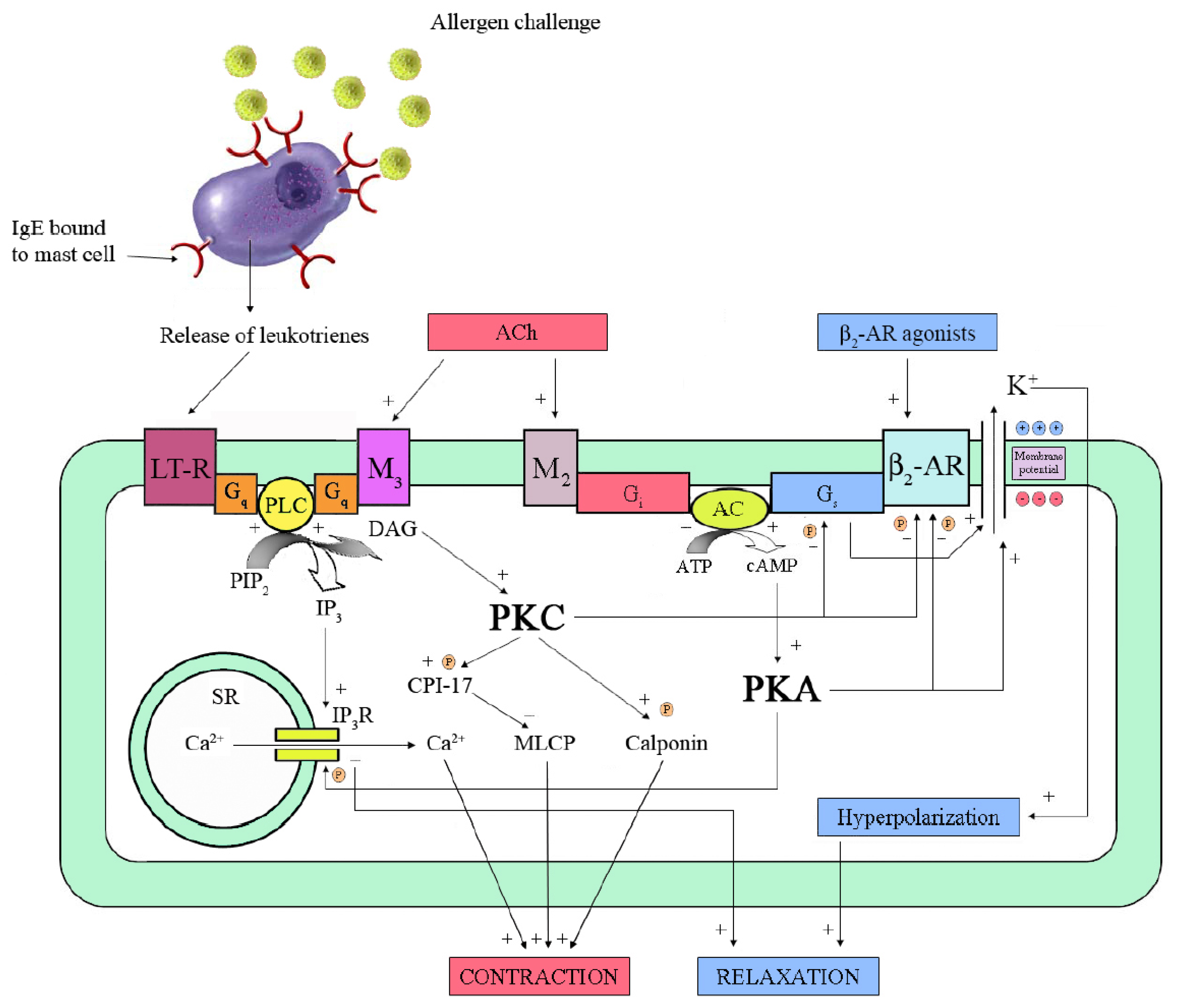
The covalent modification of β-AR represents the earliest event in the desensitization process [37]. Over a time frame of less than 30 min of exposure to an agonist in vitro, phosphorylation of β-AR takes place by PKA and β-AR kinase (βARK). The latter belongs to G-protein-coupled receptor kinases family (GRK) [73]. Both kinases cause a complete or partial loss of functional coupling between receptor and Gs subunit by phosphorylating specific sites [67]. The uncoupling between receptor and Gs subunit may be a mechanism of allergen-induced β2-AR dysfunction observed in human isolated passively sensitized bronchi [73] (Figure 2).
PKA is a cAMP-dependent enzyme which is activated by low concentrations of β2-AR agonist, such as circulating catecholamines or drugs. Any process leading to an increase in intracellular concentrations of cAMP sufficient to activate PKA will result in phosphorylation of β2-AR, even if unoccupied by agonist [37]. This underlies one of the principal mechanisms of heterologous desensitization of the β2-AR, so termed because dysfunction can be evoked by processes distinct from activation of the receptor as, for instance, those elicited by inflammatory mediators [67]. Data from our laboratory showed, in passively sensitized isolated human bronchial rings, that allergen challenge caused β2-AR dysfunction without involving cytokines [75] or mechanisms distal to cAMP formation [76]. Moreover, this dysfunction was prevented by a LTD4-receptor antagonist or a cell membrane stabilizer, but not by cetirizine or indomethacin, suggesting a central role for leukotrienes released from resident inflammatory cells [77]. The involvement of PKC in this process was subsequently shown on both cultured ASM cell and bronchial rings [78].
Figure 2.
Mechanisms of relaxation and β2-adrenoceptor (β2-AR) desensitization in airway smooth muscle (ASM). The binding of a specific agonist to β2-AR stimulates the receptor-coupled Gs-protein, which activates adenylyl cyclase (AC). The resulting increase of cyclic 3',5'-adenosine monophosphate (cAMP) activates protein kinase A (PKA), which phosphorylates inositol 1,4,5-trisphosphate receptor (IP3R) of sarcoplasmic reticulum (SR) and opens Ca2+-activated K+ (BKCa) channels, thus leading to relaxation. However, activated PKA phosphorylates β2-AR, uncoupling it from Gs-protein. Exposure of sensitized mast-cells to allergen causes release of leukotrienes (LT). Their interaction with the specific receptor, i.e., LT-R, activates a Gq-protein, which increases phospholipase C (PLC) activity. PLC catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), which produces IP3 and diacylglycerol (DAG). IP3 leads to contraction by increasing release of Ca2+ from SR while DAG activates protein kinase C (PKC). The latter phosphorylates several substrates like calponin and CPI-17, which is an inhibitor of myosin-light-chain phosphatase (MLCP). In addition, PKC phosphorylates both β2-AR and Gs-protein. A similar cascade of events seems to occur in response to acetylcholine (ACh) released by cholinergic nerves through M3-muscarinic receptors. In addition, activation of M2-muscarinic receptors inhibits AC, thus decreasing cAMP level and PKA activity. ATP: adenosine trisphosphate; ![Pharmaceuticals 03 01016 i001]() : phosphorylation sites.
: phosphorylation sites.
 : phosphorylation sites.
: phosphorylation sites.
Figure 2.
Mechanisms of relaxation and β2-adrenoceptor (β2-AR) desensitization in airway smooth muscle (ASM). The binding of a specific agonist to β2-AR stimulates the receptor-coupled Gs-protein, which activates adenylyl cyclase (AC). The resulting increase of cyclic 3',5'-adenosine monophosphate (cAMP) activates protein kinase A (PKA), which phosphorylates inositol 1,4,5-trisphosphate receptor (IP3R) of sarcoplasmic reticulum (SR) and opens Ca2+-activated K+ (BKCa) channels, thus leading to relaxation. However, activated PKA phosphorylates β2-AR, uncoupling it from Gs-protein. Exposure of sensitized mast-cells to allergen causes release of leukotrienes (LT). Their interaction with the specific receptor, i.e., LT-R, activates a Gq-protein, which increases phospholipase C (PLC) activity. PLC catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), which produces IP3 and diacylglycerol (DAG). IP3 leads to contraction by increasing release of Ca2+ from SR while DAG activates protein kinase C (PKC). The latter phosphorylates several substrates like calponin and CPI-17, which is an inhibitor of myosin-light-chain phosphatase (MLCP). In addition, PKC phosphorylates both β2-AR and Gs-protein. A similar cascade of events seems to occur in response to acetylcholine (ACh) released by cholinergic nerves through M3-muscarinic receptors. In addition, activation of M2-muscarinic receptors inhibits AC, thus decreasing cAMP level and PKA activity. ATP: adenosine trisphosphate; ![Pharmaceuticals 03 01016 i001]() : phosphorylation sites.
: phosphorylation sites.
: phosphorylation sites.
If cysteinyl-leukotrienes, released from resident or circulating inflammatory cells or even from the ASM itself, are responsible for β2-AR desensitization in asthma, then the concurrent administration of cysteinyl-LT1-R antagonists might represent a useful strategy to improve the response to β2-AR agonists in this disease. On the other hand, homologous desensitization is mediated by βARK. It describes processes that are specific to an attenuation of function of activated, i.e., occupied, receptor [79]. βARK requires high (μM/mL) concentrations of the agonist [73]. Other data suggest that a cofactor is required for βARK function. A protein, inhibiting the signaling function of βARK-phosphorylated receptors by more than 75%, was found [80] and termed β-arrestin [81]. It is now recognized that β-arrestins act as versatile adapters controlling receptor signaling, desensitization, and trafficking. Most endogenous receptors appear to signal in a balanced fashion using both β-arrestin and G-protein-mediated pathways. Some 7-transmembrane receptors are thought to be non-signaling "decoys" because of their inability to activate typical G-protein signaling pathways. It has been proposed that these receptors act to scavenge ligands or function as co-receptors [82].
Nino et al. [83] have recently examined the regulation and role of phosphodiesterase-4 (PDE4) activity in mediating the pro-asthmatic changes in ASM responsiveness that accompany its prolonged homologous desensitization. Their findings provide new evidence that prolonged exposure of ASM to β2-AR agonists results in an upregulated expression of PDE4D5 isoform due to PKA-dependent activation of Gi protein signaling. This is consistent with a potential switch in coupling of the β2-AR from Gs to Gi resulting in Gβγ-subunit-mediated activation of ERK1/2 which, in turn, leads to a phosphorylating cascade of transcription factors.
During exposure to low levels of agonist, few β2-AR on the cell are occupied [67]. Such low (≤10 nM/mL) concentrations of agonist are present in systemic circulation during exercise-induced activation of the sympathoadrenal system and in stress, such as under haemodynamic instability or respiratory insufficiency. Similar concentrations are also obtained during systemic administration of agonist, such as terbutaline, for the treatment of asthma and various catecholamines for the treatment of hypotension and heart failure [67]. Although only a β2-AR fraction is occupied, this is sufficient to increase intracellular cAMP, resulting in activation of PKA and phosphorylation of all β2-AR on the cell. Under these conditions, only the agonist-occupied receptors are phosphorylated by βARK, which contributes minimally to the process under these circumstances. Other G-protein-coupled receptors with PKA sites may also be phosphorylated, resulting in desensitization by β2-AR agonists [37].
During exposure to high concentrations of β2-AR agonists, as occurring in bronchodilator treatment of asthmatic patients, the majority of β2-AR on the cell may be occupied [67]. Under these circumstances, PKA is activated, as with low agonist exposure, and all β2-AR are phosphorylated by this kinase. In addition, all occupied receptors are phosphorylated by βARK. Such phosphorylated receptor binds β-arrestin with subsequent partial uncoupling from Gs [37].
Phosphorylation by PKA and GRKs leads to desensitization of β2-AR signaling, and is thought to be a mechanism involved with cell and organ homeostasis and tolerance to agonists. However, there is little direct evidence that these events are relevant to β2-AR physiological function, such as ASM relaxation. Recently, Wang et al. [84] have shed some light on the matter by using a transgenic mouse model expressing the human wild-type and mutated β2-AR lacking PKA and/or GRK phosphorylation sites on ASM. They showed that, at low receptor occupancy, β2-PKA(-) had enhanced agonist-promoted relaxation, while β2-GRK(-) airways were unaffected. In contrast, at saturating agonist concentrations, the greatest relaxation enhancement was with β2-GRK(-), with no evidence for additivity when PKA sites were also removed. For the full range of responses, the β2-PKA(-)/GRK(-) airways had the greatest relaxation efficiency, indicating a graded effect of GRKs as agonist concentration increased. Thus, these two mechanisms impact a relevant β2-AR physiologic function, by acting as attenuators of the acute response, and represent specific interfaces where adjunct therapy or biased ligands may improve β2-AR agonist treatment of obstructive lung disease.
5.2. Desensitization by Sequestration
After about 30 min of agonist exposure, cell surface β2-AR undergo a process leading to internalization or sequestration of the receptor to a sub-cellular compartment. The proportion of receptors at the surface that undergo sequestration appears to be highly cell-type dependent. In cells where there is little receptor reserve and sequestration results in ~60% loss of cell surface receptor, desensitization by this mechanism would be expected, as shown in lymphocytes [37]. Recent data indicate that the β2-AR of human pro-inflammatory cells is exquisitely sensitive to desensitization, whereas β2-AR-mediated relaxation of human ASM is relatively resistant. An explanation for this discrepancy is that a large β2-AR reserve exists in ASM cells for sympathomimetic bronchodilators, which protects against desensitization [85]. After agonist removal, sequestered receptors, which have not been shuttled to a degradation pathway, can recycle to the cell surface, with a time course that is about the same as for initiation of the process. Some data suggest that the pool of sequestered β2-AR undergoes phosphatases-mediated dephosphorylation [86].
5.3. Desensitization by Down-Regulation
After 3–6 h of exposure to agonist, a net loss of receptors occurs independent of compartment [37]. This is termed down-regulation and reaches a steady state at 18–24 h. The process can result in an loss of as much as 90% of receptors and is presumed to be a major component in long-term agonist-promoted desensitization of β2-AR [37]. The mechanisms responsible are both a decrease in receptor production and an increase in receptor protein degradation. The decrease in production of new receptors in the presence of agonist is primarily due to a loss of β2-AR mRNA stability [87], although a change in the rate of transcription may also play a role [88]. Destabilization of β2-AR mRNA appears to be due to cAMP-promoted binding of a protein to a destabilization sequence (AUUUA) in the 3’ non-coding region of the gene [89]. The second mechanism of down-regulation may be related to the degradation of sequestered β2-AR pool. One study suggests that PKA-mediated phosphorylation is required for full agonist-promoted down-regulation [90]. Also, tyrosine residues in the cytoplasmic tail have been implicated as necessary for agonist-promoted down-regulation [91].
5.4. The β-AR Paradox
Long-acting β2-AR agonists have been introduced as a therapeutic option in asthma [5,6] to improve symptom control. However, there is growing concern about whether control of disease may deteriorate with the regular use of these agents. Several studies have shown a temporary increase in airway responsiveness to histamine upon withdrawal of inhaled β2-AR agonists [92,93,94]. In an 8-week study, Cheung et al. [95] showed that the bronchodilator effect of salmeterol was maintained, whereas its protective effect against methacholine (MCh)-induced bronchoconstriction was significantly attenuated. This dissociation between bronchodilator and bronchoprotective effect of long-acting β2-AR agonists was further explored in mice with ablated β-AR genes (β-AR-/-) and transgenic mice overexpressing β2-AR (β2-AR-OE) [96]. Unexpectedly, β-AR-/- mice had markedly decreased bronchoconstrictive responses to MCh and other Gq-coupled receptor agonists such as thromboxane-A2 and 5-hydroxytryptamine (5-HT). In contrast, β2-AR-OE mice had enhanced constrictive responses. Inositol phosphate accumulation by Gq-coupled M3-muscarinic, thromboxane-A2, and 5-HT2 receptors was desensitized in ASM cells from β-AR-/- mice and sensitized in cells from β2-AR-OE mice. Thus, β-AR seem to regulate antithetically constrictive signals, affecting bronchomotor tone/reactivity by additional effects other than direct bronchodilation. Studies of signaling elements in these pathways identified the nodal point of this cross-talk in phospholipase C-β1, whose expression was altered by β-AR in a direction and magnitude consistent with the physiologic and cellular responses. These results establish a possible mechanism of the β-AR paradox and identify a potential asthma modifier gene (phospholipase C-β1), which may also be a therapeutic target in asthma when chronic β-AR agonists are required [96]. An alternative explanation is that regular use of β2-AR agonists may negatively impact the balance of factors contributing to airway inflammation and remodeling in asthma [97].
Two elegant studies [98,99], in ovalbumin-driven murine model of asthma, have shown that β-AR agonists and antagonist, with inverse agonist properties, may exert reciprocating effects on cellular signaling dependent on duration of administration. In particular, acute dosing with β-blockers produced bronchoconstriction, whereas the chronic administration of the same agents did exert a protective effect against MCh-induced airway narrowing [98]. Surprisingly, the bronchoprotective effect of β-blockers was associated with reduced inflammation and mucous metaplasia, up-regulation of β2-AR, and reduced expression of various spasmogenic proteins [99]. These findings in mice led to the first proof-of-concept open-label study in ten patients with mild steroid-naïve asthma who were given incremental doses of nadolol for nine weeks [100]. In eight out of the 10 subjects tested, chronic treatment with this β-blocker produced a significant, dose-dependent, decrease in airway responsiveness to MCh. However, these findings warrant further testing in future by means of larger trials [101,102].
5.5. Reversal of Desensitization
As detailed by Davies and Lefkowitz [103], corticosteroids have been shown to increase β2-AR synthesis, reverse down-regulation, and improve coupling, the last by partially restoring the fraction of receptors in the high-affinity state. A study from our laboratory indicates that short-term (3 h) exposure to beclomethasone dipropionate ablated β2-AR dysfunction induced by allergen challenge in passively sensitized human bronchi. This effect was mediated by an increase in the activity of the α-subunit of Gs protein, suggesting an action independent of gene transcription [104].
6. Genetic Polymorphisms of the β2-AR
A series of studies revealed that the gene encoding for β2-AR is polymorphic within the human population [105,106]. Such polymorphisms may be responsible for heterogeneity in the response to β2-AR agonists and/or receptor dysfunction in asthma [107]. For example, the B16 Arg/Arg homozygosity is associated with more frequent exacerbations during chronic daily dosing of salbutamol [108] or salmeterol [109]. In contrast, it seems that there is no genotypic risk for exacerbations in patients using inhaled β2-AR agonists less than once a day [109]. In asthma patients with B16 Arg/Arg and B16 Gly/Gly genotypes, combination treatment with salmeterol and inhaled corticosteroid improved airway function when compared with inhaled corticosteroid therapy alone. These findings suggest that patients should continue to be treated with long-acting β2-AR agonists plus moderate-dose inhaled corticosteroids irrespective of B16 genotype [110].
β2-AR function undergoes desensitization during persistent agonist exposure because of receptor phosphorylation by GRKs, which was found to be highly expressed in ASM [111]. The coding region is polymorphic at codon 41, where glutamine (Gln) can be substituted by leucine (Leu) (minor allele), but almost exclusively in those of African descent. So, GRK5-Leu41 represents a gain-of-function polymorphism that evokes enhanced loss-of-function of β2-AR during persistent agonist exposure, thus possibly contributing to β2-AR agonist variability in asthma treatment of African-Americans [111].
If one considers β2-AR expression as being constantly regulated by systemic catecholamines or continuosly administered β2-AR agonists, then the glutamic (Glu)27 allele may be expressed to a higher level in the lung, as it is refractory to down-regulation. More than a decade ago, a study by Hall et al. [112] addressed this point and found that asthmatics with the Glu27 polymorphism had approximately fourfold lower airway responsiveness to MCh as compared to those with Gln27 (wild type). Recently, a novel gene (arginine1) associated with acute response to inhaled β2-AR agonists in both children and adults with asthma was identified [113].
7. Role of β2-AR Agonists in the Treatment of Obstructive Lung Diseases
7.1. Symptom-Relief
7.1.1. Asthma
Short-acting β2-AR agonists administered by inhalation are the most effective therapy for rapid reversal of airflow obstruction and prompt relief of asthmatic symptoms. The most widely used are salbutamol (commonly known as albuterol in the US) and terbutaline; their action occurs in 5 min or less, peaks after 30 to 60 min and lasts 4 to 6 h [114]. With regular use of a bronchodilator (four or more times daily), the potency (as measured by the increase in one second forced expiratory volume, FEV1) does not decline, but the duration of action is slightly shortened [115]. A regular schedule of administration four times a day does not improve outcomes as compared with as-needed administration [116]. Moreover, in patients with certain genotypic variants of the β2-AR it may have a deleterious effect [117,118]. So, the short-acting β2-AR agonists are recommended for use on request to relieve symptoms or before anticipated exposure to known asthmatic triggers, especially exercise. In mild-to-moderate asthmatic patients, a cumulative bronchodilator effect of salbutamol can be seen up to inhaled doses ranging from 700 to 1,500 µg [119]. Dose-dependent sympathomimetic-type side effects, including tremor, anxiety, heart pounding, and tachycardia (but not hypertension), are common, and a small dose-dependent decrease in serum potassium and magnesium levels is detectable. However, at the usual dose, adverse effects are uncommon [14].
The decision about which of the various short-acting β2-AR agonists to use is based largely on cost and patient's or physician's preference. Salbutamol has been made available in a metered-dose inhaler free of chlorofluorocarbons (CFCs), and CFC-containing salbutamol inhalers were taken off the market on 31 December 2008. Like CFCs, the alternative propellant, hydrofluoroalkane (HFA), is inert in the human airway [120]. The HFA inhalers are equipotent to the CFC-propelled inhalers [121] and can be used with valved holding chambers (spacers) in patients with poor inhalational technique. They provide bronchodilation comparable to nebulized salbutamol when a sufficient number of puffs is administered and inhalational technique is good [122]. Levalbuterol, the purified D-rotatory isomer of salbutamol, was developed on the purpose to eliminate side effects, which some argue are limited to the S-rotatory isomer [123]. However, when delivered by metered-dose inhaler, levalbuterol has an efficacy and side-effect profile that is indistinguishable from that of the racemic mixture of molecules in salbutamol [124].
The inhaled long-acting β2-AR agonists salmeterol and formoterol have a bronchodilator potency similar to that of their short-acting counterpart. Features distinguishing the two long-acting β2-AR agonists are both practical and theoretical. The onset of action of formoterol occurs within 5 min, a period similar to that for short-acting β2-AR agonists, whereas salmeterol has a slower onset of action (15 to 20 min) [14]. For this reason, a combination formoterol-corticosteroid inhaler is recommended both for quick relief of asthmatic symptoms and, when used regularly, for long-term control [125]. Formoterol is a full agonist in its action at the β2-AR, whereas salmeterol is a partial agonist (and partial antagonist) [14]. The implication of this pharmacologic distinction, particularly as it might apply to the risk of fatal asthmatic attacks, is uncertain. Both salmeterol and formoterol display a sustained activity (>12 h), and because of their higher degree of β2-AR specificity, have fewer side effects than short-acting β2-AR agonists [126]. As much as with short-acting β2-AR agonists, regular use of long-acting β2-AR agonists results in only mild tachyphylaxis to the maximal bronchodilator effect and the duration of action [126]. In contrast, the bronchoprotective effect of long-acting β2-AR agonists (i.e., their inhibition of exercise-induced bronchoconstriction) rapidly wanes with regular use [127], a contrary pharmacologic effect that has not been fully explained. With rare exceptions [128], the quick symptom relief provided by short-acting β2-AR agonists is not impeded by regular use of long-acting β2-AR agonists [129].
7.1.2. COPD
In COPD, formoterol and salmeterol are more effective bronchodilators than their short-acting counterpart [130] and have an add-on effect to anticholinergics [131]. Both drugs improve FEV1, reduce symptoms and use of rescue medication, and improve exercise capacity and health status [132]. There is also evidence that formoterol may be used as a rescue inhaler without significant unwanted effects [133]. Though tolerance in the long-term use of long-acting β2-AR agonists does not appear to be a problem in clinical practice, concerns have been raised about the side effects, particularly cardiac dysrhythmias, in susceptible elderly populations [132]. Nevertheless, salmeterol and formoterol at usual doses (100 µg/day and 24 µg/day, respectively) are effective and safe in treating patients with COPD [134]. Higher doses may cause more untoward effects, although serious adverse events are very uncommon [132].
7.2. Disease-Control
The fact that long-acting β2-AR agonists afford sustained improvement in lung function may tempt clinicians to use them as a long-term controller medication, without concomitant use of anti-inflammatory treatment. However, this strategy results in unsuppressed airway inflammation and an unacceptably high rate of asthmatic exacerbations [135,136,137,138,139]. For these reasons, the use of long acting β2-AR agonists as single treatment for asthma control is not recommended and several formulations combining β2-AR agonist with an inhaled corticosteroid in a single device have been made available. Combinations of salmeterol with inhaled corticosteroid are generally used for regular treatment only, whereas those of salbutamol or formoterol have been suggested for both as needed and regular treatment [140].
8. The Great β2-AR Agonists Controversy
8.1. Short-Acting β2-AR Agonists
Epinephrine was the first synthetic β-agonist bronchodilator to be used clinically and was introduced in the early 1900’s [141]. As systemic side effects due to potent α- and non-selective β2-AR stimulatory effects were reduced but not eliminated by aerosol inhalation, its widespread use for the treatment of asthma was abandoned. Moreover, up to a five-fold increase in mortality among users of inhaled epinephrine was reported in 1948 [142].
Isoproterenol was introduced in 1940 into medicine as short-acting β2-AR agonist. It gradually replaced epinephrine because of its more potent β-stimulant properties and its virtual absence of α-agonist activity [143]. However, this epinephrine derivative possesses the same major disadvantage of its parent compound, namely, a relatively short duration of action (due to rapid metabolism mainly by COMT and, to a lesser extent, by MAO) and undesirable cardiac side effects and mortality [143].
The most-often-cited examples of the possible relationship between short-acting β2-AR agonists and mortality in asthma were the startling rise in asthma deaths in several countries, including the UK, Australia and New Zealand during the early 1960s [144] and in New Zealand in the late 1970s [145,146]. In investigating the earlier epidemic in the UK, Speizer et al. [144,147] found that these deaths were most often sudden, unexpected and closely correlated with the increased use of pressurized metered dose inhalers. In the late 1960s, following recognition of the potential danger from the overuse of β2-AR agonists and restrictions on their over-the-counter sale, the usage of inhaled short-acting β2-AR agonists declined. Simultaneously, the asthma death rate fell sharply, suggesting that the previously noted relationship might have been a causal one. It is noteworthy, however, that this decline in the asthma death rate occurred concomitantly with a greater reliance on corticosteroid therapy, suggesting that the latter might also have contributed to the drop in mortality due to asthma [148].
Several hypotheses have been advanced incriminating short-acting β2-AR agonists as contributing to the second epidemic of asthma deaths that occurred in New Zealand in the late 1970s and early 1980s [146]. One suggested factor was a change in prescribing practices, in that inhaled β2-AR agonists, along with sustained-release theophylline preparations, were prescribed in place of inhaled corticosteroids and cromolyn [149], possibly predisposing to fatal cardiac arrhythmias. Another proposed factor was an increased use of β2-AR agonists delivered by air-driven home nebulizers for self-treatment of severe asthma. In this case, patients could be exposed to the risk of hypoxic cardiac arrest if large doses of short-acting β2-AR agonists were delivered in the face of uncorrected hypoxaemia [150]. Two case-control studies involving asthma deaths in patients aged 5–45 years in New Zealand from 1981 to 1983 [151] and from 1977 to 1981 [152] have led to a particularly contentious hypothesis linking some of the excess in asthma mortality to prescribed fenoterol by metered-dose inhaler. In 1992, Spitzer et al. [153], by using health insurance data from Saskatchewan (Canada), found an increased risk of death or near-death from asthma in association with regular use of short acting β2-AR agonists. These last included fenoterol (odds ratio, 5.4 per canister), but also salbutamol (odds ratio, 2.4 per canister). The authors concluded that these findings could be due to adverse effects of the β2-AR agonists themselves but raised the alternative possibility that their increased use might simply be a marker of more severe disease. A 1 year clinical trial showed increased airway responsiveness and worsened asthma control during regular treatment with fenoterol added to usual therapy compared with short-acting β2-AR agonists used only as needed for symptom relief [154]. However, subsequent US and UK trials of regular versus as-needed salbutamol did not detect sustained adverse effects on asthma control [116,155]. Nevertheless, in 1997 the British Guidelines on Asthma Management increasingly advocated the use of short-acting β2-AR only as needed for symptom relief [156].
8.2. Long-Acting β2-AR Agonists
The introduction of long-acting β2-AR agonists salmeterol [157] and formoterol [158] in the 1990’s was considered a major advance in bronchodilator therapy with evidence that their use led to improved lung function and quality of life [114]. There were also potential safety advantages due to the twice daily, fixed dose usage, which reduced the risk of overuse of β2-AR agonist therapy in the situation of severe exacerbations. However, concerns about the possible risks associated with long-acting β2-AR agonists therapy were raised soon after their introduction into clinical practice. This was due to the evidence that their regular use had the potential to reduce bronchodilator sensitivity to β2-AR agonists [159,160] and could induce tolerance to their bronchoprotective effects [95], which may not be restored by concurrent use of inhaled corticosteroids [161]. It also became apparent that patients using long-acting β2-AR agonists may be at risk of severe exacerbations if the symptom control, achieved with long-acting β2-AR agonists use, led to a discontinuation of inhaled corticosteroid therapy [162].
There were also concerns about a potential risk of mortality from the Salmeterol Nationwide Surveillance Study [163]. In this UK-based study of >25,000 subjects, there was a non-significant three-fold increased risk of asthma death in subjects on salmeterol compared with regular salbutamol. This led to the US-based Salmeterol Multicenter Asthma Research Trial, a placebo-controlled study of the safety of salmeterol in adults with unstable asthma [164]. The trial was stopped after an interim analysis showing a statistically significant, four-fold increase in asthma mortality with salmeterol. By contrast, in a large UK-based case-control study there was no positive association between long-acting β2-AR agonists therapy and asthma death [165]. However, during the period of this study, ~95% of UK asthma patients receiving long-acting β2-AR agonists therapy were co-prescribed inhaled corticosteroid [166]. Due to conflicting evidence, the Food and Drug Administration (FDA) confirmed the availability of both salmeterol and formoterol, but required black-box warnings on their product labels [167].
Formoterol fumarate is currently the most widely used long-acting β2-AR worldwide. Three placebo-controlled trials showed that the higher dose (48 µg/day) of formoterol tended to be associated with more serious asthma exacerbations than a lower one [168]. However, a large phase IV trial found all doses of formoterol to be associated with fewer exacerbations than placebo, with no indication of any dose-response relationship [169].
Recently, Sears et al. [170] have reported a comprehensive meta-analysis of safety data (n = 68,004 patients) obtained in completed trials. When comparing all formoterol-exposed patients to all patients not exposed to long-acting β2-AR agonists in the whole data set, no increased risks of cardiac-related deaths or cardiac-related non-fatal serious adverse events were observed. Similarly, when examined by inhaled corticosteroids use there was no increased risk of cardiac serious adverse events with regimens of formoterol with inhaled corticosteroids vs. inhaled corticosteroids without long-acting β2-AR agonists. Noteworthy, there was a significant reduction in asthma-related serious adverse events with formoterol (>90% of which were hospitalizations) speaking against a relationship between formoterol and increased asthma mortality. The main limitation of the study by Sears et al. [170] was the lack of sufficient power to form a definitive conclusion regarding the risk of death for patients treated with formoterol.
9. Future Prospects and Conclusions
A variety of β2-AR agonists with longer half-lives are currently undergoing development, with the hope of achieving once-daily dosing. Among them, the development of indacaterol is very advanced and it is likely that this drug will be launched by the end of 2010 [171,172]. Indacaterol provided effective and sustained 24 h bronchodilator effect, with a rapid onset of action (<5 min) and a good tolerability and safety profile in asthma control [173].
Numerous studies have shown the benefit of adding a long-acting β2-AR instead of doubling or increasing the dose of inhaled corticosteroids [136,174,175] and the greater benefits to most outcomes of adding a long-acting β2-AR compared with adding a leukotriene antagonist [176]. However, both the International Asthma Treatment Guidelines [5,6] and the FDA [177] now emphasize that long-acting β2-AR agonists should not be used as monotherapy in asthma but always together with an inhaled corticosteroid.
In conclusion, either as rescue medications and therapy added to or combined with inhaled corticosteroids, short- and long-acting β2-AR agonists have proved effective in achieving a good control of asthma. In particular, they are essential in reducing daytime and especially nighttime symptoms, improving lung function, reducing the risk of exacerbations, and minimizing the required dose of inhaled corticosteroids.
References
- Chen, K.K.; Schmidt, C.F. Ephedrine and related substances. Medicine (Baltimore) 1930, 9, 1–117. [Google Scholar] [CrossRef]
- Ahlquist, R.P. A study of adrenotropic receptors. Am. J. Physiol. 1948, 153, 586–600. [Google Scholar]
- Cazzola, M.; Matera, M.G. Emerging inhaled bronchodilators: an update. Eur. Respir. J. 2009, 34, 757–769. [Google Scholar]
- Rabe, K.F.; Hurd, S.; Anzueto, A.; Barnes, P.J.; Buist, S.A.; Calverley, P.; Fukuchi, Y.; Jenkins, C.; Rodriguez-Roisin, R.; van Weel, C.; et al. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. GOLD executive summary. Am. J. Respir. Crit. Care Med. 2007, 176, 532–555. [Google Scholar] [PubMed]
- National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma - summary report 2007. J. Allergy Clin. Immunol. 2007, 120 Suppl., S94–S138. [CrossRef] [PubMed]
- Bateman, E.D.; Hurd, S.S.; Barnes, P.J.; Bousquet, J.; Drazen, J.M.; FitzGerald, M.; Gibson, P.; Ohta, K.; O'Byrne, P.; Pedersen, S.E.; et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008, 31, 143–178. [Google Scholar] [CrossRef] [PubMed]
- Doidge, J.M.; Satchell, D.G. Adrenergic and non-adrenergic inhibitory nerves in mammalian airways. J. Auton. Nerv. Syst. 1982, 5, 83–99. [Google Scholar]
- Barnes, P.J. Neural control of human airways in health and disease. Am. Rev. Respir. Dis. 1986, 134, 1289–1314. [Google Scholar]
- Belvisi, M.G. Overview of the innervation of the lung. Curr. Opin. Pharmacol. 2002, 2, 211–215. [Google Scholar]
- Groneberg, D.A.; Harrison, S.; Dinh, Q.T.; Geppetti, P.; Fischer, A. Tachykinins in the respiratory tract. Curr. Drug Targets 2006, 7, 1005–1010. [Google Scholar]
- Barnes, P.J. Neural control of airway function: new perspectives. Mol. Aspects Med. 1990, 11, 351–423. [Google Scholar]
- Barnes, P.J. Modulation of neurotransmission in airways. Physiol. Rev. 1992, 72, 699–729. [Google Scholar]
- Lees, G.M. A hitch-hiker’s guide to the galaxy of adrenoceptors. Br. J. Med. 1981, 283, 173–178. [Google Scholar]
- Hoffman, B.B.; Lefkowitz, R.J. Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists. In Goodman & Gilman’sthe Pharmacological Basis of Therapeutics, 9th; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Goodman, L.S., Gilman, A., Eds.; McGraw-Hill: New York, NY, USA, 1996; pp. 199–248. [Google Scholar]
- Barnes, P.J. Endogenous catecholamines and asthma. J. Allergy Clin. Immunol. 1986, 77, 791–795. [Google Scholar]
- Nadel, J.A.; Barnes, P.J.; Holtzman, M.J. Autonomic factors in hyperreactivity of airway smooth muscle. In Handbook of Physiology. The Respiratory System; Macklem, P.T., Mead, J., Eds.; American Physiological Society: Bethesda, MD, USA, 1986; Volume 3, pp. 693–702, Part 2. [Google Scholar]
- Rhoden, K.J.; Meldrum, L.A.; Barnes, P.J. Inhibition of cholinergic neurotransmission in human airways by β2-adrenoceptors. J. Appl. Physiol. 1988, 65, 700–705. [Google Scholar]
- Bai, T.R.; Lam, R.; Prasad, F.Y. Effects of adrenergic agonists and adenosine on cholinergic neurotransmission in human tracheal smooth muscle. Pulmon. Pharmacol. 1989, 1, 193–199. [Google Scholar]
- Barnes, P.J. Muscarinic receptor subtype in airways. Life Sci. 1993, 52, 521–528. [Google Scholar]
- Lands, A.M.; Arnold, A.; McAuliff, J.P.; Luduena, F.P.; Brown, T.G., Jr. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214, 597–598. [Google Scholar]
- Carstairs, J.R.; Nimmo, A.J.; Barnes, P.J. Autoradiographic visualization of beta-adrenoceptor subtype in human lung. Am. Rev. Respir. Dis. 1985, 132, 541–547. [Google Scholar]
- Barnes, P.J.; Nadel, J.A.; Skoogh, B.E.; Roberts, J.M. Characterization of beta adrenoceptor subtypes in canine airway smooth muscle by radioligand binding and physiological responses. J. Pharmacol. Exp. Ther. 1983, 225, 456–461. [Google Scholar]
- Hamid, Q.A.; Mak, J.C.; Sheppard, M.N.; Corrin, B.; Venter, J.C.; Barnes, P.J. Localization of beta2-adrenoceptor messenger RNA in human and rat lung using in situ hybridization: correlation with receptor autoradiography. Eur. J. Pharmacol. 1991, 206, 133–138. [Google Scholar]
- Tamaoki, J.; Yamauchi, F.; Chiyotani, A.; Yamawaki, I.; Takeuchi, S.; Konno, K. Atypical β-adrenoceptor (β3-adrenoceptor) mediated relaxation of canine isolated bronchial smooth muscle. J. Appl. Physiol. 1993, 74, 297–302. [Google Scholar]
- Martin, C.A.; Naline, E.; Bakdach, H.; Advenier, C. Beta 3-adrenoceptor agonists, BRL 37344 and SR 58611A, do not induce relaxation of human, sheep and guinea-pig airway smooth muscle in vitro. Eur. Respir. J. 1994, 7, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Emorine, L.J.; Marullo, S.; Briend-Sutren, M.M.; Patey, G.; Tate, K.; Delavier-Klutchko, C.; Strosberg, A.D. Molecular characterization of the human beta 3-adrenergic receptor. Science 1989, 245, 1118–1121. [Google Scholar]
- Goldie, R.G.; Paterson, J.W.; Spina, D.; Wale, J.L. Classification of β2-adrenoceptors in human isolated bronchus. Br. J. Pharmacol. 1984, 81, 611–615. [Google Scholar]
- Nials, A.T.; Coleman, R.A.; Johnson, M.; Magnussen, H.; Rabe, K.F.; Vardey, C.J. Effects of β-adrenoceptor agonists in human bronchial smooth muscle. Br. J. Pharmacol. 1993, 110, 1112–1116. [Google Scholar] [PubMed]
- Löfdahl, C.G.; Svedmyr, N. Effects of prenalterol in asthmatic patients. Eur. J. Clin. Pharmacol. 1982, 23, 297–302. [Google Scholar]
- Torphy, T.J.; Rinard, G.A.; Rietow, M.G.; Mayer, S.E. Functional antagonism in canine tracheal smooth muscle: inhibition by methacholine of the mechanical and biochemical responses to isoproterenol. J. Pharmacol. Exp. Ther. 1983, 227, 694–699. [Google Scholar]
- Torphy, T.J.; Zheng, C.; Peterson, S.M.; Fiscus, R.R.; Rinard, G.A.; Mayer, S.E. Inhibitory effect of methacholine on drug-induced relaxation, cyclic AMP accumulation, and cyclic AMP-dependent protein kinase activation in canine tracheal smooth muscle. J. Pharmacol. Exp. Ther. 1985, 232, 409–417. [Google Scholar]
- Gross, N.J.; Skorodin, M.S. Anticholinergic, antimuscarinic bronchodilators. Am. Rev. Respir. Dis. 1984, 129, 856–870. [Google Scholar]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar]
- Dohlman, H.G.; Thorner, J.; Caron, M.G.; Lefkowitz, R.J. Model systems for the study of seven-transmembrane-segment receptors. Annu. Rev. Biochem. 1991, 60, 653–688. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar]
- Dohlman, H.G. A scaffold makes the switch. Sci. Signal. 2008, 1. pe46.. [Google Scholar]
- Liggett, S.B.; Lefkowitz, R.J. Adrenergic receptor-coupled adenylyl cyclase systems: regulation of receptor function by phosphorylation, sequestration and downregulation. In Regulation of Cellular Signal Transduction Pathways by Desensitization and Amplification; Sibley, D., Houslay, M., Eds.; John Wiley & Sons: London, UK, 1993; pp. 71–97. [Google Scholar]
- Giembycz, M.A.; Raeburn, D. Putative substrates for cyclic nucleotide-dependent protein kinases and the control of airway smooth muscle tone. J. Auton. Pharmacol. 1991, 11, 365–398. [Google Scholar]
- Gerthoffer, U.T. Calcium dependence of myosin phosphorylation and airway smooth muscle contraction and relaxation. Am. J. Physiol. 1986, 250, C597–C604. [Google Scholar]
- Hall, I.P.; Hill, S.J. Beta-adrenoceptor stimulation inhibits histamine-stimulated inositol phospholipids hydrolysis in bovine tracheal smooth muscle. Br. J. Pharmacol. 1988, 95, 1204–1212. [Google Scholar]
- Twort, C.H.; van Breemen, C. Human airway smooth muscle in cell culture: control of the intracellular calcium store. Pulm. Pharmacol. 1989, 2, 45–53. [Google Scholar]
- Gunst, S.J.; Stropp, J.Q. Effect of Na-K adenosinetriphosphatase activity on relaxation of canine tracheal smooth muscle. J. Appl. Physiol. 1988, 64, 635–641. [Google Scholar]
- Jones, T.R.; Charette, L.; Garcia, M.L.; Kaczorowski, G.J. Selective inhibition of relaxation of guinea-pig trachea by charybdotoxin, a potent Ca(++)-activated K+ channel inhibitor. J. Pharmacol. Exp. Ther. 1990, 255, 697–706. [Google Scholar]
- Jones, T.R.; Charette, L.; Garcia, M.L.; Kaczorowski, G.J. Interaction of iberiotoxin with beta-adrenoceptor agonists and sodium nitroprusside on guinea pig trachea. J. Appl. Physiol. 1993, 74, 1879–1884. [Google Scholar]
- Tanaka, Y.; Yamashita, Y.; Yamaki, F.; Horinouchi, T.; Shigenobu, K.; Koike, K. MaxiK channel mediates beta2-adrenoceptor-activated relaxation to isoprenaline through cAMP-dependent and -independent mechanisms in guinea-pig tracheal smooth muscle. J. Smooth Muscle Res. 2003, 39, 205–219. [Google Scholar]
- Koike, K.; Yamashita, Y.; Horinouchi, T.; Yamaki, F.; Tanaka, Y. cAMP-independent mechanism is significantly involved in beta2-adrenoceptor-mediated tracheal relaxation. Eur. J. Pharmacol. 2004, 492, 65–70. [Google Scholar]
- Miura, M.; Belvisi, M.G.; Stretton, C.D.; Yacoub, M.H.; Barnes, P.J. Role of potassium channels in bronchodilator responses in human airways. Am. Rev. Respir. Dis. 1992, 146, 132–136. [Google Scholar]
- Kume, H.; Graziano, M.P.; Kotlikoff, M.I. Stimulatory and inhibitory regulation of calcium-activated potassium channels by guanine nucleotide-binding proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 11051–11055. [Google Scholar]
- Belvisi, M.G.; Patel, H.J.; Takahashi, T.; Barnes, P.J.; Giembycz, M.A. Paradoxical facilitation of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by isoprenaline. Br. J. Pharmacol. 1996, 117, 1413–1420. [Google Scholar]
- De Haas, J.R.; Terpstra, J.S.; Van der Zwaag, M.; Kockelbergh, P.G.; Roffel, A.D.F; Zaagsma, J. Facilitatory β2-adrenoceptors on cholinergic and adrenergic nerve endings of the guinea pig trachea. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, L420–L425. [Google Scholar]
- Zhang, X.Y.; Olszewski, M.A.; Robinson, N.E. β2-Adrenoceptor activation augments acetylcholine release from tracheal parasympathetic nerves. Am. J. Physiol. Lung Cell. Mol. Physiol. 1995, 268, L950–L956. [Google Scholar]
- Wessler, I.; Reinheimer, T.; Brunn, G.; Anderson, G.P.; Maclagan, J.; Rackè, K. β2-adrenoceptors mediate inhibition of [3H]-acetylcholine release from the isolated rat and guinea-pig trachea: role of the airway mucosa and prostaglandins. Br. J. Pharmacol. 1994, 113, 1221–1230. [Google Scholar] [PubMed]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar]
- Patel, H.J.; Giembycz, M.A.; Keeling, J.E.; Barnes, P.J.; Belvisi, M.G. Inhibition of cholinergic neurotransmission in guinea pig trachea by NS1619, a putative activator of large-conductance, calcium-activated potassium channels. J. Pharmacol. Exp. Ther. 1998, 286, 952–958. [Google Scholar] [PubMed]
- Brichetto, L.; Song, P.; Crimi, E.; Rehder, K.; Brusasco, V. Modulation of cholinergic responsiveness through the β-adrenoceptor signal transmission pathway in bovine trachealis. J. Appl. Physiol. 2003, 95, 735–741. [Google Scholar]
- Freund-Michel, V.C.; Birrell, M.A.; Giembycz, M.A.; Hele, D.J.; Haj-Yahia, S.; Belvisi, M.G. Beta2-agonists block tussive responses in guinea pigs via an atypical cAMP-dependent pathway. Eur. Respir. J. 2010, 35, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.F.; Chen, C.C.; Ma, H.P.; Wu, Y.C.; Chen, Y.C.; Wang, T.L. Comparison of lidocaine and bronchodilator inhalation treatments for cough suppression in patients with chronic obstructive pulmonary disease. Emerg. Med. J. 2005, 22, 429–432. [Google Scholar]
- Mulrennan, S.; Wright, C.; Thompson, R.; Goustas, P.; Morice, A. Effect of salbutamol on smoking related cough. Pulm. Pharmacol. Ther. 2004, 17, 127–131. [Google Scholar]
- Chang, A.B.; Phelan, P.D.; Carlin, J.B.; Sawyer, S.M.; Robertson, C.F. A randomised, placebo controlled trial of inhaled salbutamol and beclomethasone for recurrent cough. Arch. Dis. Child. 1998, 79, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Adamson, D.L.; Choudry, N.B.; Fuller, R.W. The effect of altering airway tone on the sensitivity of the cough reflex in normal volunteers. Eur. Respir. J. 1991, 4, 1078–1079. [Google Scholar]
- Brusasco, V.; Crimi, E.; Gherson, G.; Nardelli, R.; Oldani, V.; Francucci, B.; Della Cioppa, S.; Senn, S.; Fabbri, L. Actions other than smooth muscle relaxation may play a role in the protective effects of formoterol on the allergen-induced late asthmatic reaction. Pulm. Pharmacol. Ther. 2002, 15, 399–406. [Google Scholar]
- Johnson, S.R.; Knox, A.J. Synthetic functions of airway smooth muscle in asthma. Trends Pharmacol. Sci. 1997, 18, 288–292. [Google Scholar]
- Baroffio, M.; Crimi, E.; Brusasco, V. Airway smooth muscle as a model for new investigative drugs in asthma. Ther. Adv. Respir. Dis. 2008, 2, 129–139. [Google Scholar]
- Baroffio, M.; Barisione, G.; Crimi, E.; Brusasco, V. Noninflammatory mechanisms of airway hyper-responsiveness in bronchial asthma: an overview. Ther. Adv. Respir. Dis. 2009, 3, 163–174. [Google Scholar]
- Black, J.L.; Roth, M. Intrinsic asthma: is it intrinsic to the smooth muscle? Clin. Exp. Allergy 2009, 39, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Holden, N.S.; Wilson, S.M.; Sukkar, M.B.; Chung, K.F.; Barnes, P.J.; Newton, R.; Giembycz, M.A. Effect of beta2-adrenoceptor agonists and other cAMP-elevating agents on inflammatory gene expression in human ASM cells: a role for protein kinase A. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L505–L514. [Google Scholar]
- Lefkowitz, R.J.; Hausdorff, W.P.; Caron, M.G. Role of phosphorylation in desensitization of the beta-adrenoceptor. Trends Pharmacol. Sci. 1990, 11, 190–194. [Google Scholar]
- Bai, T.R.; Mak, J.C.W.; Barnes, P.J. A comparison of β-adrenergic receptors and in vitro relaxant responses to isoproterenol in asthmatic airway smooth muscle. Am. J. Respir. Cell. Mol. Biol. 1992, 6, 647–651. [Google Scholar] [PubMed]
- Brusasco, V.; Crimi, E.; Baroffio, M. Allergic airway inflammation and beta-adrenoceptor dysfunction. Cell. Biochem. Biophys. 2006, 44, 129–138. [Google Scholar]
- Szentivanyi, A. Beta adrenergic theory of atopic abnormality in bronchial asthma. J. Allergy 1968, 42, 203–232. [Google Scholar]
- Trian, T.; Ge, Q.; Moir, L.M.; Burgess, J.K.; Kuo, C.; King, N.J.; Reddel, H.K.; Black, J.L.; Oliver, B.G.; McParland, B.E. Rhinovirus-induced exacerbations of asthma - How is the beta2-adrenoceptor implicated? Am. J. Respir. Cell. Mol. Biol. 2009. Epub ahead of print.. [Google Scholar]
- Laporte, J.D.; Moore, P.E.; Panettieri, R.A.; Moeller, W.; Heyder, J.; Shore, S.A. Prostanoids mediate IL-1β-induced β-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am. J. Physiol. 1998, 275, L491–L501. [Google Scholar]
- Hausdorff, W.P.; Caron, M.G.; Lefkowitz, R.J. Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 1990, 4, 2881–2889. [Google Scholar]
- Song, P.; Milanese, M.; Crimi, E.; Bruzzone, S.; Zocchi, E.; Rehder, K.; Brusasco, V. G(s) protein dysfunction in allergen-challenged human isolated passively sensitized bronchi. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L209–L215. [Google Scholar]
- Milanese, M.; Riccio, A.M.; Gamalero, C.; De Giovanni, B.; Brichetto, L.; Baroffio, M.; Crimi, E.; Brusasco, V.; Canonica, G.W. A model of allergen-driven human airway contraction: beta2- pathway dysfunction without cytokine involvement. Ann. Allergy Asthma Immunol. 2005, 94, 273–278. [Google Scholar]
- Song, P.; Milanese, M.; Crimi, E.; Rehder, K.; Brusasco, V. Allergen challenge of passively sensitized human bronchi alters M2- and beta2-receptor function. Am. J. Respir. Crit. Care Med. 1997, 155, 1230–1234. [Google Scholar]
- Song, P.; Crimi, E.; Milanese, M.; Duan, J.; Rehder, K.; Brusasco, V. Anti-inflammatory agents and allergen-induced beta2-receptor dysfunction in isolated human bronchi. Am. J. Respir. Crit. Care Med. 1998, 158, 1809–1814. [Google Scholar]
- Rovati, G.E.; Baroffio, M.; Citro, S.; Brichetto, L.; Ravasi, S.; Milanese, M.; Crimi, E.; Brusasco, V. Cysteinyl-leukotrienes in the regulation of beta2-adrenoceptor function: an in vitro model of asthma. Respir. Res. 2006, 7, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Mayor, F., Jr.; Staniszewski, C.; Lefkowitz, R.J.; Caron, M.G. Purification and characterization of the beta-adrenergic receptor kinase. J. Biol. Chem. 1987, 262, 9026–9032. [Google Scholar]
- Benovic, J.L.; Kühn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar]
- Nino, G.; Hu, A.; Grunstein, J.S.; Grunstein, M. Mechanism regulating proasthmatic effects of prolonged homologous β2-adrenergic receptor desensitization in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L746–L757. [Google Scholar]
- Wang, W.C.; Mihlbachler, K.A.; Brunnett, A.C.; Liggett, S.B. Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway beta2-adrenergic receptor physiologic signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 15007–15012. [Google Scholar]
- Giembycz, M.A. An estimation of beta2-adrenoceptor reserve on human bronchial smooth muscle for some sympathomimetic bronchodilators. Br. J. Pharmacol. 2009, 158, 287–299. [Google Scholar]
- Yu, S.S.; Lefkowitz, R.J.; Hausdorff, W.P. Beta-adrenergic receptor sequestration. A potential mechanism of receptor resensitization. J. Biol. Chem. 1993, 268, 337–341. [Google Scholar] [PubMed]
- Hadcock, J.R.; Wang, H.Y.; Malbon, C.C. Agonist-induced destabilization of beta-adrenergic receptor mRNA. Attenuation of glucocorticoid-induced up-regulation of beta-adrenergic receptors. J. Biol. Chem. 1989, 264, 19928–19933. [Google Scholar] [PubMed]
- Collins, S.; Bouvier, M.; Bolanowski, M.A.; Caron, M.G.; Lefkowitz, R.J. cAMP stimulates transcription of the beta2-adrenergic receptor gene in response to short-term agonist exposure. Proc. Natl. Acad. Sci. USA 1989, 86, 4853–4857. [Google Scholar]
- Huang, L.Y.; Tholanikunnel, B.G.; Vakalopoulou, E.; Malbon, C.C. The M(r) 35,000 beta-adrenergic receptor mRNA-binding protein induced by agonists requires both an AUUUA pentamer and U-rich domains for RNA recognition. J. Biol. Chem. 1993, 268, 25769–25775. [Google Scholar]
- Bouvier, M.; Collins, S.; O'Dowd, B.F.; Campbell, P.T.; de Blasi, A.; Kobilka, B.K.; MacGregor, C.; Irons, G.P.; Caron, M.G.; Lefkowitz, R.J. Two distinct pathways for cAMP-mediated down-regulation of the beta2-adrenergic receptor. Phosphorylation of the receptor and regulation of its mRNA level. J. Biol. Chem. 1989, 264, 16786–16792. [Google Scholar] [PubMed]
- Valiquette, M.; Bonin, H.; Hnatowich, M.; Caron, M.G.; Lefkowitz, R.J.; Bouvier, M. Involvement of tyrosine residues located in the carboxyl tail of the human beta2-adrenergic receptor in agonist-induced down-regulation of the receptor. Proc. Natl. Acad. Sci. USA 1990, 87, 5089–5093. [Google Scholar]
- Kraan, J.; Koëter, G.H.; vd Mark, T.W.; Sluiter, H.J.; de Vries, K. Changes in bronchial hyperreactivity induced by 4 weeks of treatment with antiasthmatic drugs in patients with allergic asthma: a comparison between budesonide and terbutaline. J. Allergy Clin. Immunol. 1985, 76, 628–636. [Google Scholar]
- Kerrebijn, K.F.; van Essen-Zandvliet, E.E.; Neijens, H.J. Effect of long-term treatment with inhaled corticosteroids and beta-agonists on the bronchial responsiveness in children with asthma. J. Allergy Clin. Immunol. 1987, 79, 653–659. [Google Scholar]
- van Schayck, C.P.; Graafsma, S.J.; Visch, M.B.; Dompeling, E.; van Weel, C.; van Herwaarden, C.L. Increased bronchial hyperresponsiveness after inhaling salbutamol during 1 year is not caused by subsensitization to salbutamol. J. Allergy Clin. Immunol. 1990, 86, 793–800. [Google Scholar]
- Cheung, D.; Timmers, M.C.; Zwinderman, A.H.; Bel, E.H.; Dijkman, J.H.; Sterk, P.J. Long-term effects of a long-acting beta2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N. Engl. J. Med. 1992, 327, 1198–1203. [Google Scholar] [PubMed]
- McGraw, D.W.; Almoosa, K.F.; Paul, R.J.; Kobilka, B.K.; Liggett, S.B. Antithetic regulation by β-adrenergic receptors of Gq receptor signaling via phospholipase C underlies the airway β-agonist paradox. J. Clin. Invest. 2003, 112, 619–626. [Google Scholar] [PubMed]
- Barnes, P.J. Scientific rationale for using a single inhaler for asthma control. Eur. Respir. J. 2007, 29, 587–595. [Google Scholar]
- Callaerts-Vegh, Z.; Evans, K.L.; Dudekula, N.; Cuba, D.; Knoll, B.J.; Callaerts, P.F.; Giles, H.; Shardonofsky, F.R.; Bond, R.A. Effects of acute and chronic administration of beta-adrenoceptor ligands on airway function in a murine model of asthma. Proc. Natl. Acad. Sci. USA 2004, 101, 4948–4953. [Google Scholar]
- Nguyen, L.P.; Omoluabi, O.; Parra, S.; Frieske, J.M.; Clement, C.; Ammar-Aouchiche, Z.; Ho, S.B.; Ehre, C.; Kesimer, M.; Knoll, B.J.; et al. Chronic exposure to beta-blockers attenuates inflammation and mucin content in a murine asthma model. Am. J. Respir. Cell. Mol. Biol. 2008, 38, 256–262. [Google Scholar] [PubMed]
- Hanania, N.A.; Singh, S.; El-Wali, R.; Flashner, M.; Franklin, A.E.; Garner, W.J.; Dickey, B.F.; Parra, S.; Ruoss, S.; Shardonofsky, F.; et al. The safety and effects of the beta-blocker, nadolol, in mild asthma: an open-label pilot study. Pulm. Pharmacol. Ther. 2008, 21, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, B.J.; Williamson, P.A. Beta blockers for asthma: a double-edged sword. Lancet 2009, 373, 104–105. [Google Scholar]
- Hanania, N.A.; Dickey, B.F.; Bond, R.A. Clinical implications of the intrinsic efficacy of beta-adrenoceptor drugs in asthma: full, partial and inverse agonism. Curr. Opin. Pulm. Med. 2010, 16, 1–5. [Google Scholar]
- Davies, A.O.; Lefkowitz, R.J. In vitro desensitization of beta-adrenergic receptors in human neutrophils. Attenuation by corticosteroids. J. Clin. Invest. 1983, 71, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Brichetto, L.; Milanese, M.; Song, P.; Patrone, M.; Crimi, E.; Rehder, K.; Brusasco, V. Beclomethasone rapidly ablates allergen-induced beta2-adrenoceptor pathway dysfunction in human isolated bronchi. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L133–L139. [Google Scholar]
- Green, S.A.; Turki, J.; Hall, I.P.; Liggett, S.B. Implications of genetic variability of human beta2-adrenergic receptor structure. Pulm. Pharmacol. 1995, 8, 1–10. [Google Scholar]
- Liggett, S.B. Genetics of beta2-adrenergic receptor variants in asthma. Clin. Exp. Allergy 1995, Suppl. 2, 89–94. [Google Scholar] [CrossRef]
- Kazani, S.; Wechsler, M.E.; Israel, E. The role of pharmacogenomics in improving the management of asthma. J. Allergy Clin. Immunol. 2010, 125, 295–302. [Google Scholar]
- Taylor, D.R.; Drazen, J.M.; Herbison, G.P.; Yandava, C.N.; Hancox, R.J.; Town, G.I. Asthma exacerbations during long term beta-agonist use: influence of beta2-adrenoceptor polymorphism. Thorax 2000, 55, 762–767. [Google Scholar]
- Basu, K.; Palmer, C.N.; Tavendale, R.; Lipworth, B.J.; Mukhopadhyay, S. Beta2-receptor genotype predisposes to exacerbations in steroid-treated asthmatic patients taking frequent albuterol or salmeterol. J. Allergy Clin. Immunol. 2009, 124, 1188–1194. [Google Scholar]
- Wechsler, M.E.; Kunselman, S.J.; Chinchilli, V.M.; Bleecker, E.; Boushey, H.A.; Calhoun, W.J.; Ameredes, B.T.; Castro, M.; Craig, T.J.; Denlinger, L.; et al. Effect of beta2-adrenergic receptor polymorphism on response to long-acting beta2-agonist in asthma (LARGE trial): A genotype-stratified, randomised, placebo-controlled, crossover trial. Lancet 2009, 374, 1754–1764. [Google Scholar] [PubMed]
- Wang, W.C.; Mihlbachler, K.A.; Bleecker, E.R.; Weiss, S.T.; Liggett, S.B. A polymorphism of G-protein coupled receptor kinase 5 alters agonist-promoted desensitization of beta2-adrenergic receptors. Pharmacogenet. Genomics 2008, 18, 729–732. [Google Scholar]
- Hall, I.P.; Wheatley, A.; Wilding, P.; Liggett, S.B. Association of Glu 27 beta2-adrenoceptor polymorphism with lower airway reactivity in asthmatic subjects. Lancet 1995, 345, 1213–1214. [Google Scholar]
- Litonjua, A.A.; Lasky-Su, J.; Schneiter, K.; Tantisira, K.G.; Lazarus, R.; Klanderman, B.; Lima, J.J.; Irvin, C.G.; Peters, S.P.; Hanrahan, J.P.; et al. ARG1 is a novel bronchodilator response gene: screening and replication in four asthma cohorts. Am. J. Respir. Crit. Care Med. 2008, 178, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.S. β-Adrenergic bronchodilators. N. Engl. J. Med. 1995, 333, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lipworth, B.J.; Struthers, A.D.; McDevitt, D.G. Tachyphylaxis to systemic but not to airway responses during prolonged therapy with high dose inhaled salbutamol in asthmatics. Am. Rev. Respir. Dis. 1989, 140, 586–592. [Google Scholar]
- Drazen, J.M.; Israel, E.; Boushey, H.A.; Chinchilli, V.M.; Fahy, J.V.; Fish, J.E.; Lazarus, S.C.; Lemanske, R.F.; Martin, R.J.; Peters, S.P.; et al. Comparison of regularly scheduled with as-needed use of albuterol in mild asthma. Asthma Clinical Research Network. N. Engl. J. Med. 1996, 335, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Israel, E.; Drazen, J.M.; Liggett, S.B.; Boushey, H.A.; Cherniack, R.M.; Chinchilli, V.M.; Cooper, D.M.; Fahy, J.V.; Fish, J.E.; Ford, J.G.; et al. The effect of polymorphisms of the beta(2)-adrenergic receptor on the response to regular use of albuterol in asthma. Am. J. Respir. Crit. Care Med. 2000, 162, 75–80. [Google Scholar] [PubMed]
- Israel, E.; Chinchilli, V.M.; Ford, J.G.; Boushey, H.A.; Cherniack, R.; Craig, T.J.; Deykin, A.; Fagan, J.K.; Fahy, J.V.; Fish, J.; et al. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet 2004, 364, 1505–1512. [Google Scholar] [PubMed]
- Milanese, M.; Saporiti, R.; Bartolini, S.; Pellegrino, R.; Baroffio, M.; Brusasco, V.; Crimi, E. Bronchodilator effects of exercise hyperpnea and albuterol in mild-to-moderate asthma. J. Appl. Physiol. 2009, 107, 494–499. [Google Scholar]
- Hendeles, L.; Colice, G.L.; Meyer, R.J. Withdrawal of albuterol inhalers containing chlorofluorocarbon propellants. N. Engl. J. Med. 2007, 356, 1344–1351. [Google Scholar]
- Ramsdell, J.W.; Colice, G.L.; Ekholm, B.P.; Klinger, N.M. Cumulative dose response study comparing HFA-134a albuterol sulfate and conventional CFC albuterol in patients with asthma. Ann. Allergy Asthma Immunol. 1998, 81, 593–599. [Google Scholar]
- Cates, C.J.; Crilly, J.A.; Rowe, B.H. Holding chambers (spacers) versus nebulizers for beta-agonist treatment of acute asthma. Cochrane Database Syst. Rev. 2006, 2, CD000052–CD000052. [Google Scholar]
- Henderson, W.R., Jr.; Banerjee, E.R.; Chi, E.Y. Differential effects of (S)- and (R)-enantiomers of albuterol in a mouse asthma model. J. Allergy Clin. Immunol. 2005, 116, 332–340. [Google Scholar]
- Berger, W.E.; Milgrom, H.; Skoner, D.P.; Tripp, K.; Parsey, M.V.; Baumgartner, R.A. Xopenex Pediatric Asthma Group. Evaluation of levalbuterol metered dose inhaler in pediatric patients with asthma: a double-blind, randomized, placebo- and active-controlled trial. Curr. Med. Res. Opin. 2006, 22, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- O'Byrne, P.M.; Bisgaard, H.; Godard, P.P.; Pistolesi, M.; Palmqvist, M.; Zhu, Y.; Ekström, T. Bateman ED Budesonide/formoterol combination therapy as both maintenance and reliever medication in asthma. Am. J. Respir. Crit. Care Med. 2005, 171, 129–136. [Google Scholar] [PubMed]
- Pearlman, D.S.; Chervinsky, P.; LaForce, C.; Seltzer, J.M.; Southern, D.L.; Kemp, J.P.; Dockhorn, R.J.; Grossman, J.; Liddle, R.F.; Yancey, S.W.; et al. A comparison of salmeterol with albuterol in the treatment of mild-to-moderate asthma. N. Engl. J. Med. 1992, 327, 1420–1425. [Google Scholar] [PubMed]
- Nelson, J.A.; Strauss, L.; Skowronski, M.; Ciufo, R.; Novak, R.; McFadden, E.R., Jr. Effect of long-term salmeterol treatment on exercise-induced asthma. N. Engl. J. Med. 1998, 339, 141–146. [Google Scholar]
- Weinberger, M.; Abu-Hasan, M. Life-threatening asthma during treatment with salmeterol. N. Engl. J. Med. 2006, 355, 852–853. [Google Scholar]
- Smyth, E.T.; Pavord, I.D.; Wong, C.S.; Wisniewski, A.F.; Williams, J.; Tattersfield, A.E. Interaction and dose equivalence of salbutamol and salmeterol in patients with asthma. Br. Med. J. 1993, 306, 543–545. [Google Scholar]
- Appleton, S.; Poole, P.; Smith, B.; Veale, A.; Bara, A. Long-acting β2-agonists for chronic obstructive pulmonary disease patients with poorly reversible airflow limitation. Cochrane Database Syst. Rev. 2002, 3, CD001104. [Google Scholar] [PubMed]
- D'Urzo, A.D.; De Salvo, M.C.; Ramirez-Rivera, A.; Almeida, J.; Sichletidis, L.; Rapatz, G.; Kottakis, J. FOR-INT-03 Study Group. In patients with COPD, treatment with a combination of formoterol and ipratropium is more effective than a combination of salbutamol and ipratropium: a 3-week, randomized, double-blind, within-patient, multicenter study. Chest 2001, 119, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Stockley, R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J. 2005, 25, 1084–1106. [Google Scholar]
- Goldkorn, A.; Diotto, P.; Burgess, C.; Weatherall, M.; Holt, S.; Beasley, R.; Siebers, R. The pulmonary and extra-pulmonary effects of high-dose formoterol in COPD: a comparison with salbutamol. Respirology 2004, 9, 102–108. [Google Scholar]
- Sovrani, M.P.; Whale, C.I.; Tattersfield, A.E. A benefit-risk assessment of inhaled long-acting β2-agonists in the management of obstructive pulmonary disease. Drug Safety 2004, 27, 689–715. [Google Scholar]
- Lazarus, S.C.; Boushey, H.A.; Fahy, J.V.; Chinchilli, V.M.; Lemanske, R.F., Jr.; Sorkness, C.A.; Kraft, M.; Fish, J.E.; Peters, S.P.; Craig, T.; et al. Long-acting beta2-agonist monotherapy vs continued therapy with inhaled corticosteroids in patientswith persistent asthma: a randomized controlled trial. JAMA 2001, 285, 2583–2593. [Google Scholar]
- Gibson, P.G.; Powell, H.; Ducharme, F.M. Differential effects of maintenance long-acting beta-agonist and inhaled corticosteroid on asthma control and asthma exacerbations. J. Allergy Clin. Immunol. 2007, 119, 344–350. [Google Scholar]
- Woolcock, A.; Lundback, B.; Ringdal, N.; Jacques, L.A. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am. J. Respir. Crit. Care Med. 1996, 153, 1481–1488. [Google Scholar]
- Pauwels, R.A.; Löfdahl, C.G.; Postma, D.S.; Tattersfield, A.E.; O'Byrne, P.; Barnes, P.J.; Ullman, A. Effect of inhaled formoterol and budesonide on exacerbations of asthma. N. Engl. J. Med. 1997, 337, 1405–1411, [Erratum, N. Engl. J. Med.1998, 338, 139.]. [Google Scholar] [CrossRef] [PubMed]
- Giembycz, M.A.; Kaur, M.; Leigh, R.; Newton, R. A Holy Grail of asthma management: toward understanding how long-acting β2-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids. Br. J. Pharmacol. 2008, 153, 1090–1104. [Google Scholar]
- Papi, A.; Canonica, G.W.; Maestrelli, P.; Paggiaro, P.L.; Olivieri, D.; Pozzi, E.; Crimi, N.; Vignola, A.M.; Morelli, P.; Nicolini, G.; et al. Rescue use of beclomethasone and albuterol in a single inhaler for mild asthma. N. Engl. J. Med. 2007, 356, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Barger, G.; Dale, H.H. Chemical structure and sympathomimetic action of amines. J. Physiol. (Lond.) 1910, 41, 19–59. [Google Scholar] [PubMed]
- Benson, R.L.; Perlman, F. Clinical effects of epinephrine by inhalation. J. Allergy 1948, 19, 129–140. [Google Scholar]
- Davies, D.S. Metabolism of isoprenaline and other bronchodilator drugs in man and dog. Bull. Physiopathol. Respir. 1972, 8, 679–682. [Google Scholar]
- Speizer, F.E.; Doll, R.; Heaf, P. Observations on recent increase in mortality from asthma. Br. Med. J. 1968, 1, 335–339. [Google Scholar]
- Fraser, P.; Doll, R. Geographical variations in the epidemic of asthma deaths. Br. J. Prev. Soc. Med. 1971, 25, 34–36. [Google Scholar]
- Jackson, R.T. ; Beaglehole, R.; Rea, H.H; Sutherland, D.C. Mortality from asthma : a new epidemic in New Zealand. Br. Med. J. 1982, 285, 771–774. [Google Scholar]
- Speizer, F.E.; Doll, R.; Heaf, P.; Strang, L.B. Investigation into use of drugs preceding death from asthma. Br. Med. J. 1968, 1, 339–343. [Google Scholar]
- Conolly, M.E.; Davies, D.S.; Dollery, C.T.; George, C.F. Resistance to beta-adrenoceptor stimulants (a possible explanation for the rise in ashtma deaths). Br. J. Pharmacol. 1971, 43, 389–402. [Google Scholar]
- Wilson, J.D.; Sutherland, D.C.; Thomas, A.C. Has the change to beta-agonists combined with oral theophylline increased cases of fatal asthma? Lancet 1981, 1, 1235–1237. [Google Scholar] [PubMed]
- Grant, I.W.B. Asthma in New Zealand. Br. Med. J. 1983, 286, 374–377. [Google Scholar]
- Crane, J.; Flatt, A.; Jackson, R.; Ball, M.; Pearce, N.; Burgess, C.; Kwong, T.; Beasley, R. Prescribed fenoterol and death from asthma in New Zealand, 1981-83; Case-control study. Lancet 1989, 1, 917–922. [Google Scholar] [PubMed]
- Pearce, N.; Grainger, J.; Atkinson, M.; Crane, J.; Burgess, C.; Culling, C.; Windom, H.; Beasley, R. Case-control study of prescribed fenoterol and death from asthma in New Zealand, 1977-81. Thorax 1990, 45, 170–175. [Google Scholar]
- Spitzer, W.O.; Suissa, S.; Ernst, P.; Horwitz, R.I.; Habbick, B.; Cockcroft, D.; Boivin, J.F.; McNutt, M.; Buist, A.S.; Rebuck, A.S. The use of beta-agonists and the risk of death and near death from asthma. N. Engl. J. Med. 1992, 326, 501–506. [Google Scholar]
- Taylor, D.R.; Sears, M.R.; Herbison, G.P.; Flannery, E.M.; Print, C.G.; Lake, D.C.; Yates, D.M.; Lucas, M.K.; Li, Q. Regular inhaled beta agonist in asthma: effect on exacerbations and lung function. Thorax 1993, 48, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.M.; Sharp, S.J.; Vickers, M.R.; Frost, C.D.; Crompton, G.K.; Barnes, P.J.; Lee, T.H. Regular inhaled salbutamol and asthma control: the TRUST randomized trial. Lancet 2000, 355, 1675–1679. [Google Scholar]
- The British Guidelines on Asthma Management 1995 Review and Position Statement. Thorax 1997, 52 Suppl. 1, S1–S20. [CrossRef]
- Ullman, A.; Svedmyr, N. Salmeterol, a new long-acting inhaled beta2-adrenoceptor agonist: comparison with salbutamol in adult asthmatic patients. Thorax 1988, 43, 674–678. [Google Scholar]
- Faulds, D.; Hollingshead, L.M.; Goa, K.L. Formoterol. A review of its pharmacologic properties and therapeutic efficacy in reversible obstructive airways disease. Drugs 1991, 42, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Grove, A.; Lipworth, B.J. Bronchodilator subsensitivity to salbutamol after twice daily salmeterol in asthmatic patients. Lancet 1995, 346, 201–206. [Google Scholar]
- Newnham, D.M.; McDevitt, D.G.; Lipworth, B.J. Bronchodilator subsensitivity after chronic dosing with eformoterol in patients with asthma. Am. J. Med. 1994, 97, 29–37. [Google Scholar]
- Yates, D.H.; Kharitonov, S.A.; Barnes, P.J. An inhaled glucocorticoid does not prevent tolerance to the bronchoprotective effect of a long-acting β2-agonist. Am. J. Respir. Crit. Care Med. 1996, 154, 1603–1607. [Google Scholar]
- Arvidsson, P.; Larsson, S.; Löfdahl, C-G.; Melander, B.; Svedmyr, N.; Wåhlander, L. Inhaled formoterol during one year in asthma: a comparison with salbutamol. Eur. Respir. J. 1991, 4, 1168–1173. [Google Scholar] [PubMed]
- Castle, W.; Fuller, R.; Hall, J.; Palmer, J. Serevent nationwide surveillance study: comparison of salmeterol with salbutamol in asthmatic patients who require regular bronchodilator treatment. Br. Med. J. 1993, 306, 1034–1037. [Google Scholar]
- Nelson, H.S.; Weiss, S.T.; Bleecker, E.R.; Yancey, S.W.; Dorinsky, P.M. The Salmeterol Multicenter Asthma Research Trial. A comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006, 129, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.R.; Ayres, J.G.; Sturdy, P.M.; Bland, J.M.; Butland, B.K.; Peckitt, C.; Taylor, J.C.; Victor, C.R. Bronchodilator treatment and deaths from asthma: case-control study. Br. Med. J. 2005, 330, 117–123. [Google Scholar]
- Maringe, C.; Rickard, K.; DiSantostefano, R.; Davis, K.; Kiri, V. Concomitant use of long-acting β-agonists with inhaled corticosteroids among asthma patients in the UK primary care. Eur. Respir. J. 2007, 30 Suppl. 51, 3698. [Google Scholar]
- Food and Drug Administration Center for Drug Evaluation and Research. Summary Minutes of the Pulmonary-Allergy Drugs Advisory Committee. Available online: www.fda.gov/ohrms/dockets/ac/05/minutes/2005-4148M1_Final.pdf Last accessed: July 2008. Last updated: June 13, 2005..
- Mann, M.; Chowdhury, B.; Sullivan, E.; Nicklas, R; Anthracite, R.; Meyer, R.J. Serious asthma exacerbations in asthmatics treated with high-dose formoterol. Chest 2003, 124, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.; LaForce, C.; Friedman, B.; Sokol, W.; Till, D.; Della Cioppa, G.; van As, A. Formoterol, 24 µg bid, and serious asthma exacerbations: similar rates compared with formoterol, 12 µg bid, with and without extra doses taken on demand, and placebo. Chest 2006, 129, 27–38. [Google Scholar] [PubMed]
- Sears, M.R.; Ottosson, A.; Radner, F.; Suissa, S. .Long-acting β-agonists: a review of formoterol safety data from asthma clinical trials. Eur. Respir. J. 2009, 33, 21–32. [Google Scholar]
- Battram, C.; Charlton, S.J.; Cuenoud, B.; Dowling, M.R.; Fairhurst, R.A.; Farr, D.; Fozard, J.R.; Leighton-Davies, J.R.; Lewis, C.A.; McEvoy, L.; et al. In vitro and in vivo pharmacological characterization of 5-[(R)-2-(5,6-diethyl-indan-2-ylamino)-1-hydroxy-ethyl]-8-hydroxy-1H-quinolin-2-one (indacaterol), a novel inhaled β2 adrenoceptor agonist with a 24-h duration of action. J. Pharmacol. Exp. Ther. 2006, 317, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Sturton, R.G.; Trifilieff, A.; Nicholson, A.G.; Barnes, P.J. Pharmacological characterization of indacaterol, a novel once daily inhaled β2-adrenoceptor agonist, on small airways in human and rat precision-cut lung slices. J. Pharmacol. Exp. Ther. 2008, 324, 270–275. [Google Scholar] [PubMed]
- Beeh, K.M.; Derom, E.; Kanniess, F.; Cameron, R.; Higgins, M.; van As, A. Indacaterol, a novel inhaled β2-agonist, provides sustained 24 h bronchodilation in asthma. Eur. Respir. J. 2007, 29, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.A.; Löfdahl, C.G.; Postma, D.S.; Tattersfield, A.E.; O’Byrne, P.; Barnes, P.J.; Ullman, A. Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (FACET) International Study Group. N. Engl. J. Med. 1997, 337, 1405–1411. [Google Scholar] [PubMed]
- Price, D.; Dutchman, D.; Mawson, A.; Bodalia, B.; Duggan, S.; Todd, P. Early asthma control and maintenance with eformoterol following reduction of inhaled corticosteroid dose. Thorax 2002, 57, 791–798. [Google Scholar]
- Currie, G.P.; Lee, D.K.; Srivastava, P. Long-acting bronchodilator or leukotriene modifier as add-on therapy to inhaled corticosteroids in persistent asthma? Chest 2005, 128, 2954–2962. [Google Scholar] [PubMed]
- FDA Public Health Advisory. Serevent Diskus (salmeterol xinafoate inhalation powder), Advair Diskus (fluticasone propionate & salmeterol inhalation powder), Foradil Aerolizer (formoterol fumarate inhalation powder). Date last updated: May 15, 2006.. Available online: www.fda.gov/cder/drug/advisory/LABA.htm October 30, 2008.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Barisione, G.; Baroffio, M.; Crimi, E.; Brusasco, V. Beta-Adrenergic Agonists. Pharmaceuticals 2010, 3, 1016-1044. https://doi.org/10.3390/ph3041016
AMA Style
Barisione G, Baroffio M, Crimi E, Brusasco V. Beta-Adrenergic Agonists. Pharmaceuticals. 2010; 3(4):1016-1044. https://doi.org/10.3390/ph3041016
Chicago/Turabian StyleBarisione, Giovanni, Michele Baroffio, Emanuele Crimi, and Vito Brusasco. 2010. "Beta-Adrenergic Agonists" Pharmaceuticals 3, no. 4: 1016-1044. https://doi.org/10.3390/ph3041016