Metallo-β-Lactamases and Aptamer-Based Inhibition

Abstract
: An evolution of antibiotic-resistant bacteria has resulted in the need for new antibiotics. β-Lactam based drugs are the most predominantly prescribed antibiotics to combat bacterial infections; however, production of β-lactamases, which catalyze the hydrolysis of the β-lactam bond of this class of antibiotics, by pathogenic bacteria such as Bacillus cereus, are rendering them useless. Some inhibitors of β-lactamases have been found, but there are no inhibitors against a class of β-lactamases known as metallo-β-lactamases, and it has been reported that the number of bacteria that produce metallo-β-lactamases is on the rise. Finding inhibitors of metallo-β-lactamases is thus an urgent necessity. One way to approach the problem is by employing the combinatorial method SELEX. The SELEX method is significant in discovering and producing new classes of inhibitors, as well as providing insight into the development of these inhibitors and paves the way for future aptamer applications that further novel drug discovery.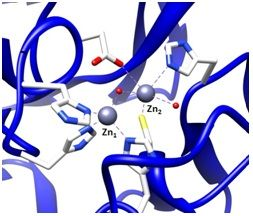
1. Introduction
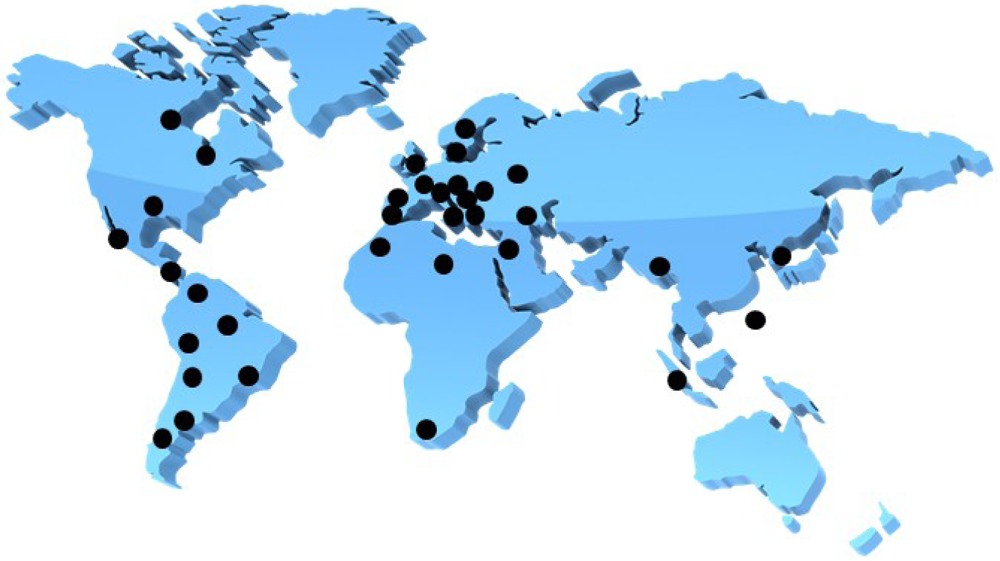
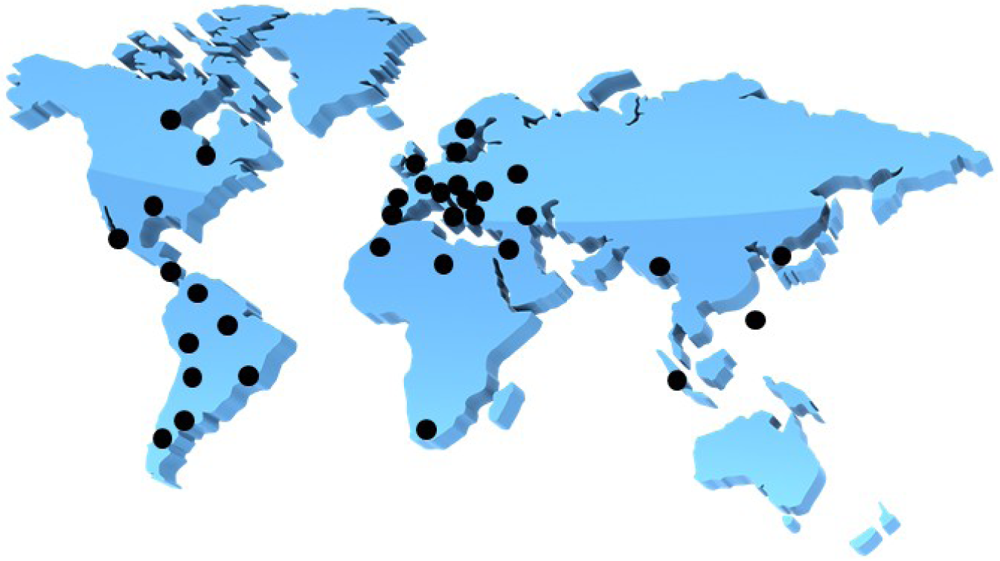
A male patient who lived in Sweden often traveled to India and was found to be infected with an extended-spectrum β-lactamase (ESBL)-producing Klebsiella pneumoniae [1]. The β-lactamase isolate from New Dehli has been named NDM-1 or blaNDM-1, which stands for New Dehli Metallo-β-lactamase. The emergence of the NDM-1 plasmid is a disturbing development because it carries resistance to not only β-lactams, but also macrolides, aminoglycosides, rifampicin, sulfamethoxazole and aztreonam [1]. This multi-drug resistant Klebsiella pneumoniae containing NDM-1 has thusly been named a ‘superbug’. NDM-1 is mainly found in K. pneumoniae, but recently other bacteria such as Enterobacteriaceae and Acinetobacter baumannii display NDM-1 activity as a result of the genetic plasticity of the plasmid that carries the genes responsible for producing the resistant mechanisms [1]. NDM-1 has spread throughout the world, including the USA, UK, Canada, Australia, continental Europe, Eastern Europe, the Middle East, Africa and South East Asia (Figure 1) [1,2]. To overcome the issue of bacterial antibiotic resistance, one solution is to discover inhibitors of metallo-β-lactamases (MBLs). Industries and academia have therefore focused intensively on discovering inhibitors of MBLs; however, no compounds are hitherto approaching phase 1 clinical trials. Hence, it is an exceedingly pressing issue when we consider the more than eight years typically necessary to approve new inhibitors as being safe enough to appear in pharmaceutical markets. The recent discovery of aptamer-based inhibitors of MBL from Bacillus cereus is interesting enough to catch our attention. In this review, we will discuss β-lactamases, development of aptamers against MBL from B. cereus, and aptamer-based inhibition tests of MBL.
2. β-Lactamases
β-Lactamases (β-lactam hydrolyases, EC 3.5.2.6) are highly efficient enzymes that inactivate β-lactam antibiotics by catalyzing the hydrolysis of the four-membered β-lactam ring of the antibiotics. So far, more than 700 β-lactamases have been identified, and are classified into four groups, A–D [3]. The class A, C, and D β-lactamases use a serine residue in the active site as a nucleophile to attack substrates, and these enzymes are called serine-β-lactamases [4,5]. The class A β-lactamases tend to attack penicillins, while the class C β-lactamases have a tendency to attack cephalosporins [4,5]. In the case of class D β-lactamases, the serine in the active site prefers to hydrolyze oxacillin [4,5]. However, the β-lactamases in the class B require one or two zinc ions for their full catalytic activity, and these enzymes are therefore called metallo-β-lactamases (MBLs) [4,5]. The enzymes have a wide range of substrate specificity [6].
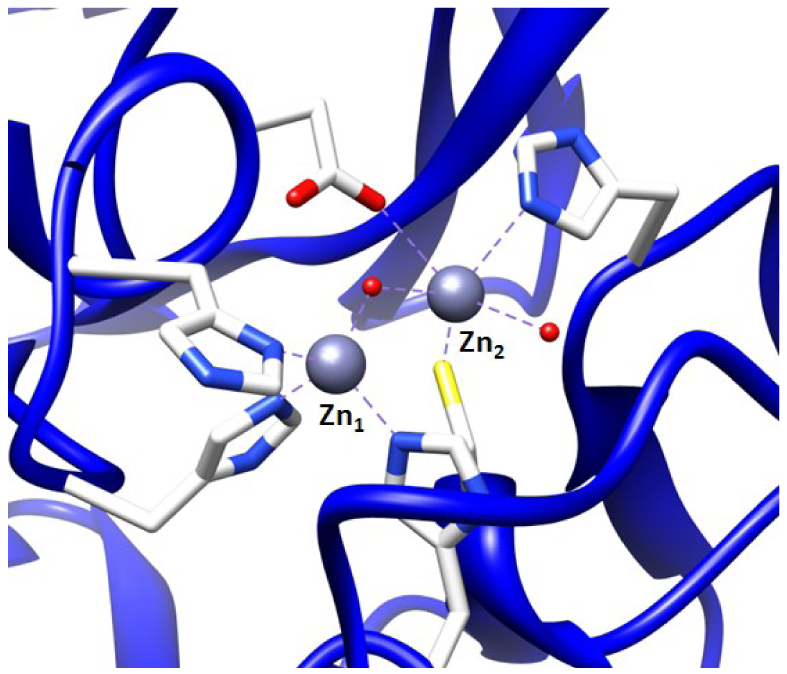
The first MBL was found in an innocuous strain of Bacillus cereus, and MBL-mediated resistance has rapidly spread to pathogenic bacteria [4]. These MBLs can be categorized into three subgroups, B1–B3. The B1 enzymes require one or two zinc ion(s) for the activity, where the tightly bound zinc is referred to as Zn1 and the less tightly bound zinc is called as Zn2 [4,5]. In the case of Zn1, a tetrahedral geometry is coordinated by three histidines and one solvent molecule [4]. A distorted trigonal bipyramidal geometry is the coordination of Zn2 by three amino acids (His, Cys, Asp), a water molecule and a solvent (e.g., H2O and glycerol) that also serves as a ligand to Zn1 [4]. For the B2 enzymes, a salient feature is that the B2 enzymes require only one zinc ion for their full activity [4]. Lastly, the B3 enzymes require two zinc ions for the full activity, but the difference between B1 and B3 enzymes is that the Zn2 ion in B3 is coordinated to another His in place of Cys [4]. The most notable plasmid- and chromosome-mediated MBLs today are listed in Table 1, along with their classification and the bacteria they have been identified in. It should be mentioned here that typically plasmid-encoded β-lactamases that are more transferable may confer resistance to expanded-spectrum β-lactams, whereas chromosome-encoded β-lactamases may confer narrow-spectrum β-lactams. This infers that the plasmid-encoded β-lactamases draw more attention than chromosome-encoded β-lactamases.
To protect β-lactam antibiotics, inhibitors of β-lactamases are needed. Mechanism-based irreversible inhibitors, such as clavulanic acid, sulbactam, and tazobactam, have been used to inactivate serine-β-lactamases, which are metal-independent-β-lactamases [5]. The co-administration of an antibiotic with one of these inhibitors has been commercially applied to cure β-lactam antibiotic resistant bacterial infections. It has been demonstrated that some potential inhibitors for serine-β-lactamase have the ability to inactivate MBLs [7]. However, none of the commercially available inhibitors of metal independent-β-lactamases are effective against MBLs.
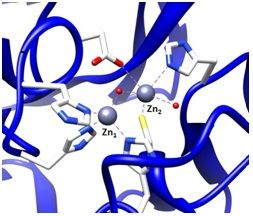
To find inhibitors of the metallo-β-lactamase BcII from B. cereus, whose active site is shown in Figure 2, the combinatorial approach SELEX was employed. Through combinatorial chemistry a wide variety of compounds can be effectively tested for promising drug activity. Combinatorial methods have been widely adopted by biotechnology and pharmaceutical companies over the past ten years.
3. SELEX Technology
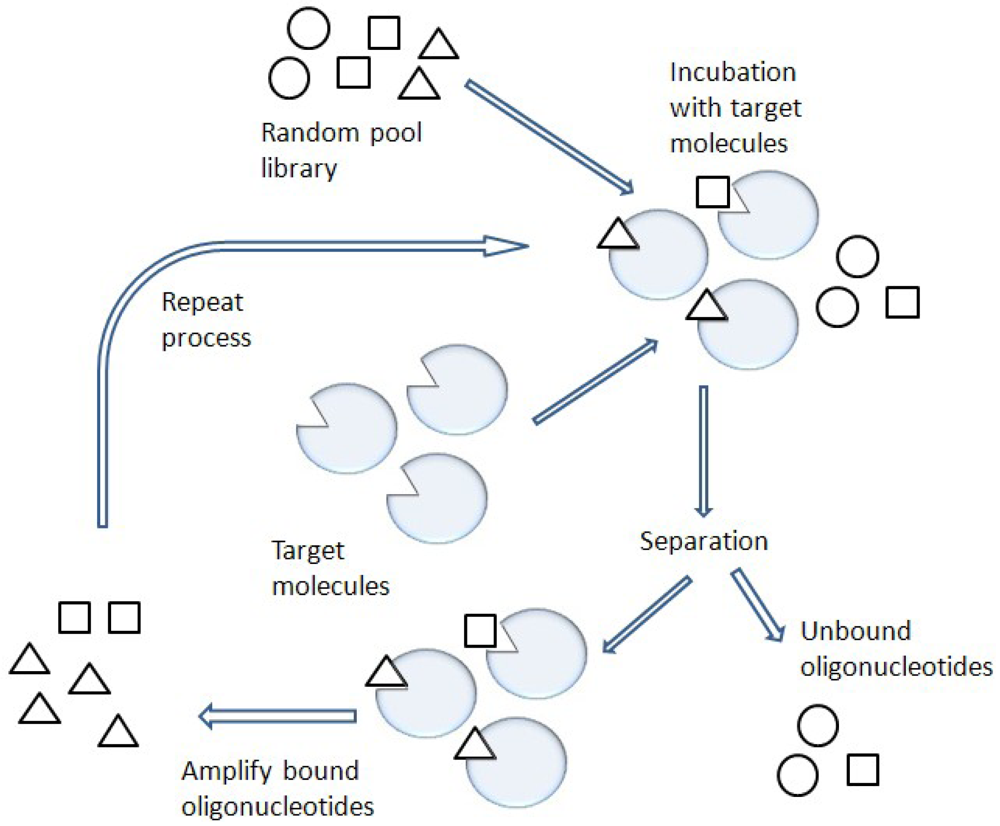
One of the most promising combinatorial chemistry techniques is known as SELEX [8] (Figure 3). This SELEX method is also known as in vitro selection or in vitro evolution, allowing the simultaneous screening of a large number of nucleic acid molecules.
Functional nucleic acid molecules are selected from the mainly non-functional pool of oligonucleotides by column chromatography or other selection techniques such as a gel shift assay [9-11]. The functional nucleic acids are called aptamers, which are usually short single-stranded (ss) nucleic acids such as ssDNA and RNA [9]. Many of the selected aptamers display affinities for their target comparable to those observed for monoclonal antibodies. However, unlike antibodies, facile modification of the selected aptamers can improve their binding to target molecules and enhance the stability of the aptamers against nuclease activity under physiological conditions [12].
The application of aptamers has been significant in the medical and pharmaceutical research fields. A recent example of a commercial product developed using SELEX technology is an aptamer against vascular endothelial growth factor (VEGF). In fact, this is the only commercially available aptamer-based therapy. The anti-VEGF aptamer blocks vessel growth and inhibits neovascularization [13,14] with very high affinity (dissociation constant, Kd = 50 pM) [15] (this aptamer affinity is as good as the antibody affinity (Kd = 54 pM) for VEGF [16]). The aptamer tightly binds to abnormally over-expressed VEGF and thus the binding reaction from VEGF to a receptor cannot proceed. This aptamer, known as Macugen®, was approved by the Food and Drug Administration (FDA) in 2004 and is currently used to cure age-related macular degeneration [17,18].
Although the anti-VEGF aptamer is based on RNA SELEX, recently we reported that ssDNA SELEX for the B. cereus metallo-β-lactamase was used to find ssDNA aptamers which act as inhibitors of the enzyme, thus providing the possibility of an antibacterial drug against this specific β-lactam resistant bacterial infection [19]. Other potential drug candidates using ssDNA SELEX technology have been developed. For example, thrombin, a protein that serves as essential role in regulation of the coagulation pathway in human, has been targeted for the development of ssDNA aptamers; an ssDNA aptamer of thrombin has been identified and shows a very promising anticoagulant drug activity [20,21]. Anti-inflammatory aptamers for L-selectin [20], viral infection prevention aptamers for Hemagglutinin from the influenza virus [22], and anti-progressive renal disease aptamers for platelet-derived growth factor [23] have been developed using ssDNA SELEX technology as well. Except for the aptamers of B. cereus metallo-β-lactamase, other aptamers functions by tightly binding to target molecules and interfering with the target molecules' next binding step. In the following section, introducing true enzymatic inhibition to B. cereus metallo-β-lactamase in an aptamer-based inhibition manner will draw our attention as to how we can screen for enzyme inhibiting aptamers.
4. SELEX for B. Cereus Metallo-β-Lactamase Using ssDNA
Previously, the metallo-β-lactamase BcII from B. cereus, a class B MBL, as a target molecule to find aptamers using SELEX technology has been studied. B. cereus was chosen because it is a pathogen that causes food poisoning and because the three-dimensional X-ray structure of BcII has been resolved (Protein Data Bank entry code: 1BC2), which increases the chances of improving our understanding of binding between potential inhibitors and BcII. This project was very successful, and this work was recently published [19]. This study had three aims, which were: (a) to find aptamers that inhibit BcII; (b) to determine inhibition values of the found aptamers by kinetic analyses, and explore the binding relationship between the aptamers and metal ions in the active site of the enzyme; (c) and to test the specificity of the aptamers and the growth inhibition of B. cereus by the aptamers in combination with an existing antibiotic.
4.1. Identification of Aptamers that Can Inhibit the Target Enzyme
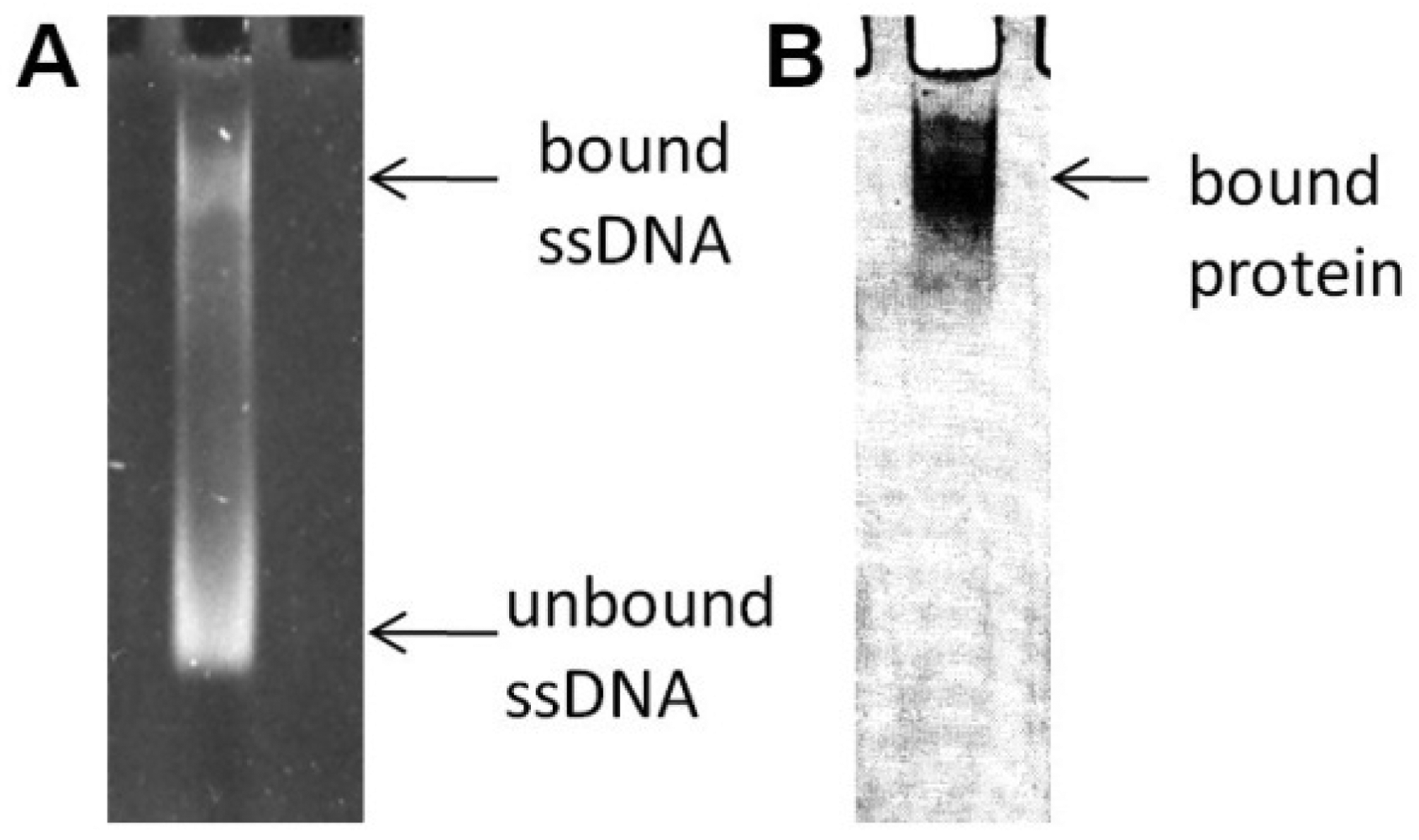
In the previously reported literature [19], the SELEX method was used to screen aptamers against the B. cereus MBL. Randomly degenerated ssDNA and the target enzyme were incubated for an appropriate period of time. A complex formation between ssDNA and the MBL was confirmed on a native gel, as shown in Figure 4. In an effort to confirm the complex, the sample was loaded in two different gel lanes on the same native gel. The gel was soaked in an ethidium bromide solution to illuminate the bound ssDNA to the enzyme after completion of the electrophoresis; the same gel was stained in Coomassie Blue to identify the existence of the enzyme. In the native gel, any non-complexed enzyme would not enter the gel due to its high isoelectric point value, but the complex migrates toward the gel bottom because it contains anionic ssDNA. During the iterative process of SELEX, to sequester tightly bound aptamers from both unbound and non-specific bound ssDNAs, more stringent selection was exerted by an increasing NaCl concentration. A typical gel after loading the sample under such conditions clearly showed two bands by ethidium bromide staining; that is, the enzyme:ssDNA complex and the unbound ssDNA (Figure 4).
The purpose was to identify aptamers that inhibit BcII, thus it was of great importance that enzyme inhibition assays be carried out during the SELEX rounds [19]. To test whether ssDNA could bind and inhibit BcII, the selected ssDNA pools were subjected to inhibition assays and only one oligonucleotide was identified after DNA sequencing, which was 30 residues in length. To understand the binding between the aptamer and the target molecule, it is necessary to recognize the importance of secondary structures formed by the ssDNA. The secondary structure of the found sequence was predicted by MFold, a secondary structural nucleic acid prediction program [25]. This program uses energy rules to predict optimal secondary structures for an RNA molecule as well as an ssDNA molecule [26,27]. By using the MFold program, two different secondary structures of the aptamer (a 30 residue ssDNA) were predicted. The two different structures commonly contained a 10 residue stem-loop structure, therefore this sequence was considered to be important to enzyme inhibition. Hence, further investigation was carried out using the 10 residue ssDNA.
4.2. The Determination of IC50 and Kinetic Parameters
The IC50value, the concentration of inhibitor necessary to inhibit 50% of the enzyme's activity, for the aptamers were determined by measuring the rate of enzymatic hydrolysis of a substrate [19]. The IC50 of the 30-oligonucleotide ssDNA was 1.2 nM, and the IC50 of the 10 residue ssDNA was also 1.2 nM [19]. These results indicate that the 10 residue ssDNA was the crucial sequence for enzyme inhibition. The remainder of the sequence was tested to determine whether other portions of the 30 residue ssDNA contributed to the inhibition, but no inhibition was found [19]. This data provides evidence to support the idea that the 10 residue ssDNA is responsible for the observed inhibition.
The steady state enzyme kinetic data of the inhibition of BcII were obtained by various concentrations of the 10 and 30 residue ssDNAs [19]. The inhibition data showed reversible, mixed inhibitions for both the 10 and 30 residue ssDNAs, with dissociation constants for the enzyme-inhibitor complex (Ki) and the enzyme-substrate-inhibitor (Ki′) in the nanomolar range, demonstrating that inhibition by those ssDNA aptamers is very effective. Previously, possible inhibitors had been developed by rational-drug approach with Ki values in the micromolar range [7]. This comparison strongly suggests that the findings (10 and 30 residue ssDNAs) are of great value in further investigating the binding of the aptamers to the target molecule.
4.3. The Test of the Effect of the Aptamer Binding to the Enzyme by Co2+ Reconstitution
It is possible to prepare an apoenzyme of a metallo-β-lactamase and reconstitute the original enzymatic activity by the addition of cobalt (II) sulfate [28]. Given that the Co2+-reconstituted MBL can provide a signature feature in the UV-Vis range, the reconstituted enzyme is then useful in investigating the binding relationship between the metal ion and the ssDNA aptamer. The preparation of Co2+-reconstitution was successful in BcII from B. cereus and thus was used for UV-Vis spectroscopy [19]. The observation of the visible electronic spectra of the Co2+ reconstituted enzyme in the absence and the presence of the 10 residue ssDNA showed significant differences [19].The changes in the spectrum observed by addition of the ssDNA at the cysteine-Co2+ charge transfer band were observed particularly at a wavelength of 347 nm. Similar changes were observed in the presence of an excess of substrates such as cephalosporin C [28,29]. Therefore, the change at 347 nm indicates that the cysteine thiol group located in the active site of the enzyme, where the cysteine thiol group serves as a ligand to the zinc ion at the Zn2 position (Figure 2), was affected by the ssDNA addition. Thus, the observation of the change of active site by the addition of the ssDNA aptamer supports the proposal that the ssDNA aptamer's binding to the enzyme elicits inhibition of the metallo-β-lactamase.
4.4. Specificity
One of the most important aspects of inhibitor design is that the inhibitors should be specific to the enzyme. To test the specificity, porcine carboxypeptidase A, a zinc metalloenzyme, and B. cereus serine-β-lactamase were used [19]. The addition of ssDNA aptamers (both 10 and 30 residue ssDNAs) to porcine carboxypeptidase A exerted no influence on its enzymatic activity, thus suggesting that the ssDNA aptamer do not affect all zinc binding sites indiscriminately [19]. The fact that addition of the ssDNA aptamer to the B. cereus serine-β-lactamase had no impact on the enzyme activity [19] provides additional evidence for the specificity of the ssDNA aptamer to the metallo-β-lactamase. In addition, the observation that the Ki value of the Co2+-reconstituted enzyme by the 10 residue ssDNA was almost 4 orders of magnitude higher than that of the normal metallo (Zn2+)-β-lactamase further demonstrates the exquisite specificity of the ssDNA aptamer in that the DNA is able to distinguish between the zinc and cobalt forms of the enzyme, even though both enzyme forms possess the same enzymatic activity.
4.5. Inhibition Tests in Bacteria
To explore the possibility that co-administration of the found aptamer and a β-lactam antibiotic have the capacity to negatively impact the growth of β-lactam antibiotic resistant bacteria, the combination of the 10 residue ssDNA with a β-lactam antibiotic, cephalexin, was applied to B. cereus 5/B/6, which produces the MBL [19]. The bacterial growth was suppressed over a 20 h period at 30 °C. As control experiments, either the antibiotic alone or the aptamer alone was used, and those had no effect on the bacterial growth; however, to obtain cell death, in the presence of 5 μM cephalexin, 75 μM of the 10 residue aptamer was needed [19]. This indicates that some nuclease activities may degrade the nucleotide aptamer under cell culture conditions. This observation suggests that modification of the aptamer is necessary to retain its strong potency as an enzyme inhibitor.
5. Future Studies
In this review, we describe the potential of an aptamers-based method using SELEX technology to discover novel inhibitors of the metallo-β-lactamase BcII from Bacillus cereus. Although we should mention that there is a possibility that the aptamers-based inhibitors to BcII would be limited to chromosome-encoded β-lactamases that may structurally differ from plasmid-encoded β-lactamases, there has been success in finding potential inhibitors to BcII in an aptamers-based inhibition fashion. To further improve the inhibition for those ssDNA aptamers, modifications of the aptamers are likely needed so that they can survive degradation by nucleases and have more potential as novel drug candidates. For example, phosphorothioate modification, which takes place at a non-bridging oxygen in the phosphate backbone at the 5′-and/or 3′-end of the aptamers, would be a good choice. This modification leads to resistance to degradation from endo- and exonuclease cleavage. Another issue in developing oligonucleotide therapeutics is the relatively short half-lives of the aptamers in the body. To overcome this problem, conjugation to polyethylene glycol (PEG) increases the molecular weight of the aptamer and extends its elimination half-life by slowing renal filtration and distribution from the central compartment. This PEG conjugation has emerged as an effective strategy to extend the circulating half-life [30]. For instance, the use of 40 kDa PEG conjugated to an aptamer was reported to extend the serum half-life time to a significant extent [31]. These strategies to optimize aptamer therapeutics are important in combination with SELEX technology to identify a new class of drug candidates that may be used in the battle against the proliferation of antibiotic-resistant bacteria.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Class | Bacteria | Encoding |
|---|---|---|---|
| IMP | B1 | Pseudomonas aeruginosa, Acinetobacter baumannii, Serratia marcesens, Enterobacter aerogenes | Plasmid |
| VIM | B1 | Pseudomonas aeruginosa, Acinetobacter baumannii, Klebsiella pneumoniae, Enterobacter aerogenes | Plasmid |
| SPM | B1 | Pseudomonas aeruginosa | Plasmid |
| GIM | B1 | Pseudomonas aeruginosa | Plasmid |
| AIM | B3 | Pseudomonas aeruginosa | Plasmid |
| SIM | B1 | Pseudomonas aeruginosa, Acinetobacter baumannii | Plasmid |
| NDM | unknown | Klebsiella pneumoniae | Plasmid |
| DIM | B1 | Pseudomonas stutzeri | Plasmid |
| KHM | unknown | Citrobacter freundii | Plasmid |
| BcII | B1 | Bacillus cereus | Chromosome |
| Bla2 | B1 | Bacillus anthracis | Chromosome |
| CcrA | B1 | Bacteroides fragilis | Chromosome |
| ImiS | B2 | Aeromonas sobria | Chromosome |
| L1 | B3 | Stenotrophomonas maltophilia | Chromosome |
References
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar]
- Walsh, T.R. Emerging carbapenemases: a global perspective. Int. J. Antimicrob. Agents 2010, 36 Suppl. 3, S8–S14. [Google Scholar]
- Perez, F.; Endimiani, A.; Hujer, K.M.; Bonomo, R.A. The continuing challenge of ESBLs. Curr. Opin. Pharmacol. 2007, 7, 459–469. [Google Scholar]
- Walsh, C.; Wright, G. Introduction: Antibiotic resistance. Chem. Rev. 2005, 105, 391–394. [Google Scholar]
- Crowder, M.W.; Spencer, J.; Vila, A.J. Metallo-beta-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 2006, 39, 721–728. [Google Scholar]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-beta-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar]
- Buynak, J.D.; Chen, H.; Vogeti, L.; Gadhachanda, V.R.; Buchanan, C.A.; Palzkill, T.; Shaw, R.W.; Spencer, J.; Walsh, T.R. Penicillin-derived inhibitors that simultaneously target both metallo- and serine-beta-lactamases. Bioorg. Med. Chem. Lett. 2004, 14, 1299–1304. [Google Scholar]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar]
- Joyce, G.F. Amplification, mutation and selection of catalytic RNA. Gene 1989, 82, 83–87. [Google Scholar]
- Gold, L.; Polisky, B.; Uhlenbeck, O.; Yarus, M. Diversity of oligonucleotide functions. Annu. Rev. Biochem. 1995, 64, 763–797. [Google Scholar]
- Jayasena, S.D. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Maberley, D. Pegaptanib for neovascular age-related macular degeneration. Issues Emerg. Health Technol. 2005, 76, 1–4. [Google Scholar]
- Chapman, J.A.; Beckey, C. Pegaptanib: A novel approach to ocular neovascularization. Ann. Pharmacother. 2006, 40, 1322–1326. [Google Scholar]
- Lee, J.H.; Canny, M.D.; Erkendez, A.D.; Krilleke, D.; Ng, Y.S.; Shima, D.T.; Pardi, A.; Jucker, F. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc. Natl. Acad. Sci. USA 2005, 102, 18902–18907. [Google Scholar]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; Koo, H.; Mangalampalli, V.R.; Ludwig, D.L.; Tonra, J.R.; Hicklin, D.J. Anti-vascular endothelial growth factor receptor-1 antagonist antibody as a therapeutic agent for cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar]
- Tucker, C.E.; Chen, L.S.; Judkins, M.B.; Farmer, J.A.; Gill, S.C.; Drolet, D.W. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 203–212. [Google Scholar]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar]
- Kim, S.K.; Sims, C.L.; Wozniak, S.E.; Drude, S.H.; Whitson, D.; Shaw, R.W. Antibiotic resistance in bacteria: novel metalloenzyme inhibitors. Chem. Biol. Drug Des. 2009, 74, 343–348. [Google Scholar]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar]
- Griffin, L.C.; Tidmarsh, G.F.; Bock, L.C.; Toole, J.J.; Leung, L.L. In vivo anticoagulant properties of a novel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood 1993, 81, 3271–3276. [Google Scholar]
- Fukuda, K.; Vishinuvardhan, D.; Sekiya, S.; Kakiuchi, N.; Shimotohno, K.; Kumar, P.K.; Nishikawa, S. Specific RNA aptamers to NS3 protease domain of hepatitis C virus. Nucleic Acids Symp. Ser. 1997, 37, 237–238. [Google Scholar]
- Pietras, K.; Ostman, A.; Sjoquist, M.; Buchdunger, E.; Reed, R.K.; Heldin, C.H.; Rubin, K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001, 61, 2929–2934. [Google Scholar]
- Kim, S.K. Inhibition of Metallo-Beta-Lactamase by Rational and Combinatorial Approaches. Ph.D. Dissertation., Texas Tech University, Lubbock, TX, USA, 2002. [Google Scholar]
- Zuker, M. On finding all suboptimal foldings of an RNA molecule. Science 1989, 244, 48–52. [Google Scholar]
- Turner, D.H.; Sugimoto, N.; Freier, S.M. RNA structure prediction. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 167–192. [Google Scholar]
- Allawi, H.T.; SantaLucia, J., Jr. Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry 1997, 36, 10581–10594. [Google Scholar]
- Bicknell, R.; Schaffer, A.; Waley, S.G.; Auld, D.S. Changes in the coordination geometry of the active-site metal during catalysis of benzylpenicillin hydrolysis by Bacillus cereus beta-lactamase II. Biochemistry 1986, 25, 7208–7215. [Google Scholar]
- Bicknell, R.; Waley, S.G. Cryoenzymology of Bacillus cereus beta-lactamase II. Biochemistry 1985, 24, 6876–6887. [Google Scholar]
- Fishburn, C.S. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J. Pharm. Sci. 2008, 97, 4167–4183. [Google Scholar]
- Zeuzem, S.; Welsch, C.; Herrmann, E. Pharmacokinetics of peginterferons. Semin. Liver. Dis. 2003, 23, 23–28. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schlesinger, S.R.; Lahousse, M.J.; Foster, T.O.; Kim, S.-K. Metallo-β-Lactamases and Aptamer-Based Inhibition. Pharmaceuticals 2011, 4, 419-428. https://doi.org/10.3390/ph4020419
Schlesinger SR, Lahousse MJ, Foster TO, Kim S-K. Metallo-β-Lactamases and Aptamer-Based Inhibition. Pharmaceuticals. 2011; 4(2):419-428. https://doi.org/10.3390/ph4020419
Chicago/Turabian StyleSchlesinger, Sara R., Mieke J. Lahousse, Taylor O. Foster, and Sung-Kun Kim. 2011. "Metallo-β-Lactamases and Aptamer-Based Inhibition" Pharmaceuticals 4, no. 2: 419-428. https://doi.org/10.3390/ph4020419