Synthesis, Screening and Pharmacokinetic Evaluation of Potential Prodrugs of Bupropion. Part One: In Vitro Development

Abstract
:
1. Introduction

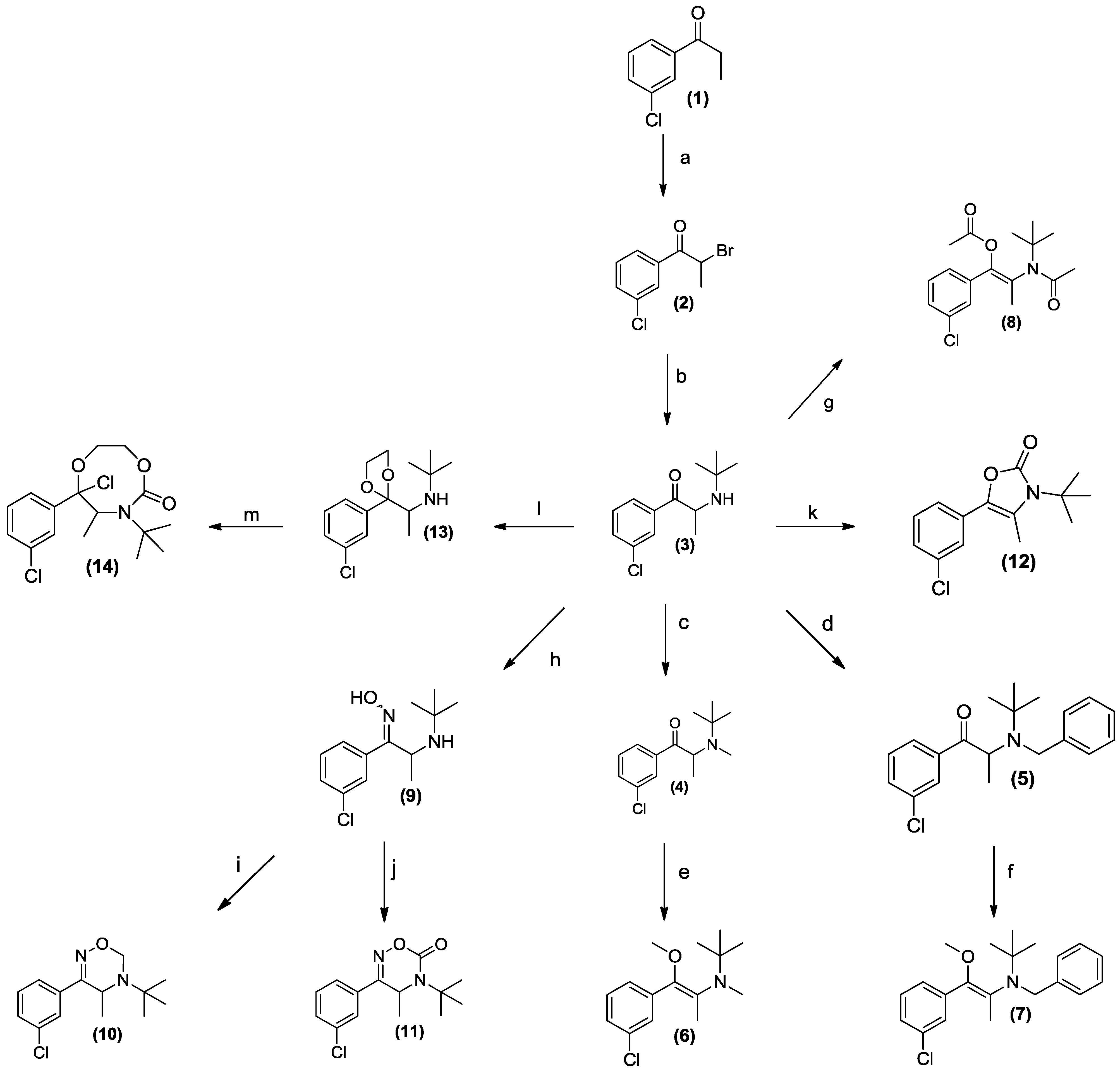
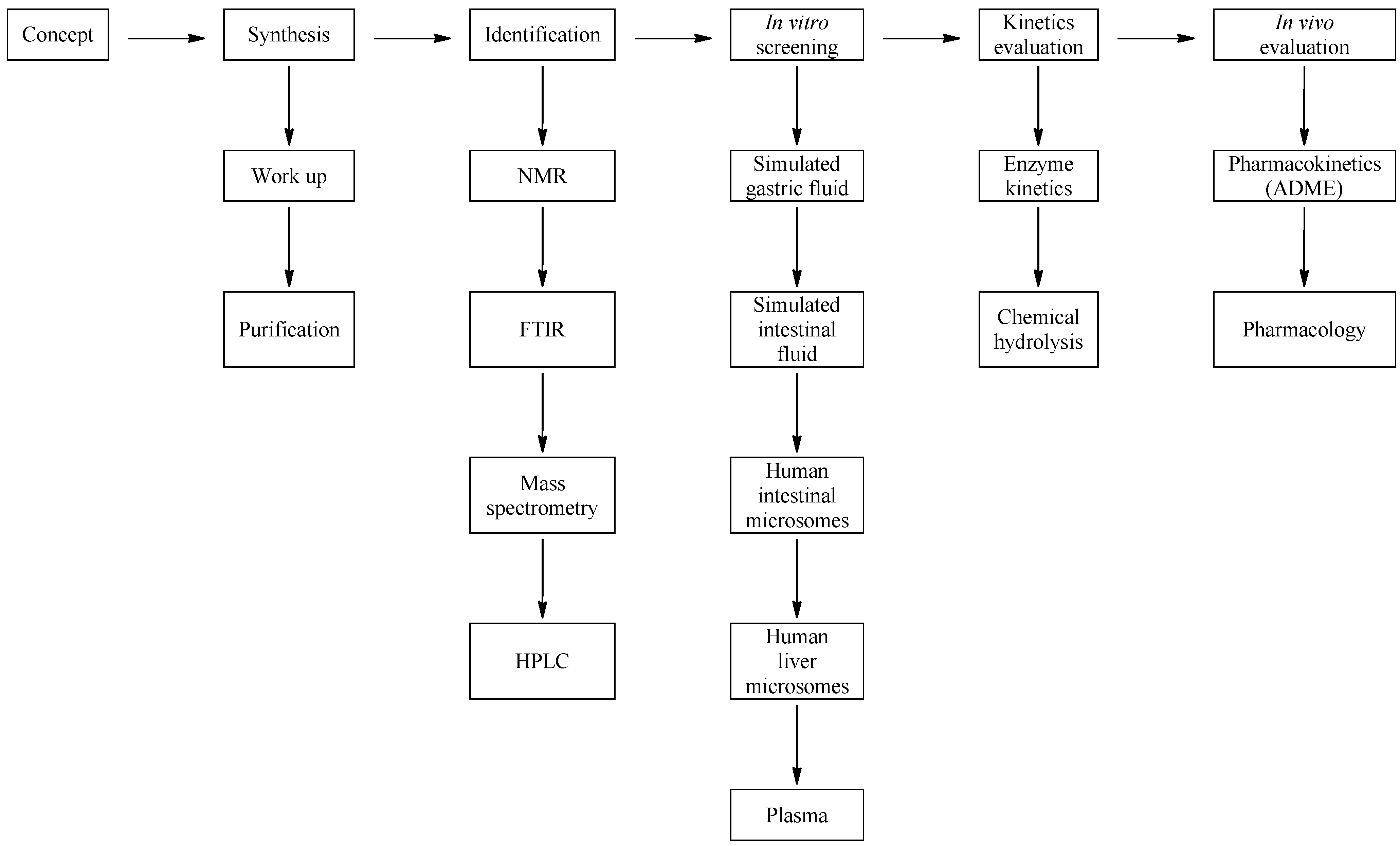
2. Experimental Section
2.1. Instrumentation
2.2. Materials
2.3. Media Preparations
2.4. Chromatographic Method for Screening Potential Prodrugs
2.5. Mass Spectrometer Conditions for Detection of Potential Prodrugs
2.6. Metworks Software
2.7. Determination of Microsomal Metabolic Stability
2.8. Determination of Michaelis-Menten Kinetic Parameters
2.9. Synthesis
2.10. LCMS Method Validation
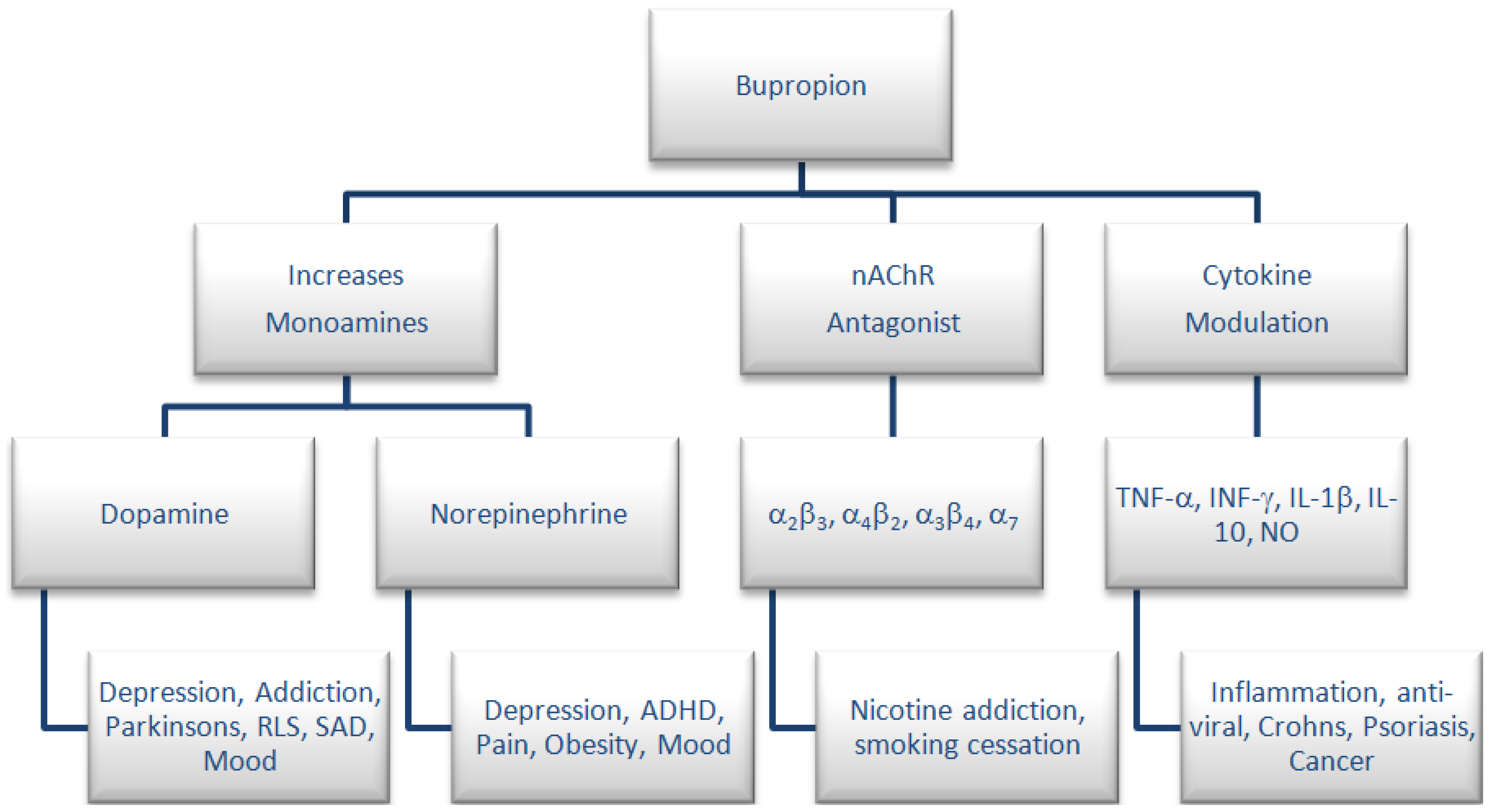
2.11. Neuropharmacology of N-Methyl Bupropion and Bupropion
3. Results and Discussion
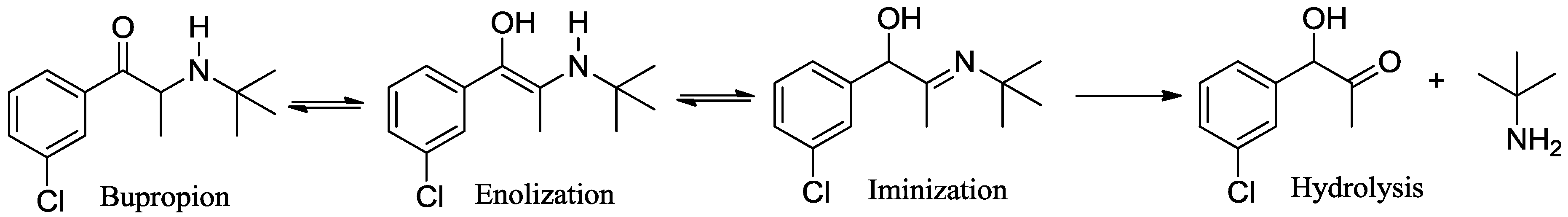
3.1. Chemistry

3.2. Development of a Screening Tool to Evaluate Drug Metabolic Stability

3.3. Media Selection
3.4. Chromatographic Methodology
3.5. Mass Spectrometric Method
3.6. LCMS Method Validation
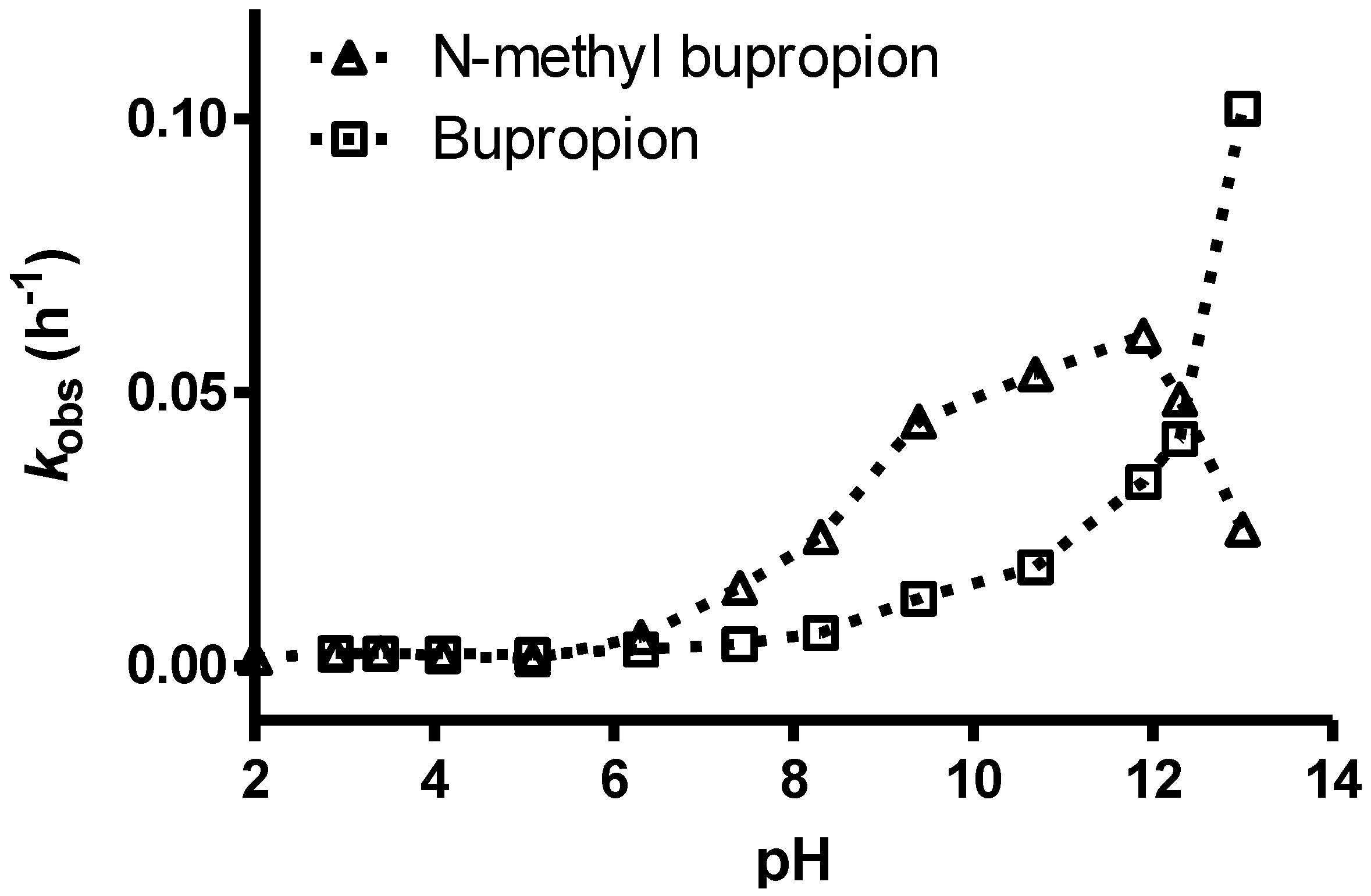
3.7. Screening Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prodrug tested | Primary product detected in media | ||||
|---|---|---|---|---|---|
| SGF | SIF | HIM | HLM | PHP | |
 | <LOD | <LOD |  | | <LOD |
 | <LOD | <LOD | | | <LOD |
 | <LOD | <LOD | | | <LOD |
 | <LOD | <LOD | | | <LOD |
 | <LOD | <LOD | <LOD | <LOD | <LOD |
 | <LOD | <LOD | <LOD | <LOD | <LOD |
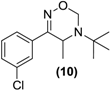 | <LOD | <LOD | | | <LOD |
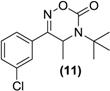 | <LOD | <LOD | <LOD | <LOD | <LOD |
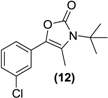 | <LOD | <LOD | <LOD | <LOD | <LOD |
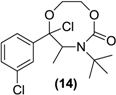 | <LOD | <LOD | <LOD | <LOD | <LOD |




| N-methylbupropion | Target | Bupropion | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) | Ki (μM) | nH | IC50 (μM) | Ki (μM) | nH | |
| 39.3 | 12.3 | 0.957 | Muscarinic non -selective, central | 86.5 | 27 | 1.42 |
| 71.6 | 49.2 | 0.836 | Nicotinic acetylcholine | >100 | - | - |
| 3.71 | 2.95 | 0.924 | Dopamine active transporter (DAT) | <1 | - | - |
| 57.9 | 54.4 | 0.764 | Norepinephrine transporter (NET) | 19.9 | 19.8 | 0.835 |
| >100 | - | - | Serotonin transporter (SERT) | >100 | - | - |
4. Conclusions
Author Contributions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Stahl, S.M.; Pradko, J.F.; Haight, B.R.; Modell, J.G.; Rockett, C.B.; Learned-Coughlin, S. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim. Care Compan. J. Clin. Psychiatry 2004, 6, 159–166. [Google Scholar] [CrossRef]
- Slemmer, J.E.; Martin, B.R.; Damaj, M.I. Bupropion is a nicotinic antagonist. J. Pharm. Exp. Ther. 2000, 295, 321–327. [Google Scholar]
- Brustolim, D.; Ribeiro-dos-Santos, R.; Kast, R.E.; Altschuler, E.L.; Soares, M.B. A new chapter opens in anti-inflammatory treatments: The antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. Int. Immunopharmacol. 2006, 6, 903–907. [Google Scholar]
- Goldstein, M.G. Bupropion sustained release and smoking cessation. J. Clin. Psychiatry 1998, 59, 66–72. [Google Scholar] [CrossRef]
- Hurt, R.D.; Sachs, D.P.; Glover, E.D.; Offord, K.P.; Johnston, J.A.; Dale, L.C.; Khayrallah, M.A.; Schroeder, D.R.; Glover, P.N.; Sullivan, C.R.; et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N. Engl. J. Med. 1997, 337, 1195–1202. [Google Scholar] [CrossRef]
- Altschuler, E.L.; Kast, R.E. Bupropion in psoriasis and atopic dermatitis: Decreased tumor necrosis factor-alpha? Psychosom. Med. 2003, 65, 719. [Google Scholar] [CrossRef]
- Altschuler, E.L.; Kast, R.E. Bupropion for fatigue and as a tumor necrosis factor-alpha lowering agent in primary biliary cirrhosis. Med. Hypotheses 2005, 64, 118–119. [Google Scholar]
- Kast, R.E. Anti- and pro-inflammatory considerations in antidepressant use during medical illness: Bupropion lowers and mirtazapine increases circulating tumor necrosis factor-alpha levels. Gen. Hosp. Psychiatry 2003, 25, 495–496. [Google Scholar] [CrossRef]
- Kast, R.E. Evidence of a mechanism by which etanercept increased tnf-alpha in multiple myeloma: New insights into the biology of tnf-alpha giving new treatment opportunities—The role of bupropion. Leuk Res. 2005, 29, 1459–1463. [Google Scholar] [CrossRef]
- Kast, R.E.; Altschuler, E.L. Remission of crohn’s disease on bupropion. Gastroenterology 2001, 121, 1260–1261. [Google Scholar] [CrossRef]
- Kast, R.E.; Altschuler, E.L. Tumor necrosis factor-alpha in hepatitis b: Potential role for bupropion. J. Hepatol. 2003, 39, 131–133. [Google Scholar] [CrossRef]
- Kast, R.E.; Altschuler, E.L. Combination of bupropion, paroxetine and quetiapine as adjuvant treatment for multiple myeloma. Med. Hypotheses 2004, 62, 817–818. [Google Scholar] [CrossRef]
- Kast, R.E.; Altschuler, E.L. Bone density loss in crohn’s disease: Role of tnf and potential for prevention by bupropion. Gut 2004, 53, 1056. [Google Scholar]
- Kast, R.E.; Altschuler, E.L. Proposal for using small molecule tumor necrosis factor-alpha lowering agents, possibly bupropion, in aplastic anemia. Med. Hypotheses 2005, 65, 374–376. [Google Scholar] [CrossRef]
- Kast, R.E.; Altschuler, E.L. Anti-apoptosis function of tnf-alpha in chronic lymphocytic leukemia: Lessons from crohn’s disease and the therapeutic potential of bupropion to lower tnf-alpha. Arch. Immunol. Ther. Exp. (Warsz.) 2005, 53, 143–147. [Google Scholar]
- Ansari, A. The efficacy of newer antidepressants in the treatment of chronic pain: A review of current literature. Harv. Rev. Psychiatry 2000, 7, 257–277. [Google Scholar] [CrossRef]
- Davidson, J.R.; France, R.D. Bupropion in chronic low back pain. J. Clin. Psychiatry 1994, 55, 362. [Google Scholar]
- Pinsker, W. Treatment of headache with bupropion. Headache 1998, 38, 58. [Google Scholar]
- Simeon, J.G.; Ferguson, H.B.; van Wyck Fleet, J. Bupropion effects in attention deficit and conduct disorders. Can. J. Psychiatry 1986, 31, 581–585. [Google Scholar]
- Kim, S.W.; Shin, I.S.; Kim, J.M.; Yang, S.J.; Shin, H.Y.; Yoon, J.S. Bupropion may improve restless legs syndrome: A report of three cases. Clin. Neuropharmacol. 2005, 28, 298–301. [Google Scholar]
- First drug for seasonal depression. Available online: http://permanent.access.gpo.gov/lps1609/www.fda.gov/fdac/departs/2006/506_upd.html#depression (accessed on 30 April 2014).
- FDA approves antidepressant drug for treatment of seasonal depression. Mayo Clin Womens Healthsource 2006, 10, 3.
- Kiptoo, P.K.; Paudel, K.S.; Hammell, D.C.; Pinninti, R.R.; Chen, J.; Crooks, P.A.; Stinchcomb, A.L. Transdermal delivery of bupropion and its active metabolite, hydroxybupropion: A prodrug strategy as an alternative approach. J. Pharm. Sci. 2009, 98, 583–594. [Google Scholar] [CrossRef]
- Hamad, M.O.; Kiptoo, P.K.; Stinchcomb, A.L.; Crooks, P.A. Synthesis and hydrolytic behavior of two novel tripartate codrugs of naltrexone and 6[beta]-naltrexol with hydroxybupropion as potential alcohol abuse and smoking cessation agents. Bio Med. Chem. 2006, 14, 7051–7061. [Google Scholar] [CrossRef]
- Santamaria, A.; Arias, H.R. Neurochemical and behavioral effects elicited by bupropion and diethylpropion in rats. Behav. Brain Res. 2010, 211, 132–139. [Google Scholar] [CrossRef]
- O’Byrne, P.M.; Williams, R.; Walsh, J.J.; Gilmer, J.F. The aqueous stability of bupropion. J. Pharm. Biomed. Anal. 2010, 53, 376–381. [Google Scholar] [CrossRef]
- Kalendra, D.M.; Sickles, B.R. Diminished reactivity of ortho-substituted phenacyl bromides toward nucleophilic displacement. J. Org. Chem. 2003, 68, 1594–1596. [Google Scholar] [CrossRef]
- Simplicio, A.; Clancy, J.; Gilmer, J. Prodrugs for amines. Molecules 2008, 13, 519–547. [Google Scholar]
- Mantyla, A.; Rautio, J.; Nevalainen, T.; Vepsalainen, J.; Juvonen, R.; Kendrick, H.; Garnier, T.; Croft, S.L.; Jarvinen, T. Synthesis and antileishmanial activity of novel buparvaquone oxime derivatives. Bioorg. Med. Chem. 2004, 12, 3497–3502. [Google Scholar] [CrossRef]
- Kumpulainen, H.; Mahonen, N.; Laitinen, M.L.; Jaurakkajarvi, M.; Raunio, H.; Juvonen, R.O.; Vepsalainen, J.; Jarvinen, T.; Rautio, J. Evaluation of hydroxyimine as cytochrome p450-selective prodrug structure. J. Med. Chem. 2006, 49, 1207–1211. [Google Scholar] [CrossRef]
- Patel, J.P.; Repta, A.J. Enol esters as potential prodrugs i. Stability and enzyme-mediated hydrolysis of alpha-acetoxystyrene. Int. J. Pharm. 1980, 5, 329–333. [Google Scholar] [CrossRef]
- Gudmundsson, O.S. Case Study: Ximelagatran: A Double Prodrug of Melagatran. In Prodrugs; Springer: New York, NY, USA, 2007; pp. 1395–1402. [Google Scholar]
- Fager, G.; Cullberg, M.; Eriksson.-Lepkowska, M.; Frison, L.; Eriksson, U.G. Pharmacokinetics and pharmacodynamics of melagatran, the active form of the oral direct thrombin inhibitor ximelagatran, are not influenced by acetylsalicylic acid. Eur J. Clin. Pharmacol. 2003, 59, 283–289. [Google Scholar] [CrossRef]
- Gasparro, D.M.; Almeida, D.R.P.; Pisterzi, L.F.; Juhasz, J.R.; Viskolcz, B.; Penke, B.; Csizmadia, I.G. Reaction profiling of the mao-b catalyzed oxidative deamination of amines in Alzheimer’s disease. J. Mol. Struct. THEOCHEM 2003, 666–667, 527–536. [Google Scholar] [CrossRef]
- Saari, W.S.; Halczenko, W.; Cochran, D.W.; Dobrinska, M.R.; Vincek, W.C.; Titus, D.C.; Gaul, S.L.; Sweet, C.S. 3-hydroxy-alpha-methyltyrosine progenitors: Synthesis and evaluation of some (2-oxo-1,3-dioxol-4-yl)methyl esters. J. Med. Chem. 1984, 27, 713–717. [Google Scholar] [CrossRef]
- Walker, R.B.; Fitz, L.D.; Williams, L.M.; McDaniel, Y.M. The effect on ephedrine prodrugs on locomotor activity in rats. Gen. Pharmacol. 1996, 27, 109–111. [Google Scholar] [CrossRef]
- Mu, L.; Qi, J.A.; Zhang, Q.D. Synthesis of oxazolidines from ephedrines as potential prodrugs. Yao Xue Xue Bao 1992, 27, 336–344. [Google Scholar]
- Peter, G.M.W.; Theodora, W.G. Protection for the Amino Group. In Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley: Hoboken, NJ, USA, 2006; pp. 696–926. [Google Scholar]
- Pitman, I.H. Pro-drugs of amides, imides, and amines. Med. Res. Rev. 1981, 1, 189–214. [Google Scholar]
- Patel, J.P.; Repta, A.J. Enol esters as potential pro-drugs ii. In vitro aqueous stability and enzyme-mediated hydrolysis of several enol esters of acetophenone. Int. J. Pharm. 1981, 9, 29–47. [Google Scholar] [CrossRef]
- Repta, A.J.; Patel, J.P. Enol esters as potential prodrugs iii. Stability and enzyme-mediated hydrolysis of enol esters of 6′-acetylpapaverin. Int. J. Pharm. 1982, 10, 29–42. [Google Scholar] [CrossRef]
- Repta, A.J.; Hageman, M.J.; Patel, J.P. Enol esters as potential prodrugs. Iv. Enhanced delivery of the quaternary species coralyne to rat brain using 6′-acetylpapaverin and its enol esters as prodrugs. Int. J. Pharm. 1982, 10, 239–248. [Google Scholar] [CrossRef]
- Langmead, C.J.; Watson, J.; Reavill, C. Muscarinic acetylcholine receptors as cns drug targets. Pharmacol. Ther. 2008, 117, 232–243. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O'Byrne, P.M.; Williams, R.; Walsh, J.J.; Gilmer, J.F. Synthesis, Screening and Pharmacokinetic Evaluation of Potential Prodrugs of Bupropion. Part One: In Vitro Development. Pharmaceuticals 2014, 7, 595-620. https://doi.org/10.3390/ph7050595
O'Byrne PM, Williams R, Walsh JJ, Gilmer JF. Synthesis, Screening and Pharmacokinetic Evaluation of Potential Prodrugs of Bupropion. Part One: In Vitro Development. Pharmaceuticals. 2014; 7(5):595-620. https://doi.org/10.3390/ph7050595
Chicago/Turabian StyleO'Byrne, Paul Matthew, Robert Williams, John J. Walsh, and John F. Gilmer. 2014. "Synthesis, Screening and Pharmacokinetic Evaluation of Potential Prodrugs of Bupropion. Part One: In Vitro Development" Pharmaceuticals 7, no. 5: 595-620. https://doi.org/10.3390/ph7050595