Antiepileptic Drug Discovery and Development: What Have We Learned and Where Are We Going?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Improvements in Currently Marketed AEDs
2.1. 1st Generation: Flupirtin; 2nd Generation: Retigabine

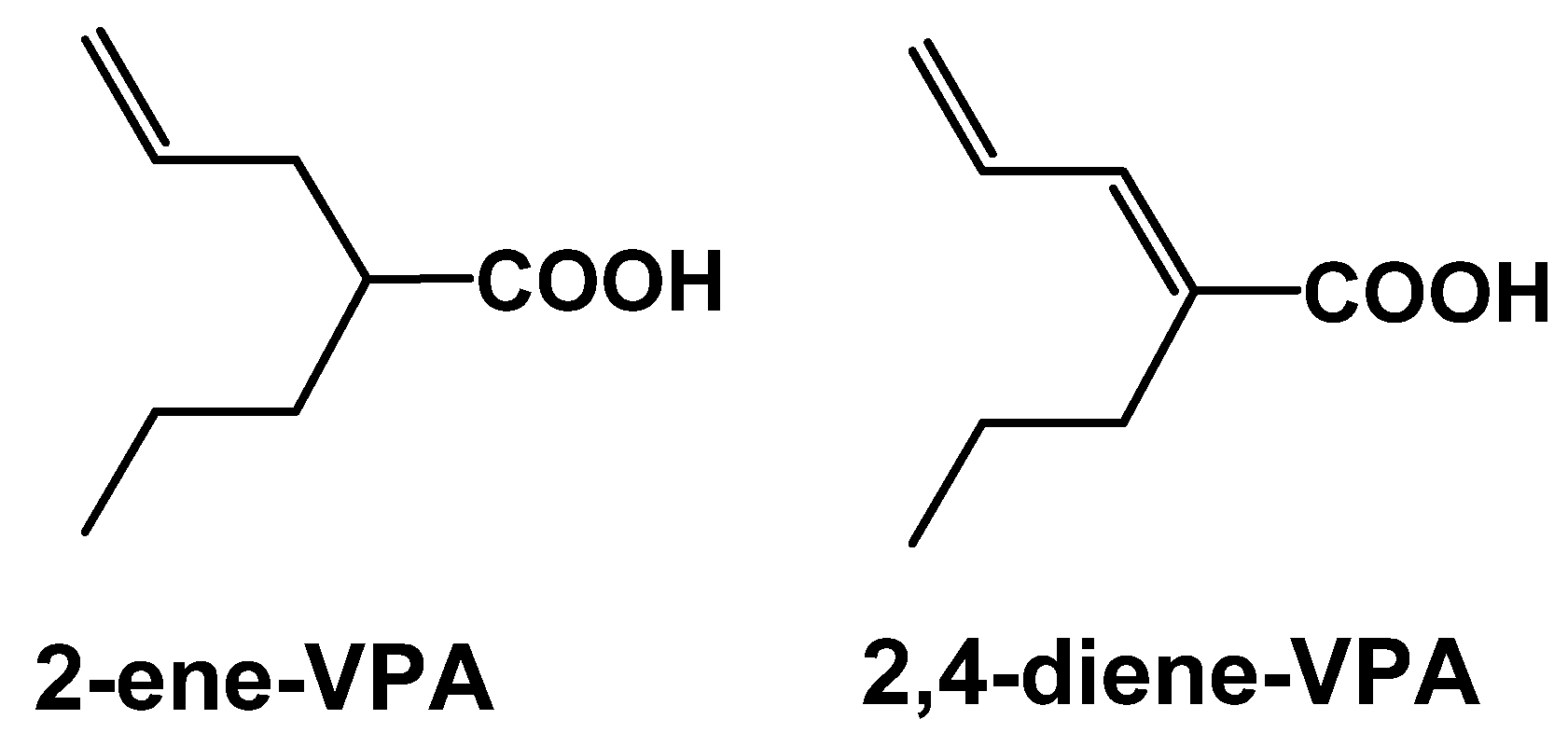
2.2. 1st Generation: Valproic acid (VPA); 2nd Generation: valnoctamide (VCD), propylisopropylacetamide (PID), 2,2,3,3-tetramethylcyclopropylcarboxcylic acid (TMCA), N-methyl-2,2,3,3-tetramethylcyclo-propanecarboxamide (MTMCD), 2,2,3,3 tetramethylcyclopropanecarbonylurea (TMCU), valrocemide


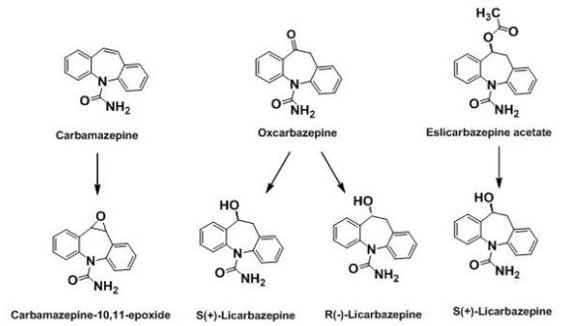
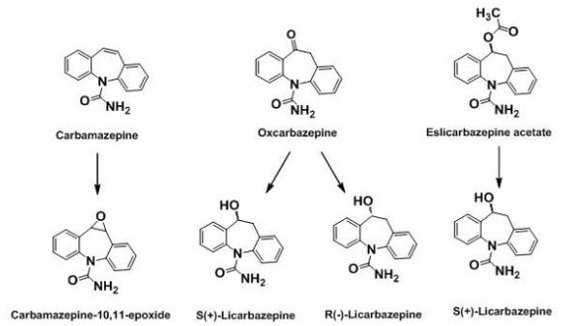
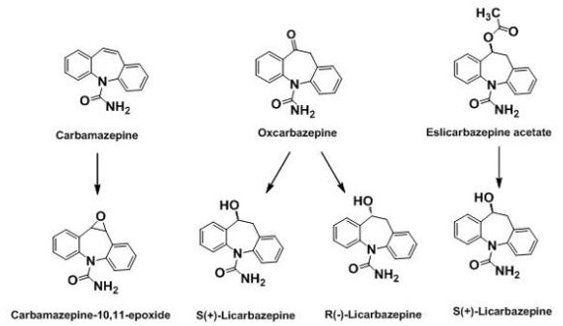
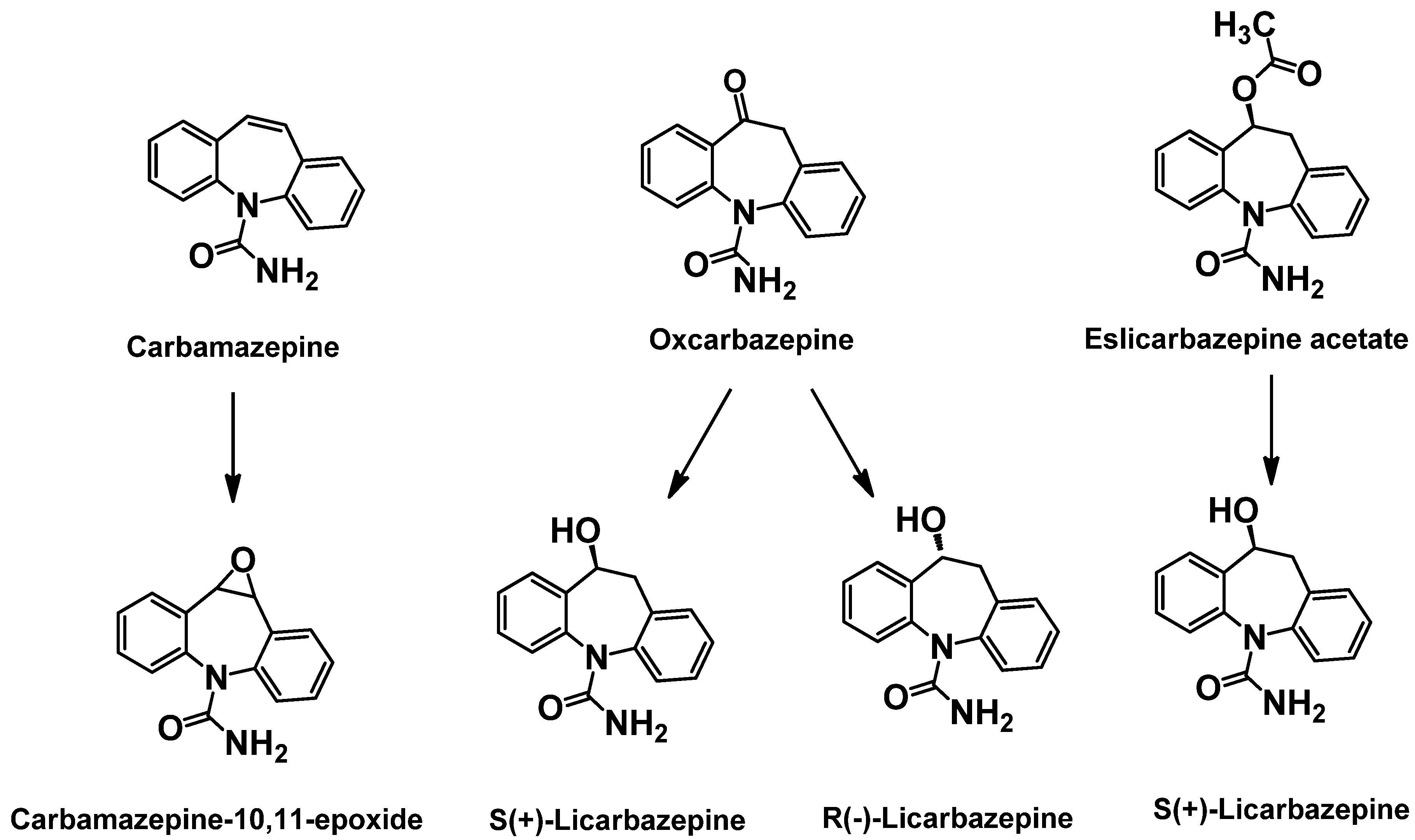
2.3. 1st Generation: Carbamazepine; 2nd Generation: Oxcarbazepine; 3rd Generation: Eslicarbazepine acetate (ESL)

3. Mechanism Based Drug Discovery
3.1. Molecular Target: SV2A—1st Generation: Levetiracetam; 2nd Generation: Brivaracetam

3.2. Molecular Target: α2δ voltage-gated calcium channel subunit—1st Generation: Gabapentin; 2nd Generation: Pregabalin

3.3. Molecular Target: M-current (KCNQ2/Q3, Kv7.2/Kv7.3)—ICA-105665 (Icagen)
4. Conclusions and Future Directions
References
- Fisher, R.S.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J., Jr. Epileptic seizures and epilepsy: Definitions proposed by the international league against epilepsy (ilae) and the international bureau for epilepsy (ibe). Epilepsia 2005, 46, 470–472. [Google Scholar]
- Epilepsy, A. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981, 22, 489–501. [Google Scholar]
- Kwan, P.; Sander, J.W. The natural history of epilepsy: An epidemiological view. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1376–1381. [Google Scholar]
- French, J.A.; Kanner, A.M.; Bautista, J.; Abou-Khalil, B.; Browne, T.; Harden, C.L.; Theodore, W.H.; Bazil, C.; Stern, J.; Schachter, S.C.; Bergen, D.; Hirtz, D.; Montouris, G.D.; Nespeca, M.; Gidal, B.; Marks, W.J., Jr.; Turk, W.R.; Fischer, J.H.; Bourgeois, B.; Wilner, A.; Faught, R.E., Jr.; Sachdeo, R.C.; Beydoun, A.; Glauser, T.A. Efficacy and tolerability of the new antiepileptic drugs i: Treatment of new onset epilepsy: Report of the therapeutics and technology assessment subcommittee and quality standards subcommittee of the american academy of neurology and the american epilepsy society. Neurology 2004, 62, 1252–1260. [Google Scholar] [PubMed]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar]
- Mohanraj, R.; Brodie, M.J. Measuring the efficacy of antiepileptic drugs. Seizure 2003, 12, 413–443. [Google Scholar]
- Rogawski, M.A.; Loscher, W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. [Google Scholar]
- Seaman, C.A.; Sheridan, P.H.; Engel, J.; Molliere, M.; Narang, P.K.; Nice, F.J. Flupirtine. In New anticonvulsant drugs; Meldrum, B.S., Porter, R.J., Eds.; Libbey: London, UK, 1986; pp. 135–146. [Google Scholar]
- Seydel, J.K.; Schaper, K.J.; Coats, E.A.; Cordes, H.P.; Emig, P.; Engel, J.; Kutscher, B.; Polymeropoulos, E.E. Synthesis and quantitative structure-activity relationships of anticonvulsant 2,3,6-triaminopyridines. J. Med. Chem. 1994, 37, 3016–3022. [Google Scholar]
- Rostock, A.; Tober, C.; Rundfeldt, C.; Bartsch, R.; Engel, J.; Polymeropoulos, E.E.; Kutscher, B.; Loscher, W.; Honack, D.; White, H.S.; Wolf, H.H. D-23129: A new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996, 23, 211–223. [Google Scholar]
- Srivastava, A.K.; White, H.S. Retigabine decreases behavioral and electrographic seizures in the lamotrogine-resistant amygdal kindled rat model of pharmacoresistant epilepsy. Epilepsia 2005, 46 (Suppl. 8), 217–218. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Partiot, A.; Sachdeo, R.; Nohria, V.; Alves, W.M. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology 2007, 68, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the ninth eilat conference (eilat ix). Epilepsy Res. 2009, 83, 1–43. [Google Scholar]
- Meunier, H.; Carraz, G.; Neunier, Y.; Eymard, P.; Aimard, M. Pharmacodynamic properties of n-dipropylacetic acid. Therapie 1963, 18, 435–438. [Google Scholar]
- Emrich, H.M.; von Zerssen, D.; Kissling, W.; Moller, H.J. Therapeutic effect of valproate in mania. Am. J. Psychiatry 1981, 138, 256. [Google Scholar]
- Calabresi, P.; Galletti, F.; Rossi, C.; Sarchielli, P.; Cupini, L.M. Antiepileptic drugs in migraine: From clinical aspects to cellular mechanisms. Trends Pharmacol.Sci. 2007, 28, 188–195. [Google Scholar]
- Terbach, N.; Williams, R.S. Structure-function studies for the panacea, valproic acid. Biochem. Soc. Trans. 2009, 37, 1126–1132. [Google Scholar]
- Bialer, M.; Yagen, B. Valproic acid: Second generation. Neurotherapeutics 2007, 4, 130–137. [Google Scholar]
- Isoherranen, N.; White, H.S.; Klein, B.D.; Roeder, M.; Woodhead, J.H.; Schurig, V.; Yagen, B.; Bialer, M. Pharmacokinetic-pharmacodynamic relationships of (2s,3s)-valnoctamide and its stereoisomer (2r,3s)-valnoctamide in rodent models of epilepsy. Pharm. Res. 2003, 20, 1293–1301. [Google Scholar]
- Isoherranen, N.; Yagen, B.; Woodhead, J.H.; Spiegelstein, O.; Blotnik, S.; Wilcox, K.S.; Finnell, R.H.; Bennett, G.D.; White, H.S.; Bialer, M. Characterization of the anticonvulsant profile and enantioselective pharmacokinetics of the chiral valproylamide propylisopropyl acetamide in rodents. Br. J. Pharmacol. 2003, 138, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Sobol, E.; Bialer, M.; Yagen, B. Tetramethylcyclopropyl analogue of a leading antiepileptic drug, valproic acid. Synthesis and evaluation of anticonvulsant activity of its amide derivatives. J. Med. Chem. 2004, 47, 4316–4326. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.; Bialer, M. Pharmacokinetic analysis and antiepileptic activity of n-valproyl derivatives of gaba and glycine. Pharm. Res. 1995, 12, 905–910. [Google Scholar]
- Isoherranen, N.; Woodhead, J.H.; White, H.S.; Bialer, M. Anticonvulsant profile of valrocemide (tv1901): A new antiepileptic drug. Epilepsia 2001, 42, 831–836. [Google Scholar]
- Bialer, M.; Johannessen, S.I.; Kupferberg, H.J.; Levy, R.H.; Loiseau, P.; Perucca, E. Progress report on new antiepileptic drugs: A summary of the sixth eilat conference (eilat vi). Epilepsy Res. 2002, 51, 31–71. [Google Scholar]
- Barcs, G.; Walker, E.B.; Elger, C.E.; Scaramelli, A.; Stefan, H.; Sturm, Y.; Moore, A.; Flesch, G.; Kramer, L.; D'Souza, J. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia 2000, 41, 1597–1607. [Google Scholar]
- Almeida, L.; Soares-da-Silva, P. Safety, tolerability, and pharmacokinetic profile of bia 2-093, a novel putative antiepileptic, in a rising multiple-dose study in young healthy humans. J. Clin. Pharmacol. 2004, 44, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Elger, C.; Halasz, P.; Maia, J.; Almeida, L.; Soares-da-Silva, P. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial-onset seizures: A randomized, double-blind, placebo-controlled, parallel-group phase iii study. Epilepsia 2009, 50, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.J.; Noyer, M.; Verloes, R.; Gobert, J.; Wulfert, E. Ucb l059, a novel anti-convulsant drug: Pharmacological profile in animals. Eur. J. Pharmacol. 1992, 222, 193–203. [Google Scholar]
- Loscher, W.; Honack, D. Profile of ucb l059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur. J. Pharmacol. 1993, 232, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.J.; Hirsch, E.; Boehrer, A.; Noyer, M.; Marescaux, C. Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Res. 1995, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wulfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur. J. Pharmacol. 1998, 353, 191–206. [Google Scholar]
- Noyer, M.; Gillard, M.; Matagne, A.; Henichart, J.P.; Wulfert, E. The novel antiepileptic drug levetiracetam (ucb l059) appears to act via a specific binding site in cns membranes. Eur. J. Pharmacol. 1995, 286, 137–146. [Google Scholar]
- Fuks, B.; Gillard, M.; Michel, P.; Lynch, B.; Vertongen, P.; Leprince, P.; Klitgaard, H.; Chatelain, P. Localization and photoaffinitylabelling of the levetiracetam binding site in rat brain and certain cell lines. Eur. J. Pharmacol. 2003, 478, 11–19. [Google Scholar]
- Custer, K.L.; Austin, N.S.; Sullivan, J.M.; Bajjalieh, S.M. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J. Neurosci. 2006, 26, 1303–1313. [Google Scholar]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein sv2a is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar]
- Kaminski, R.M.; Matagne, A.; Leclercq, K.; Gillard, M.; Michel, P.; Kenda, B.; Talaga, P.; Klitgaard, H. Sv2a protein is a broad-spectrum anticonvulsant target: Functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology 2008, 54, 715–720. [Google Scholar]
- Kenda, B.M.; Matagne, A.C.; Talaga, P.E.; Pasau, P.M.; Differding, E.; Lallemand, B.I.; Frycia, A.M.; Moureau, F.G.; Klitgaard, H.V.; Gillard, M.R.; Fuks, B.; Michel, P. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549. [Google Scholar]
- Matagne, A.; Margineanu, D.G.; Kenda, B.; Michel, P.; Klitgaard, H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, sv2a. Br. J. Pharmacol. 2008, 154, 1662–1671. [Google Scholar] [PubMed]
- French, J.A.; Brodsky, A.; von Rosensteil, P. Efficacy and tolerability of 5, 20 and 50 mg/day brivaracetam (ucb 34714) as adjunctive treatment in adults with refractory partial-onset seizures. Epilepsy Res. 2007, 49, 400. [Google Scholar]
- Su, T.Z.; Lunney, E.; Campbell, G.; Oxender, D.L. Transport of gabapentin, a gamma-amino acid drug, by system l alpha-amino acid transporters: A comparative study in astrocytes, synaptosomes, and cho cells. J. Neurochem. 1995, 64, 2125–2131. [Google Scholar] [PubMed]
- Suman-Chauhan, N.; Webdale, L.; Hill, D.R.; Woodruff, G.N. Characterisation of [3h]gabapentin binding to a novel site in rat brain: Homogenate binding studies. Eur. J. Pharmacol. 1993, 244, 293–301. [Google Scholar]
- Gee, N.S.; Brown, J.P.; Dissanayake, V.U.; Offord, J.; Thurlow, R.; Woodruff, G.N. The novel anticonvulsant drug, gabapentin (neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem. 1996, 271, 5768–5776. [Google Scholar] [PubMed]
- Bryans, J.S.; Davies, N.; Gee, N.S.; Dissanayake, V.U.; Ratcliffe, G.S.; Horwell, D.C.; Kneen, C.O.; Morrell, A.I.; Oles, R.J.; O'Toole, J.C.; Perkins, G.M.; Singh, L.; Suman-Chauhan, N.; O'Neill, J.A. Identification of novel ligands for the gabapentin binding site on the alpha2delta subunit of a calcium channel and their evaluation as anticonvulsant agents. J. Med. Chem. 1998, 41, 1838–1845. [Google Scholar]
- Belliotti, T.R.; Capiris, T.; Ekhato, I.V.; Kinsora, J.J.; Field, M.J.; Heffner, T.G.; Meltzer, L.T.; Schwarz, J.B.; Taylor, C.P.; Thorpe, A.J.; Vartanian, M.G.; Wise, L.D.; Zhi-Su, T.; Weber, M.L.; Wustrow, D.J. Structure-activity relationships of pregabalin and analogues that target the alpha(2)-delta protein. J. Med. Chem. 2005, 48, 2294–2307. [Google Scholar]
- Dooley, D.J.; Mieske, C.A.; Borosky, S.A. Inhibition of k(+)-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci.Lett. 2000, 280, 107–110. [Google Scholar]
- Fink, K.; Dooley, D.J.; Meder, W.P.; Suman-Chauhan, N.; Duffy, S.; Clusmann, H.; Gothert, M. Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology 2002, 42, 229–236. [Google Scholar]
- Brown, J.T.; Randall, A. Gabapentin fails to alter p/q-type Ca2+ channel-mediated synaptic transmission in the hippocampus in vitro. Synapse 2005, 55, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, J.; Van Minh, A.T.; Heblich, F.; Nieto-Rostro, M.; Watschinger, K.; Striessnig, J.; Wratten, J.; Davies, A.; Dolphin, A.C. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc. Natl. Acad. Sci. USA 2008, 105, 3628–3633. [Google Scholar]
- Stefani, A.; Spadoni, F.; Bernardi, G. Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacology 1998, 37, 83–91. [Google Scholar]
- Yonekawa, W.D.; Kapetanovic, I.M.; Kupferberg, H.J. The effects of anticonvulsant agents on 4-aminopyridine induced epileptiform activity in rat hippocampus in vitro. Epilepsy Res. 1995, 20, 137–150. [Google Scholar]
- Rundfeldt, C. The new anticonvulsant retigabine (d-23129) acts as an opener of k+ channels in neuronal cells. Eur. J. Pharmacol. 1997, 336, 243–249. [Google Scholar]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. Kcnq2 and kcnq3 potassium channel subunits: Molecular correlates of the m-channel. Science 1998, 282, 1890–1893. [Google Scholar]
- Main, M.J.; Cryan, J.E.; Dupere, J.R.; Cox, B.; Clare, J.J.; Burbidge, S.A. Modulation of kcnq2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 2000, 58, 253–262. [Google Scholar]
- Wickenden, A.D.; Yu, W.; Zou, A.; Jegla, T.; Wagoner, P.K. Retigabine, a novel anti-convulsant, enhances activation of kcnq2/q3 potassium channels. Mol. Pharmacol. 2000, 58, 591–600. [Google Scholar] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; McHarg, M.L.; Gagnon, D.; Rosales, T.O.; Peiffer, A.; Anderson, V.E.; Leppert, M. A novel potassium channel gene, kcnq2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [PubMed]
- Cooper, E.C.; Harrington, E.; Jan, Y.N.; Jan, L.Y. M channel kcnq2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001, 21, 9529–9540. [Google Scholar]
- Turski, W.A.; Cavalheiro, E.A.; Bortolotto, Z.A.; Mello, L.M.; Schwarz, M.; Turski, L. Seizures produced by pilocarpine in mice: A behavioral, electroencephalographic and morphological analysis. Brain Res. 1984, 321, 237–253. [Google Scholar]
- Aiken, S.P.; Lampe, B.J.; Murphy, P.A.; Brown, B.S. Reduction of spike frequency adaptation and blockade of m-current in rat ca1 pyramidal neurones by linopirdine (dup 996), a neurotransmitter release enhancer. Br. J. Pharmacol. 1995, 115, 1163–1168. [Google Scholar]
- Wickenden, A.D.; Krajewski, J.L.; London, B.; Wagoner, P.K.; Wilson, W.A.; Clark, S.; Roeloffs, R.; McNaughton-Smith, G.; Rigdon, G.C. N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ica-27243): A novel, selective kcnq2/q3 potassium channel activator. Mol. Pharmacol. 2008, 73, 977–986. [Google Scholar] [PubMed]
- Rigdon, G.C. ICA-105665, AntiepilepticDrug Trials X; Coral Gables: FL, USA, 2009. [Google Scholar]
- Binnie, C.D.; Kasteleijn-Nolst Trenite, D.G.; De Korte, R. Photosensitivity as a model for acute antiepileptic drug studies. Electroencephalogr Clin. Neurophysiol. 1986, 63, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kasteleijn-Nolst Trenite, D.G.; Marescaux, C.; Stodieck, S.; Edelbroek, P.M.; Oosting, J. Photosensitive epilepsy: A model to study the effects of antiepileptic drugs. Evaluation of the piracetam analogue, levetiracetam. Epilepsy Res. 1996, 25, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kasteleijn-Nolst, Trenité D.; Abou-Khalil, B.; French, J.; Krauss, G.; Rigdon, G.; Moore, E.; Hetherington, S. Clinical Development of ICA-105665, A Novel KCNQ Agonist as a Potential Treatment for Epilepsy, Tenth Eilat Conference on New Antiepileptic Drugs Eilat Israel, April 26, 2010. Eilat Israel, 2010.
- Meldrum, B.S.; Rogawski, M.A. Molecular targets for antiepileptic drug development. Neurotherapeutics 2007, 4, 18–61. [Google Scholar]
- Ogiwara, I.; Miyamoto, H.; Morita, N.; Atapour, N.; Mazaki, E.; Inoue, I.; Takeuchi, T.; Itohara, S.; Yanagawa, Y.; Obata, K.; Furuichi, T.; Hensch, T.K.; Yamakawa, K. Na(v)1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an scn1a gene mutation. J. Neurosci. 2007, 27, 5903–5914. [Google Scholar] [PubMed]
- Osaka, H.; Ogiwara, I.; Mazaki, E.; Okamura, N.; Yamashita, S.; Iai, M.; Yamada, M.; Kurosawa, K.; Iwamoto, H.; Yasui-Furukori, N.; Kaneko, S.; Fujiwara, T.; Inoue, Y.; Yamakawa, K. Patients with a sodium channel alpha 1 gene mutation show wide phenotypic variation. Epilepsy Res. 2007, 75, 46–51. [Google Scholar]
- Lossin, C.; Rhodes, T.H.; Desai, R.R.; Vanoye, C.G.; Wang, D.; Carniciu, S.; Devinsky, O.; George, A.L., Jr. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel scn1a. J. Neurosci. 2003, 23, 11289–11295. [Google Scholar]
- Guerrini, R.; Dravet, C.; Genton, P.; Belmonte, A.; Kaminska, A.; Dulac, O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998, 39, 508–512. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gerlach, A.C.; Krajewski, J.L. Antiepileptic Drug Discovery and Development: What Have We Learned and Where Are We Going? Pharmaceuticals 2010, 3, 2884-2899. https://doi.org/10.3390/ph3092884
Gerlach AC, Krajewski JL. Antiepileptic Drug Discovery and Development: What Have We Learned and Where Are We Going? Pharmaceuticals. 2010; 3(9):2884-2899. https://doi.org/10.3390/ph3092884
Chicago/Turabian StyleGerlach, Aaron C., and Jeffrey L. Krajewski. 2010. "Antiepileptic Drug Discovery and Development: What Have We Learned and Where Are We Going?" Pharmaceuticals 3, no. 9: 2884-2899. https://doi.org/10.3390/ph3092884