Hedgehog Pathway Blockade Inhibits Melanoma Cell Growth in Vitro and in Vivo


 ,
,
Abstract
:1. Introduction
2. Experimental
2.1. Human Melanoma Cell Lines
2.2. Data Mining of mRNA Microarray of Melanoma Cell Lines and qRT-PCR Confirmation
2.3. Data Mining of mRNA Microarray of Melanoma Cell Lines and qRT-PCR Confirmation, mRNA Microarray of Metastatic Melanoma Specimens
2.4. Statistical Analysis
2.5. In Vitro Cell Proliferation Assays of Cyclopamine, NVP-LDE-225, and Vemurafenib
2.6. Cell Cycle and Apoptosis Analysis
2.7. siRNA Assays
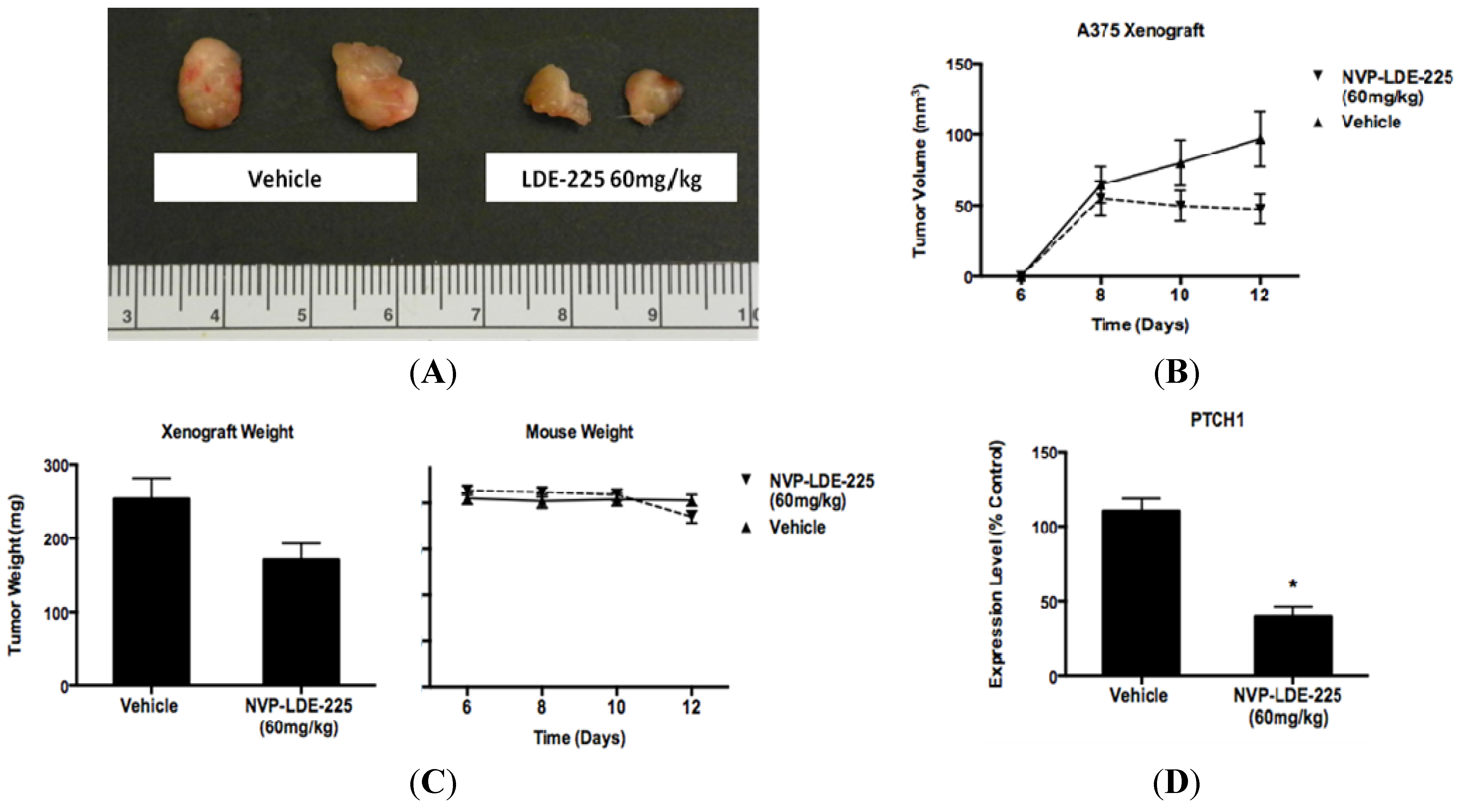
2.8. Xenograft Assay of A375 Cells
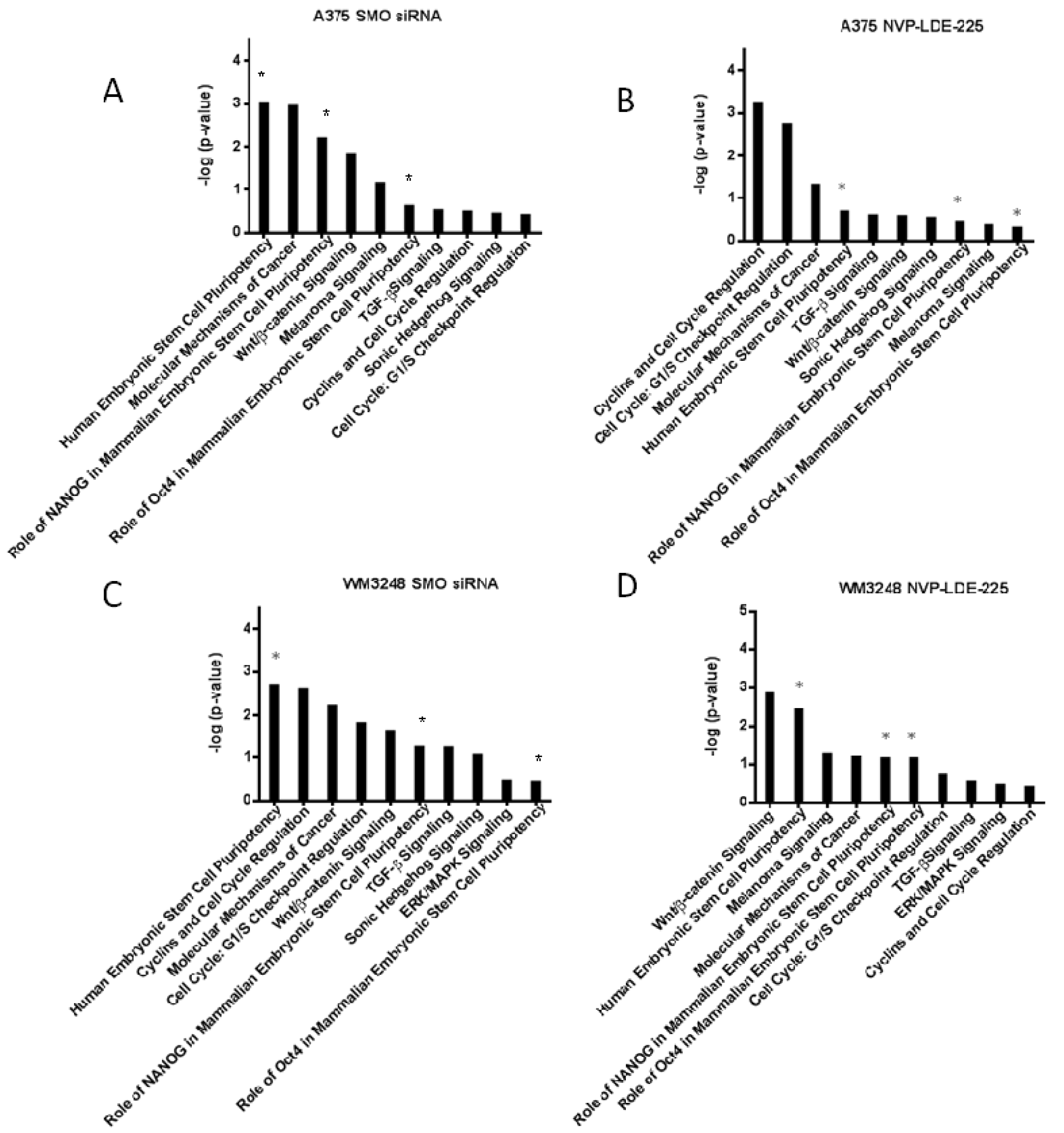
2.9. Pathway Analysis of SMO Inhibition Transcriptional Profile
3. Results and Discussion
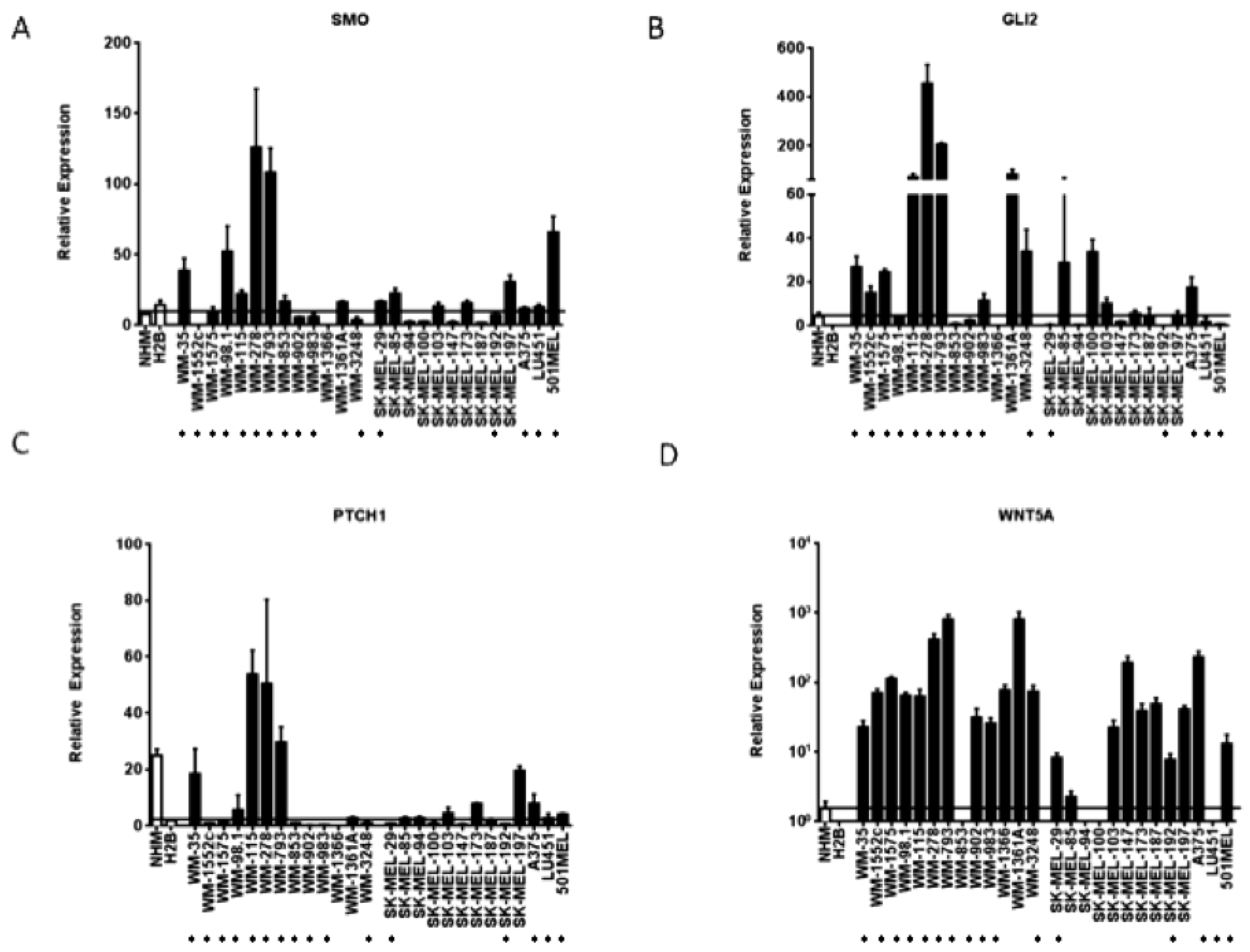
3.1. Expression of Hedgehog Pathway Members is Elevated in Melanoma Cell Lines compared to Melanocytes


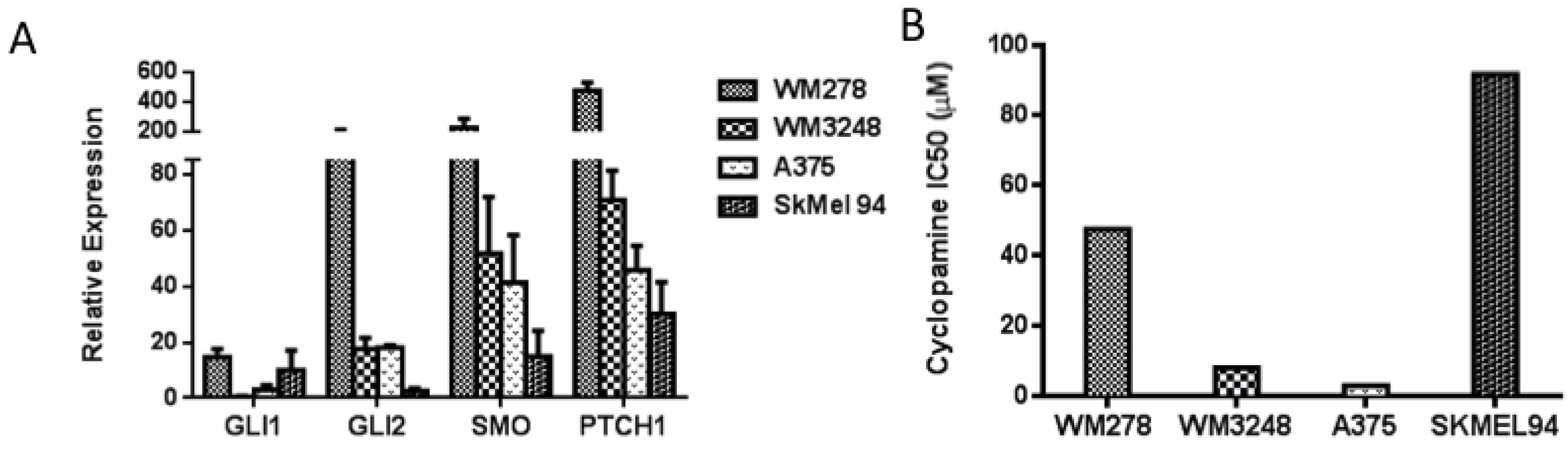
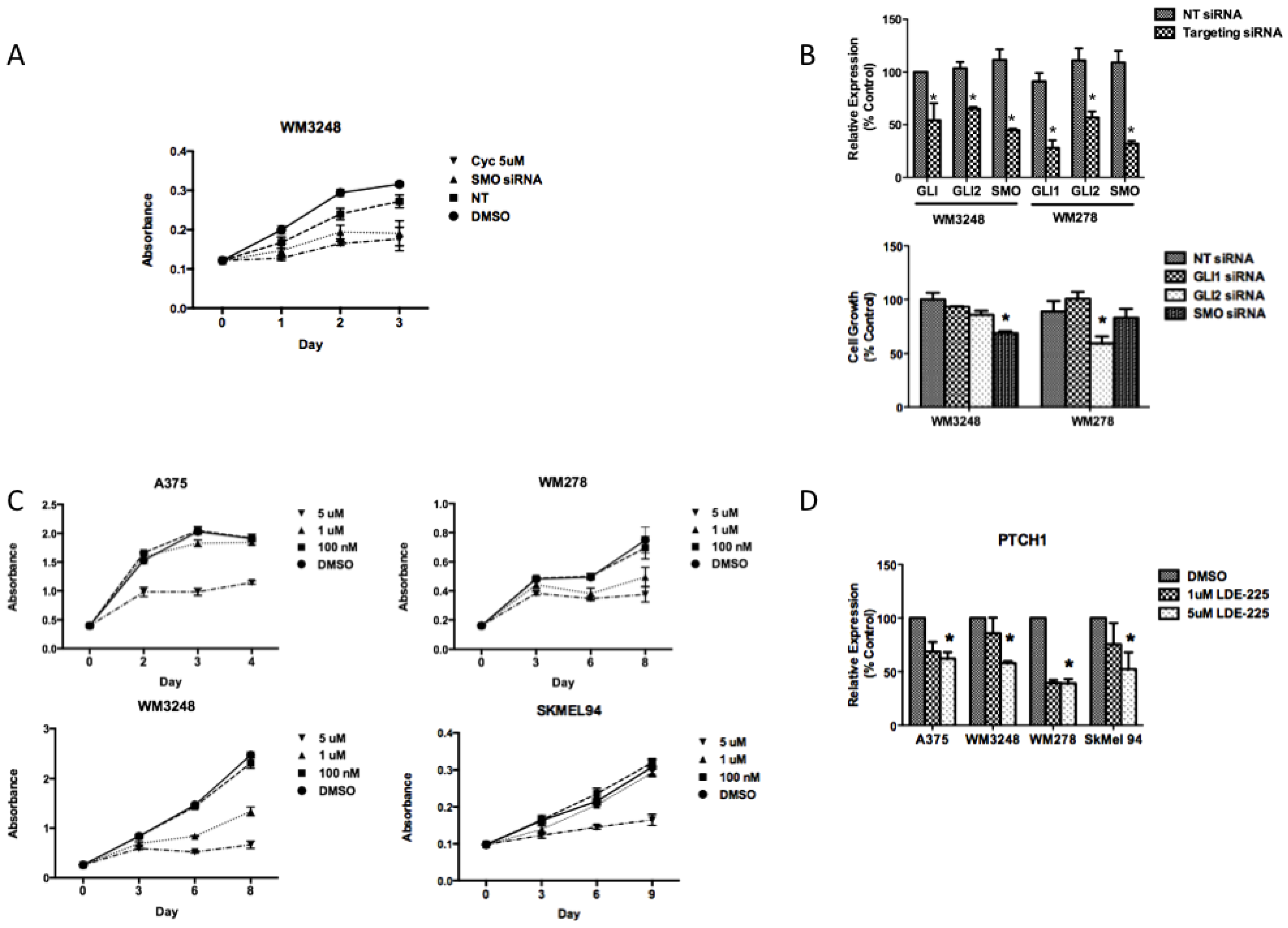
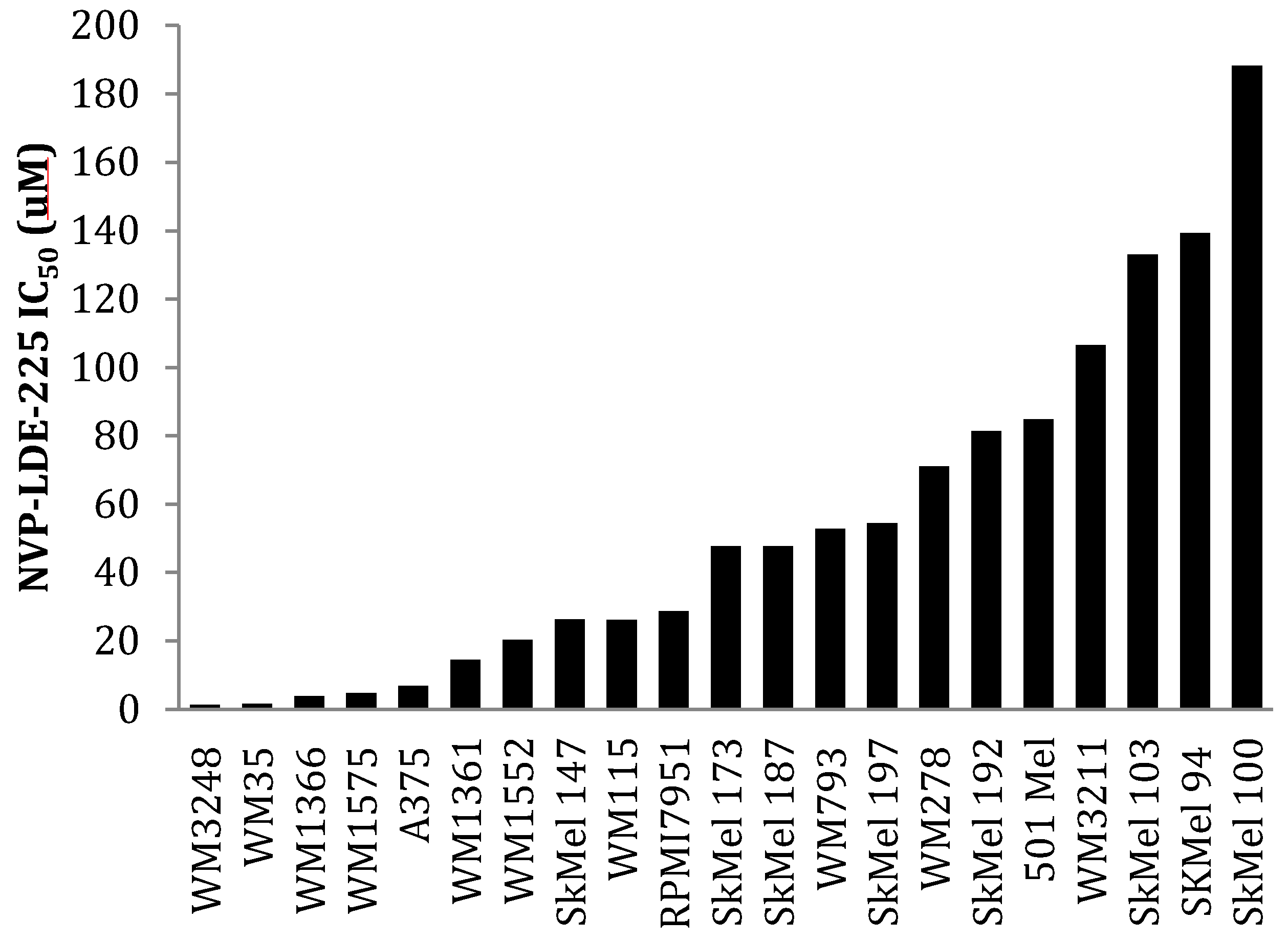
3.2. SMO Inhibition Decreases Melanoma Cell Proliferation in Vitro


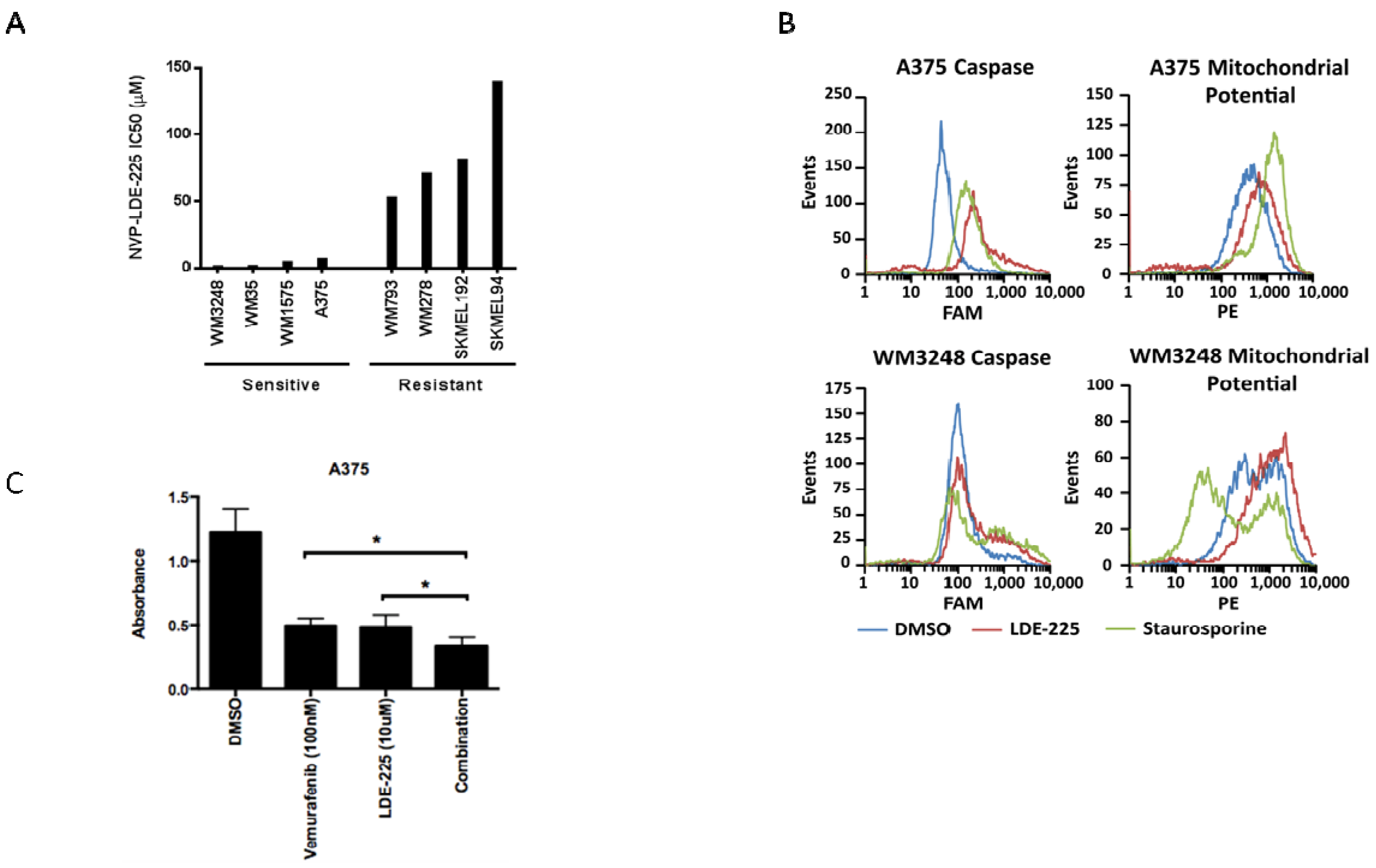
3.3. NVP-LDE-225 Inhibits Melanoma Cell Proliferation and Induces Apoptosis in Vitro


3.4. NVP-LDE-225 Inhibits Melanoma Growth in Vivo

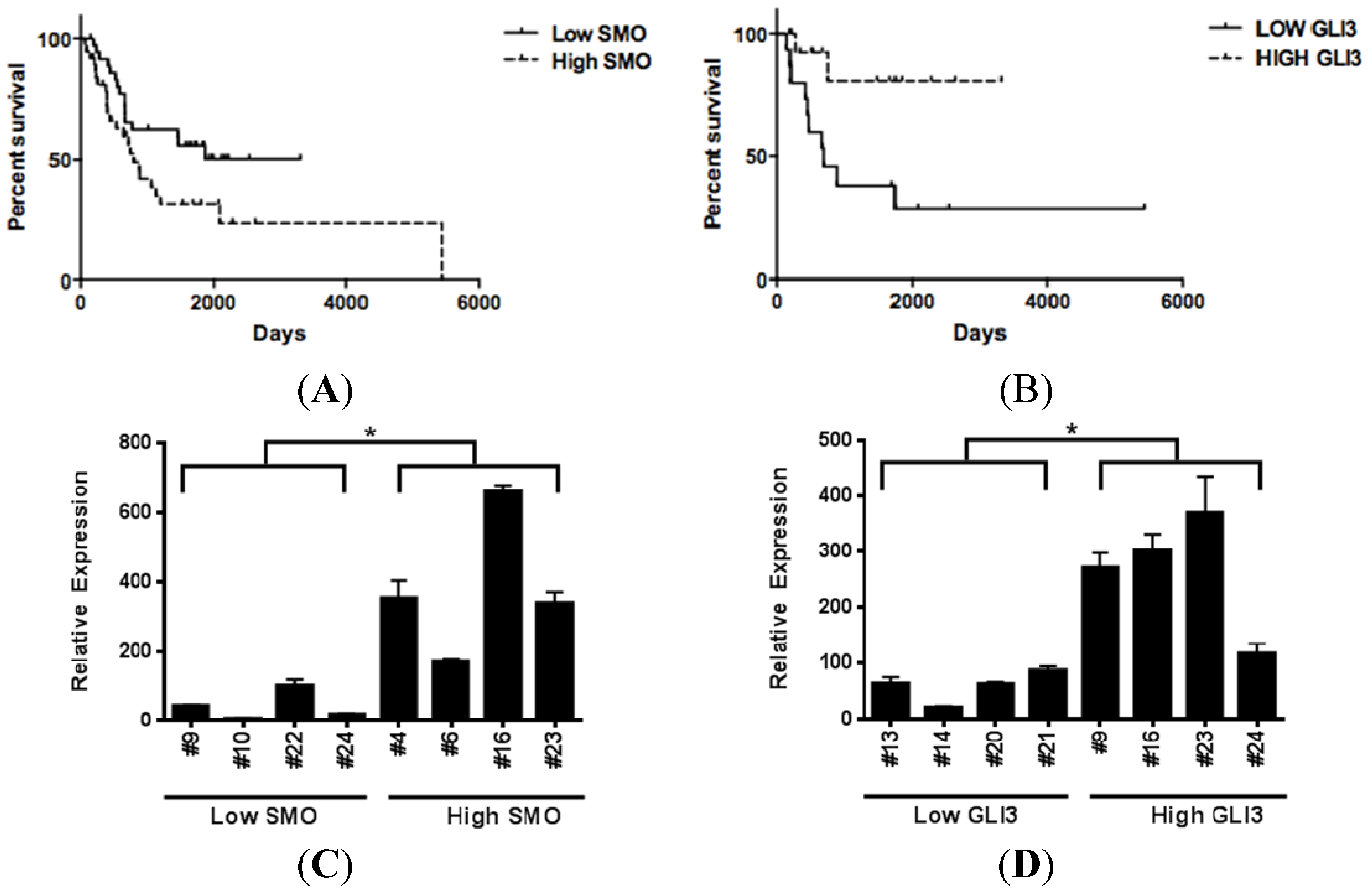
3.5. Increased SMO Levels are Associated with Decreased Survival, while Elevated GLI3 Levels are Associated with Better Survival in Metastatic Melanoma Patients

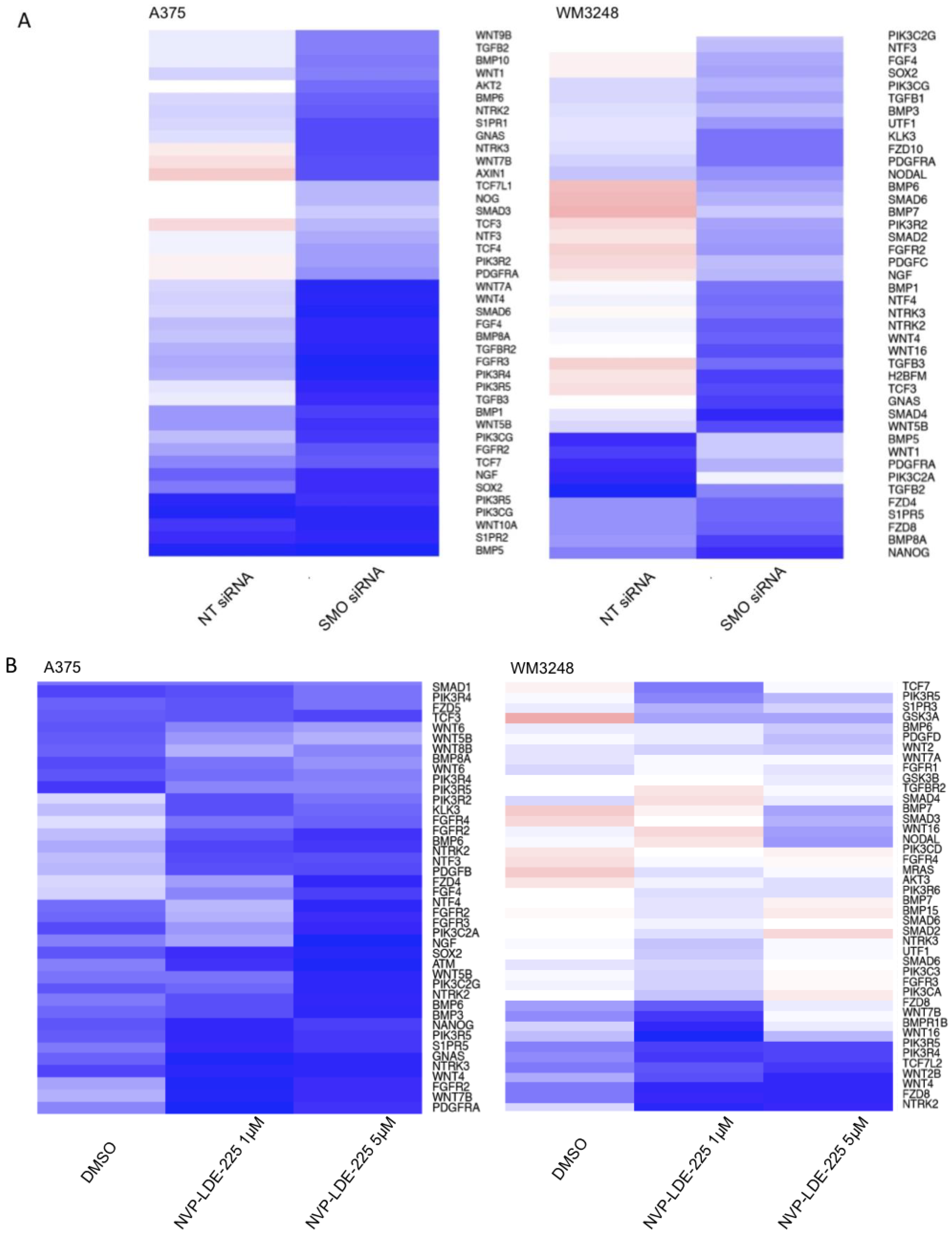
3.6. SMO Inhibition Induces a Programmed Cell Death Gene Expression Signature and Downregulates Embryonic Stem Cell Pluripotency



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A375 | WM3248 | |
|---|---|---|
| SMO siRNA | -Death Receptor Signaling Pathway | -Myc Mediated Apoptosis Signaling |
| NVP-LDE-225 | -Apoptosis Signaling | -Death Receptor Signaling Pathway |
| -Myc Mediated Apoptosis Signaling | -Myc Mediated Apoptosis Signaling |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. SEER Stat Fact Sheets: Melanoma of the Skin. Available online: http://seer.cancer.gov/statfacts/html/melan.html/ (accessed on 29 January 2013).
- Garbe, C.; McLeod, G.R.; Buettner, P.G. Time trends of cutaneous melanoma in Queensland, Australia and Central Europe. Cancer 2000, 89, 1269–1278. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Neyman, N.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Available online: http://seer.cancer.gov/csr/1975_2009_pops09/ (accessed on 31 October 2003).
- Bedikian, A.Y.; Millward, M.; Pehamberger, H.; Conry, R.; Gore, M.; Trefzer, U.; Pavlick, A.C.; de Conti, R.; Hersh, E.M.; Hersey, P.; et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J. Clin. Oncol. 2006, 24, 4738–4345. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Solit, D.; Sawyers, C.L. Drug discovery: How melanomas bypass new therapy. Nature 2010, 468, 902–903. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef]
- Zhang, K.; Wong, P.; Zhang, L.; Jacobs, B.; Borden, E.C.; Aster, J.C.; Bedogni, B. A Notch1-neuregulin1 autocrine signaling loop contributes to melanoma growth. Oncogene 2012, 31, 4609–4618. [Google Scholar] [CrossRef]
- Huynh, C.; Poliseno, L.; Segura, M.F.; Medicherla, R.; Haimovic, A.; Menendez, S.; Shang, S.; Pavlick, A.; Shao, Y.; Darvishian, F.; et al. The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One 2011, 6, e25264. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef]
- Imbimbo, B.P. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr. Top. Med. Chem. 2008, 8, 54–61. [Google Scholar] [CrossRef]
- Okamoto, R.; Tsuchiya, K.; Nemoto, Y.; Akiyama, J.; Nakamura, T.; Kanai, T.; Watanabe, M. Requirement of Notch activation during regeneration of the intestinal epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G23–G35. [Google Scholar]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–900. [Google Scholar] [CrossRef]
- Alexaki, V.I.; Javelaud, D.; van Kempen, L.C.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juàrez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef]
- Das, S.; Harris, L.G.; Metge, B.J.; Liu, S.; Riker, A.I.; Samant, R.S.; Shevde, L.A. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J. Biol. Chem. 2009, 284, 22888–22897. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef]
- Jalili, A.; Mertz, K.D.; Romanov, J.; Wagner, C.; Kalthoff, F.; Stuetz, A.; Pathria, G.; Gschaider, M.; Stingl, G.; Wagner, S.N. NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo. PLoS One 2013, 8, e69064. [Google Scholar]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef]
- Rose, A.E.; Poliseno, L.; Wang, J.; Clark, M.; Pearlman, A.; Wang, G.; de Miera, E.C.; Medicherla, R.; Christos, P.J.; Shapiro, R.; et al. Integrative genomics identifies molecular alterations that challenge the linear model of melanoma progression. Cancer Res. 2011, 71, 2561–2571. [Google Scholar] [CrossRef]
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef]
- Schreck, K.C.; Taylor, P.; Marchionni, L.; Gopalakrishnan, V.; Bar, E.E.; Gaiano, N.; Eberhart, C.G. The Notch target Hes1 directly modulates Gli1 expression and Hedgehog signaling: a potential mechanism of therapeutic resistance. Clin. Cancer Res. 2010, 16, 6060–6670. [Google Scholar] [CrossRef]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51–ra70. [Google Scholar] [CrossRef]
- Metcalfe, C.; de Sauvage, F.J. Hedgehog fights back: Mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011, 71, 5057–5061. [Google Scholar] [CrossRef]
- Li, C.; Chi, S.; Xie, J. Hedgehog signaling in skin cancers. Cell. Signal. 2011, 23, 1235–1243. [Google Scholar] [CrossRef]
- Mas, C.; Ruiz, I.; Altaba, A. Small molecule modulation of HH-GLI signaling: current leads, trials and tribulations. Biochem. Pharmacol. 2010, 80, 712–723. [Google Scholar] [CrossRef]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O'Reilly, K.E.; De Miera, E.V.-S.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog Pathway Blockade Inhibits Melanoma Cell Growth in Vitro and in Vivo. Pharmaceuticals 2013, 6, 1429-1450. https://doi.org/10.3390/ph6111429
O'Reilly KE, De Miera EV-S, Segura MF, Friedman E, Poliseno L, Han SW, Zhong J, Zavadil J, Pavlick A, Hernando E, et al. Hedgehog Pathway Blockade Inhibits Melanoma Cell Growth in Vitro and in Vivo. Pharmaceuticals. 2013; 6(11):1429-1450. https://doi.org/10.3390/ph6111429
Chicago/Turabian StyleO'Reilly, Kathryn E., Eleazar Vega-Saenz De Miera, Miguel F. Segura, Erica Friedman, Laura Poliseno, Sung Won Han, Judy Zhong, Jiri Zavadil, Anna Pavlick, Eva Hernando, and et al. 2013. "Hedgehog Pathway Blockade Inhibits Melanoma Cell Growth in Vitro and in Vivo" Pharmaceuticals 6, no. 11: 1429-1450. https://doi.org/10.3390/ph6111429