Opportunities to Target Specific Contractile Abnormalities with Smooth Muscle Protein Kinase Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
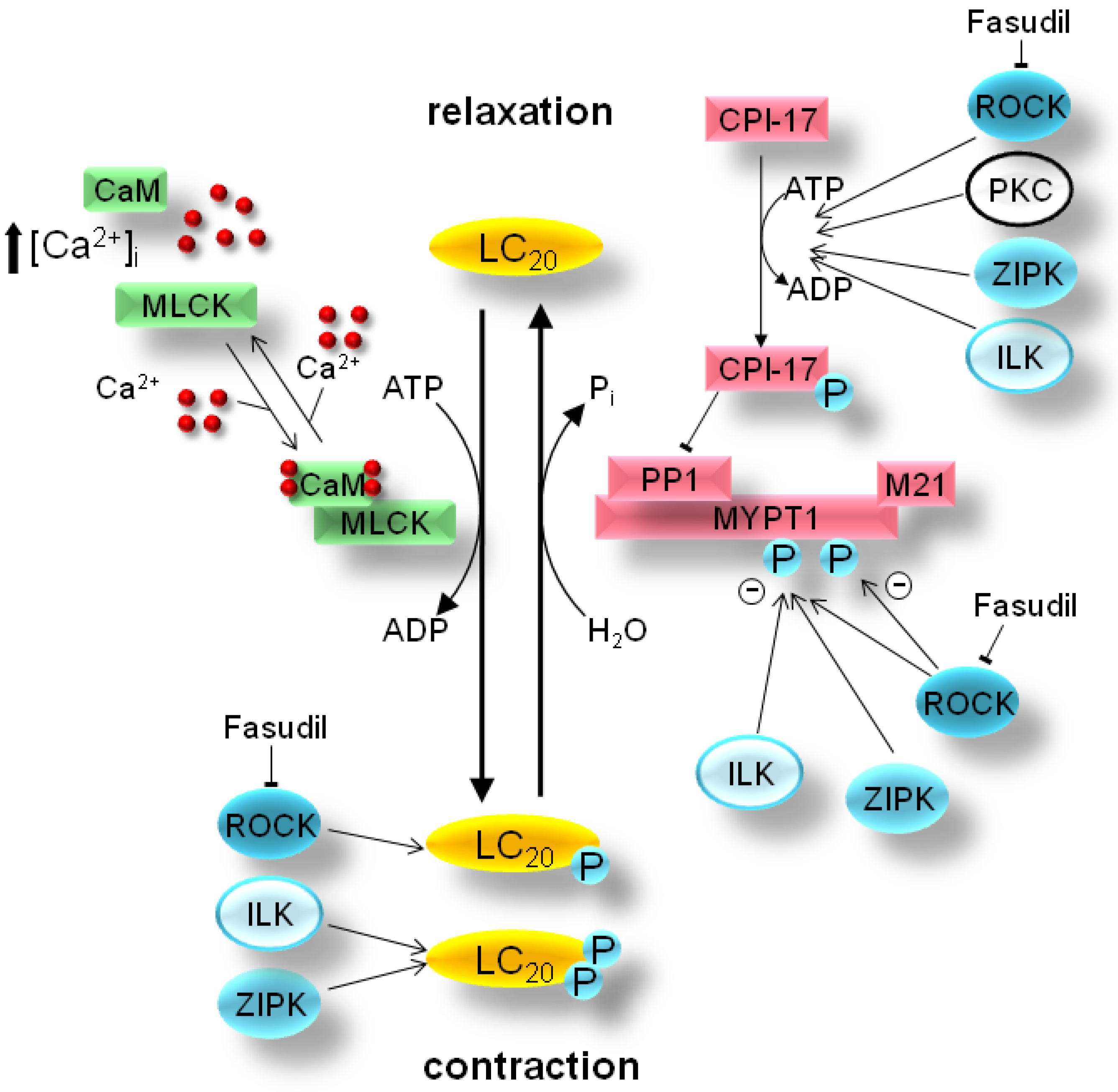
2. Mechanism of Smooth Muscle Contraction

3. Pathology of Smooth Muscle Contraction
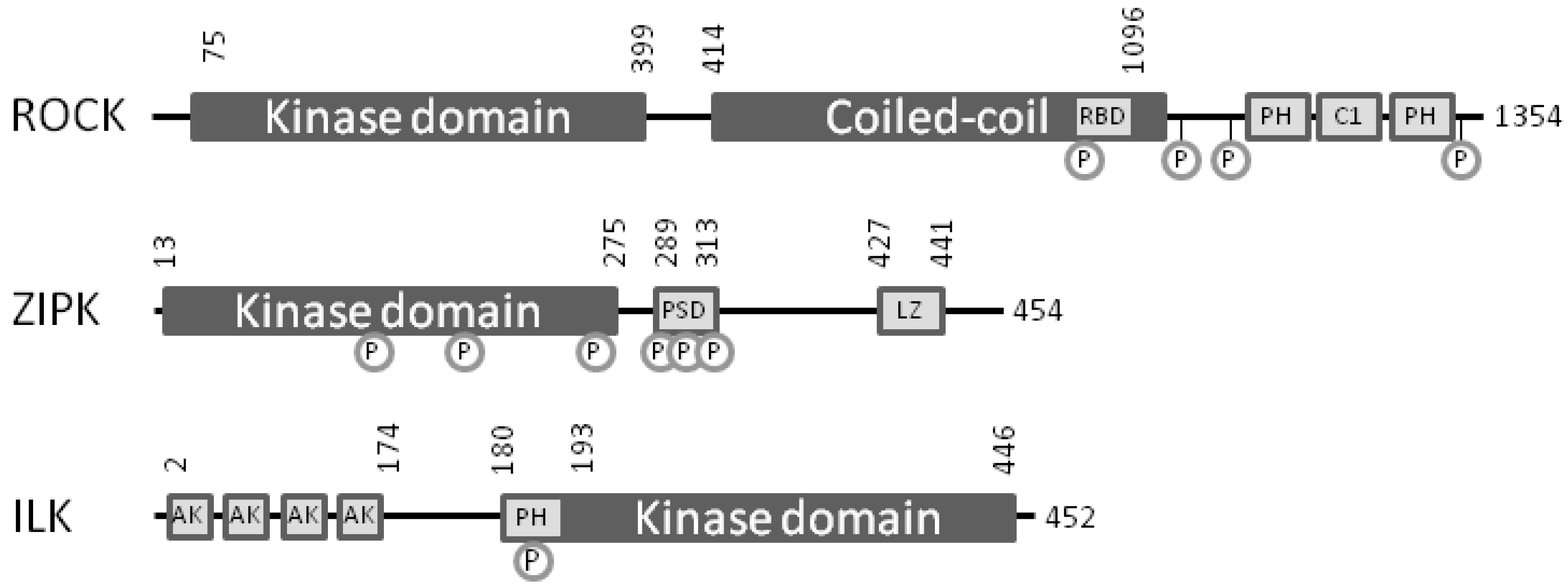
4. Bench-to-Bedside Success: The Story of the ROCK-Inhibitor Fasudil


5. Zipper-Interacting Protein Kinase

6. Integrin-Linked Protein Kinase

7. Conclusions and Outlook
Acknowledgements
References and Notes
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [PubMed]
- Gerthoffer, W.T. Signal-transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G849–G853. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006, 68, 345–374. [Google Scholar]
- Kim, H.R.; Appel, S.; Vetterkind, S.; Gangopadhyay, S.S.; Morgan, K.G. Smooth muscle signalling pathways in health and disease. J. Cell. Mol. Med. 2008, 12, 2165–2180. [Google Scholar]
- Chong, D.Y.; Michel, T. Pharmacology of vascular tone. In Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy; Golan, D.E., Tashjian, A.H., Jr., Armstrong, E.J., Armstrong, A.W., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; pp. 367–386. [Google Scholar]
- Matsumura, F.; Hartshorne, D.J. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008, 369, 149–156. [Google Scholar]
- Bradley, A.B.; Morgan, K.G. Alterations in cytoplasmic calcium sensitivity during porcine coronary artery contractions as detected by aequorin. J. Physiol. 1987, 385, 437–448. [Google Scholar]
- Wilson, D.P.; Sutherland, C.; Borman, M.A.; Deng, J.T.; MacDonald, J.A.; Walsh, M.P. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem. J. 2005, 392, 641–648. [Google Scholar]
- Harnett, K.M.; Biancani, P. Calcium-dependent and calcium-independent contractions in smooth muscles. Am. J. Med. 2003, 115, 24–30. [Google Scholar]
- Ikebe, M.; Hartshorne, D.J. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J. Biol. Chem. 1985, 260, 10027–10031. [Google Scholar]
- Murata-Hori, M.; Suizu, F.; Iwasaki, T.; Kikuchi, A.; Hosoya, H. ZIP kinase identified as a novel myosin regulatory light chain kinase in HeLa cells. FEBS Lett. 1999, 451, 81–84. [Google Scholar]
- Niiro, N.; Ikebe, M. Zipper-interacting protein kinase induces Ca(2+)-free smooth muscle contraction via myosin light chain phosphorylation. J. Biol. Chem. 2001, 276, 29567–29574. [Google Scholar]
- Borman, M.A.; MacDonald, J.A.; Haystead, T.A. Staurosporine inhibition of zipper-interacting protein kinase contractile effects in gastrointestinal smooth muscle. Biochem. Cell Biol. 2007, 85, 111–120. [Google Scholar]
- Hirano, K. Current topics in the regulatory mechanism underlying the Ca2+ sensitization of the contractile apparatus in vascular smooth muscle. J. Pharmacol. Sci. 2007, 104, 109–115. [Google Scholar]
- Seto, M.; Yano, K.; Sasaki, Y.; Azuma, H. Intimal hyperplasia enhances myosin phosphorylation in rabbit carotid artery. Exp. Mol. Pathol. 1993, 58, 1–13. [Google Scholar]
- Bulter, W.E.; Peterson, J.W.; Zervas, N.T.; Morgan, K.G. Intracellular calcium, myosin light chain phosphorylation, and contractile force in experimental cerebral vasospasm. Neurosurgery 1996, 38, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Seto, M.; Sasaki, Y.; London, S.; Luo, Z.; Mayberg, M. The time course of myosin light-chain phosphorylation in blood-induced vasospasm. Neurosurgery 1995, 36, 1178–1183. [Google Scholar]
- Katsumata, N.; Shimokawa, H.; Seto, M.; Kozai, T.; Yamawaki, T.; Kuwata, K.; Egashira, K.; Ikegaki, I.; Asano, T.; Sasaki, Y.; Takeshita, A. Enhanced myosin light chain phosphorylations as a central mechanism for coronary artery spasm in a swine model with interleukin-1beta. Circulation 1997, 96, 4357–4363. [Google Scholar]
- Hosseini, J.M.; Goldhill, J.M.; Bossone, C.; Pineiro-Carrero, V.; Shea-Donohue, T. Progressive alterations in circular smooth muscle contractility in TNBS-induced colitis in rats. Neurogastroenterol. Motil. 1999, 11, 347–356. [Google Scholar]
- Ozaki, H.; Hori, M.; Kinoshita, K.; Ohama, T. Intestinal dysmotility in inflammatory bowel disease: Mechanisms of the reduced activity of smooth muscle contraction. Inflammopharmacology 2005, 13, 103–111. [Google Scholar]
- Vrees, M.D.; Pricolo, V.E.; Potenti, F.M.; Cao, W. Abnormal motility in patients with ulcerative colitis: The role of inflammatory cytokines. Arch. Surg. 2002, 137, 439–446. [Google Scholar]
- Akiho, H.; Deng, Y.; Blennerhassett, P.; Kanbayashi, H.; Collins, S.M. Mechanisms underlying the maintenance of muscle hypercontractility in a model of postinfective gut dysfunction. Gastroenterology 2005, 129, 131–141. [Google Scholar]
- Cook, T.A.; Brading, A.F.; Mortensen, N.J. Abnormal contractile properties of rectal smooth muscle in chronic ulcerative colitis. Aliment. Pharmacol. Ther. 2000, 14, 1287–1294. [Google Scholar]
- Olson, M.F. Applications for ROCK kinase inhibition. Curr. Opin. Cell Biol. 2008, 20, 242–248. [Google Scholar]
- Schmandke, A.; Schmandke, A.; Strittmatter, S.M. ROCK and Rho: Biochemistry and neuronal functions of Rho-associated protein kinases. Neuroscientist 2007, 13, 454–469. [Google Scholar]
- Lowery, D.M.; Clauser, K.R.; Hjerrild, M.; Lim, D.; Alexander, J.; Kishi, K.; Ong, S.E.; Gammeltoft, S.; Carr, S.A.; Yaffe, M.B. Proteomic screen defines the Polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO J. 2007, 26, 2262–2273. [Google Scholar]
- Kawano, Y.; Fukata, Y.; Oshiro, N.; Amano, M.; Nakamura, T.; Ito, M.; Matsumura, F.; Inagaki, M.; Kaibuchi, K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J. Cell Biol. 1999, 147, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar]
- Eto, M.; Wong, L.; Yazawa, M.; Brautigan, D.L. Inhibition of myosin/moesin phosphatase by expression of the phosphoinhibitor protein CPI-17 alters microfilament organization and retards cell spreading. Cell Motil. Cytoskeleton 2000, 46, 222–234. [Google Scholar]
- Doggrell, S.A. Rho-kinase inhibitors show promise in pulmonary hypertension. Expert Opin. Investig. Drugs 2005, 14, 1157–1159. [Google Scholar]
- Huentelman, M.J.; Stephan, D.A.; Talboom, J.; Corneveaux, J.J.; Reiman, D.M.; Gerber, J.D.; Barnes, C.A.; Alexander, G.E.; Reiman, E.M.; Bimonte-Nelson, H.A. Peripheral delivery of a ROCK inhibitor improves learning and working memory. Behav. Neurosci. 2009, 123, 218–223. [Google Scholar]
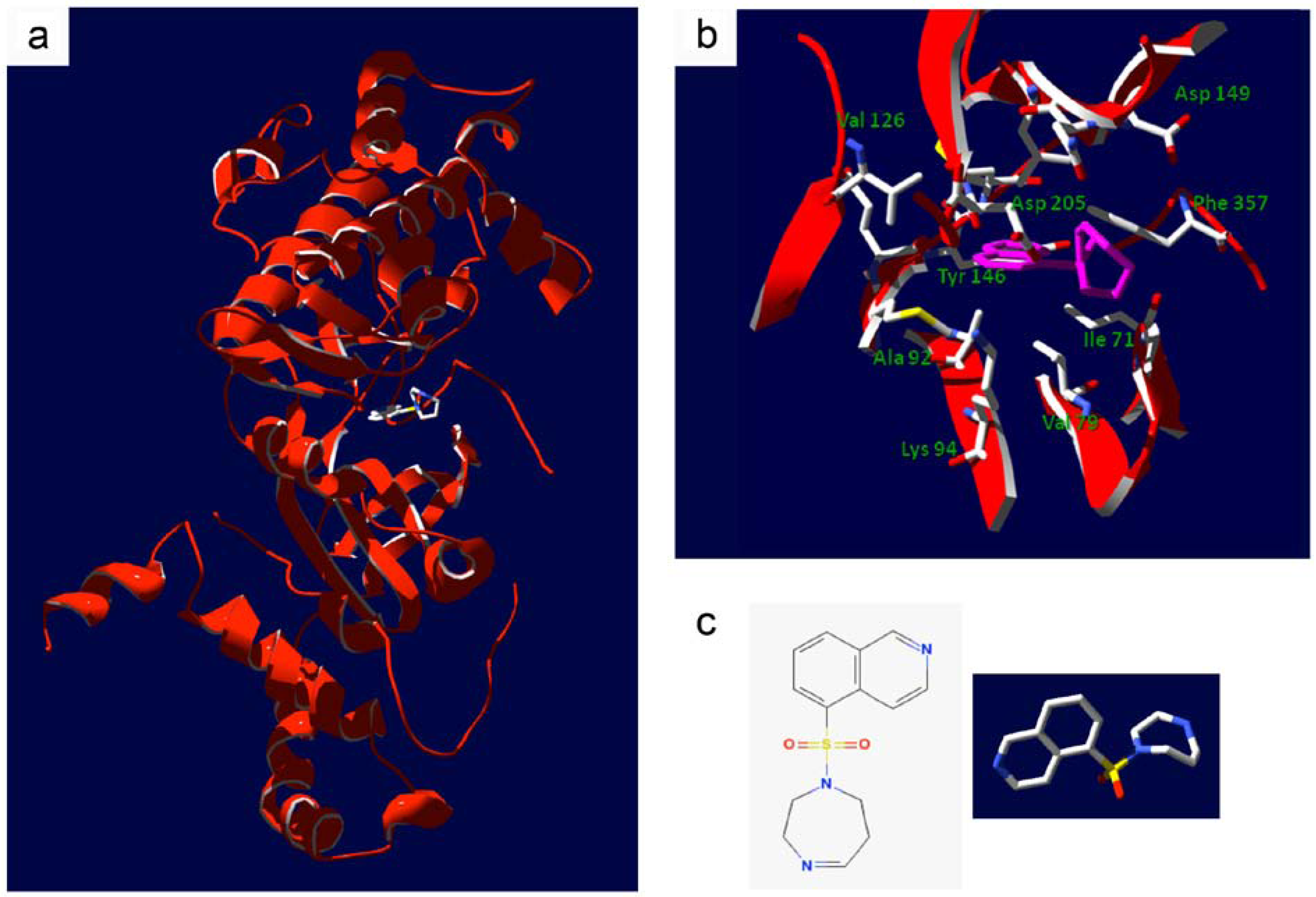
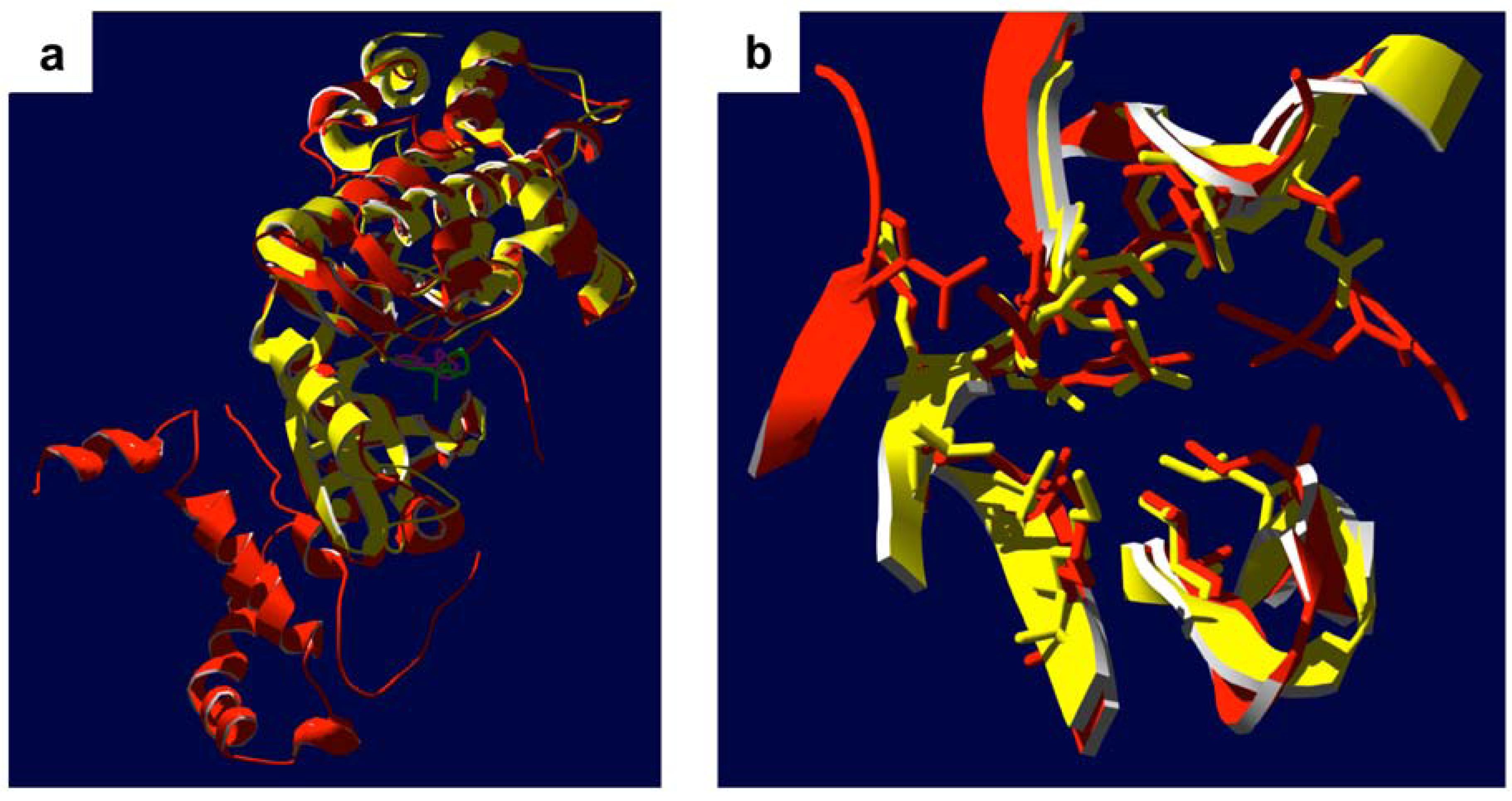
- Breitenlechner, C.; Gassel, M.; Hidaka, H.; Kinzel, V.; Huber, R.; Engh, R.A.; Bossemeyer, D. Protein kinase A in complex with Rho-kinase inhibitors Y-27632, Fasudil, and H-1152P: structural basis of selectivity. Structure 2003, 11, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kasa, M.; Amano, M.; Kaibuchi, K.; Hakoshima, T. Molecular mechanism for the regulation of rho-kinase by dimerization and its inhibition by fasudil. Structure 2006, 14, 589–600. [Google Scholar]
- Takayasu, M.; Suzuki, Y.; Shibuya, M.; Asano, T.; Kanamori, M.; Okada, T.; Kageyama, N.; Hidaka, H. The effects of HA compound calcium antagonists on delayed cerebral vasospasm in dogs. J. Neurosurg. 1986, 65, 80–85. [Google Scholar]
- Vicari, R.M.; Chaitman, B.; Keefe, D.; Smith, W.B.; Chrysant, S.G.; Tonkon, M.J.; Bittar, N.; Weiss, R.J.; Morales-Ballejo, H.; Thadani, U. Efficacy and safety of fasudil in patients with stable angina: A double-blind, placebo-controlled, phase 2 trial. J. Am. Coll. Cardiol. 2005, 46, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.; Ohtomo, E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar]
- Nohria, A.; Grunert, M.E.; Rikitake, Y.; Noma, K.; Prsic, A.; Ganz, P.; Liao, J.K.; Creager, M.A. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ. Res. 2006, 99, 1426–1432. [Google Scholar]
- Noma, K.; Higashi, Y.; Jitsuiki, D.; Hara, K.; Kimura, M.; Nakagawa, K.; Goto, C.; Oshima, T.; Yoshizumi, M.; Chayama, K. Smoking activates rho-kinase in smooth muscle cells of forearm vasculature in humans. Hypertension 2003, 41, 1102–1105. [Google Scholar]
- Noma, K.; Goto, C.; Nishioka, K.; Jitsuiki, D.; Umemura, T.; Ueda, K.; Kimura, M.; Nakagawa, K.; Oshima, T.; Chayama, K.; et al. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J. Am. Coll. Cardiol. 2007, 49, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kondoh, Y.; Satoh, Y.; Watahiki, Y.; Yokoyama, E.; Yuya, H.; Hirata, Y.; Shishido, F.; Hatazawa, J.; Kanno, I.; et al. Effects of fasudil hydrochloride on cerebral blood flow in patients with chronic cerebral infarction. Clin. Neuropharmacol. 1993, 16, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, K.; Ito, A.; Fukumoto, Y.; Matoba, T.; Shiose, A.; Nishida, T.; Masuda, M.; Morita, S.; Shimokawa, H. Usefulness of fasudil, a Rho-kinase inhibitor, to treat intractable severe coronary spasm after coronary artery bypass surgery. J. Cardiovasc. Pharmacol. 2004, 44, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Mohri, M.; Shimokawa, H.; Hirakawa, Y.; Masumoto, A.; Takeshita, A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J. Am. Coll. Cardiol. 2003, 41, 15–19. [Google Scholar]
- Eldawoody, H.; Shimizu, H.; Kimura, N.; Saito, A.; Nakayama, T.; Takahashi, A.; Tominaga, T. Fasudil, a Rho-kinase inhibitor, attenuates induction and progression of cerebral aneurysms: Experimental study in rats using vascular corrosion casts. Neurosci. Lett. 2010, 470, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Martin-McNulty, B.; da Cunha, V.; Vincelette, J.; Lu, X.; Feng, Q.; Halks-Miller, M.; Mahmoudi, M.; Schroeder, M.; Subramanyam, B.; et al. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation 2005, 111, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Ikesugi, K.; Nishio, M.; Sugimoto, M.; Sasoh, M.; Hidaka, H.; Uji, Y. The effect of the Rho-associated protein kinase inhibitor, HA-1077, in the rabbit ocular hypertension model induced by water loading. Curr. Eye. Res. 2009, 34, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Hirooka, Y.; Masumoto, A.; Ito, K.; Kimura, Y.; Inokuchi, K.; Tagawa, T.; Shimokawa, H.; Takeshita, A.; Sunagawa, K. Rho-kinase inhibitor improves increased vascular resistance and impaired vasodilation of the forearm in patients with heart failure. Circulation 2005, 111, 2741–2747. [Google Scholar]
- Bussemaker, E.; Herbrig, K.; Pistrosch, F.; Palm, C.; Passauer, J. Role of rho-kinase in the regulation of vascular tone in hypertensive renal transplant recipients. Atherosclerosis 2009, 207, 567–572. [Google Scholar]
- Masumoto, A.; Hirooka, Y.; Shimokawa, H.; Hironaga, K.; Setoguchi, S.; Takeshita, A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001, 38, 1307–1310. [Google Scholar]
- Sasaki, Y.; Suzuki, M.; Hidaka, H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol. Ther. 2002, 93, 225–232. [Google Scholar]
- Nichols, R.; Dzamko, N.; Hutti, J.; Cantley, L.; Deak, M.; Moran, J.; Bamborough, P.; Reith, A.; Alessi, D. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem. J. 2009, 23, 47–60. [Google Scholar]
- Lohn, M.; Plettenburg, O.; Ivashchenko, Y.; Kannt, A.; Hofmeister, A.; Kadereit, D.; Schaefer, M.; Linz, W.; Kohlmann, M.; Herbert, J.M.; et al. Pharmacological characterization of SAR407899, a novel Rho-kinase inhibitor. Hypertension 2009, 54, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, J.S.; Badejo, A.M., Jr.; Casey, D.B.; Murthy, S.N.; Kadowitz, P.J. Analysis of pulmonary vasodilator responses to SB-772077-B [4-(7-((3-amino-1-pyrrolidinyl)carbonyl)-1-ethyl-1H-imidazo(4,5-c)pyridin- 2-yl)-1,2,5-oxadiazol-3-amine], a novel aminofurazan-based Rho kinase inhibitor. J. Pharmacol. Exp. Ther. 2009, 330, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Ihara, E.; Moffat, L.D.; Ostrander, J.M.; Walsh, M.P.; MacDonald, J.A. Characterization of protein kinase pathways responsible for Ca2+ sensitization in rat ileal longitudinal smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G699–G710. [Google Scholar]
- Kim, N.; Cao, W.; Song, I.S.; Kim, C.Y.; Harnett, K.M.; Cheng, L.; Walsh, M.P.; Biancani, P. Distinct kinases are involved in contraction of cat esophageal and lower esophageal sphincter smooth muscles. Am. J. Physiol. Cell Physiol. 2004, 287, C384–C394. [Google Scholar]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [PubMed]
- Ihara, E.; MacDonald, J.A. The regulation of smooth muscle contractility by zipper-interacting protein kinase. Can. J. Physiol. Pharmacol. 2007, 85, 79–87. [Google Scholar]
- Haystead, T. A. ZIP kinase, a key regulator of myosin protein phosphatase 1. Cell Signal. 2005, 17, 1313–1322. [Google Scholar]
- Komatsu, S.; Ikebe, M. ZIP kinase is responsible for the phosphorylation of myosin II and necessary for cell motility in mammalian fibroblasts. J. Cell Biol. 2004, 165, 243–254. [Google Scholar]
- Borman, M.A.; MacDonald, J.A.; Muranyi, A.; Hartshorne, D.J.; Haystead, T.A. Smooth muscle myosin phosphatase-associated kinase induces Ca2+ sensitization via myosin phosphatase inhibition. J. Biol. Chem. 2002, 277, 23441–23446. [Google Scholar]
- MacDonald, J.A.; Borman, M.A.; Muranyi, A.; Somlyo, A.V.; Hartshorne, D.J.; Haystead, T.A. Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 2419–2424. [Google Scholar]
- Kawai, T.; Matsumoto, M.; Takeda, K.; Sanjo, H.; Akira, S. ZIP kinase, a novel serine/threonine kinase which mediates apoptosis. Mol. Cell Biol. 1998, 18, 1642–1651. [Google Scholar]
- Kogel, D.; Plottner, O.; Landsberg, G.; Christian, S.; Scheidtmann, K.H. Cloning and characterization of Dlk, a novel serine/threonine kinase that is tightly associated with chromatin and phosphorylates core histones. Oncogene 1998, 17, 2645–2654. [Google Scholar]
- Endo, A.; Surks, H.K.; Mochizuki, S.; Mochizuki, N.; Mendelsohn, M.E. Identification and characterization of zipper-interacting protein kinase as the unique vascular smooth muscle myosin phosphatase-associated kinase. J. Biol. Chem. 2004, 279, 42055–42061. [Google Scholar]
- MacDonald, J.A.; Eto, M.; Borman, M.A.; Brautigan, D.L.; Haystead, T.A. Dual Ser and Thr phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by MYPT-associated kinase. FEBS Lett. 2001, 493, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Ihara, E.; Edwards, E.; Borman, M.A.; Wilson, D.P.; Walsh, M.P.; MacDonald, J.A. Inhibition of zipper-interacting protein kinase function in smooth muscle by a myosin light chain kinase pseudosubstrate peptide. Am. J. Physiol. Cell Physiol. 2007, 292, C1951–C1959. [Google Scholar]
- Ihara, E.; Moffat, L.; Borman, M.A.; Amon, J.E.; Walsh, M.P.; MacDonald, J.A. Ca2+-independent contraction of longitudinal ileal smooth muscle is potentiated by a zipper-interacting protein kinase pseudosubstrate peptide. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G361–G370. [Google Scholar]
- Filippakopoulos, P.; Rellos, P.; Eswaran, J.; Fedorov, O.; Berridge, G.; Niesen, F.; Bracher, F.; Huber, K.; Pike, A.C.W.; Roos, A.; Von Delft, F.; et al. Crystal structure of human death associated protein kinase 3 (Dapk3) in complex with a beta-carboline ligand. PDB Deposition 2009, 3BHY. [Google Scholar]
- Shohat, G.; Spivak-Kroizman, T.; Cohen, O.; Bialik, S.; Shani, G.; Berrisi, H.; Eisenstein, M.; Kimchi, A. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J. Biol. Chem. 2001, 276, 47460–47467. [Google Scholar]
- Shani, G.; Henis-Korenblit, S.; Jona, G.; Gileadi, O.; Eisenstein, M.; Ziv, T.; Admon, A.; Kimchi, A. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 2001, 20, 1099–1113. [Google Scholar]
- Stull, J.T.; Tansey, M.G.; Tang, D.C.; Word, R.A.; Kamm, K.E. Phosphorylation of myosin light chain kinase: A cellular mechanism for Ca2+ desensitization. Mol. Cell. Biochem. 1993, 127, 229–237. [Google Scholar]
- Sato, N.; Kamada, N.; Muromoto, R.; Kawai, T.; Sugiyama, K.; Watanabe, T.; Imoto, S.; Sekine, Y.; Ohbayashi, N.; Ishida, M.; et al. Phosphorylation of threonine-265 in Zipper-interacting protein kinase plays an important role in its activity and is induced by IL-6 family cytokines. Immunol. Lett. 2006, 103, 127–134. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Ray, P.S.; Arif, A.; Brady, A.K.; Kinter, M.; Fox, P.L. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol. Cell 2008, 32, 371–382. [Google Scholar]
- Graves, P.R.; Winkfield, K.M.; Haystead, T.A. Regulation of zipper-interacting protein kinase activity in vitro and in vivo by multisite phosphorylation. J. Biol. Chem. 2005, 280, 9363–9374. [Google Scholar] [PubMed]
- Shani, G.; Marash, L.; Gozuacik, D.; Bialik, S.; Teitelbaum, L.; Shohat, G.; Kimchi, A. Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol. Cell Biol. 2004, 24, 8611–8626. [Google Scholar]
- Ohbayashi, N.; Okada, K.; Kawakami, S.; Togi, S.; Sato, N.; Ikeda, O.; Kamitani, S.; Muromoto, R.; Sekine, Y.; Kawai, T.; et al. Physical and functional interactions between ZIP kinase and UbcH5. Biochem. Biophys. Res. Commun. 2008, 372, 708–712. [Google Scholar]
- Okamoto, M.; Takayama, K.; Shimizu, T.; Ishida, K.; Takahashi, O.; Furuya, T. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J. Med. Chem. 2009, 52, 7323–7327. [Google Scholar]
- Okamoto, M.; Takayama, K.; Shimizu, T.; Muroya, A.; Furuya, T. Structure-activity relationship of novel DAPK inhibitors identified by structure-based virtual screening. Bioorg. Med. Chem. 2010, 18, 2728–2734. [Google Scholar]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Peptides targeting protein kinases: Strategies and implications. Physiology 2006, 21, 411–418. [Google Scholar]
- Shiga, K.; Takayama, K.; Futaki, S.; Hutti, J.E.; Cantley, L.C.; Ueki, K.; Ono, Y.; Mukai, H. Development of an intracellularly acting inhibitory peptide selective for PKN. Biochem. J. 2010, 425, 445–543. [Google Scholar]
- Kemp, B.E.; Parker, M.W.; Hu, S.; Tiganis, T.; House, C. Substrate and pseudosubstrate interactions with protein kinases: Determinants of specificity. Trends Biochem. Sci. 1994, 19, 440–444. [Google Scholar]
- Hannigan, G.E.; Leung-Hagesteijn, C.; Fitz-Gibbon, L.; Coppolino, M.G.; Radeva, G.; Filmus, J.; Bell, J.C.; Dedhar, S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 1996, 379, 91–96. [Google Scholar]
- Deng, J.T.; Van Lierop, J.E.; Sutherland, C.; Walsh, M.P. Ca2+-independent smooth muscle contraction—A novel function for integrin-linked kinase. J. Biol. Chem. 2001, 276, 16365–16373. [Google Scholar]
- McDonald, P.C.; Fielding, A.B.; Dedhar, S. Integrin-linked kinase-essential roles in physiology and cancer biology. J. Cell Sci. 2008, 121, 3121–3132. [Google Scholar]
- Ho, B.; Bendeck, M.P. Integrin linked kinase (ILK) expression and function in vascular smooth muscle cells. Cell Adh. Migr. 2009, 3, 174–176. [Google Scholar]
- Lal, H.; Verma, S.K.; Foster, D.M.; Golden, H.B.; Reneau, J.C.; Watson, L.E.; Singh, H.; Dostal, D.E. Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci. 2009, 14, 2307–2334. [Google Scholar]
- Hannigan, G.; Troussard, A.A.; Dedhar, S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 2005, 5, 51–63. [Google Scholar]
- Boudeau, J.; Miranda-Saavedra, D.; Barton, G.J.; Alessi, D.R. Emerging roles of pseudokinases. Trends Cell Biol. 2006, 16, 443–452. [Google Scholar]
- Fukuda, K.; Gupta, S.; Chen, K.; Wu, C.; Qin, J. The pseudoactive site of ILK is essential for its binding to alpha-Parvin and localization to focal adhesions. Mol. Cell 2009, 36, 819–830. [Google Scholar]
- Yau, C.Y.F.; Wheeler, J.J.; Sutton, K.L.; Hedley, D.W. Inhibition of integrin-linked kinase by a selective small molecule inhibitor, QLT0254, inhibits the PI3K/PKB/mTOR, Stat3, and FKHR pathways and tumor growth, and enhances Gemcitabine-induced apoptosis in human orthotopic primary pancreatic cancer xenografts. Cancer Res. 2005, 65, 1497–1504. [Google Scholar] [CrossRef]
- Persad, S.; Attwell, S.; Gray, V.; Mawji, N.; Deng, J.T.; Leung, D.; Yan, J.; Sanghera, J.; Walsh, M.P.; Dedhar, S. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: Critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 2001, 276, 27462–27469. [Google Scholar]
- Muranyi, A.; MacDonald, J.A.; Deng, J.T.; Wilson, D.P.; Haystead, T.A.; Walsh, M.P.; Erdodi, F.; Kiss, E.; Wu, Y.; Hartshorne, D.J. Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem. J. 2002, 366, 211–216. [Google Scholar]
- Deng, J.T.; Sutherland, C.; Brautigan, D.L.; Eto, M.; Walsh, M.P. Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem. J. 2002, 367, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Persad, S.; Attwell, S.; Gray, V.; Delcommenne, M.; Troussard, A.; Sanghera, J.; Dedhar, S. Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3207–3212. [Google Scholar]
- Younes, M.N.; Kim, S.; Yigitbasi, O.G.; Yazici, Y.D.; Jasser, S.A.; Bucana, C.D.; El-Naggar, A.K.; Mills, G.B.; Myers, J.N. Integrin-linked kinase is a potential therapeutic target for anaplastic thyroid cancer. Mol. Cancer Ther. 2005, 4, 1146–1156. [Google Scholar]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar] [PubMed]
- Nakamura, K.; Koga, Y.; Sakai, H.; Homma, K.; Ikebe, M. cGMP-dependent relaxation of smooth muscle is coupled with the change in the phosphorylation of myosin phosphatase. Circ. Res. 2007, 101, 712–722. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ulke-Lemée, A.; MacDonald, J.A. Opportunities to Target Specific Contractile Abnormalities with Smooth Muscle Protein Kinase Inhibitors. Pharmaceuticals 2010, 3, 1739-1760. https://doi.org/10.3390/ph3061739
Ulke-Lemée A, MacDonald JA. Opportunities to Target Specific Contractile Abnormalities with Smooth Muscle Protein Kinase Inhibitors. Pharmaceuticals. 2010; 3(6):1739-1760. https://doi.org/10.3390/ph3061739
Chicago/Turabian StyleUlke-Lemée, Annegret, and Justin A. MacDonald. 2010. "Opportunities to Target Specific Contractile Abnormalities with Smooth Muscle Protein Kinase Inhibitors" Pharmaceuticals 3, no. 6: 1739-1760. https://doi.org/10.3390/ph3061739