Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics

Abstract
:1. Introduction

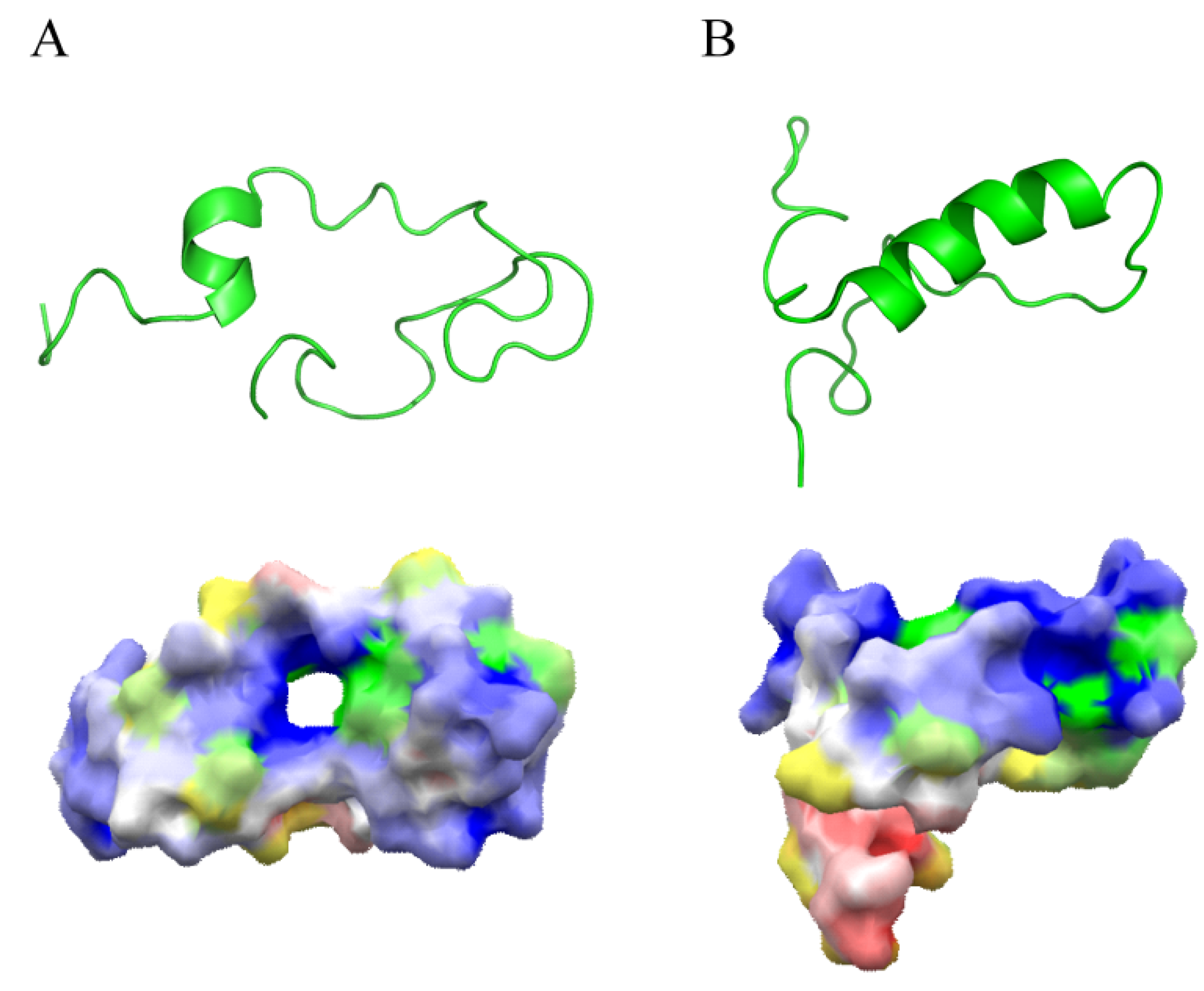{kind=link}
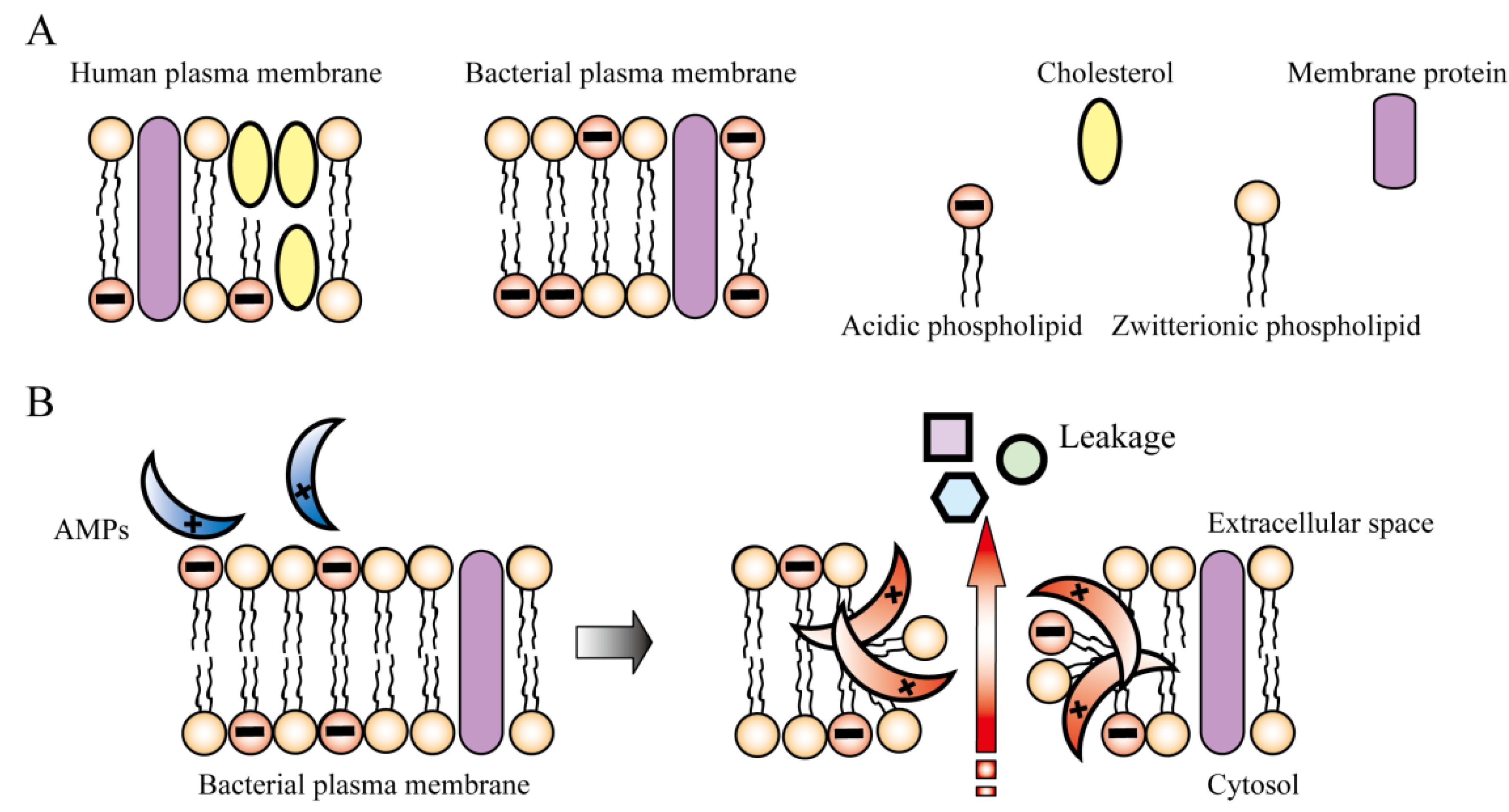{kind=link}
{kind=link}
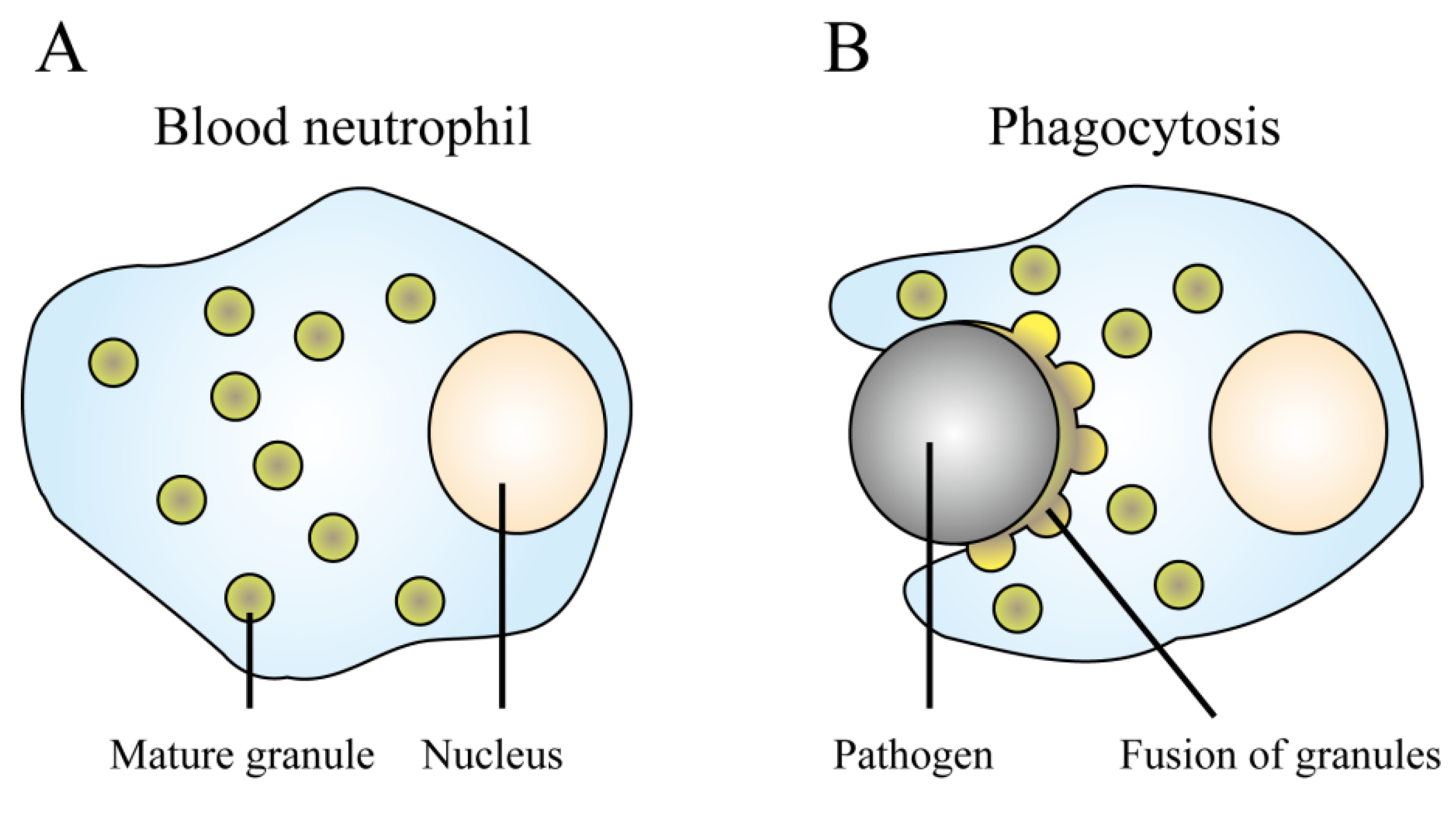{kind=link}
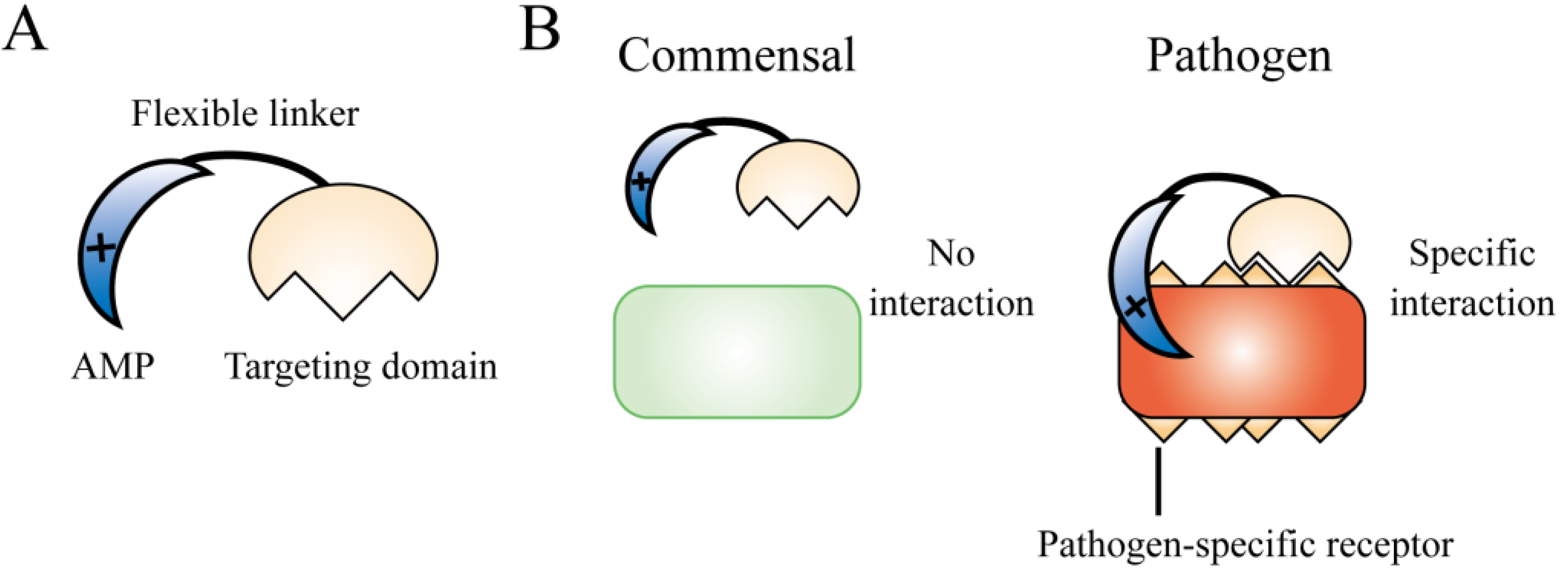{kind=link}
{kind=link}
| Antimicrobial peptide | Sequence | Origin | Description |
|---|---|---|---|
| Magainin 2 | GIGKFLHSAKKFGKAFVGEIMNS | X. laevis | First AMP isolated from X. laevis |
| Lactoferricin | GRRRRSVQWCA | Homo sapiens | AMP derived from lactoferrin |
| Buforin II | TRSSRAGLQFPVGRVHRLLRK | Bufo gargarizans | AMP derived from histone H2A |
| Drosocin | GKPRPYSPRPTSHPRPIRV | Drosophila melanogaster | The Thr residue is O-glycosylated. |
| Pyrrhocoricin | VDKGSYLPRPTPPRPIYNRN | Pyrrhocoris apterus | Inducible AMP of a sap-sucking insect |
| Apidaecin | GNNRPVYIPQPRPPHPRL | Apis mellifera | Isolated from the lymph fluid of honeybees |
| Lasioglossin-III | VNWKKILGKIIKVVK | Lasioglossum laticeps | AMP derived from bee venom |
| HNP1 | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils | Human defensins stored in azurophil granules |
| HNP2 | CYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils | |
| HNP3 | DCYCRIPACIAGERRYGTCIYQGRLWAFCC | Neutrophils | |
| HNP4 | VCSCRLVFCRRTELRVGNCLIGGVSFTYCCTRV | Neutrophils | |
| HBD1 | DHYNCVSSGGQCLYSACPIFTKIQGTCYRGKAKCCK | Epithelial cells | Human defensins secreted by epithelial cells |
| HBD2 | TCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP | Epithelial cells | |
| HBD3 | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK | Epithelial cells | |
| HBD4 | ELDRICGYGTARCRKKCRSQEYRIGRCPNTYACCLRK | Epithelial cells | |
| RTD1 | GFCRCLCRRGVCRCICTR | Primate | Premature stop codons in the human θ-defensin sequence |
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ | A. mellifera | Peptide antibiotic with toxicity to human cells |
| Gramicidin S | VOrnLdFPVOrnLdFP | Bacillus brevis | Peptide antibiotic with toxicity to human cells |
| Adepantin 1 | GIGKHVGKALKGLKGLLKGLGES | Artificial | Predicted by AMPad to have low hemolytic activity |
| R5L | PLCRCRVRPYRCRCVG | Artificial | Designed to mimic the LPS-binding sites of LBP, cyclic |
| Oncocin | VDKPPYLPRPRPPRRIYNR | Artificial | Proline-rich, Gram-selective AMP |
| M8G2 | TFFRLFNRGGGKNLRIIRKGIHIIKKY | Artificial | Designed using STAMP technology to target Streptococcus mutans |
| Clavanin A | VFQFLGKIIHHVGNFVHGFSHVF | Styela clava | Histidine-rich, pH-dependent AMP |
| AAP2 | FHFFHHFFHFFHHF | Artificial | Acid-activated AMP based on clavanin A |
| Protease-activated AMP | DDAEAVGPEAFADEDLDEGFIKAFPKRRWQWRMKKLG | Artificial | Protease-activated AMP based on lactoferricin |
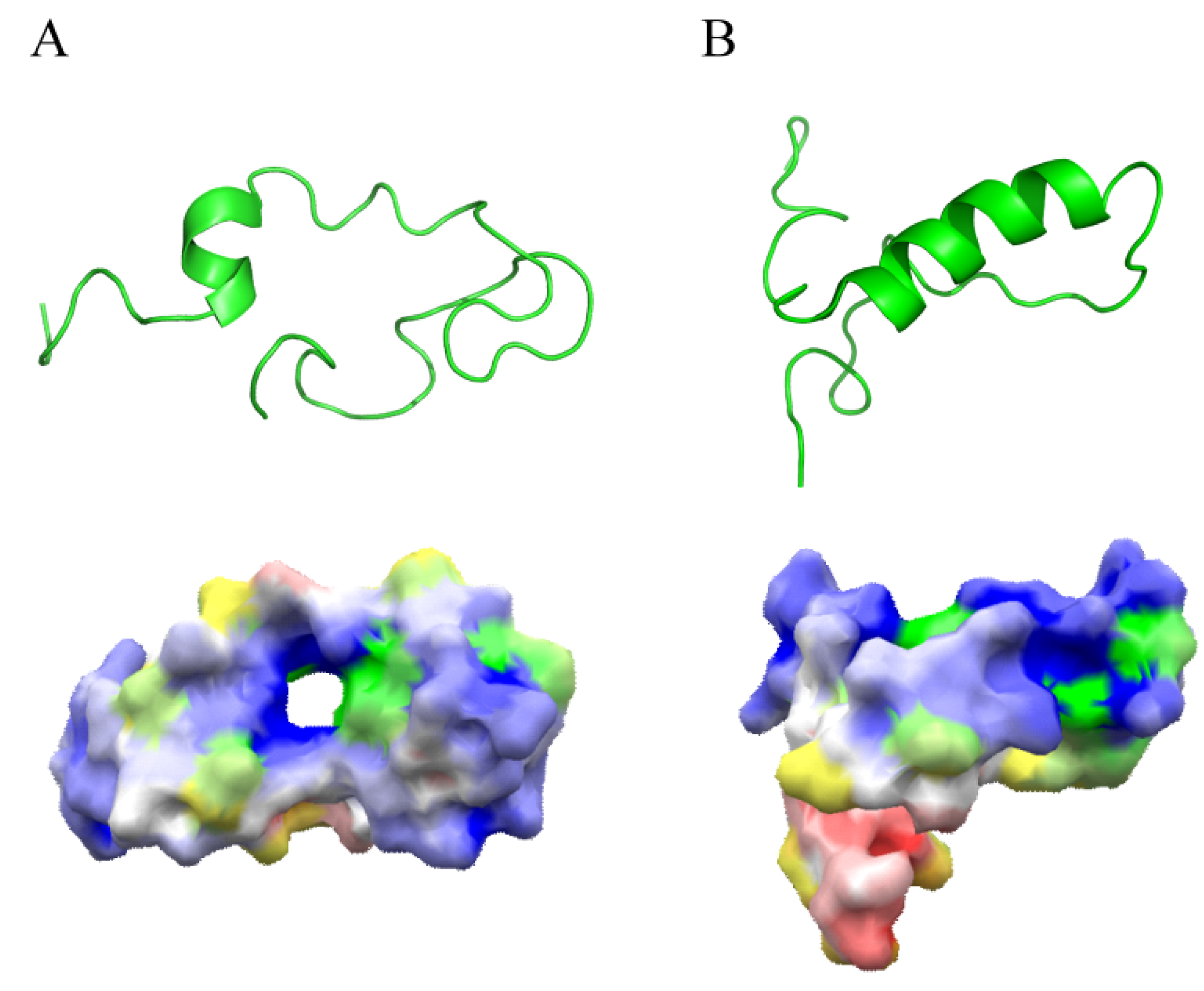
2. Mechanism of Action of AMPs

3. Improvement of AMPs for Clinical Use
3.1. Hemolytic Activity
3.2. Rapid Turnover in the Human Body
3.3. Reduced Activity due to Salt Sensitivity
3.4. High Cost of Production
4. Temporal and Spacial Regulation of AMPs in Nature
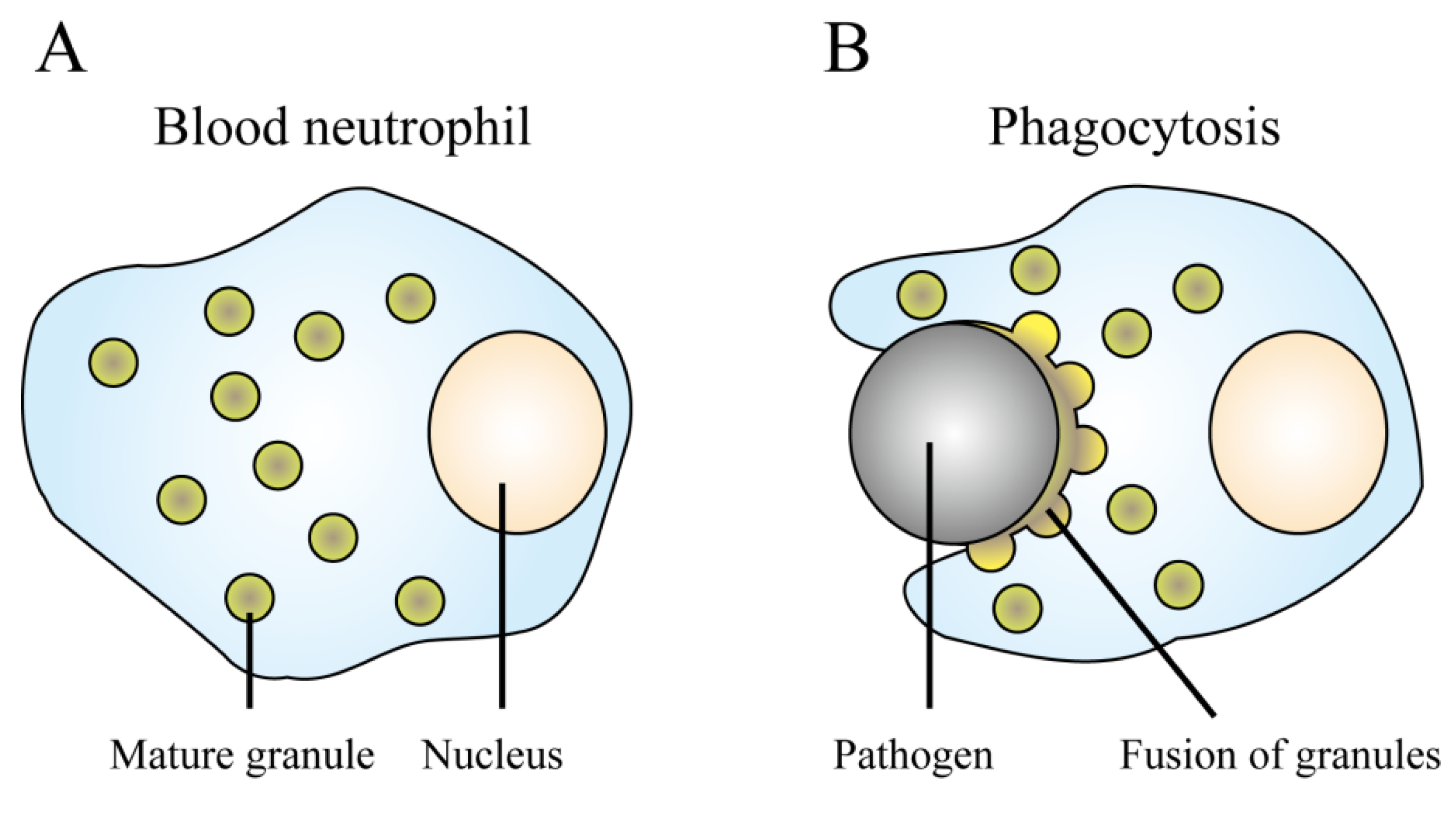
4.1. Neutrophils
4.2. Epithelial Cells
5. Design of Molecular-Targeted AMPs
5.1. Bacterium-Selective AMPs
5.2. Gram Nature-Selective AMPs
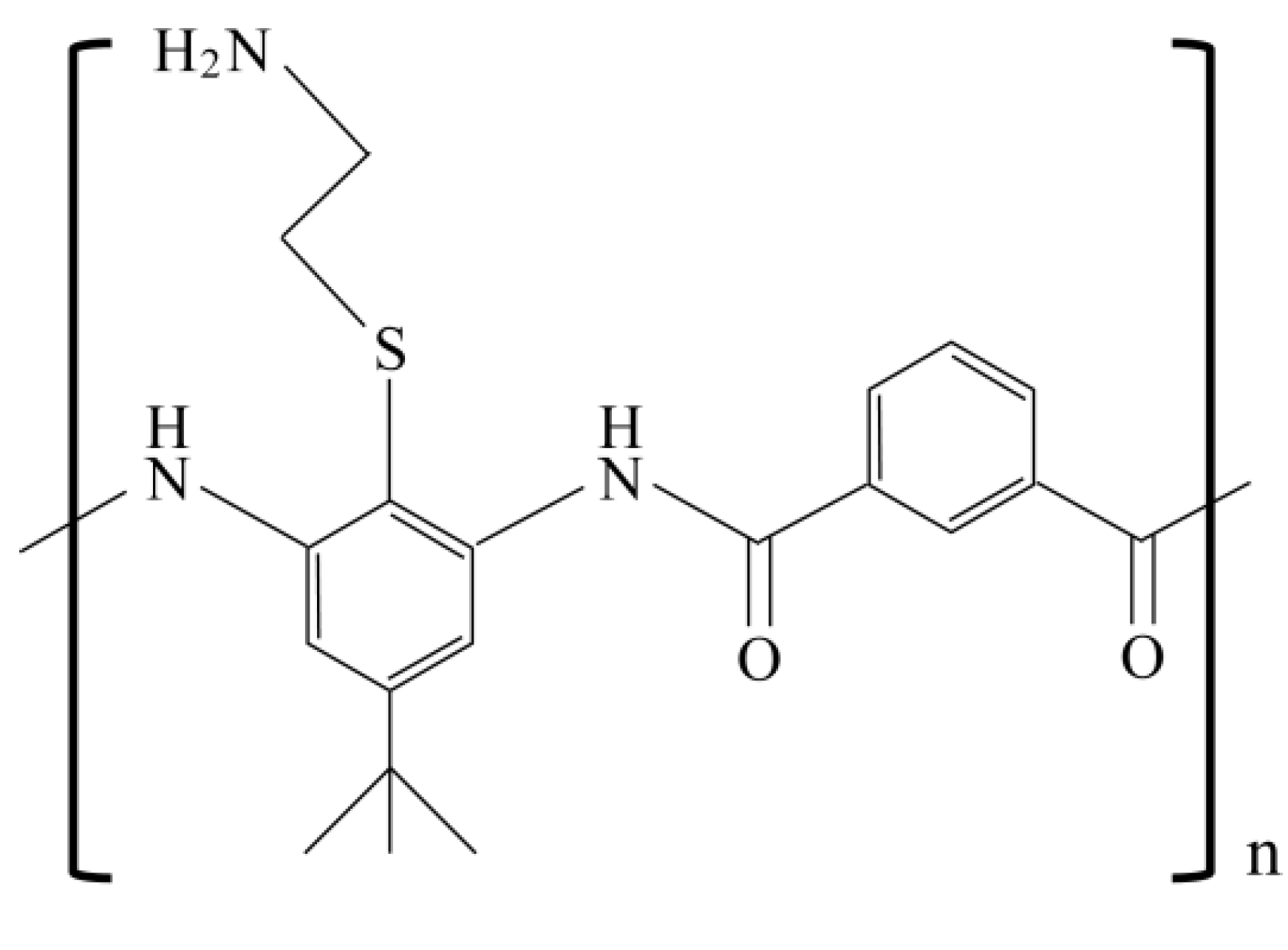
5.3. STAMP Technology

5.4. Environment-Sensing AMPs
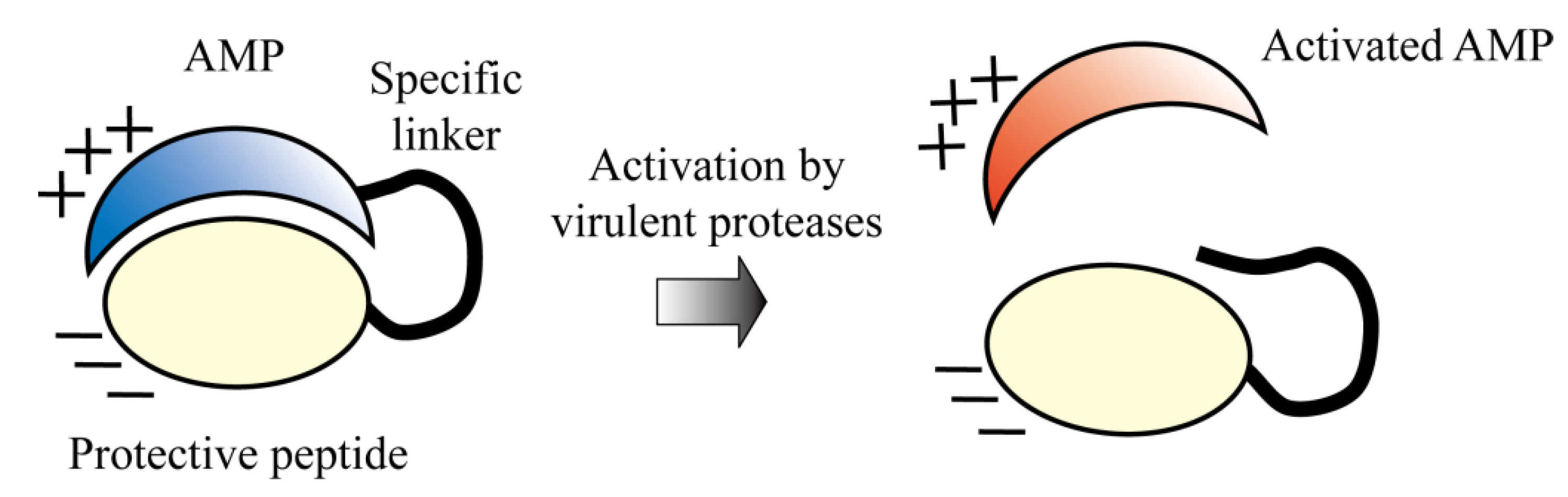
5.5. Protease-Activated AMPs
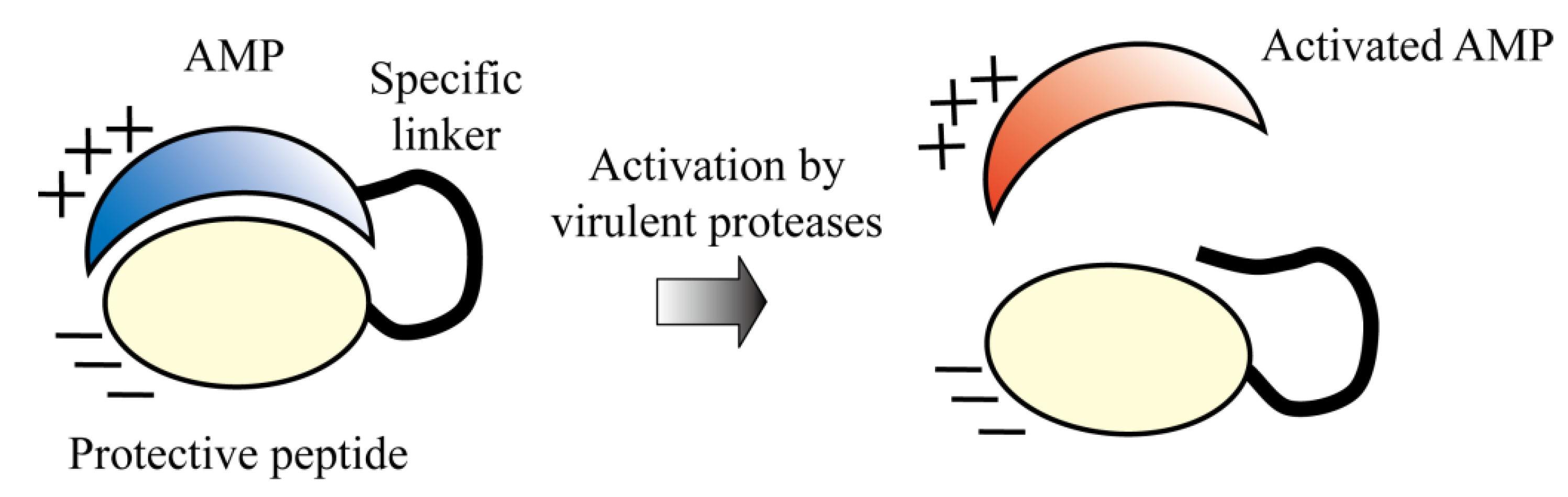
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Demain, A.L.; Sanchez, S. Microbial drug discovery: 80 years of progress. J. Antibiot. 2009, 62, 5–16. [Google Scholar] [CrossRef]
- Schatz, A.; Bugie, E.; Waksman, S.A. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Exp. Biol. Med. 1944, 55, 66–69. [Google Scholar] [CrossRef]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Morris, S.; Bai, G.H.; Suffys, P.; Portillo-Gomez, L.; Fairchok, M.; Rouse, D. Molecular mechanisms of multiple drug resistance in clinical isolates of Mycobacterium tuberculosis. J. Infect. Dis. 1995, 171, 954–960. [Google Scholar] [CrossRef]
- Cohen, M.L. Epidemiology of drug resistance: Implications for a post-antimicrobial era. Science 1992, 257, 1050–1055. [Google Scholar]
- Aloush, V.; Navon-Venezia, S.; Seigman-Igra, Y.; Cabili, S.; Carmeli, Y. Multidrug-resistant Pseudomonas aeruginosa: Risk factors and clinical impact. Antimicrob. Agents Chemother. 2006, 50, 43–48. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Aritaka, N.; Hanaki, H.; Kawasaki, S.; Hosoda, Y.; Hori, S.; Fukuchi, Y.; Kobayashi, I. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 1997, 350, 1670–1673. [Google Scholar] [CrossRef]
- Manchanda, V.; Sanchaita, S.; Singh, N. Multidrug resistant Acinetobacter. J. Glob. Infect. Dis. 2010, 2, 291–304. [Google Scholar] [CrossRef]
- Lee, M.; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B.; et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518. [Google Scholar] [CrossRef]
- Ament, P.W.; Jamshed, N.; Horne, J.P. Linezolid: Its role in the treatment of gram-positive, drug-resistant bacterial infections. Am. Fam. Physician 2002, 65, 663–670. [Google Scholar]
- Colca, J.R.; McDonald, W.G.; Waldon, D.J.; Thomasco, L.M.; Gadwood, R.C.; Lund, E.T.; Cavey, G.S.; Mathews, W.R.; Adams, L.D.; Cecil, E.T.; et al. Cross-linking in the living cell locates the site of action of oxazolidinone antibiotics. J. Biol. Chem. 2003, 278, 21972–21979. [Google Scholar] [CrossRef]
- Sanchez Garcia, M.; de la Torre, M.A.; Morales, G.; Pelaez, B.; Tolon, M.J.; Domingo, S.; Candel, F.J.; Andrade, R.; Arribi, A.; Garcia, N.; et al. Clinical outbreak of linezolid-resistant Staphylococcus aureus in an intensive care unit. JAMA 2010, 303, 2260–2264. [Google Scholar] [CrossRef]
- Seo, M.D.; Won, H.S.; Kim, J.H.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial peptides for therapeutic applications: A review. Molecules 2012, 17, 12276–12286. [Google Scholar] [CrossRef]
- Kosciuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzalkowska, N.; Jozwik, A.; Horbanczuk, J.; Krzyzewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus. skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Soravia, E.; Martini, G.; Zasloff, M. Antimicrobial properties of peptides from Xenopus granular gland secretions. FEBS Lett. 1988, 228, 337–340. [Google Scholar] [CrossRef]
- Giuliani, A.; Pirri, G.; Nicoletto, S.F. Antimicrobial peptides: An overview of a promising class of therapeutics. Cent. Eur. J. Biol. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Structural congruence among membrane-active host defense polypeptides of diverse phylogeny. Biochim. Biophys. Acta 2006, 1758, 1373–1386. [Google Scholar]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Park, S.C.; Park, Y.; Hahm, K.S. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Hiratani, T.; Uchida, K.; Yamaguch, H. Antifungal spectrum and fungicidal mechanism of an N-terminal peptide of bovine lactoferrin. J. Infect. Chemother. 1996, 1, 185–189. [Google Scholar] [CrossRef]
- Yamauchi, K.; Tomita, M.; Giehl, T.J.; Ellison, R.T., 3rd. Antibacterial activity of lactoferrin and a pepsin-derived lactoferrin peptide fragment. Infect. Immun. 1993, 61, 719–728. [Google Scholar]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sonksen, C.P.; Ludvigsen, S.; Raventos, D.; Buskov, S.; Christensen, B.; de Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- Helmerhorst, E.J.; Reijnders, I.M.; van't Hof, W.; Veerman, E.C.; Nieuw Amerongen, A.V. A critical comparison of the hemolytic and fungicidal activities of cationic antimicrobial peptides. FEBS Lett. 1999, 449, 105–110. [Google Scholar] [CrossRef]
- Aoki, W.; Kuroda, K.; Ueda, M. Next generation of antimicrobial peptides as molecular targeted medicines. J. Biosci. Bioeng. 2012, 114, 365–370. [Google Scholar] [CrossRef]
- Jenssen, H.; Aspmo, S.I. Serum stability of peptides. Methods Mol. Biol. 2008, 494, 177–186. [Google Scholar] [CrossRef]
- Bals, R.; Goldman, M.J.; Wilson, J.M. Mouse beta-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect. Immun. 1998, 66, 1225–1232. [Google Scholar]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.; Fujii, N.; Miyajima, K. Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2. Biochemistry 1995, 34, 3423–3429. [Google Scholar] [CrossRef]
- Ganz, T. Defensins and host defense. Science 1999, 286, 420–421. [Google Scholar] [CrossRef]
- Bellamy, W. Antifungal properties of lactoferricin B, a peptide derived from the N-terminal region of bovine lactoferrin. Lett. Appl. Microbiol. 1994, 18, 230–233. [Google Scholar] [CrossRef]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef]
- Nibbering, P.H.; Ravensbergen, E.; Welling, M.M.; van Berkel, L.A.; van Berkel, P.H.; Pauwels, E.K.; Nuijens, J.H. Human lactoferrin and peptides derived from its N terminus are highly effective against infections with antibiotic-resistant bacteria. Infect. Immun. 2001, 69, 1469–1476. [Google Scholar] [CrossRef]
- Dijkshoorn, L.; Brouwer, C.P.; Bogaards, S.J.; Nemec, A.; van den Broek, P.J.; Nibbering, P.H. The synthetic N-terminal peptide of human lactoferrin, hLF(1-11), is highly effective against experimental infection caused by multidrug-resistant Acinetobacte. baumannii. Antimicrob. Agents Chemother. 2004, 48, 4919–4921. [Google Scholar] [CrossRef]
- Bellamy, W.; Wakabayashi, H.; Takase, M.; Kawase, K.; Shimamura, S.; Tomita, M. Killing of Candida albicans by lactoferricin B, a potent antimicrobial peptide derived from the N-terminal region of bovine lactoferrin. Med. Microbiol. Immunol. 1993, 182, 97–105. [Google Scholar]
- Bellamy, W.; Takase, M.; Wakabayashi, H.; Kawase, K.; Tomita, M. Antibacterial spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J. Appl. Bacteriol. 1992, 73, 472–479. [Google Scholar] [CrossRef]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Mani, R.; Cady, S.D.; Tang, M.; Waring, A.J.; Lehrer, R.I.; Hong, M. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2006, 103, 16242–16247. [Google Scholar] [CrossRef]
- Hwang, P.M.; Zhou, N.; Shan, X.; Arrowsmith, C.H.; Vogel, H.J. Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry 1998, 37, 4288–4298. [Google Scholar] [CrossRef]
- Hunter, H.N.; Demcoe, A.R.; Jenssen, H.; Gutteberg, T.J.; Vogel, H.J. Human lactoferricin is partially folded in aqueous solution and is better stabilized in a membrane mimetic solvent. Antimicrob. Agents Chemother. 2005, 49, 3387–3395. [Google Scholar] [CrossRef]
- Jing, W.; Svendsen, J.S.; Vogel, H.J. Comparison of NMR structures and model-membrane interactions of 15-residue antimicrobial peptides derived from bovine lactoferricin. Biochem. Cell Biol. 2006, 84, 312–326. [Google Scholar] [CrossRef]
- Strom, M.B.; Haug, B.E.; Rekdal, O.; Skar, M.L.; Stensen, W.; Svendsen, J.S. Important structural features of 15-residue lactoferricin derivatives and methods for improvement of antimicrobial activity. Biochem. Cell Biol. 2002, 80, 65–74. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Uyterhoeven, E.T.; Butler, C.H.; Ko, D.; Elmore, D.E. Investigating the nucleic acid interactions and antimicrobial mechanism of buforin II. FEBS Lett. 2008, 582, 1715–1718. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; O, I.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Papo, N.; Shai, Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 2006, 281, 1636–1643. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Koehler, T.; Jacobsen, F.; Daigeler, A.; Goertz, O.; Langer, S.; Kesting, M.; Steinau, H.; Eriksson, E.; Hirsch, T. Host defense peptides in wound healing. Mol. Med. 2008, 14, 528–537. [Google Scholar]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expet. Opin. Invest. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef]
- Quinn, K.; Henriques, M.; Parker, T.; Slutsky, A.S.; Zhang, H. Human neutrophil peptides: A novel potential mediator of inflammatory cardiovascular diseases. Am. J. Physiol. Heart C 2008, 295, 1817–1824. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef]
- Nizet, V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 2006, 8, 11–26. [Google Scholar]
- Peschel, A.; Collins, L.V. Staphylococcal resistance to antimicrobial peptides of mammalian and bacterial origin. Peptides 2001, 22, 1651–1659. [Google Scholar] [CrossRef]
- Peschel, A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002, 10, 179–186. [Google Scholar] [CrossRef]
- Andra, J.; Goldmann, T.; Ernst, C.M.; Peschel, A.; Gutsmann, T. Multiple peptide resistance factor (MprF)-mediated resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol. J. Biol. Chem. 2011, 286, 18692–18700. [Google Scholar]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Bjorck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol. 2004, 172, 1169–1176. [Google Scholar]
- Sperandio, B.; Regnault, B.; Guo, J.; Zhang, Z.; Stanley, S.L., Jr.; Sansonetti, P.J.; Pedron, T. Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 2008, 205, 1121–1132. [Google Scholar] [CrossRef]
- Eilers, B.; Mayer-Scholl, A.; Walker, T.; Tang, C.; Weinrauch, Y.; Zychlinsky, A. Neutrophil antimicrobial proteins enhance Shigella flexneri adhesion and invasion. Cell. Microbiol. 2010, 12, 1134–1143. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Malmsten, M.; Kasetty, G.; Pasupuleti, M.; Alenfall, J.; Schmidtchen, A. Highly selective end-tagged antimicrobial peptides derived from PRELP. PLoS One 2011, 6, e16400. [Google Scholar]
- Maloy, W.L.; Kari, U.P. Structure-activity studies on magainins and other host defense peptides. Biopolymers 1995, 37, 105–122. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic alpha-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Dennison, S.R.; Phoenix, D.A. Influence of C-terminal amidation on the efficacy of modelin-5. Biochemistry 2011, 50, 1514–1523. [Google Scholar] [CrossRef]
- Hwang, H.; Hyun, S.; Kim, Y.; Yu, J. Reduction of helical content by insertion of a disulfide bond leads to an antimicrobial peptide with decreased hemolytic activity. Chem. Med. Chem. 2013, 8, 59–62. [Google Scholar]
- McInnes, C.; Kondejewski, L.H.; Hodges, R.S.; Sykes, B.D. Development of the structural basis for antimicrobial and hemolytic activities of peptides based on gramicidin S and design of novel analogs using NMR spectroscopy. J. Biol. Chem. 2000, 275, 14287–14294. [Google Scholar]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Kay, C.M.; Hancock, R.E.; Hodges, R.S. Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin S analogs. J. Biol. Chem. 1996, 271, 25261–25268. [Google Scholar]
- Jiang, Z.; Kullberg, B.J.; van der Lee, H.; Vasil, A.I.; Hale, J.D.; Mant, C.T.; Hancock, R.E.; Vasil, M.L.; Netea, M.G.; Hodges, R.S. Effects of hydrophobicity on the antifungal activity of alpha-helical antimicrobial peptides. Chem. Biol. Drug Des. 2008, 72, 483–495. [Google Scholar] [CrossRef]
- Kreil, G. D-Amino acids in animal peptides. Annu. Rev. Biochem. 1997, 66, 337–345. [Google Scholar] [CrossRef]
- Yin, L.M.; Lee, S.; Mak, J.S.; Helmy, A.S.; Deber, C.M. Differential binding of L- vs. d-isomers of cationic antimicrobial peptides to the biofilm exopolysaccharide alginate. Protein Pept. Lett. 2013, 20, 843–847. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Feder, R.; Gaidukov, L.; Carmeli, Y.; Mor, A. Antibacterial properties of dermaseptin S4 derivatives with in vivo activity. Antimicrob. Agents Chemother. 2002, 46, 689–694. [Google Scholar] [CrossRef]
- Stromstedt, A.A.; Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob. Agents Chemother. 2009, 53, 593–602. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One 2010, 5, e12684. [Google Scholar]
- Meng, H.; Kumar, K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J. Am. Chem. Soc. 2007, 129, 15615–15622. [Google Scholar] [CrossRef]
- Berhanu, W.M.; Ibrahim, M.A.; Pillai, G.G.; Oliferenko, A.A.; Khelashvili, L.; Jabeen, F.; Mirza, B.; Ansari, F.L.; Ul-Haq, I.; El-Feky, S.A.; et al. Similarity analysis, synthesis, and bioassay of antibacterial cyclic peptidomimetics. Beilstein. J. Org. Chem. 2012, 8, 1146–1160. [Google Scholar] [CrossRef]
- Tew, G.N.; Liu, D.; Chen, B.; Doerksen, R.J.; Kaplan, J.; Carroll, P.J.; Klein, M.L.; DeGrado, W.F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [Google Scholar]
- Mensa, B.; Kim, Y.H.; Choi, S.; Scott, R.; Caputo, G.A.; DeGrado, W.F. Antibacterial mechanism of action of arylamide foldamers. Antimicrob. Agents Chemother. 2011, 55, 5043–5053. [Google Scholar] [CrossRef]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar]
- Tam, J.P.; Lu, Y.A.; Yang, J.L. Correlations of cationic charges with salt sensitivity and microbial specificity of cystine-stabilized beta-strand antimicrobial peptides. J. Biol. Chem. 2002, 277, 50450–50456. [Google Scholar]
- Park, I.Y.; Cho, J.H.; Kim, K.S.; Kim, Y.B.; Kim, M.S.; Kim, S.C. Helix stability confers salt resistance upon helical antimicrobial peptides. J. Biol. Chem. 2004, 279, 13896–13901. [Google Scholar]
- Yu, H.Y.; Tu, C.H.; Yip, B.S.; Chen, H.L.; Cheng, H.T.; Huang, K.C.; Lo, H.J.; Cheng, J.W. Easy strategy to increase salt resistance of antimicrobial peptides. Antimicrob. Agents Chemother. 2011, 55, 4918–4921. [Google Scholar] [CrossRef]
- Chu, H.L.; Yu, H.Y.; Yip, B.S.; Chih, Y.H.; Liang, C.W.; Cheng, H.T.; Cheng, J.W. Boosting salt resistance of short antimicrobial peptides. Antimicrob. Agents Chemother. 2013. [Google Scholar] [CrossRef]
- Mishra, B.; Basu, A.; Saravanan, R.; Xiang, L.; Yang, L.K.; Leong, S.S.J. Lasioglossin-III: Antimicrobial characterization and feasibility study for immobilization applications. RSC Adv. 2013, 3, 9534–9543. [Google Scholar] [CrossRef]
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef]
- Bogomolovas, J.; Simon, B.; Sattler, M.; Stier, G. Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein Expr. Purif. 2009, 64, 16–23. [Google Scholar] [CrossRef]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.E.; Hancock, R.E.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef]
- Vidovic, V.; Prongidi-Fix, L.; Bechinger, B.; Werten, S. Production and isotope labeling of antimicrobial peptides in Escherichia coli by means of a novel fusion partner that enables high-yield insoluble expression and fast purification. J. Pept. Sci. 2009, 15, 278–284. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.H.; Hwang, S.W.; Lee, W.J.; Yoon, H.K.; Lee, H.S.; Hong, S.S. High-level expression of antimicrobial peptide mediated by a fusion partner reinforcing formation of inclusion bodies. Biochem. Biophys. Res. Commun. 2000, 277, 575–580. [Google Scholar] [CrossRef]
- Kim, J.M.; Jang, S.A.; Yu, B.J.; Sung, B.H.; Cho, J.H.; Kim, S.C. High-level expression of an antimicrobial peptide histonin as a natural form by multimerization and furin-mediated cleavage. Appl. Microbiol. Biotechnol. 2008, 78, 123–130. [Google Scholar]
- Lee, J.H.; Minn, I.; Park, C.B.; Kim, S.C. Acidic peptide-mediated expression of the antimicrobial peptide buforin II as tandem repeats in Escherichia coli. Protein Expr. Purif. 1998, 12, 53–60. [Google Scholar] [CrossRef]
- Kim, H.K.; Chun, D.S.; Kim, J.S.; Yun, C.H.; Lee, J.H.; Hong, S.K.; Kang, D.K. Expression of the cationic antimicrobial peptide lactoferricin fused with the anionic peptide in Escherichia coli. Appl. Microbiol. Biotechnol. 2006, 72, 330–338. [Google Scholar] [CrossRef]
- Agerberth, B.; Gudmundsson, G.H. Host antimicrobial defence peptides in human disease. Curr. Top. Microbiol. Immunol. 2006, 306, 67–90. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef]
- Putsep, K.; Carlsson, G.; Boman, H.G.; Andersson, M. Deficiency of antibacterial peptides in patients with morbus Kostmann: An observation study. Lancet 2002, 360, 1144–1149. [Google Scholar] [CrossRef]
- Diamond, G.; Beckloff, N.; Ryan, L.K. Host defense peptides in the oral cavity and the lung: Similarities and differences. J. Dent. Res. 2008, 87, 915–927. [Google Scholar] [CrossRef]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef]
- Desgranges, S.; Le Prieult, F.; Daly, A.; Lydon, J.; Brennan, M.; Rai, D.K.; Subasinghage, A.P.; Hewage, C.M.; Cryan, S.A.; Greene, C.; et al. In vitro activities of synthetic host defense propeptides processed by neutrophil elastase against cystic fibrosis pathogens. Antimicrob. Agents Chemother. 2011, 55, 2487–2489. [Google Scholar] [CrossRef]
- Dale, B.A.; Fredericks, L.P. Antimicrobial peptides in the oral environment: Expression and function in health and disease. Curr. Issues Mol. Biol. 2005, 7, 119–133. [Google Scholar]
- Gorr, S.-U. Antimicrobial peptides of the oral cavity. Periodontol. 2000 2009, 51, 152–180. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Cole, A.M.; Hong, T.; Boo, L.M.; Nguyen, T.; Zhao, C.; Bristol, G.; Zack, J.A.; Waring, A.J.; Yang, O.O.; Lehrer, R.I. Retrocyclin: A primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc. Natl. Acad. Sci. USA 2002, 99, 1813–1818. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Invest. 1985, 76, 1427–1435. [Google Scholar] [CrossRef]
- Ganz, T.; Lehrer, R.I. Defensins. Pharmacol. Ther. 1995, 66, 191–205. [Google Scholar] [CrossRef]
- Rice, W.G.; Ganz, T.; Kinkade, J.M., Jr.; Selsted, M.E.; Lehrer, R.I.; Parmley, R.T. Defensin-rich dense granules of human neutrophils. Blood 1987, 70, 757–765. [Google Scholar]
- Cowland, J.B.; Borregaard, N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J. Leukoc. Biol. 1999, 66, 989–995. [Google Scholar]
- Yount, N.Y.; Wang, M.S.; Yuan, J.; Banaiee, N.; Ouellette, A.J.; Selsted, M.E. Rat neutrophil defensins. Precursor structures and expression during neutrophilic myelopoiesis. J. Immunol. 1995, 155, 4476–4484. [Google Scholar]
- Faurschou, M.; Sorensen, O.E.; Johnsen, A.H.; Askaa, J.; Borregaard, N. Defensin-rich granules of human neutrophils: Characterization of secretory properties. Biochim. Biophys. Acta 2002, 1591, 29–35. [Google Scholar]
- Ganz, T. Extracellular release of antimicrobial defensins by human polymorphonuclear leukocytes. Infect. Immun. 1987, 55, 568–571. [Google Scholar]
- Ganz, T.; Oren, A.; Lehrer, R.I. Defensins: Microbicidal and cytotoxic peptides of mammalian host defense cells. Med. Microbiol. Immunol. 1992, 181, 99–105. [Google Scholar]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Sengelov, H.; Kjeldsen, L.; Borregaard, N. Control of exocytosis in early neutrophil activation. J. Immunol. 1993, 150, 1535–1543. [Google Scholar]
- Joly, S.; Maze, C.; McCray, P.B., Jr.; Guthmiller, J.M. Human beta-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J. Clin. Microbiol. 2004, 42, 1024–1029. [Google Scholar]
- Dale, B.A.; Kimball, J.R.; Krisanaprakornkit, S.; Roberts, F.; Robinovitch, M.; O’Neal, R.; Valore, E.V.; Ganz, T.; Anderson, G.M.; Weinberg, A. Localized antimicrobial peptide expression in human gingiva. J. Periodontal Res. 2001, 36, 285–294. [Google Scholar]
- Sawaki, K.; Mizukawa, N.; Yamaai, T.; Fukunaga, J.; Sugahara, T. Immunohistochemical study on expression of alpha-defensin and beta-defensin-2 in human buccal epithelia with candidiasis. Oral Dis. 2002, 8, 37–41. [Google Scholar] [CrossRef]
- Chung, W.O.; Dale, B.A. Innate immune response of oral and foreskin keratinocytes: Utilization of different signaling pathways by various bacterial species. Infect. Immun. 2004, 72, 352–358. [Google Scholar] [CrossRef]
- Liu, L.; Roberts, A.A.; Ganz, T. By IL-1 signaling, monocyte-derived cells dramatically enhance the epidermal antimicrobial response to lipopolysaccharide. J. Immunol. 2003, 170, 575–580. [Google Scholar]
- Garcia, J.R.; Krause, A.; Schulz, S.; Rodriguez-Jimenez, F.J.; Kluver, E.; Adermann, K.; Forssmann, U.; Frimpong-Boateng, A.; Bals, R.; Forssmann, W.G. Human beta-defensin 4: A novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J. 2001, 15, 1819–1821. [Google Scholar]
- Sorensen, O.E.; Cowland, J.B.; Theilgaard-Monch, K.; Liu, L.; Ganz, T.; Borregaard, N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J. Immunol. 2003, 170, 5583–5589. [Google Scholar]
- Arvola, T.; Laiho, K.; Torkkeli, S.; Mykkanen, H.; Salminen, S.; Maunula, L.; Isolauri, E. Prophylactic Lactobacillus GG reduces antibiotic-associated diarrhea in children with respiratory infections: A randomized study. Pediatrics 1999, 104, e64. [Google Scholar] [CrossRef]
- Doron, S.I.; Hibberd, P.L.; Gorbach, S.L. Probiotics for prevention of antibiotic-associated diarrhea. J. Clin. Gastroenterol. 2008, 42, 58–63. [Google Scholar] [CrossRef]
- Danna, P.L.; Urban, C.; Bellin, E.; Rahal, J.J. Role of Candida in pathogenesis of antibiotic-associated diarrhoea in elderly inpatients. Lancet 1991, 337, 511–514. [Google Scholar] [CrossRef]
- Giannella, R.A. Antibiotic-associated diarrhea and Clostridium difficile colitis: An update. Rev. Esp. Enferm. Dig. 2001, 93, 535–543. [Google Scholar]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar] [CrossRef]
- Umeyama, M.; Kira, A.; Nishimura, K.; Naito, A. Interactions of bovine lactoferricin with acidic phospholipid bilayers and its antimicrobial activity as studied by solid-state NMR. Biochim. Biophys. Acta 2006, 1758, 1523–1528. [Google Scholar]
- Katsu, T.; Kuroko, M.; Morikawa, T.; Sanchika, K.; Fujita, Y.; Yamamura, H.; Uda, M. Mechanism of membrane damage induced by the amphipathic peptides gramicidin S and melittin. Biochim. Biophys. Acta 1989, 983, 135–141. [Google Scholar] [CrossRef]
- Steiner, H.; Hultmark, D.; Engstrom, A.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Chou, H.T.; Kuo, T.Y.; Chiang, J.C.; Pei, M.J.; Yang, W.T.; Yu, H.C.; Lin, S.B.; Chen, W.J. Design and synthesis of cationic antimicrobial peptides with improved activity and selectivity against Vibrio spp. Int. J. Antimicrob. Agents 2008, 32, 130–138. [Google Scholar] [CrossRef]
- Juretic, D.; Vukicevic, D.; Petrov, D.; Novkovic, M.; Bojovic, V.; Lucic, B.; Ilic, N.; Tossi, A. Knowledge-based computational methods for identifying or designing novel, non-homologous antimicrobial peptides. Eur. Biophys. J. 2011, 40, 371–385. [Google Scholar] [CrossRef]
- Juretic, D.; Vukicevic, D.; Ilic, N.; Antcheva, N.; Tossi, A. Computational design of highly selective antimicrobial peptides. J. Chem. Inf. Model. 2009, 49, 2873–2882. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef]
- Gaynes, R.; Edwards, J.R. Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis. 2005, 41, 848–854. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Hooper, D.C. Hospital-acquired infections due to gram-negative bacteria. N. Engl. J. Med. 2010, 362, 1804–1813. [Google Scholar] [CrossRef]
- Su, Y.; Waring, A.J.; Ruchala, P.; Hong, M. Structures of beta-hairpin antimicrobial protegrin peptides in lipopolysaccharide membranes: Mechanism of gram selectivity obtained from solid-state nuclear magnetic resonance. Biochemistry 2011, 50, 2072–2083. [Google Scholar] [CrossRef]
- Muhle, S.A.; Tam, J.P. Design of Gram-negative selective antimicrobial peptides. Biochemistry 2001, 40, 5777–5785. [Google Scholar] [CrossRef]
- Scott, M.G.; Rosenberger, C.M.; Gold, M.R.; Finlay, B.B.; Hancock, R.E. An alpha-helical cationic antimicrobial peptide selectively modulates macrophage responses to lipopolysaccharide and directly alters macrophage gene expression. J. Immunol. 2000, 165, 3358–3365. [Google Scholar]
- Casteels, P.; Ampe, C.; Jacobs, F.; Vaeck, M.; Tempst, P. Apidaecins: Antibacterial peptides from honeybees. EMBO J. 1989, 8, 2387–2391. [Google Scholar]
- Bulet, P.; Dimarcq, J.L.; Hetru, C.; Lagueux, M.; Charlet, M.; Hegy, G.; van Dorsselaer, A.; Hoffmann, J.A. A novel inducible antibacterial peptide of Drosophila carries an O-glycosylated substitution. J. Biol. Chem. 1993, 268, 14893–14897. [Google Scholar]
- Knappe, D.; Piantavigna, S.; Hansen, A.; Mechler, A.; Binas, A.; Nolte, O.; Martin, L.L.; Hoffmann, R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): A novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 2010, 53, 5240–5247. [Google Scholar] [CrossRef]
- Shamova, O.; Brogden, K.A.; Zhao, C.; Nguyen, T.; Kokryakov, V.N.; Lehrer, R.I. Purification and properties of proline-rich antimicrobial peptides from sheep and goat leukocytes. Infect. Immun. 1999, 67, 4106–4111. [Google Scholar]
- He, J.; Yarbrough, D.K.; Kreth, J.; Anderson, M.H.; Shi, W.; Eckert, R. Systematic approach to optimizing specifically targeted antimicrobial peptides against Streptococcus mutans. Antimicrob. Agents Chemother. 2010, 54, 2143–2151. [Google Scholar] [CrossRef]
- Eckert, R.; Qi, F.; Yarbrough, D.K.; He, J.; Anderson, M.H.; Shi, W. Adding selectivity to antimicrobial peptides: Rational design of a multidomain peptide against Pseudomonas spp. Antimicrob. Agents Chemother. 2006, 50, 1480–1488. [Google Scholar] [CrossRef]
- Banas, J.A. Virulence properties of Streptococcus mutans. Front. Biosci. 2004, 9, 1267–1277. [Google Scholar] [CrossRef]
- Loesche, W.J. Role of Streptococcus mutans in human dental decay. Microbiol. Rev. 1986, 50, 353–380. [Google Scholar]
- Eckert, R.; He, J.; Yarbrough, D.K.; Qi, F.; Anderson, M.H.; Shi, W. Targeted killing of Streptococcus mutans by a pheromone-guided “smart” antimicrobial peptide. Antimicrob. Agents Chemother. 2006, 50, 3651–3657. [Google Scholar] [CrossRef]
- Li, L.N.; Guo, L.H.; Lux, R.; Eckert, R.; Yarbrough, D.; He, J.; Anderson, M.; Shi, W.Y. Targeted antimicrobial therapy against Streptococcus mutans establishes protective non-cariogenic oral biofilms and reduces subsequent infection. Int. J. Oral Sci. 2010, 2, 66–73. [Google Scholar] [CrossRef]
- Svensater, G.; Sjogreen, B.; Hamilton, I.R. Multiple stress responses in Streptococcus mutans and the induction of general and stress-specific proteins. Microbiology 2000, 146, 107–117. [Google Scholar]
- Lee, I.H.; Zhao, C.; Cho, Y.; Harwig, S.S.; Cooper, E.L.; Lehrer, R.I. Clavanins, alpha-helical antimicrobial peptides from tunicate hemocytes. FEBS Lett. 1997, 400, 158–162. [Google Scholar]
- Li, L.; He, J.; Eckert, R.; Yarbrough, D.; Lux, R.; Anderson, M.; Shi, W. Design and characterization of an acid-activated antimicrobial peptide. Chem. Biol. Drug Des. 2009, 75, 127–132. [Google Scholar]
- Tsuboi, R.; Matsuda, K.; Ko, I.J.; Ogawa, H. Correlation between culture medium pH, extracellular proteinase activity, and cell growth of Candida albicans in insoluble stratum corneum-supplemented media. Arch. Dermatol. Res. 1989, 281, 342–345. [Google Scholar] [CrossRef]
- Wegscheid-Gerlach, C.; Gerber, H.D.; Diederich, W.E. Proteases of Plasmodium falciparum as potential drug targets and inhibitors thereof. Curr. Top. Med. Chem. 2010, 10, 346–367. [Google Scholar] [CrossRef]
- Aoki, W.; Kitahara, N.; Miura, N.; Morisaka, H.; Yamamoto, Y.; Kuroda, K.; Ueda, M. Candida albicans possesses Sap7 as a pepstatin A-insensitive secreted aspartic protease. PLoS One 2012, 7, e32513. [Google Scholar]
- Aoki, W.; Kitahara, N.; Miura, N.; Morisaka, H.; Yamamoto, Y.; Kuroda, K.; Ueda, M. Comprehensive characterization of secreted aspartic proteases encoded by a virulence gene family in Candida albicans. J. Biochem. 2011, 150, 431–438. [Google Scholar] [CrossRef]
- Aoki, W.; Kitahara, N.; Miura, N.; Morisaka, H.; Kuroda, K.; Ueda, M. Design of a novel antimicrobial peptide activated by virulent proteases. Chem. Biol. Drug Des. 2012, 80, 725–733. [Google Scholar] [CrossRef]
- Aoki, W.; Kitahara, N.; Miura, N.; Morisaka, H.; Kuroda, K.; Ueda, M. Profiling of adhesive properties of the agglutinin-like sequence (ALS) protein family, a virulent attribute of Candida albicans. FEMS Immunol. Med. Microbiol. 2012, 65, 121–124. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, T.; Tatsukami, Y.; Kitahara, N.; Morisaka, H.; Kuroda, K.; Ueda, M. Time-course proteomic profile of Candida albicans during adaptation to a fetal serum. Pathog. Dis. 2013, 67, 67–75. [Google Scholar] [CrossRef]
- Aoki, W.; Tatsukami, Y.; Kitahara, N.; Matsui, K.; Morisaka, H.; Kuroda, K.; Ueda, M. Elucidation of potentially virulent factors of Candida albicans during serum adaptation by using quantitative time-course proteomics. J. Proteomics 2013, in press. [Google Scholar]
- Aoki, W.; Ueda, M. New developments in the pathogenesis of Candida albicans: Characterization of secreted aspartic proteases and profiling of survival mechanism in blood. In Candida albicans: Symptoms, Causes and Treatment Options; Nova Science Publishers: New York, NY, USA, 2013; in press. [Google Scholar]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta 1788, 1680–1686. [Google Scholar]
- Bulmus, V.; Ding, Z.; Long, C.J.; Stayton, P.S.; Hoffman, A.S. Site-specific polymer-streptavidin bioconjugate for pH-controlled binding and triggered release of biotin. Bioconjug. Chem. 2000, 11, 78–83. [Google Scholar] [CrossRef]
- Kim, J.J.; Park, K. Modulated insulin delivery from glucose-sensitive hydrogel dosage forms. J. Control. Release 2001, 77, 39–47. [Google Scholar]
- Fogueri, L.R.; Singh, S. Smart polymers for controlled delivery of proteins and peptides: A review of patents. Recent Pat. Drug Deliv. Formul. 2009, 3, 40–48. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aoki, W.; Ueda, M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals 2013, 6, 1055-1081. https://doi.org/10.3390/ph6081055
Aoki W, Ueda M. Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics. Pharmaceuticals. 2013; 6(8):1055-1081. https://doi.org/10.3390/ph6081055
Chicago/Turabian StyleAoki, Wataru, and Mitsuyoshi Ueda. 2013. "Characterization of Antimicrobial Peptides toward the Development of Novel Antibiotics" Pharmaceuticals 6, no. 8: 1055-1081. https://doi.org/10.3390/ph6081055