“Specificity Determinants” Improve Therapeutic Indices of Two Antimicrobial Peptides Piscidin 1 and Dermaseptin S4 Against the Gram-negative Pathogens Acinetobacter baumannii and Pseudomonas aeruginosa

Abstract
:
1. Introduction
2. Experimental
2.1. Peptide Synthesis and Purification
2.2. Analytical RP-HPLC and Temperature Profiling of Peptides
2.3. Characterization of Helical Structure
2.4. Determination of Peptide Amphipathicity
2.5. Gram-Negative Bacteria Strains Used in This Study
2.6. Measurement of Antimicrobial Activity (MIC)
2.7. Measurement of Hemolytic Activity (HC50)
2.8. Calculation of Therapeutic Index (HC50/MIC Ratio)
3. Results and Discussion
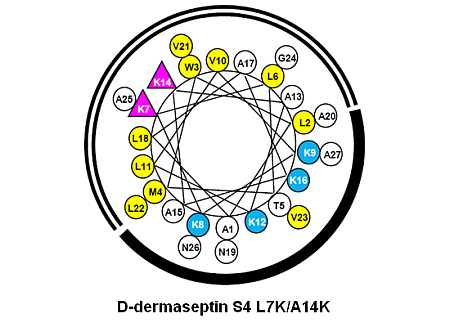
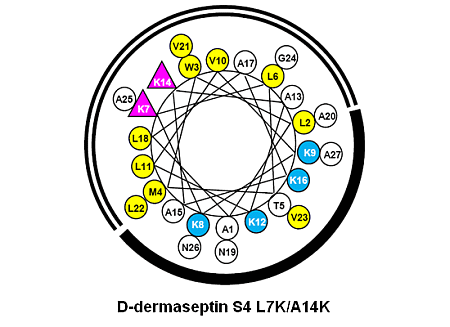
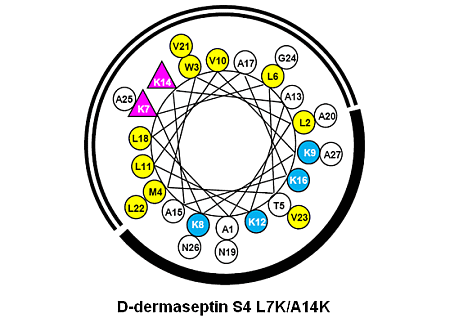
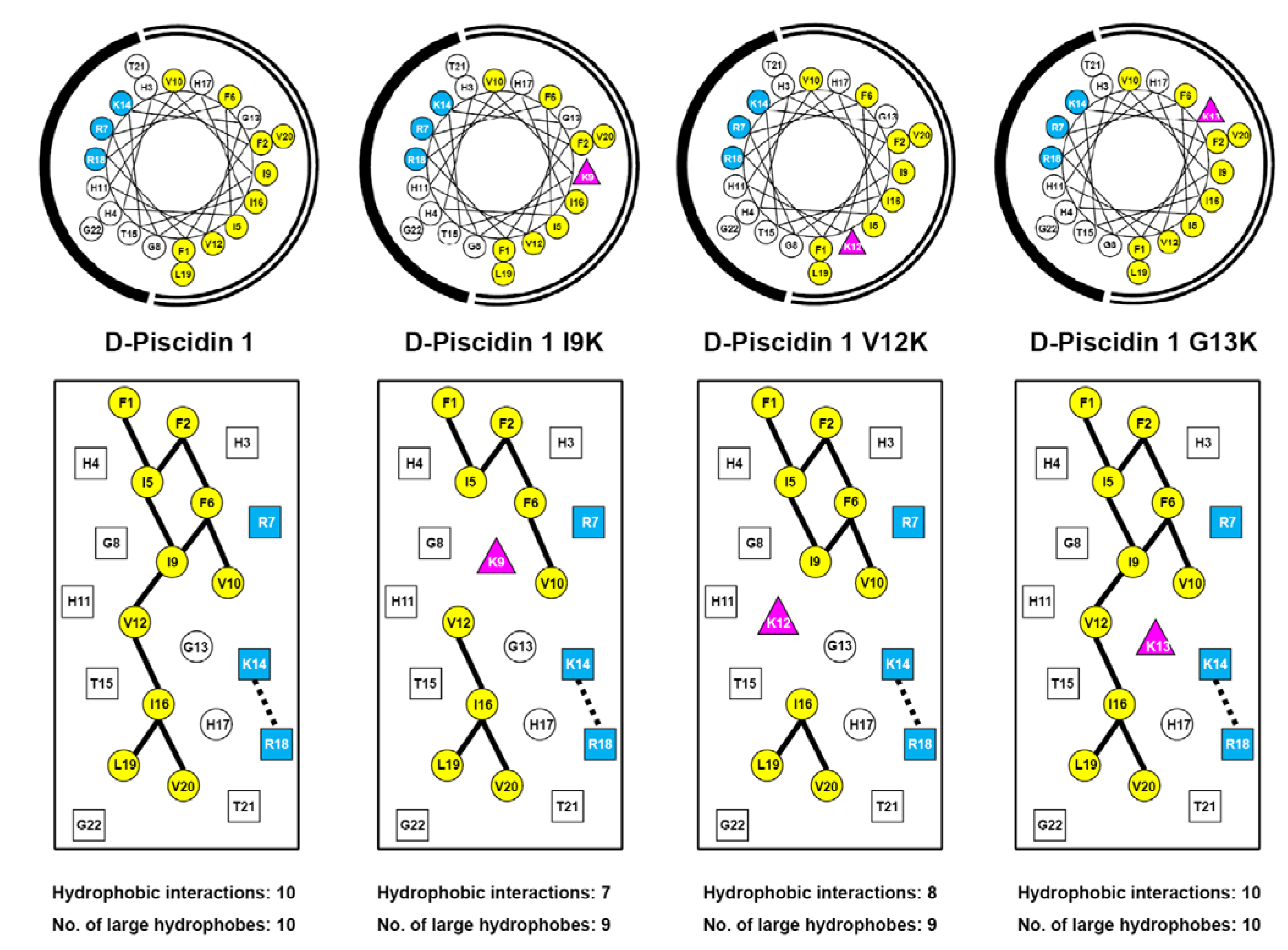
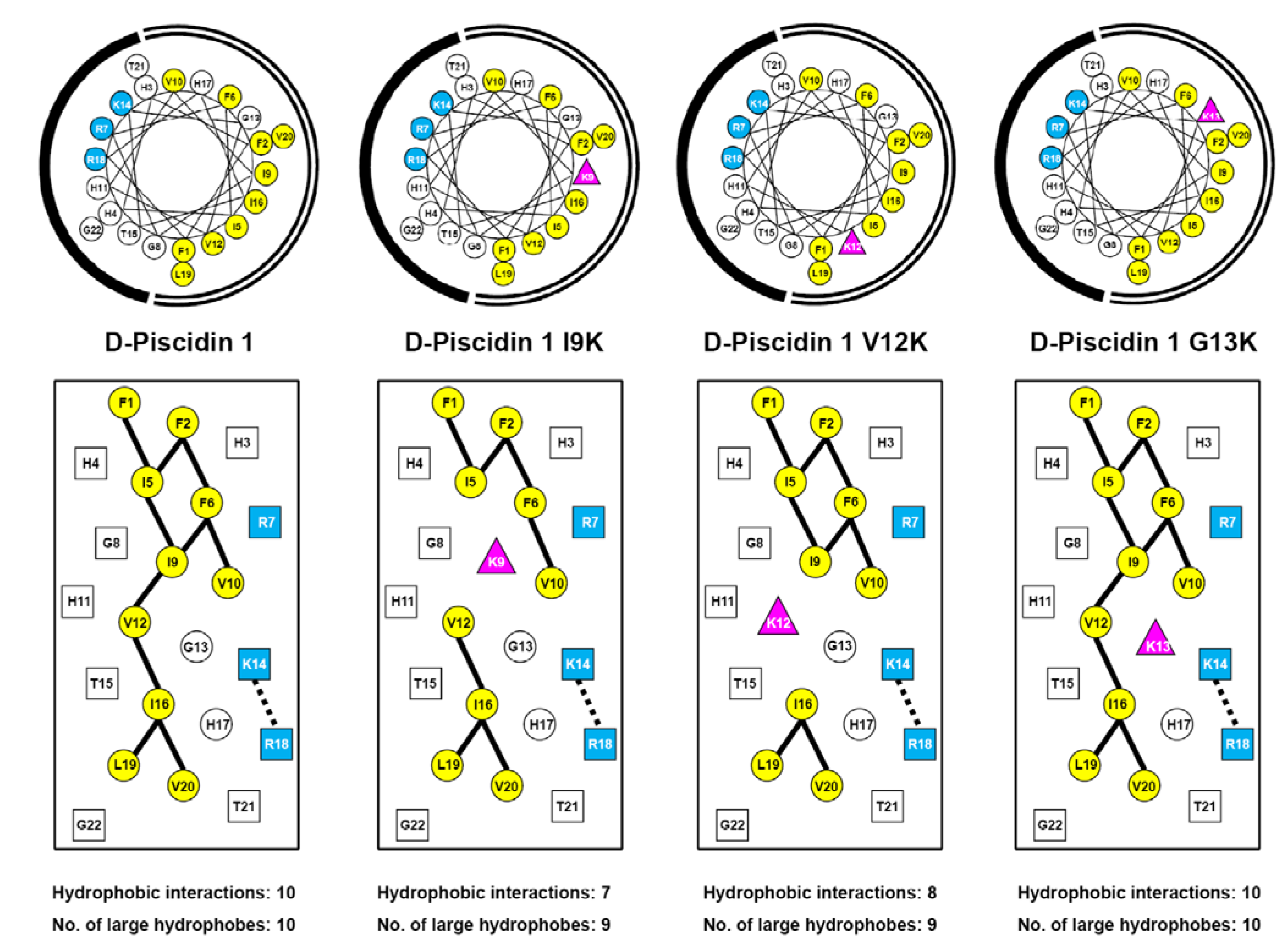
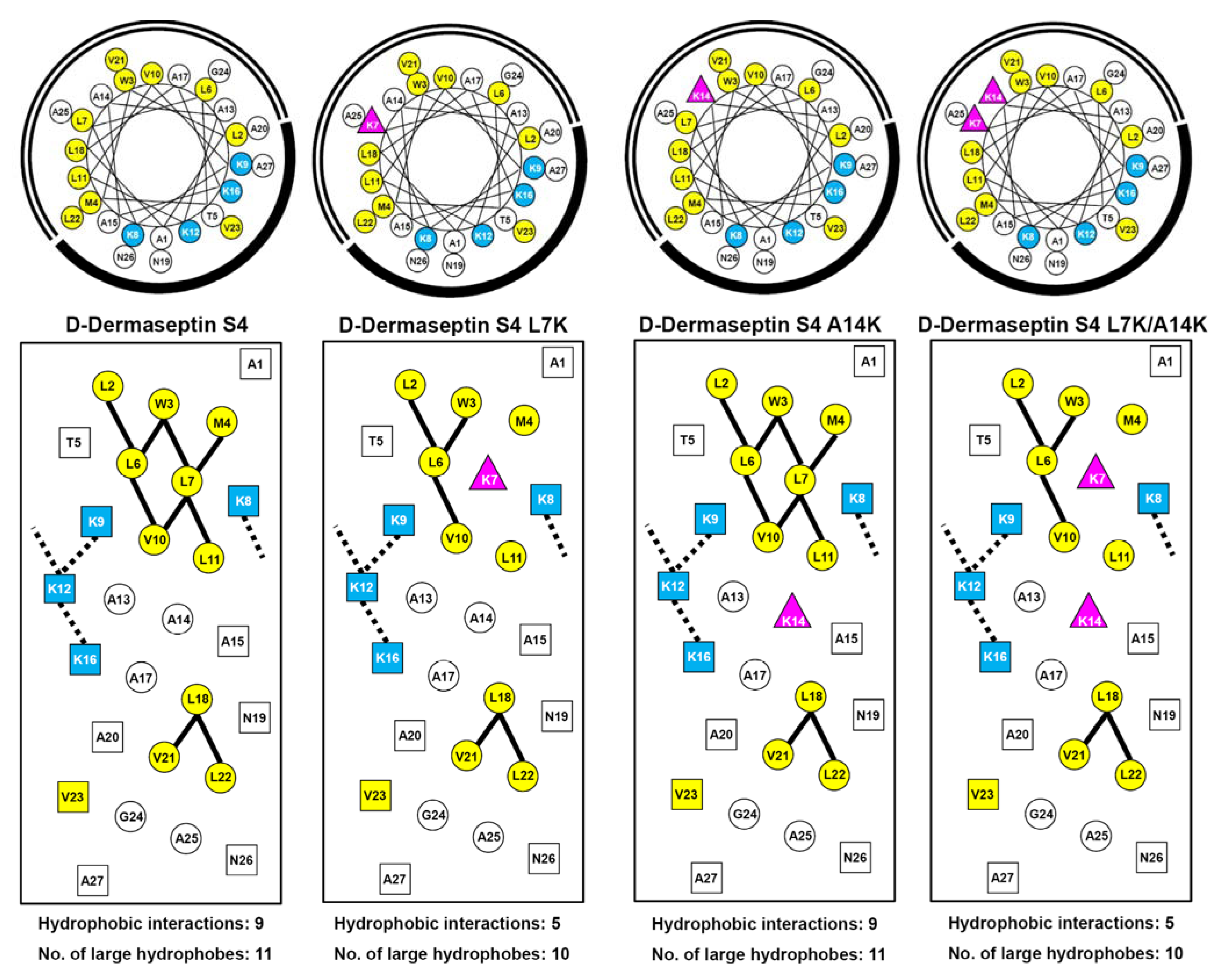
3.1. Peptide Design and Specificity Determinant(s)

{kind=link}
{kind=link}
{kind=link}
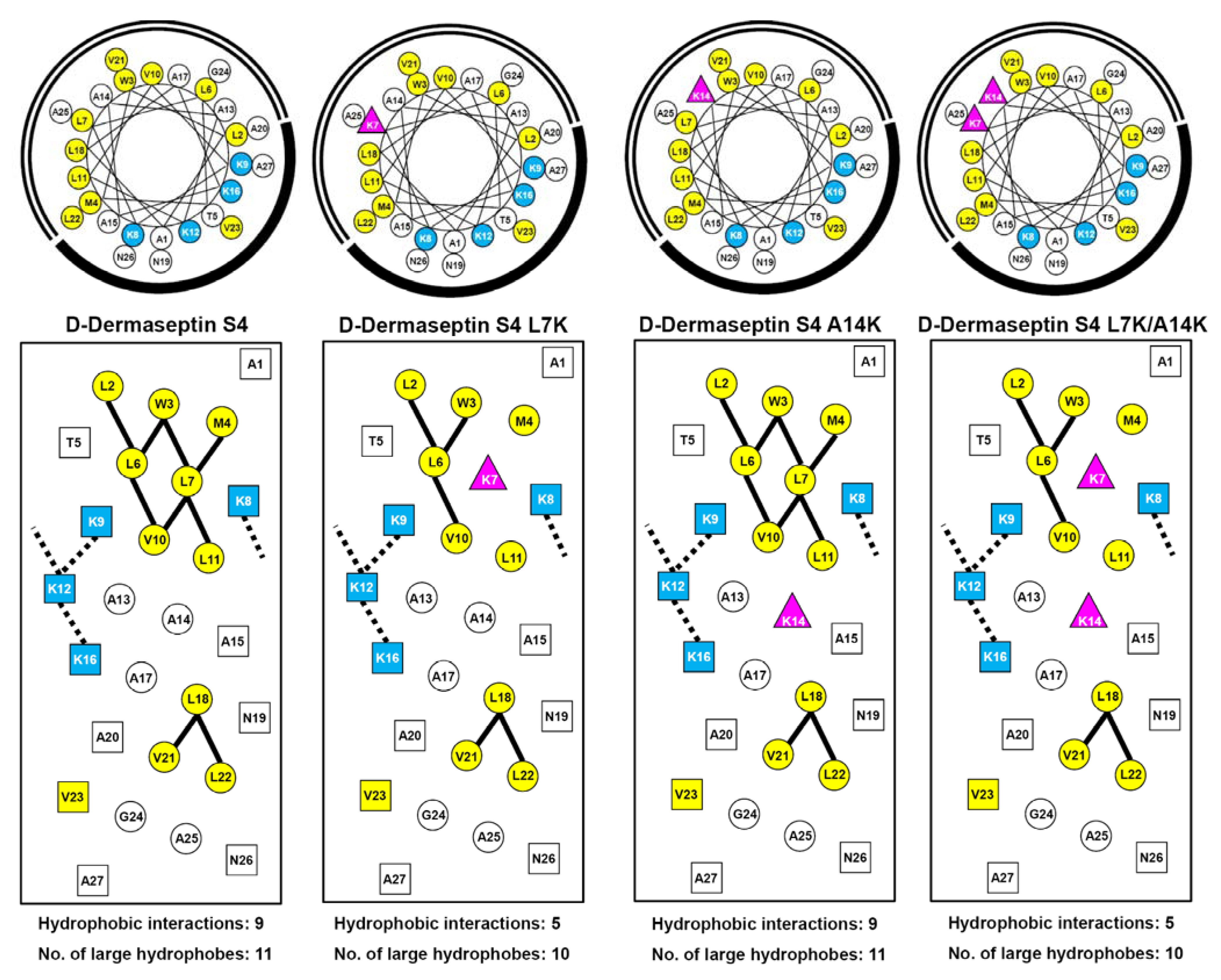{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name a | Length | Sequence b | MW |
|---|---|---|---|
| D-Piscidin 1 | 22 | NH2-FFHHIFRGIVHVGKTIHRLVTG-amide | 2571 |
| D-Piscidin 1 G8P c | 22 | NH2-FFHHIFRPIVHVGKTIHRLVTG-amide | 2611 |
| D-Piscidin 1 I9K | 22 | NH2-FFHHIFRGKVHVGKTIHRLVTG-amide | 2586 |
| D-Piscidin 1 V12K | 22 | NH2-FFHHIFRGIVHKGKTIHRLVTG-amide | 2600 |
| D-Piscidin 1 G13K | 22 | NH2-FFHHIFRGIVHVKKTIHRLVTG-amide | 2642 |
| D-Dermaseptin S4 | 27 | NH2-ALWMTLLKKVLKAAAKALNAVLVGANA-amide | 2778 |
| D-Dermaseptin S4 L7K | 27 | NH2-ALWMTLKKKVLKAAAKALNAVLVGANA-amide | 2794 |
| D-Dermaseptin S4 A14K | 27 | NH2-ALWMTLLKKVLKAKAKALNAVLVGANA-amide | 2837 |
| D-Dermaseptin S4 L7K, A14K | 27 | NH2-ALWMTLKKKVLKAKAKALNAVLVGANA-amide | 2851 |
| Control C d | 18 | Ac-ELEKGGLEGEKGGKELEK-amide | - |
3.2. Peptide Hydrophobicity
| Peptide name | Net charge | Hydrophobicity | Benign | 50% TFE | PAd | Amphipathicity e | ||
|---|---|---|---|---|---|---|---|---|
| tRa (min) | [θ]222b | %Helix c | [θ]222b | %Helix c | ||||
| d-Piscidin 1 | +3 | 76.4 | 100 | <1 | 36,200 | 100 | 0.78 | 5.32 |
| d-Piscidin 1 I9K | +4 | 65.4 | −300 | <1 | 20,950 | 58 | 1.29 | 4.24 |
| d-Piscidin 1 V12K | +4 | 65.9 | −200 | <1 | 16,000 | 44 | 0.69 | 4.81 |
| d-Piscidin 1 G13K | +4 | 74.6 | −250 | <1 | 34,050 | 94 | 0.95 | 5.27 |
| d-Dermaseptin S4 | +4 | 124.4 | 28,900 | 75 | 38,400 | 100 | 12.61 | 3.58 (5.48) |
| d-Dermaseptin S4 L7K | +5 | 95.1 | 1,950 | 5 | 27,250 | 71 | 4.80 | 2.64 (4.12) |
| d-Dermaseptin S4 L7K,A14K | +6 | 78.6 | 2360 | 6 | 36,042 | 94 | 2.29 | 2.42 (3.76) |
3.3. Amphipathicity
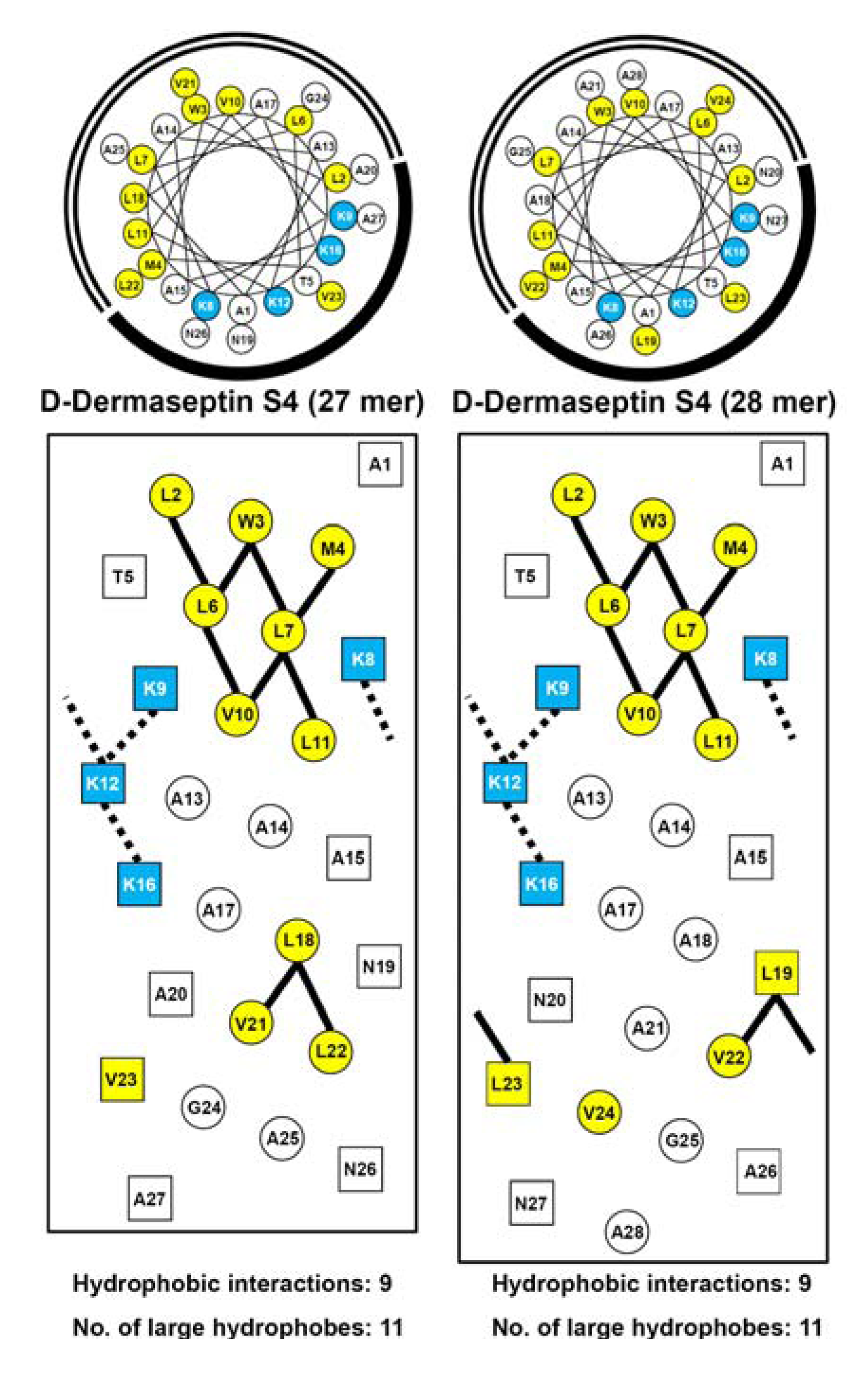
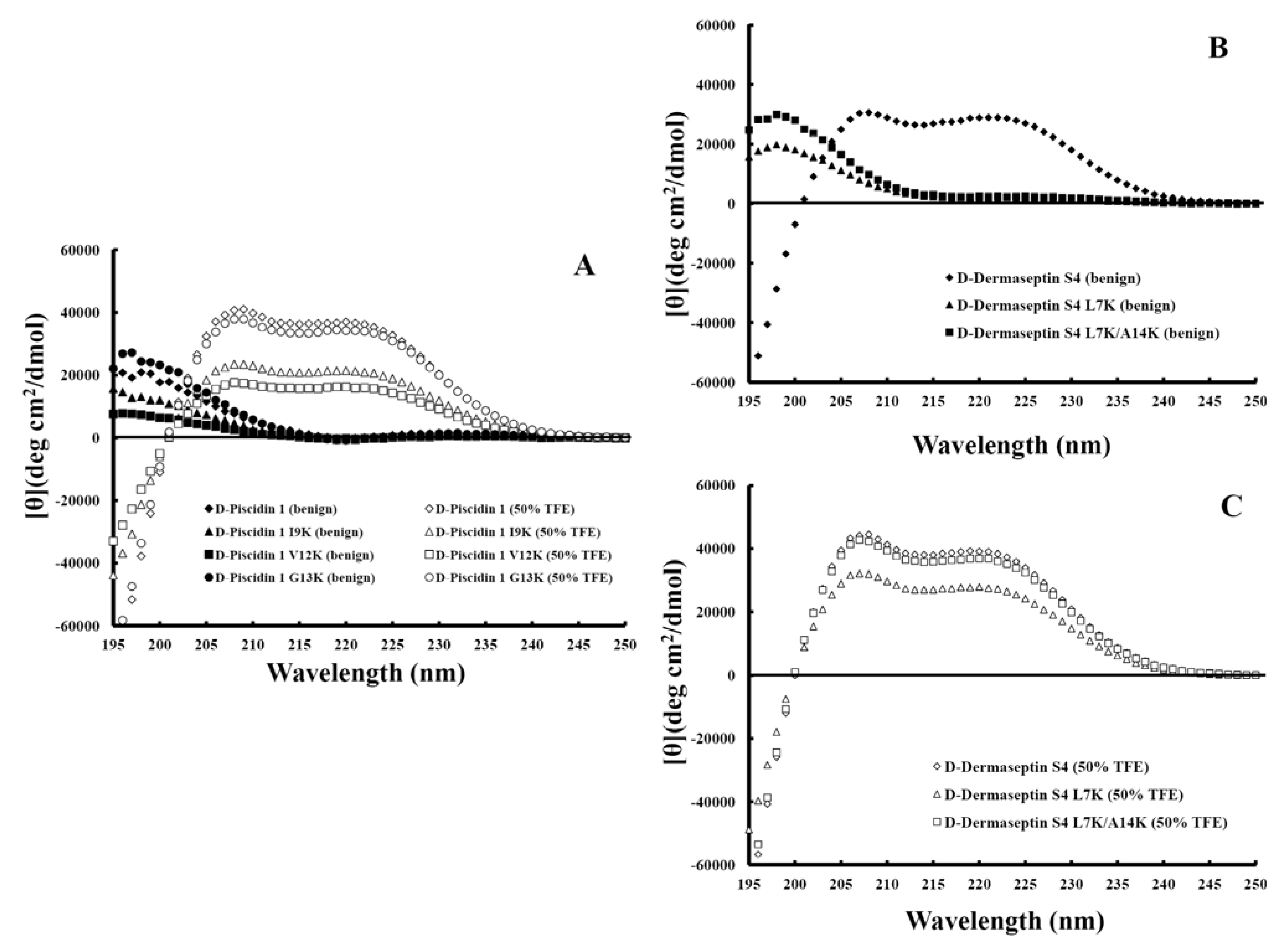
3.4. Secondary Structure of Peptides
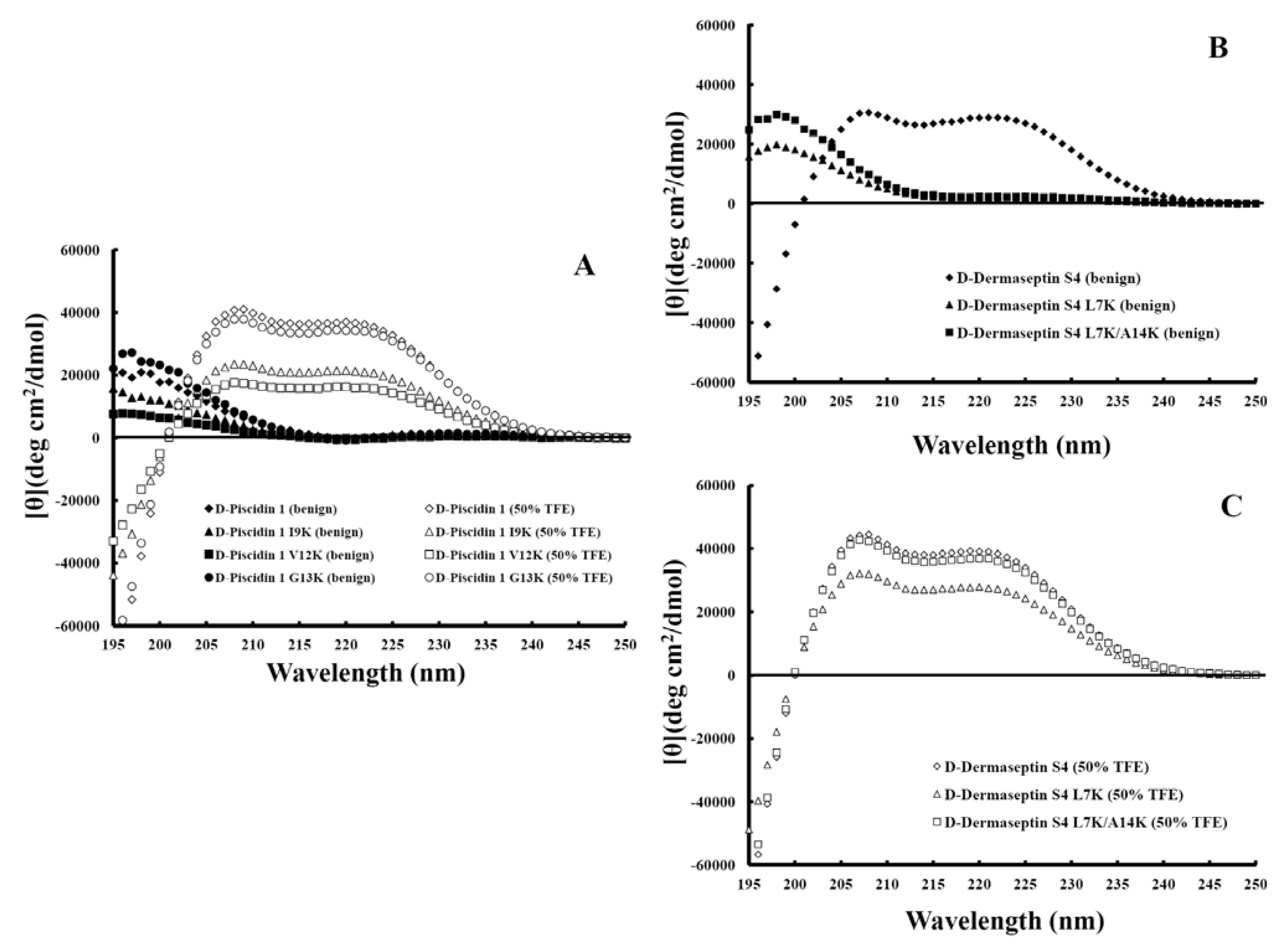
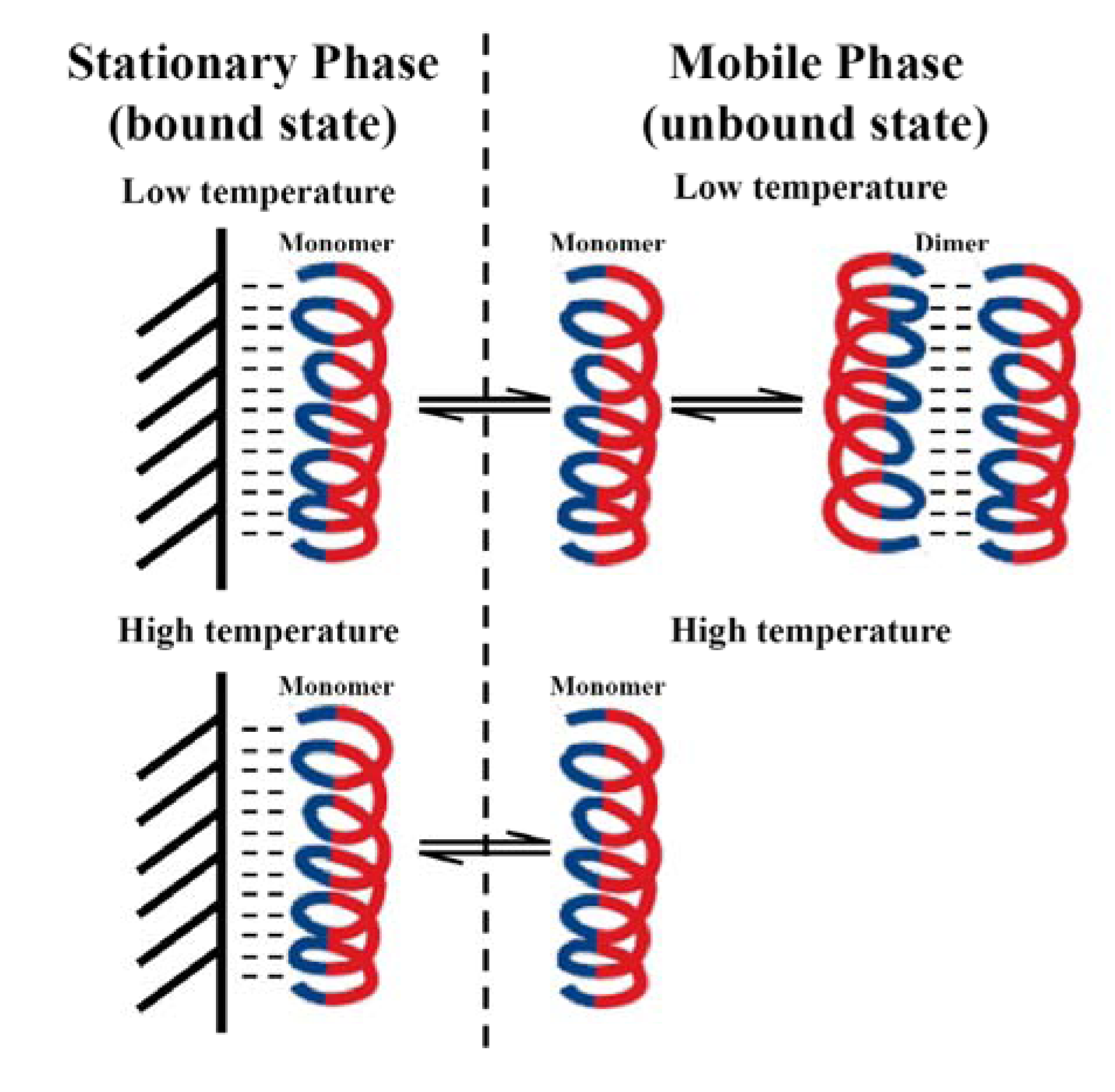
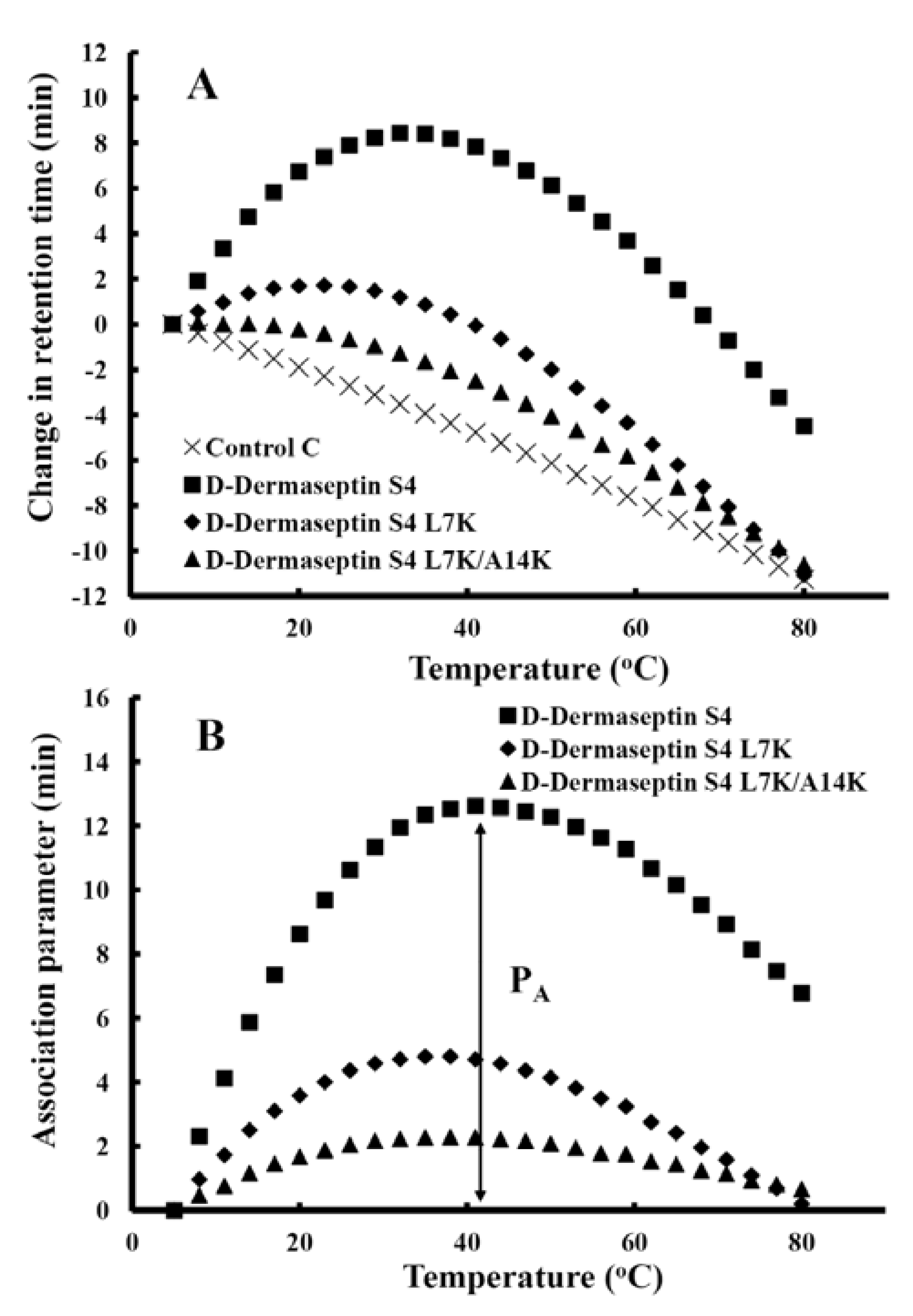
3.5. Peptide Self-Association
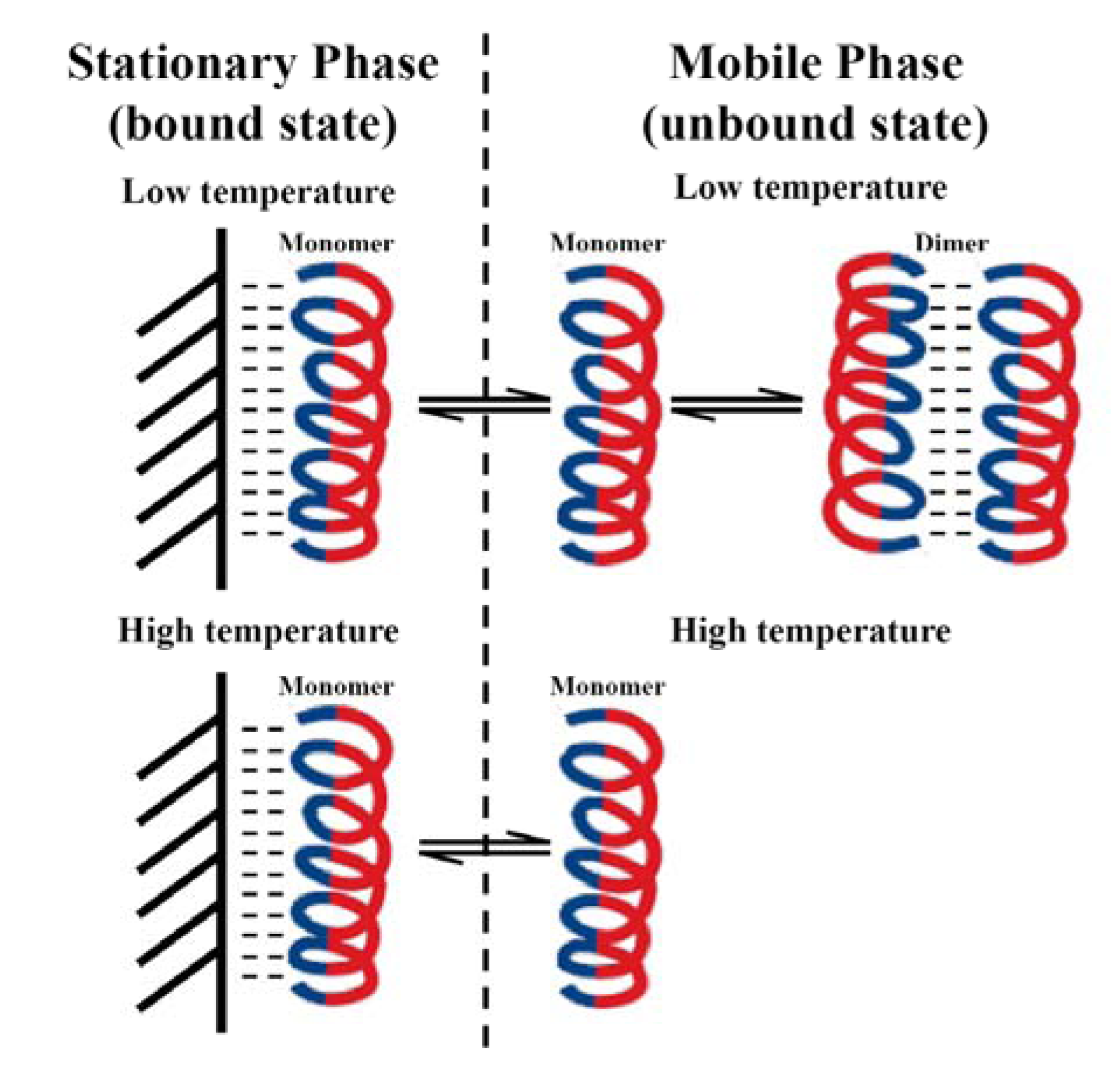
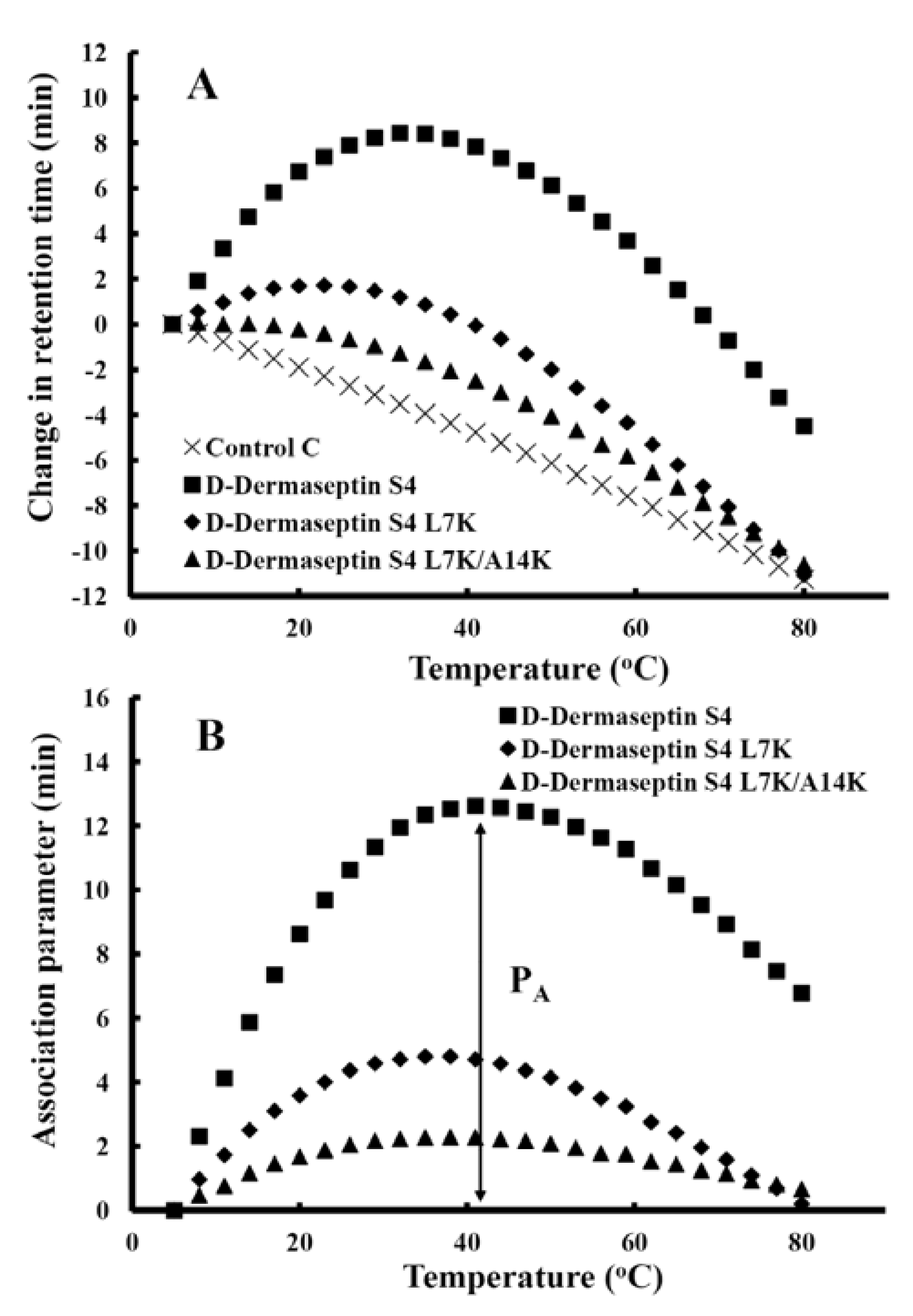
3.6. Antibacterial Activity
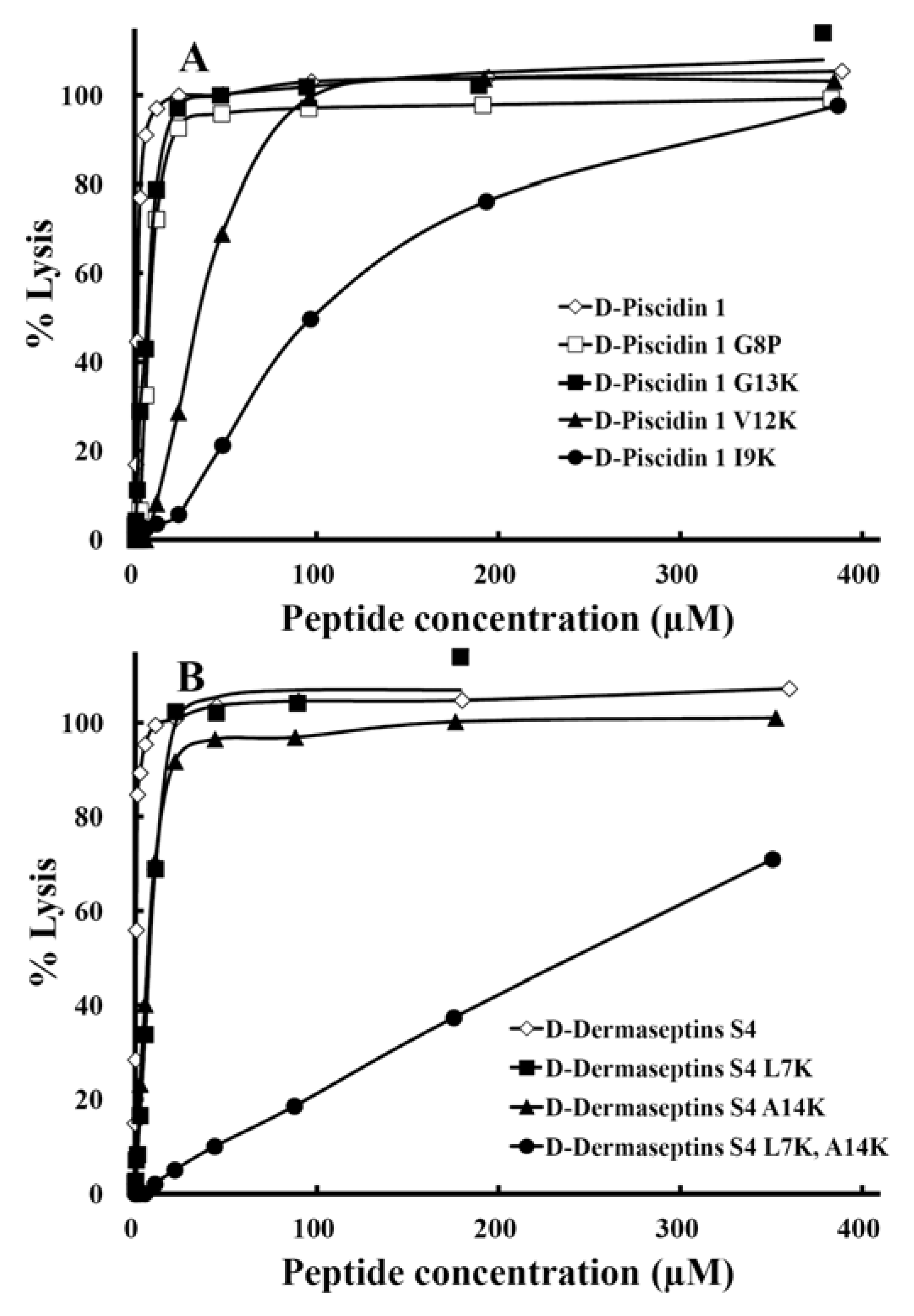
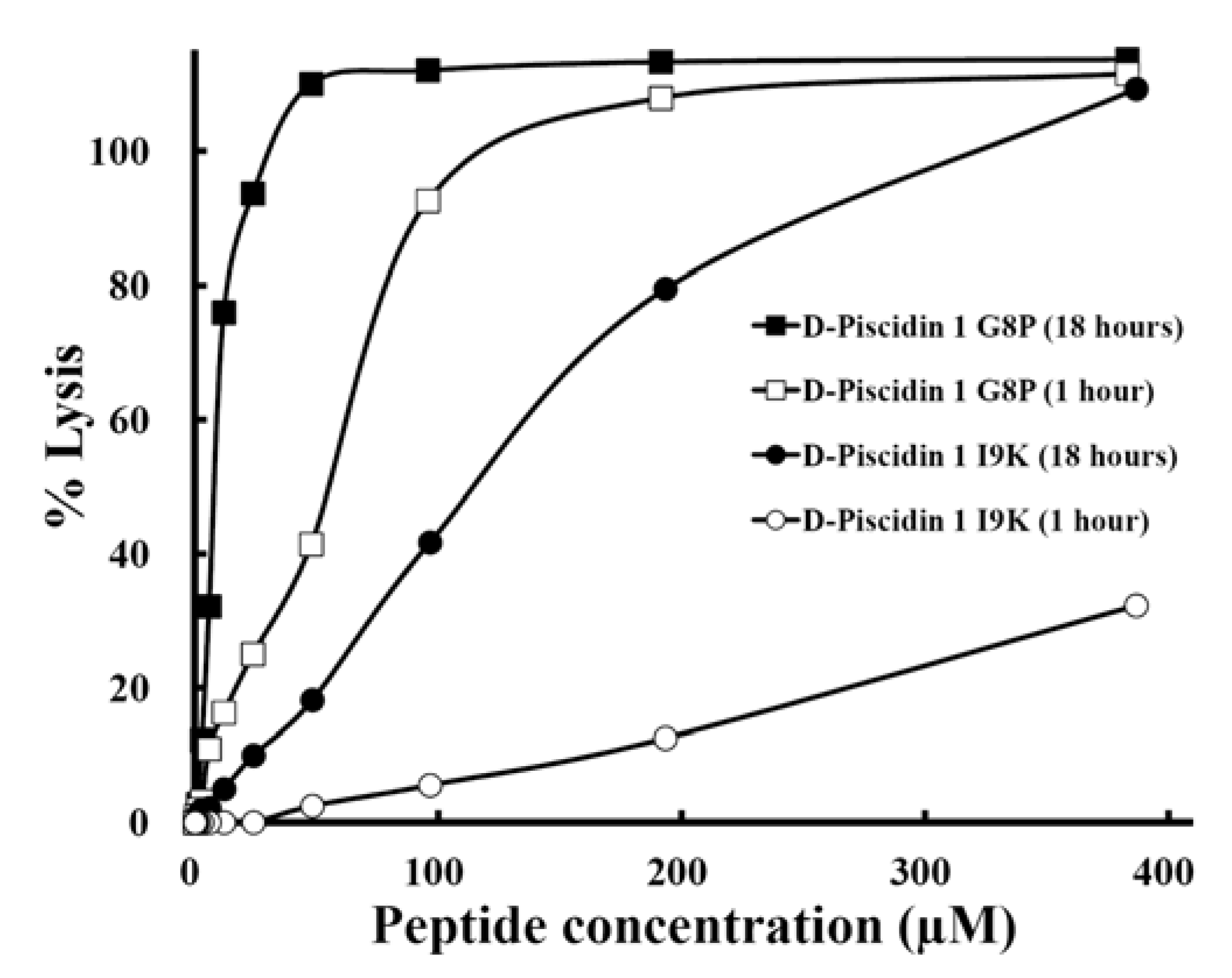
3.7. Hemolytic Activity
| Strain | MIC a (µM) | Fold c | ||||||||||||
| ATCC17978 | ATCC19606 | 649 | 689 | 759 | 821 | 884 | 899 | 964 | 985 | 1012 | GM b | |||
| Peptide | Fatal meningitis | Urine | Blood | Groin | Gluteus | Urine | Axilla | Perineum | Throat | Pleural fluid | Sputum | |||
| D-Piscidin 1 | 3.0 | 3.0 | 3.0 | 1.5 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 1.5 | 6.1 | 2.8 | 1.0 | |
| D-Piscidin 1 G13K | 5.9 | 5.9 | 3.0 | 5.9 | 5.9 | 5.9 | 5.9 | 5.9 | 5.9 | 3.0 | 5.9 | 5.2 | 0.5 | |
| D-Piscidin 1 V12K | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 1.5 | 3.0 | 2.8 | 1.0 | |
| D-Piscidin 1 I9K | 3.0 | 1.5 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 6.0 | 3.0 | 0.9 | |
| D-Dermaseptin S4 | 2.8 | 2.8 | 1.4 | 1.4 | 1.4 | 2.8 | 2.8 | 1.4 | 1.4 | 0.7 | 2.8 | 1.8 | 1.0 | |
| D-Dermaseptin S4 L7K | 0.7 | 0.4 | 0.7 | 0.7 | 0.4 | 0.4 | 1.4 | 0.4 | 2.8 | 0.7 | 1.4 | 0.7 | 2.6 | |
| D-Dermaseptin S4 L7K,A14K | 0.7 | 0.7 | 0.7 | 0.7 | 1.4 | 0.7 | 1.4 | 1.4 | 2.7 | 0.7 | 2.7 | 1.1 | 1.6 | |
| Strain | MIC a (µM) | Fold c | |||||||
| PAO1 | PAK | PA14 | CP204 | M2 | WR5 | GM b | |||
| Peptide | Human wound | — | — | Cystic fibrosis patient | Burn mouse model | Burn patient | |||
| D-Piscidin 1 | 24.3 | 12.2 | 24.3 | 24.3 | 24.3 | 12.2 | 19.3 | 1.0 | |
| D-Piscidin 1 G13K | 23.7 | 11.8 | 23.7 | 47.3 | 11.8 | 23.7 | 21.1 | 0.9 | |
| D-Piscidin 1 V12K | 24.0 | 6.0 | 24.0 | 24.0 | 24.0 | 12.0 | 17.0 | 1.1 | |
| D-Piscidin 1 I9K | 48.3 | 12.1 | 24.2 | 48.3 | 48.3 | 24.2 | 30.5 | 0.6 | |
| D-Dermaseptin S4 | 11.3 | 11.3 | 22.5 | 11.3 | 11.3 | 11.3 | 12.6 | 1.0 | |
| D-Dermaseptin S4 L7K | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | 2.8 | 4.5 | |
| D-Dermaseptin S4 L7K,A14K | 5.5 | 1.4 | 5.5 | 1.4 | 21.9 | 11.0 | 4.9 | 2.6 | |
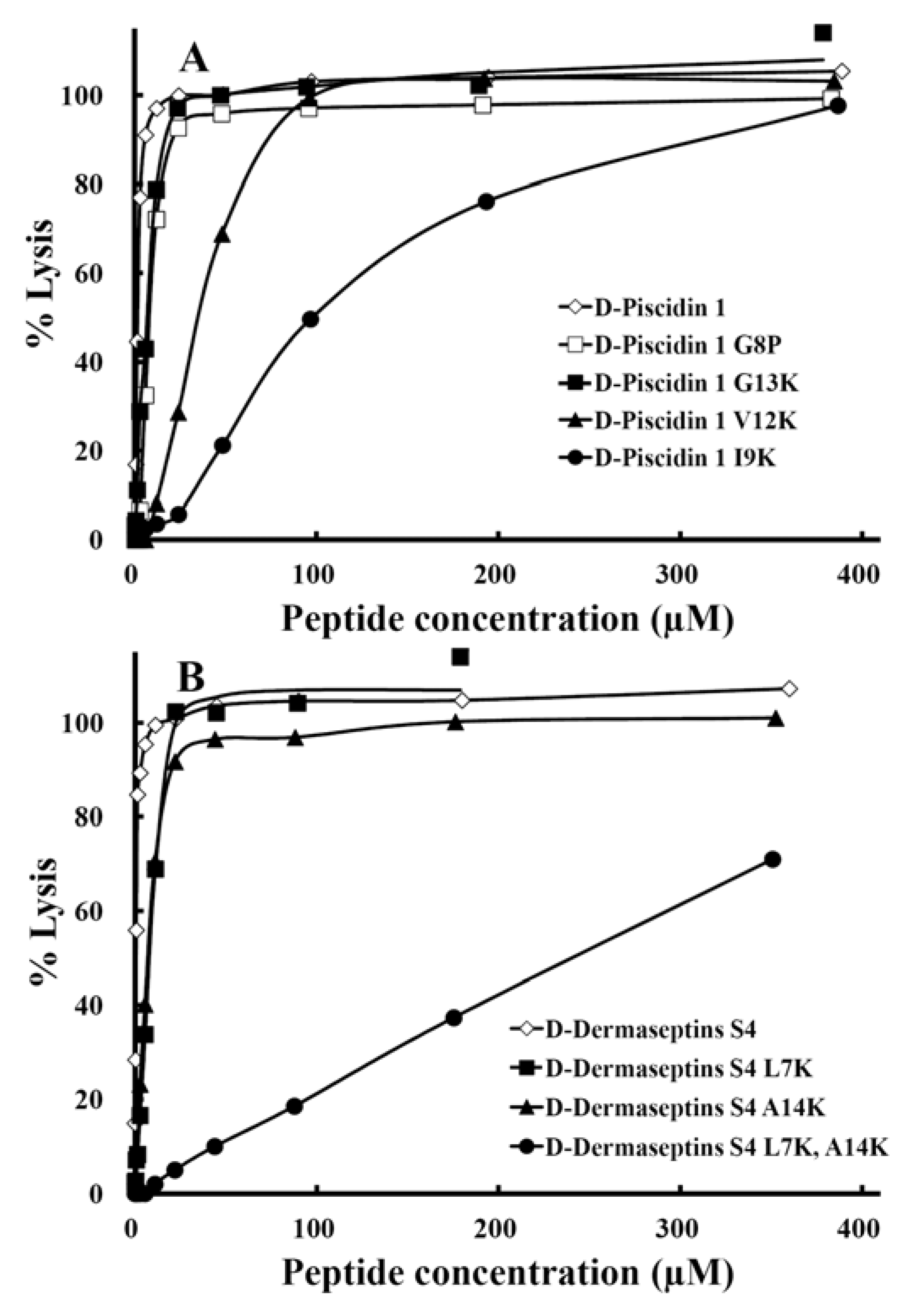
| Peptide Name | Hemolytic activity | Antimicrobial activity | ||||||
|---|---|---|---|---|---|---|---|---|
| Acinetobacter baumannii | Pseudomonas aeruginosa | |||||||
| HC50a (µM) | Fold b | MICGMc (µM) | T. I. d | Fold e | MICGMc (µM) | T. I. d | Fold e | |
| d-Piscidin 1 | 1.8 | 1.0 | 2.8 | 0.6 | 1.0 | 19.3 | 0.1 | 1.0 |
| d-Piscidin 1 G13K | 7.0 | 3.9 | 5.2 | 1.3 | 2.2 | 21.1 | 0.3 | 3.0 |
| d-Piscidin 1 V12K | 35 | 19 | 2.8 | 13 | 22 | 17.0 | 2.1 | 21 |
| d-Piscidin 1 I9K | 98 | 54 | 3.0 | 33 | 55 | 30.5 | 3.2 | 32 |
| d-Dermaseptin S4 | 0.6 | 1.0 | 1.8 | 0.3 | 1.0 | 12.6 | 0.05 | 1.0 |
| d-Dermaseptin S4 L7K | 8.6 | 14 | 0.7 | 12 | 40 | 2.8 | 3.1 | 62 |
| d-Dermaseptin S4 L7K,A14K | 241 | 402 | 1.1 | 219 | 730 | 4.9 | 49 | 980 |

3.8. Therapeutic Index
3.9. Mechanism of AMP Interaction with Membranes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. The Use of Essential Drugs. Sixth Report of the WHO Expert Committee; WHO Technical Report Series, No. 850; World Health Organization: Geneva, Switzerland, 1995. [Google Scholar]
- U.S. Congress. Impacts of Antibiotic-Resistant Bacteria; Office of Technology Assessment: Washington, DC, USA, 1995. [Google Scholar]
- House of Lords, UK. Science and Technology 7th Report: Resistance to Antibiotics and Other Antimicrobial Agents; House of Lords: London, UK, 1998. [Google Scholar]
- Garza-Gonzalez, E.; Llaca-Diaz, J.M.; Bosques-Padilla, F.J.; Gonzalez, G.M. Prevalence of multidrug-resistant bacteria at a tertiary-care teaching hospital in Mexico: Special focus on Acinetobacter baumannii. Chemotherapy 2010, 56, 275–279. [Google Scholar] [CrossRef]
- Bland, J.M.; de Lucca, A.J.; Jacks, T.J.; Vigo, C.B. All-D-cecropin B: Synthesis, conformation, lipopolysaccharide binding, and antibacterial activity. Mol. Cell. Biochem. 2001, 218, 105–111. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Pike, C.J.; Weinstein, S.L.; Velazquez, P.; Cotman, C.W. All-D-enantiomers of beta-amyloid exhibit similar biological properties to all-L-beta-amyloids. J. Biol. Chem. 1997, 272, 7431–7436. [Google Scholar]
- De Lucca, A.J.; Bland, J.M.; Vigo, C.B.; Jacks, T.J.; Peter, J.; Walsh, T.J. D-cecropin B: Proteolytic resistance, lethality for pathogenic fungi and binding properties. Med. Mycol. 2000, 38, 301–308. [Google Scholar] [CrossRef]
- Elmquist, A.; Langel, U. In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem. 2003, 384, 387–393. [Google Scholar]
- Hamamoto, K.; Kida, Y.; Zhang, Y.; Shimizu, T.; Kuwano, K. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with d-amino acid substitutions. Microbiol. Immunol. 2002, 46, 741–749. [Google Scholar] [CrossRef]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of d-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Wade, D.; Boman, A.; Wahlin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Matsumoto, H.; Hashimoto, K.; Teraguchi, S.; Takase, M.; Hayasawa, H. N-Acylated and D enantiomer derivatives of a nonamer core peptide of lactoferricin B showing improved antimicrobial activity. Antimicrob. Agents Chemother. 1999, 43, 1267–1269. [Google Scholar]
- Chen, Y.; Vasil, A.I.; Rehaume, L.; Mant, C.T.; Burns, J.L.; Vasil, M.L.; Hancock, R.E.; Hodges, R.S. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des. 2006, 67, 162–173. [Google Scholar] [CrossRef]
- Gura, T. Innate immunity. Ancient system gets new respect. Science 2001, 291, 2068–2071. [Google Scholar] [CrossRef]
- Levy, O. Antimicrobial proteins and peptides of blood: Templates for novel antimicrobial agents. Blood 2000, 96, 2664–2672. [Google Scholar]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar]
- Jiang, Z.; Vasil, A.I.; Gera, L.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides to target Gram-negative pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of charge, “specificity determinants”, total hydrophobicity, hydrophobe type and location as design parameters to improve the therapeutic ratio. Chem. Biol. Drug Des. 2011, 77, 225–240. [Google Scholar] [CrossRef]
- Mor, A.; Nguyen, V.H.; Delfour, A.; Migliore-Samour, D.; Nicolas, P. Isolation, amino acid sequence and synthesis of dermaseptin, a novel antimicrobial peptide of amphibian skin. Biochemistry 1991, 30, 8824–8830. [Google Scholar] [CrossRef]
- Mor, A.; Amiche, M.; Nicolas, P. Structure, synthesis, and activity of Dermaseptin b, a novel vertebrate defensive peptide from frog skin: Relationship with adenoregulin. Biochemistry 1994, 33, 6642–6650. [Google Scholar] [CrossRef]
- Mor, A.; Hani, K.; Nicolas, P. The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J. Biol. Chem. 1994, 269, 31635–31641. [Google Scholar]
- Belaid, A.; Aouni, M.; Khelifa, R.; Trabelsi, A.; Jemmali, M.; Hani, K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J. Med. Virol. 2002, 66, 229–234. [Google Scholar] [CrossRef]
- Lorin, C.; Saidi, H.; Belaid, A.; Zairi, A.; Baleux, F.; Hocini, H.; Belec, L.; Hani, K.; Tangy, F. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology 2005, 334, 264–275. [Google Scholar] [CrossRef]
- Feder, R.; Dagan, A.; Mor, A. Structure-activity relationship study of antimicrobial dermaseptin S4 showing the consequences of peptide oligomerization on selective cytotoxicity. J. Biol. Chem. 2000, 275, 4230–4238. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Feder, R.; Gaidukov, L.; Carmeli, Y.; Mor, A. Antibacterial properties of dermaseptin S4 derivatives with in vivo activity. Antimicrob. Agents Chemother. 2002, 46, 689–694. [Google Scholar] [CrossRef]
- Feder, R.; Nehushtai, R.; Mor, A. Affinity driven molecular transfer from erythrocyte membrane to target cells. Peptides 2001, 22, 1683–1690. [Google Scholar] [CrossRef]
- Kustanovich, I.; Shalev, D.E.; Mikhlin, M.; Gaidukov, L.; Mor, A. Structural requirements for potent versus selective cytotoxicity for antimicrobial dermaseptin S4 derivatives. J. Biol. Chem. 2002, 277, 16941–16951. [Google Scholar] [CrossRef]
- Noga, E.J.; Silphaduang, U.; Park, N.G.; Seo, J.K.; Stephenson, J.; Kozlowicz, S. Piscidin 4, a novel member of the piscidin family of antimicrobial peptides. Comp. Biochem. Physiol. 2009, 152, 299–305. [Google Scholar]
- Silphaduang, U.; Noga, E.J. Antimicrobials: Peptide antibiotics in mast cells of fish. Nature 2001, 414, 268–269. [Google Scholar] [CrossRef]
- Noga, E.J.; Fan, Z.; Silphaduang, U. Histone-like proteins from fish are lethal to the parasitic dinoflagellate Amyloodinium ocellatum. Parasitology 2001, 123, 57–65. [Google Scholar]
- Noga, E.J.; Silphaduang, U. Piscidins: A novel family of peptide antibiotics from fish. Drug News Perspect. 2003, 16, 87–92. [Google Scholar] [CrossRef]
- Lauth, X.; Shike, H.; Burns, J.C.; Westerman, M.E.; Ostland, V.E.; Carlberg, J.M.; van Olst, J.C.; Nizet, V.; Taylor, S.W.; Shimizu, C.; et al. Discovery and characterization of two isoforms of moronecidin, a novel antimicrobial peptide from hybrid striped bass. J. Biol. Chem. 2002, 277, 5030–5039. [Google Scholar] [CrossRef]
- Chinchar, V.G.; Bryan, L.; Silphadaung, U.; Noga, E.; Wade, D.; Rollins-Smith, L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology 2004, 323, 268–275. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.K.; Lim, S.S.; Zhu, W.L.; Ko, H.; Shin, S.Y.; Hahm, K.S.; Kim, Y. Solution structure and cell selectivity of piscidin 1 and its analogues. Biochemistry 2007, 46, 3653–3663. [Google Scholar] [CrossRef]
- Menousek, J.; Mishra, B.; Hanke, M.L.; Heim, C.E.; Kielian, T.; Wang, G. Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int. J. Antimicrob. Agents 2012, 39, 402–406. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Hodges, R.S. Preparative reversed-phase high-performance liquid chromatography collection efficiency for an antimicrobial peptide on columns of varying diameters (1mm to 9.4mm I.D.). J. Chromatogr. 2007, 1140, 112–120. [Google Scholar] [CrossRef]
- Lee, D.L.; Mant, C.T.; Hodges, R.S. A novel method to measure self-association of small amphipathic molecules: Temperature profiling in reversed-phase chromatography. J. Biol. Chem. 2003, 278, 22918–22927. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef]
- Jiang, Z.; Hggins, M.P.; Whitehurst, J.; Kisich, K.O.; Voskuil, M.I.; Hodges, R.S. Anti-tuberculosis activity of alpha-helical antimicrobial peptides: De novo designed L- and d-enantiomers versus L- and d-LL-37. Protein Pept. Lett. 2011, 18, 241–252. [Google Scholar] [CrossRef]
- Jiang, Z.; Kullberg, B.J.; van der Lee, H.; Vasil, A.I.; Hale, J.D.; Mant, C.T.; Hancock, R.E.W.; Vasil, M.L.; Netea, M.G.; Hodges, R.S. Effects of Hydrophobicity on the Antifungal Activity of a-Helical Antimicrobial Peptides. Chem. Biol. Drug Des. 2008, 72, 483–495. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef]
- Carver, T.; Bleasby, A. The design of Jemboss: A graphical user interface to EMBOSS. Bioinformatics (Oxford, England) 2003, 19, 1837–1843. [Google Scholar] [CrossRef]
- Kovacs, J.M.; Mant, C.T.; Hodges, R.S. Determination of intrinsic hydrophilicity/ hydrophobicity of amino acid side chains in peptides in the absence of nearest-neighbor or conformational effects. Biopolymers 2006, 84, 283–297. [Google Scholar] [CrossRef]
- Mant, C.T.; Kovacs, J.M.; Kim, H.M.; Pollock, D.D.; Hodges, R.S. Intrinsic amino acid side-chain hydrophilicity/hydrophobicity coefficients determined by reversed-phase high-performance liquid chromatography of model peptides: Comparison with other hydrophilicity/hydrophobicity scales. Biopolymers 2009, 92, 573–595. [Google Scholar] [CrossRef]
- Holloway, B.W. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 1955, 13, 572–581. [Google Scholar] [CrossRef]
- Bjorn, M.J.; Vasil, M.L.; Sadoff, J.C.; Iglewski, B.H. Incidence of exotoxin production by Pseudomonas species. Infect. Immun. 1977, 16, 362–366. [Google Scholar]
- Pavlovskis, O.R.; Pollack, M.; Callahan, L.T., 3rd; Iglewski, B.H. Passive protection by antitoxin in experimental Pseudomonas aeruginosa burn infections. Infect. Immun. 1977, 18, 596–602. [Google Scholar]
- Frost, L.S.; Paranchych, W. Composition and molecular weight of pili purified from Pseudomonas aeruginosa K. J. Bacteriol. 1977, 131, 259–269. [Google Scholar]
- Watts, T.H.; Kay, C.M.; Paranchych, W. Dissociation and characterization of pilin isolated from Pseudomonas aeruginosa strains PAK and PAO. Can. J. Biochem. 1982, 60, 867–872. [Google Scholar] [CrossRef]
- Rahme, L.G.; Ausubel, F.M.; Cao, H.; Drenkard, E.; Goumnerov, B.C.; Lau, G.W.; Mahajan-Miklos, S.; Plotnikova, J.; Tan, M.W.; Tsongalis, J.; et al. Plants and animals share functionally common bacterial virulence factors. Proc. Natl. Acad. Sci. USA 2000, 97, 8815–8821. [Google Scholar] [CrossRef]
- Stieritz, D.D.; Holder, I.A. Experimental studies of the pathogenesis of infections due to Pseudomonas aeruginosa: Description of a burned mouse model. J. Infect. Dis. 1975, 131, 688–691. [Google Scholar] [CrossRef]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem. 2008, 283, 18636–18645. [Google Scholar] [CrossRef]
- Shalev, D.E.; Rotem, S.; Fish, A.; Mor, A. Consequences of N-acylation on structure and membrane binding properties of dermaseptin derivative K4-S4-(1–13). J. Biol. Chem. 2006, 281, 9432–9438. [Google Scholar] [CrossRef]
- Mant, C.T.; Chen, Y.; Hodges, R.S. Temperature profiling of polypeptides in reversed-phase liquid chromatography. I. Monitoring of dimerization and unfolding of amphipathic alpha-helical peptides. J. Chromatogr. 2003, 1009, 29–43. [Google Scholar] [CrossRef]
- Mant, C.T.; Tripet, B.; Hodges, R.S. Temperature profiling of polypeptides in reversed-phase liquid chromatography. II. Monitoring of folding and stability of two-stranded alpha-helical coiled-coils. J. Chromatogr. 2003, 1009, 45–59. [Google Scholar] [CrossRef]
- Ghosh, J.K.; Shaool, D.; Guillaud, P.; Ciceron, L.; Mazier, D.; Kustanovich, I.; Shai, Y.; Mor, A. Selective cytotoxicity of dermaseptin S3 toward intraerythrocytic Plasmodium falciparum and the underlying molecular basis. J. Biol. Chem. 1997, 272, 31609–31616. [Google Scholar]
- Hodges, R.S.; Jiang, Z.; Whitehurst, J.; Mant, C.T. Development of antimicrobial peptides as therapeutic agents. In Development of Therapeutic Agents, Handbook in Pharmaceutical Sciences; Gad, S.C., Ed.; John Wiley and Sons: Cary, NC, USA, 2012; pp. 285–357. [Google Scholar]
- Blazyk, J.; Wiegand, R.; Klein, J.; Hammer, J.; Epand, R.M.; Epand, R.F.; Maloy, W.L.; Kari, U.P. A novel linear amphipathic beta-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 2001, 276, 27899–27906. [Google Scholar]
- Chekmenev, E.Y.; Vollmar, B.S.; Forseth, K.T.; Manion, M.N.; Jones, S.M.; Wagner, T.J.; Endicott, R.M.; Kyriss, B.P.; Homem, L.M.; Pate, M.; et al. Investigating molecular recognition and biological function at interfaces using piscidins, antimicrobial peptides from fish. Biochim. Biophys. Acta 2006, 1758, 1359–1372. [Google Scholar] [CrossRef]
- Gibson, B.W.; Tang, D.Z.; Mandrell, R.; Kelly, M.; Spindel, E.R. Bombinin-like peptides with antimicrobial activity from skin secretions of the Asian toad, Bombina orientalis. J. Biol. Chem. 1991, 266, 23103–23111. [Google Scholar]
- Conlon, J.M.; Sonnevend, A.; Davidson, C.; Smith, D.D.; Nielsen, P.F. The ascaphins: A family of antimicrobial peptides from the skin secretions of the most primitive extant frog, Ascaphus truei. Biochem. Biophys. Res. Commun. 2004, 320, 170–175. [Google Scholar] [CrossRef]
- Shin, S.Y.; Hahm, K.S. A short alpha-helical antimicrobial peptide with antibacterial selectivity. Biotechnol. Lett. 2004, 26, 735–739. [Google Scholar] [CrossRef]
- Tencza, S.B.; Douglass, J.P.; Creighton, D.J., Jr.; Montelaro, R.C.; Mietzner, T.A. Novel antimicrobial peptides derived from human immunodeficiency virus type 1 and other lentivirus transmembrane proteins. Antimicrob. Agents Chemother. 1997, 41, 2394–2398. [Google Scholar]
- Baumann, G.; Mueller, P. A molecular model of membrane excitability. J. Supramol. Struct. 1974, 2, 538–557. [Google Scholar] [CrossRef]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Rahmanpour, A.; Ghahremanpour, M.M.; Mehrnejad, F.; Moghaddam, M.E. Interaction of Piscidin-1 with zwitterionic versus anionic membranes: A comparative molecular dynamics study. J. Biomol. Struct. Dyn. 2013, 31, 1393–1403. [Google Scholar] [CrossRef]
- De Angelis, A.A.; Grant, C.V.; Baxter, M.K.; McGavin, J.A.; Opella, S.J.; Cotten, M.L. Amphipathic antimicrobial piscidin in magnetically aligned lipid bilayers. Biophys. J. 2011, 101, 1086–1094. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, Z.; Vasil, A.I.; Vasil, M.L.; Hodges, R.S. “Specificity Determinants” Improve Therapeutic Indices of Two Antimicrobial Peptides Piscidin 1 and Dermaseptin S4 Against the Gram-negative Pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals 2014, 7, 366-391. https://doi.org/10.3390/ph7040366
Jiang Z, Vasil AI, Vasil ML, Hodges RS. “Specificity Determinants” Improve Therapeutic Indices of Two Antimicrobial Peptides Piscidin 1 and Dermaseptin S4 Against the Gram-negative Pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals. 2014; 7(4):366-391. https://doi.org/10.3390/ph7040366
Chicago/Turabian StyleJiang, Ziqing, Adriana I. Vasil, Michael L. Vasil, and Robert S. Hodges. 2014. "“Specificity Determinants” Improve Therapeutic Indices of Two Antimicrobial Peptides Piscidin 1 and Dermaseptin S4 Against the Gram-negative Pathogens Acinetobacter baumannii and Pseudomonas aeruginosa" Pharmaceuticals 7, no. 4: 366-391. https://doi.org/10.3390/ph7040366