The Medicinal Chemistry of Imidazotetrazine Prodrugs

School of Pharmacy, University of Bradford, Bradford BD7 1DP, UK
*
Authors to whom correspondence should be addressed.
Pharmaceuticals 2014, 7(7), 797-838; https://doi.org/10.3390/ph7070797
Submission received: 3 April 2014
/
Revised: 17 June 2014
/
Accepted: 18 June 2014
/
Published: 10 July 2014
(This article belongs to the Special Issue Prodrugs: from Design to Clinic)
Abstract
:Temozolomide (TMZ) is the standard first line treatment for malignant glioma, reaching “blockbuster” status in 2010, yet it remains the only drug in its class. The main constraints on the clinical effectiveness of TMZ therapy are its requirement for active DNA mismatch repair (MMR) proteins for activity, and inherent resistance through O6-methyl guanine-DNA methyl transferase (MGMT) activity. Moreover, acquired resistance, due to MMR mutation, results in aggressive TMZ-resistant tumour regrowth following good initial responses. Much of the attraction in TMZ as a drug lies in its PK/PD properties: it is acid stable and has 100% oral bioavailability; it also has excellent distribution properties, crosses the blood-brain barrier, and there is direct evidence of tumour localisation. This review seeks to unravel some of the mysteries of the imidazotetrazine class of compounds to which TMZ belongs. In addition to an overview of different synthetic strategies, we explore the somewhat unusual chemical reactivity of the imidazotetrazines, probing their mechanisms of reaction, examining which attributes are required for an active drug molecule and reviewing the use of this combined knowledge towards the development of new and improved anti-cancer agents.
| 1. Introduction | 798 | |
| 2. Mechanism of Action | 799 | |
| 3. Elucidation of Prodrug Activation | 800 | |
| 3.1. The Chemistry of MTZ and ETZ | 801 | |
| 4. Kinetic Considerations | 805 | |
| 4.1 Prodrug Activation Kinetics | 805 | |
| 4.2. Ultimate Electrophile Lifetime | 807 | |
| 5. Temozolomide Co-crystals | 807 | |
| 6. Synthesis of Temozolomide and the Imidazotetrazine Core | 810 | |
| 7. Synthesis of Structural Analogues | 817 | |
| 7.1. Alternative Cores | 817 | |
| 7.2. 6- and 8-Analogues | 819 | |
| 7.3. 3-Analogues | 822 | |
| 8. Design of MGMT/MMR-Independent Anti-Cancer Agents | 825 | |
| 8.1. Introduction to NGP Analogues | 826 | |
| 8.2. Synthesis of Novel N-Linked Imidazotetrazine Dimers | 828 | |
| 8.3. Properties and Activity of N-Linked Compounds | 829 | |
| 9. Conclusions | 931 | |
1. Introduction
Imidazotetrazines are a class of bicyclic aromatic heterocycles, exemplified by the DNA methylating agent, temozolomide (Temodar®, Temodal®, TMZ, 1a). Notable previous reviews of the imidazotetrazines have focused on the development of TMZ [1,2] and the clinical properties and significance of TMZ [3,4]. The last detailed analysis of imidazotetrazine chemistry appeared in 1990 [5]. TMZ entered clinical trials in 1985 and since its release to the market by Schering-Plough in 1999 [6], has been widely used in combination with radiotherapy as the standard first-line treatment for malignant glioma (glioblastoma multiforme, grade IV astrocytoma). Somewhat serendipitously, as a drug it has excellent features: it is a stable solid which is also acid stable, allowing for 100% oral bioavailability, so administration is straightforward in comparison with many anticancer drugs. Absorption is rapid, with peak plasma concentration achieved within 0.33–2 h. Biodistribution is excellent (Vd 17 Lm−2) [7], and PET studies using 11C-labelled prodrug have provided direct evidence of blood-brain barrier penetration [8]. In 2010, TMZ reached blockbuster drug status, yet despite this impressive record it remains the only member of its class in the clinic. In this review we will explore the reasons for this and what makes TMZ special, along with surveying current efforts to achieve new effective drugs in this class.
The early history of imidazotetrazinone pharmacology indicated a considerable activity cliff with only a very small number of analogues exhibiting anticancer activity in vitro or in vivo. The 3-haloethyl analogues had good activity, the 3-methyl was of moderate effectiveness but other analogues, as exemplified by the 3-ethyl, were inactive (Table 1) [9]. The solution to this conundrum and the vision to design active new analogues arose from detailed definition of the mechanism of prodrug activation using kinetic NMR experiments. Indeed, TMZ entered clinical trials before the final details of prodrug activation chemistry were elucidated.

Table 1.
Activity of various TMZ analogues against TLX5 lymphoma implanted s.c. in CBA/CA mice. Adapted with permission from reference [9]. Copyright (1987) American Association for Cancer Research.
 | |||
| R | Dose mg/kg/day | T/C b % | |
| 1a | CH3 (TMZ) | 160 | 151 |
| 80 a | 154 | ||
| 40 a | 181 | ||
| 1b | (CH2)2Cl (MTZ) | 40 | 458 |
| 16 a | 302 | ||
| 1c | CH2CH3 (ETZ) | 640 | 123 |
| 80 a | 111 | ||
| 1d | (CH2)2Br | 160 | 137 |
| 1e | (CH2)2CH3 | 320 | 103 |
| 1f | (CH2)2OCH3 | 320 | 98 |
| 1g | (CH2)3Cl | 320 | 108 |
| 1h | CH2CHClCH2Cl | 320 | 103 |
| 1j | CH2CH=CH2 | 320 | 97 |
| 1k | CH(CH3)CH2CH3 | 320 | 83 |
| 1m | (CH2)5CH3 | 320 | 103 |
a Divided dose schedules; b treated versus control.
2. Mechanism of Action
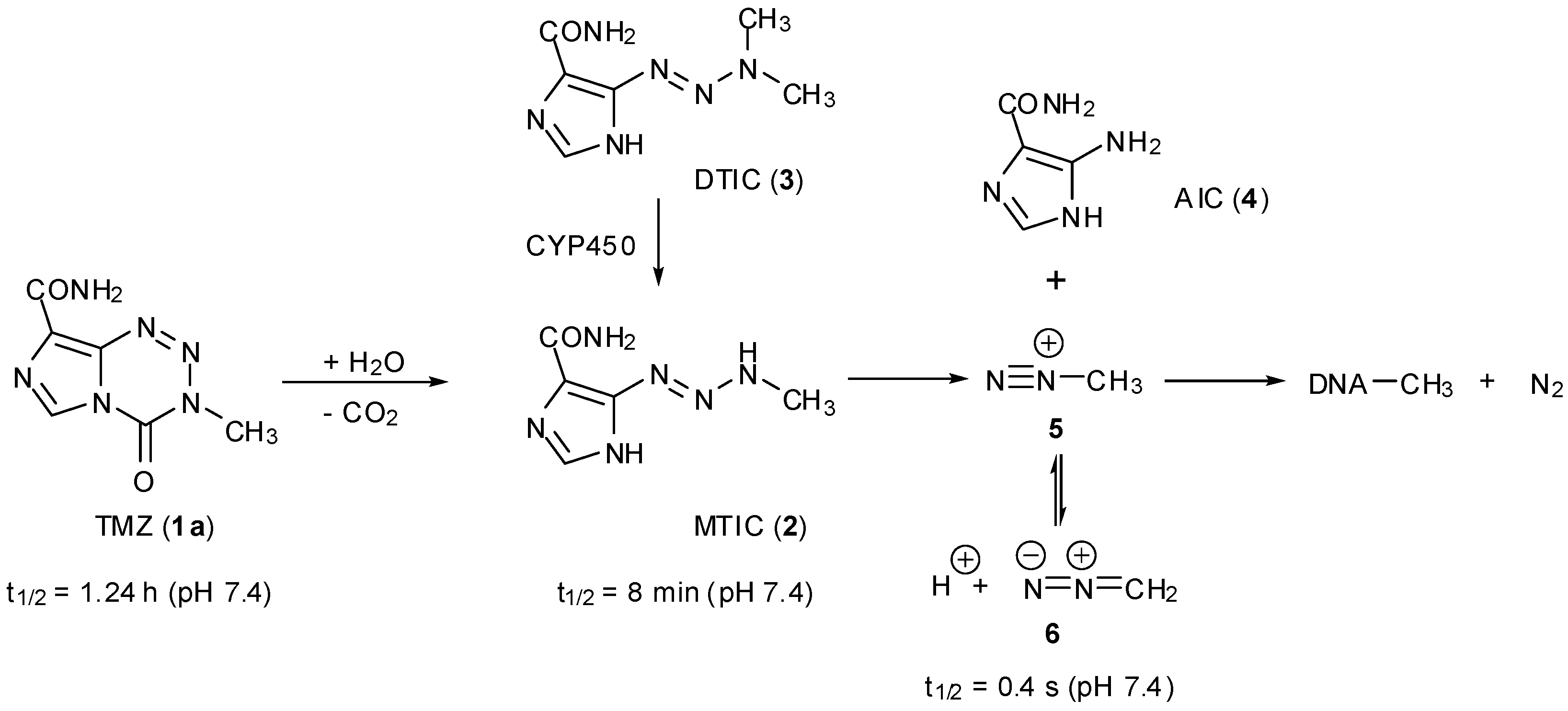
TMZ (1a) is in fact a prodrug of a prodrug and its aqueous chemistry is typical of imidazotetrazine compounds (Scheme 1). Under neutral or alkaline conditions it undergoes hydrolytic ring opening, under purely chemical control, to give the open chain triazene MTIC (2) as the first significant intermediate. The prodrug dacarbazine (DTIC, 3), which is used to treat a number of cancers including malignant melanoma, also shares this intermediate but in contrast requires hepatic demethylation via CYP450 oxidation and loss of formaldehyde to release MTIC [10]. This activated intermediate fragments to liberate AIC (4) and methyl diazonium ions 5 which then react with nucleophilic sites on DNA; it is the interaction of this methylated DNA with various DNA repair pathways that elicits the cell killing response.
Scheme 1.
Prodrug activation of TMZ.

Methylation of DNA occurs at a number of nucleophilic sites, the majority of which appears at guanine-N7 (70%) with a further 9% at adenine-N3 [1]. This N-methylation, however, is of no therapeutic value since the lesions are efficiently handled by base excision repair (BER) enzymes [11]; the anti-cancer activity of TMZ is solely attributed to the small fraction (5%) of methylation which occurs at guanine-O6.
The therapeutic effect of TMZ is dependent on the presence of DNA mismatch repair (MMR) proteins. O6-Methylguanine results in a mispairing during replication as it preferentially forms a wobble base pair with thymine. This is recognised by MMR which triggers a response; that is, T is identified, removed but then replaced with another T. Futile cycles of excision/repair result in depletion of dTTP, ultimately leading to long-lived strand breaks and cell death [1,11,12]. The GO6 methyl adduct can, however, be repaired by O6-methylguanine-DNA methyltransferase (MGMT), resulting in active resistance to TMZ in tumours in which MGMT is proficient. Acquired resistance to TMZ can be brought about by mutations in MMR, in particular the MSH6 subunit that recognises GO6 modifications, or enhanced MGMT activity through promoter hypomethylation [13]. The action of MGMT is not enzymatic; the protein is consumed stoichiometrically during the repair process and as such, inactivation of the protein can have a beneficial effect. With tumour cells high in MGMT levels, pre-treatment with inactivators such as O-benzylguanine has been shown to enhance the in vitro and in vivo activity of TMZ [14]. Several combination studies with inhibitors of alternative repair processes have also been carried out and are reviewed elsewhere [11].
It is evident that in order for a tumour to be responsive to TMZ treatment, it must be deficient in MGMT whilst also proficient in MMR; absence of the latter results in a tolerant phenotype whilst presence of MGMT impedes therapy regardless of MMR status.
3. Elucidation of Prodrug Activation
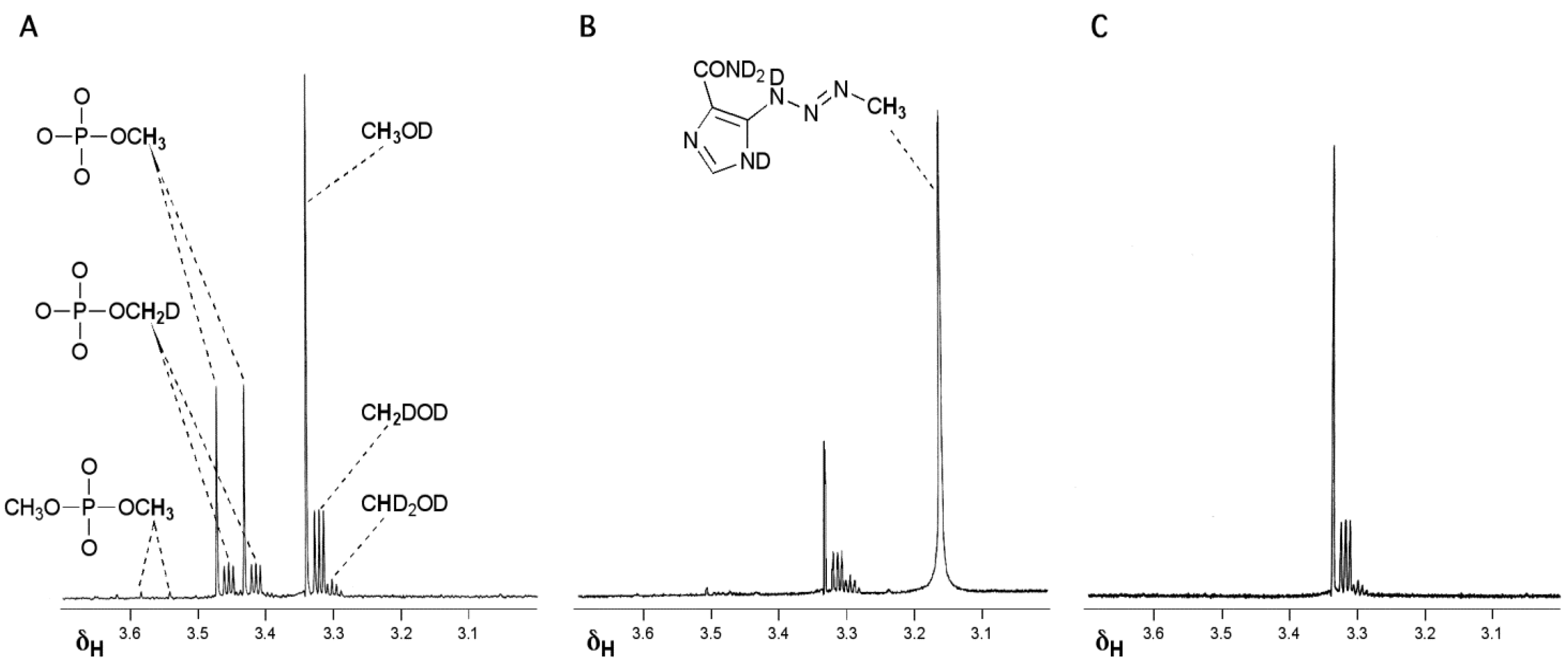
The key experiment attempted to identify intermediates in the prodrug activation pathway by time-dependent NMR spectroscopy [15,16,17]. Although the experiment was too insensitive to detect any reactive intermediates directly, the NMR data proved crucial to understanding the prodrug activation mechanism. When TMZ was reacted in a phosphate buffer/D2O system at pD = 7.8, designed to mimic physiological pH, the methyl group bearing products (methanol and methyl phosphate) showed significant loss of 1H signal and the emergence of complex multiplet signals typical of 1H–2H geminal coupling in the products (Figure 1A). It appeared that during the reaction, all three 1Hs of the TMZ methyl became freely exchangeable with the solvent so that the 1H/2H ratio in the final products of methyl group transfer reflected the relative proportions of 1H and 2H in the H2O/D2O solvent mixture. Furthermore, at high pH it proved possible to simultaneously observe MTIC (2) which did not exhibit methyl group 1H/2H exchange as well as its reaction products where similar 1H/2H exchange was evident (Figure 1B). The final experiment to confirm the diazonium intermediate involved condensing diazomethane (6) into the H2O/D2O systems whereupon the products of 1H/2H exchange again appeared (Figure 1C); indeed this process was similar to the published synthesis of isotopically-labelled diazomethane [18]. These experiments established the position of methyldiazonium as the ultimate electrophile in the prodrug activation mechanism of TMZ (Scheme 1).
Figure 1.
Reaction of TMZ, MTIC and CH2N2 in D2O systems. (A) Products of reaction of TMZ with phosphate buffer pD = 7.8 showing deuterium incorporation into methanol and methyl phosphate; (B) MTIC in Na2CO3 (10% in D2O) showing the intact isotopic configuration of the MTIC methyl and deuterium incorporation into the methanol product of hydrolysis; (C) Products of condensing diazomethane into Na2CO3 (10% in D2O) [15,16,17].
Figure 1.
Reaction of TMZ, MTIC and CH2N2 in D2O systems. (A) Products of reaction of TMZ with phosphate buffer pD = 7.8 showing deuterium incorporation into methanol and methyl phosphate; (B) MTIC in Na2CO3 (10% in D2O) showing the intact isotopic configuration of the MTIC methyl and deuterium incorporation into the methanol product of hydrolysis; (C) Products of condensing diazomethane into Na2CO3 (10% in D2O) [15,16,17].

3.1. The Chemistry of MTZ and ETZ
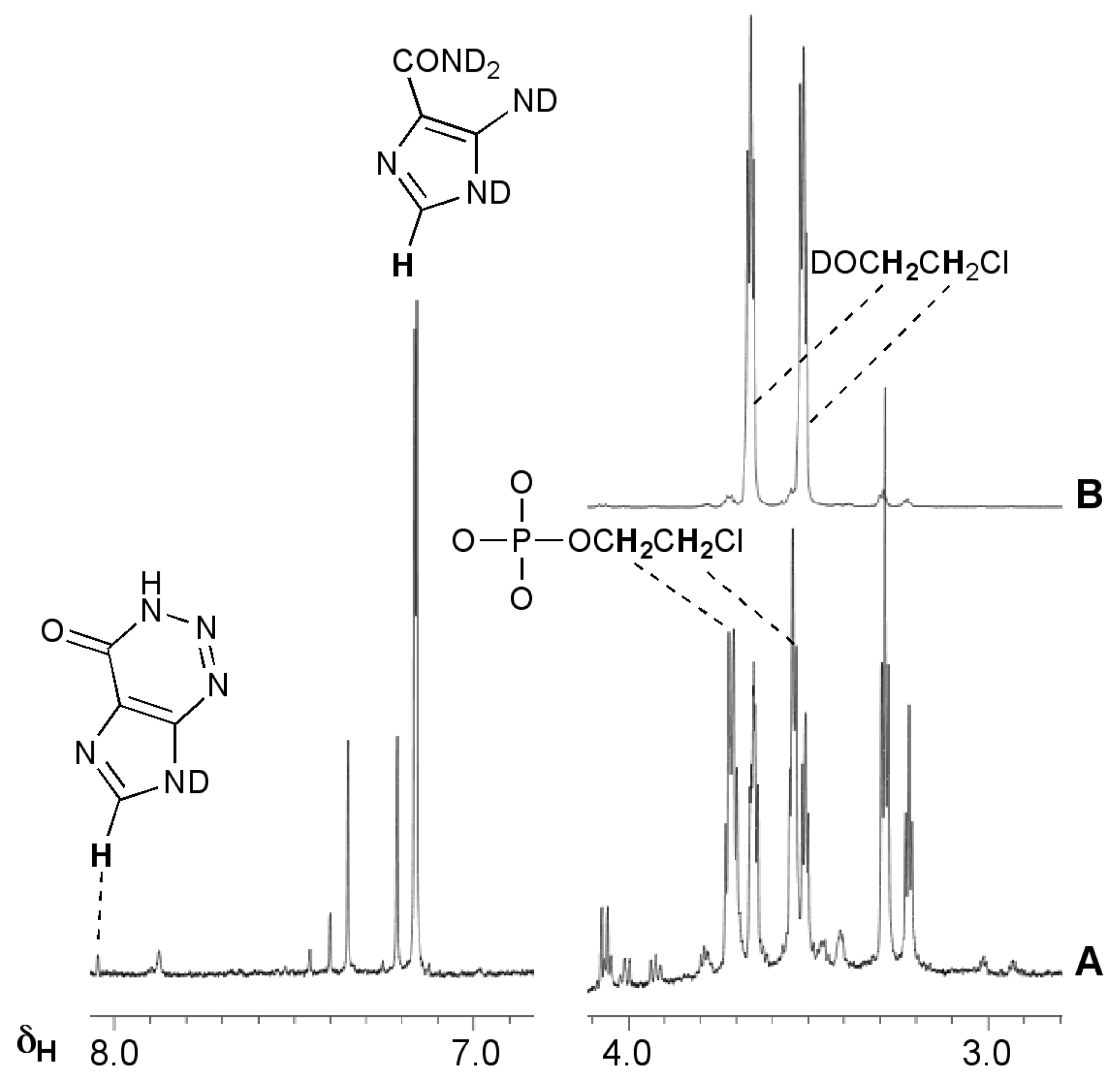
The realisation that diazonium ions were formed by triazenes allowed explanation of the activity and inactivity of other imidazotetrazine analogues. MTZ as the more potent (but myelosuppressive), first-generation imidazotetrazine prodrug was shown to be activated predominantly through the chloroethyltriazene analogue of MTIC [19,20]. The chloroethyldiazonium ions 5b that would be derived this way have a complex aqueous chemistry involving elimination and hydrolysis pathways that was extensively investigated for reactive intermediates generated by the chemically-related diazonium prodrugs, the chloroethyl nitrosoureas [21]. NMR experiments with MTZ confirmed the complexity of this chemistry in the context of the imidazotetrazines (Figure 2, Scheme 2). The 1H-NMR spectra were more complex than for TMZ and the reaction took longer to reach completion. As expected, chloroethanol (7) and alkylated phosphates 8 were the principal aliphatic products; in the aromatic region, the major product was AIC (4) with a trace of 2-azahypoxanthine (AHX, 9), the product of intramolecular trapping of the TMZ precursor diazo-IC 10, and at least two other modified imidazoles. In contrast to TMZ, there was no evidence of deuterium incorporation into any products (Figure 2, Scheme 2c), neither was there evidence of elimination to vinyl chloride 11 (Scheme 2d). These observations are significant as they suggest a role for the chlorine atom in diazonium ion stability, for example by formation of a full or partial chloronium ion 12. Other DNA reactivity considerations also point to the involvement of a chloronium intermediate [22].
Figure 2.
Reaction of MTZ with phosphate buffer pD = 7.8, 37 °C. (A) 1H-NMR spectrum after 14 days; (B) The same sample spiked with authentic chloroethanol (7).
Figure 2.
Reaction of MTZ with phosphate buffer pD = 7.8, 37 °C. (A) 1H-NMR spectrum after 14 days; (B) The same sample spiked with authentic chloroethanol (7).

Interestingly, reaction in lower alcohols and acetonitrile at elevated temperatures followed a retro-cycloaddition route, as evidenced by detection of degradation products of diazo-IC, i.e., AHX, and chloroethyl isocyanate (13b) (Scheme 2b). The trace of AHX produced in the aqueous buffer system showed that this route, although not preferred, exists in aqueous systems, giving credence to early reports of MTZ as a carbamoylating agent [23]. Incidentally, the trifluoroethyl analogue only reacted by the retro-cycloaddition route in aqueous systems (vide infra, Scheme 4).
Scheme 2.
The aqueous chemistry of MTZ.

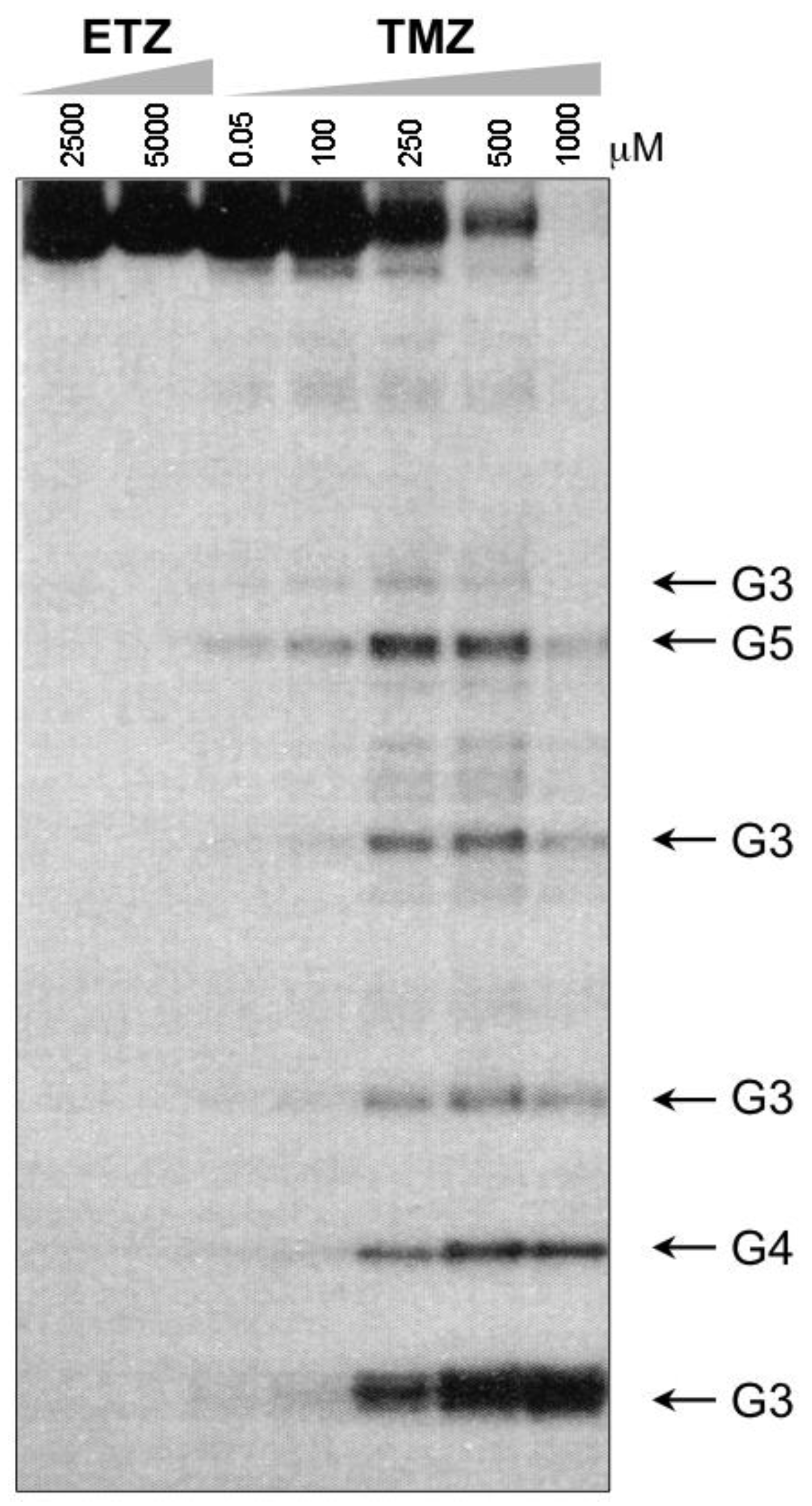
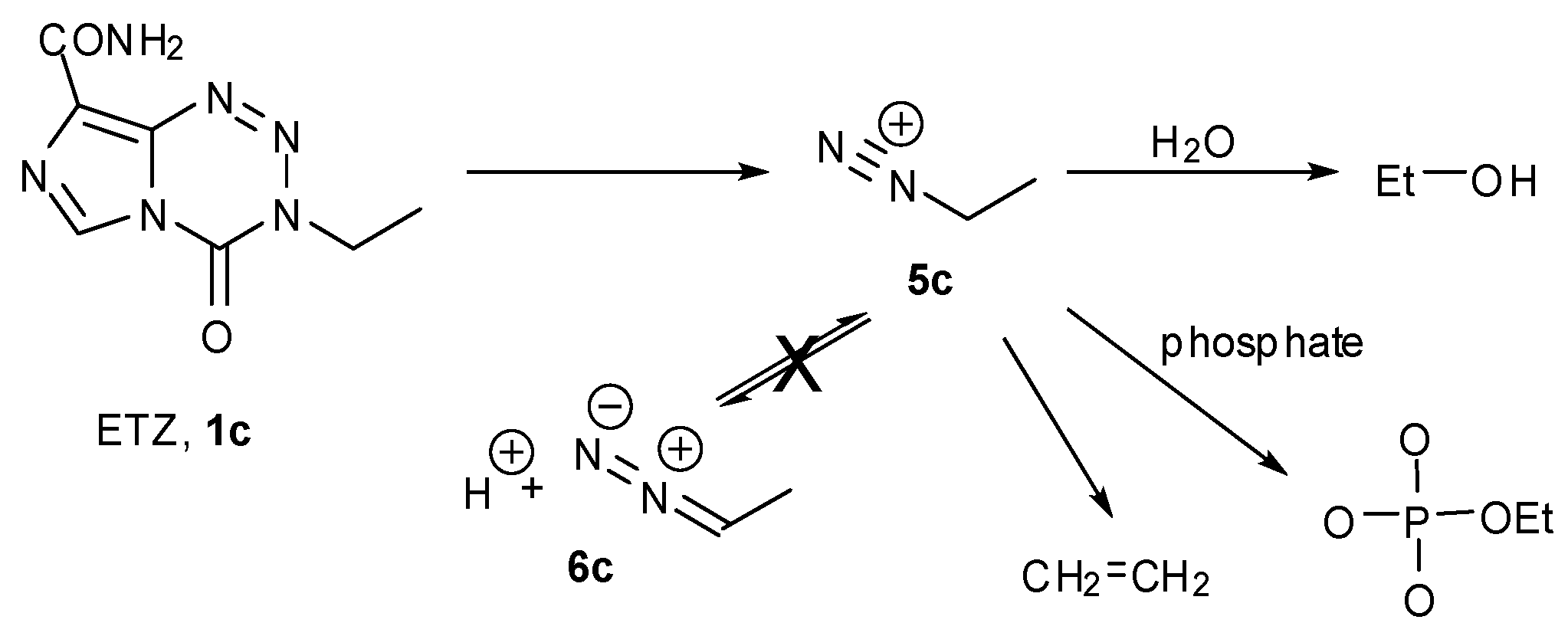
A great anomaly of the early compounds was the inactivity of the ethyl and higher alkyl analogues (vide supra, Table 1). In reaction with DNA it was impossible to detect GN7 alkylation sites, even at very high dose (5 mM) of ethazolastone (ETZ, 1c) and there was no in vitro or in vivo antitumour activity (Figure 3) [24]. The reason for this becomes apparent when the fates of ethyldiazonium ions are considered (Scheme 3) [25].
Figure 3.
Comparison of reaction of ETZ and TMZ with DNA, showing the unreactivity of ETZ up to 5 mM, sites of GN7 alkylation produced in the 276-bpBamHI-Sal1 fragment of pBR322, by the piperidine cleavage method. Adapted from reference [24].
Figure 3.
Comparison of reaction of ETZ and TMZ with DNA, showing the unreactivity of ETZ up to 5 mM, sites of GN7 alkylation produced in the 276-bpBamHI-Sal1 fragment of pBR322, by the piperidine cleavage method. Adapted from reference [24].

Scheme 3.
The aqueous chemistry of ETZ.

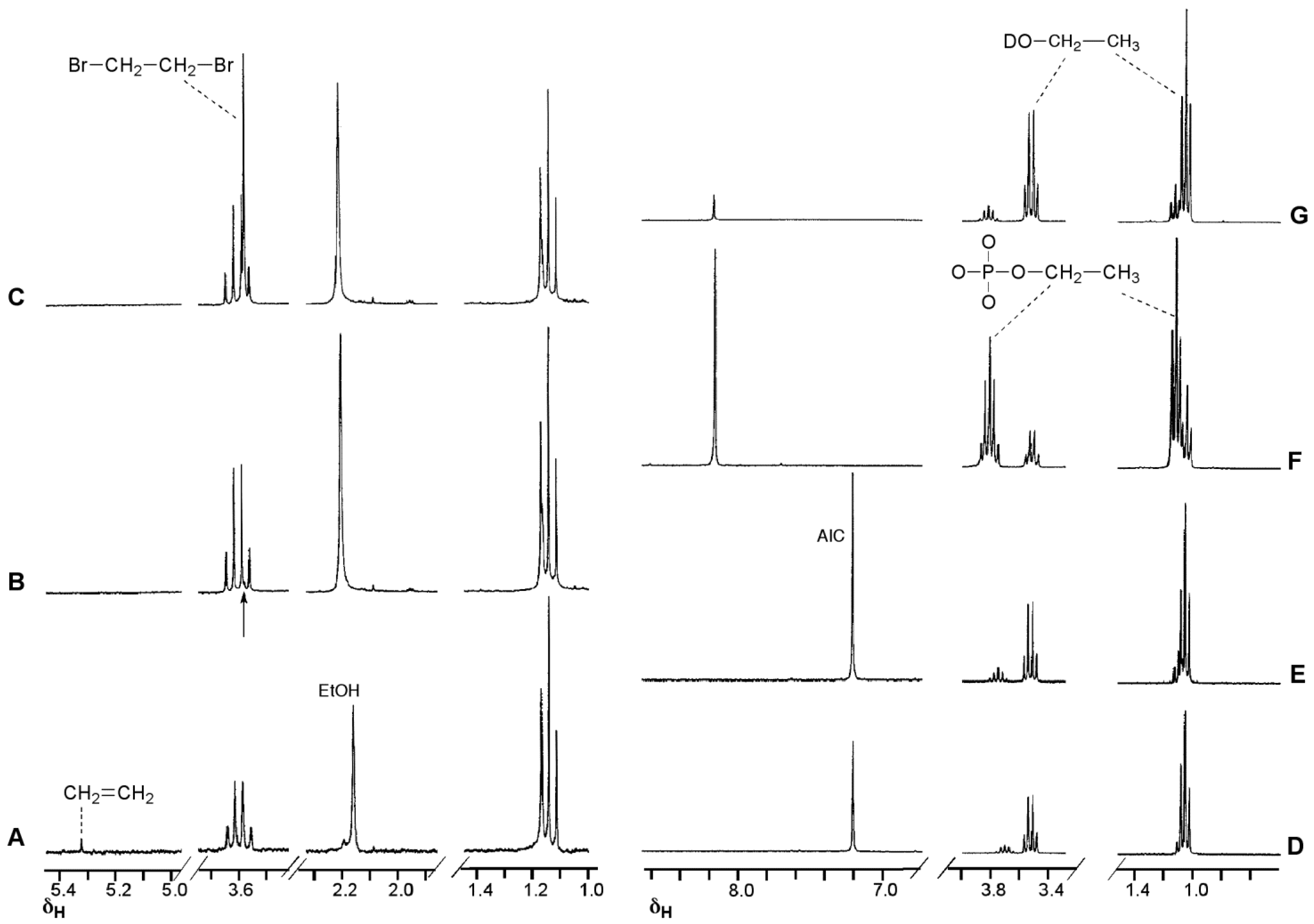
The NMR experiment in deuterated buffer was relatively uncomplicated (Figure 4). In this case the experiment was conducted in a two phase D2O/CDCl3 system. Deuterium incorporation was not detected in any products so, as with MTZ, the diazoalkane equilibration with 6c did not occur. Ethanol and ethylphosphate were confirmed as the major reaction products in the D2O layer. In the CDCl3 layer, a singlet peak appeared at 5.3 ppm in the 1H spectrum. Reaction with Br2 and subsequent spiking with authentic dibromoethane identified the by-product as ethene: a result of elimination of ethyldiazonium 5c. This implies that the fate of ethyldiazonium is either immediate nucleophilic substitution or rapid elimination to ethene without time for diazoethane equilibration. In the former case, the incipient electrophile may be too short lived to effectively locate a reaction site on DNA, while the latter route results in drug inactivation by elimination.
Figure 4.
1H-NMR investigation of the reaction of ETZ with phosphate buffer (pH 7.8) in a two phase system (D2O/CDCl3). Spectra A–C CDCl3 layer, spectra D–G, D2O layer. (A) At the end of the imidazotetrazine hydrolysis reaction identifying the putative ethene peak; (B) after addition of Br2, the arrow indicates the position of a new peak; (C) sample of spectrum B after adding authentic dibromoethane; (D) at the end of the imidazotetrazine hydrolysis reaction; (E–G) sample of spectrum D after sequentially spiking with authentic AIC, ethylphosphate and ethanol (note the shift in the AIC signal after adding the acidic ethyl phosphate).
Figure 4.
1H-NMR investigation of the reaction of ETZ with phosphate buffer (pH 7.8) in a two phase system (D2O/CDCl3). Spectra A–C CDCl3 layer, spectra D–G, D2O layer. (A) At the end of the imidazotetrazine hydrolysis reaction identifying the putative ethene peak; (B) after addition of Br2, the arrow indicates the position of a new peak; (C) sample of spectrum B after adding authentic dibromoethane; (D) at the end of the imidazotetrazine hydrolysis reaction; (E–G) sample of spectrum D after sequentially spiking with authentic AIC, ethylphosphate and ethanol (note the shift in the AIC signal after adding the acidic ethyl phosphate).

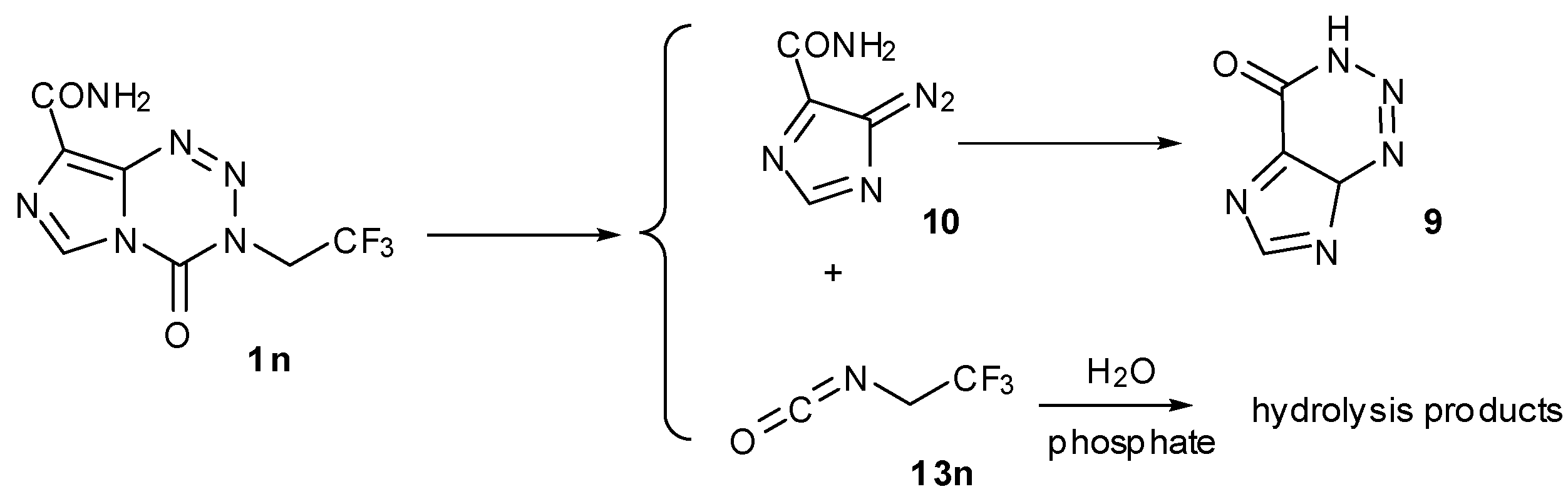
The need to block elimination led first to analogues lacking hydrogen atoms in the β-position of the side chain such as the 3-furfuryl and 3-trifluoroethyl 1n (Scheme 4) [25]. Of these, the trifluoroethyl analogue showed reasonable activity in the NCI60 cell line panel, with mean GI50 = 10.7 µM (log range = 2.35) compared with MTZ (79 µM, log range = 0.96), with a distinctly different pharmacological profile from MTZ (p = −0.347). Interestingly, a sample dissolved in DMSO showed clean retro-cycloaddition to give AHX and trifluoroethylcarbamic acid on standing overnight at room temperature. In a capricious turn of fate, investigation of the aqueous prodrug activation mechanism by 1H-, 31P- and 19F-NMR showed a complex mixture of products that included AHX and a mixture of at least nine fluorine containing compounds of which trifluoroethanol and trifluoroethylamine were components [25]. The aliphatic portion of the product mixture spectra (1H and 19F) could be replicated by reaction of trifluoroethylisocyanate (13n) with buffer. This is a contrast to MTZ where heating in a non-aqueous system was required to effect the retro-cycloaddition and indicates that the balance between retro-cycloaddition and hydrolysis at C4 is determined by the electron withdrawing effect of the 3-substituent.
Scheme 4.
The aqueous fate of the 3-trifluoroethylanalogue 1n.

4. Kinetic Considerations
The data in Scheme 1 highlight two kinetic parameters key to the design of new agents. The first is the rate of addition of water (or hydroxide ion) to initiate the ring-opening reaction. The prodrug itself must be sufficiently stable to allow uptake and distribution without significant hydrolysis. The second is the reactivity of the latent electrophile.
4.1. Prodrug Activation Kinetics
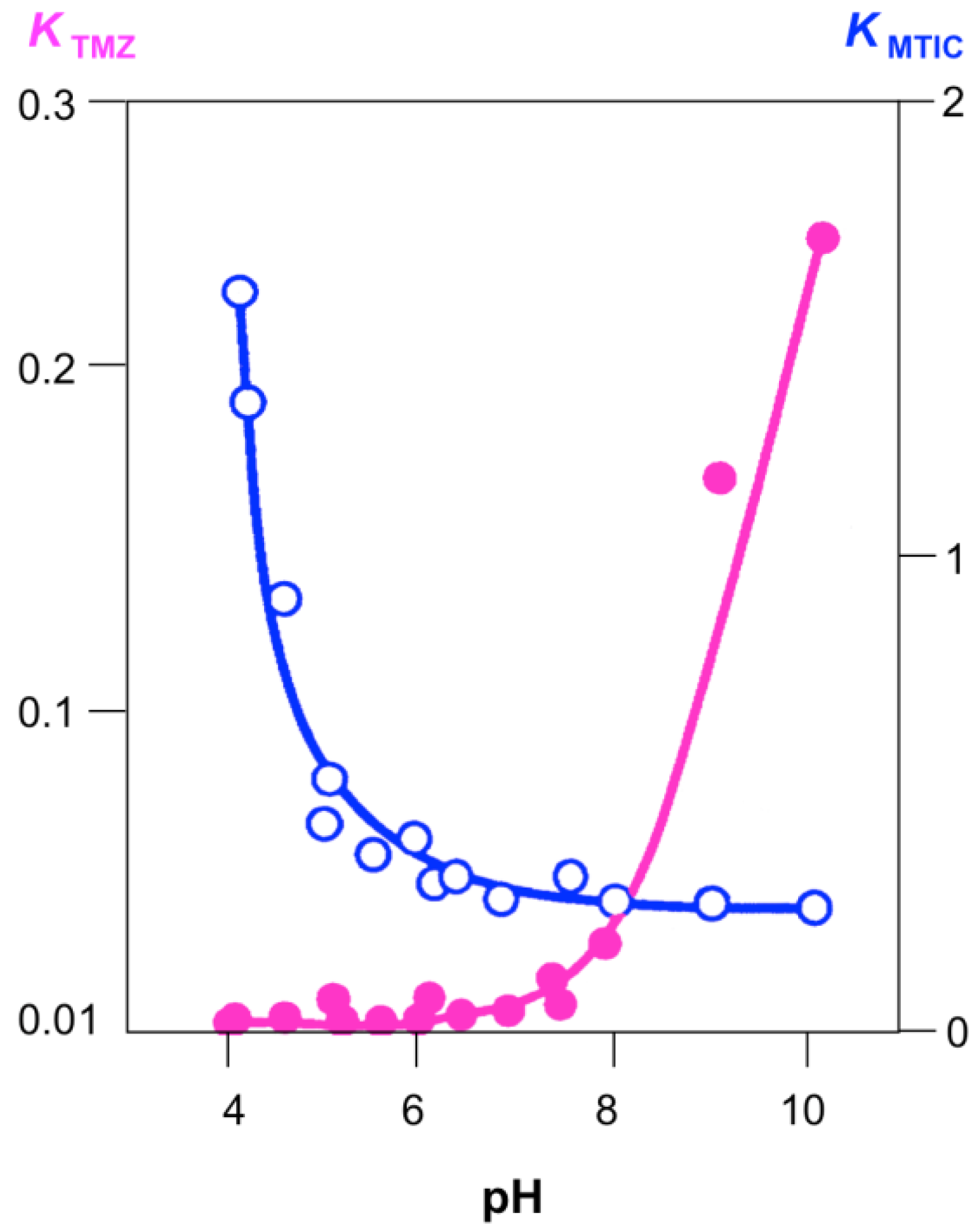
An important property of the imidazotetrazines is the contrast of robust acid stability with rapid alkaline hydrolysis (Figure 5); the slow hydrolysis at low pH confers acid stability and the convenience of oral dosing [16]. The pH-dependence of this reaction also influences distribution of the prodrug around the body: hydrolysis kinetics at pH 7.4 match closely the uptake rate (peak plasma concentration after 30 min) and metabolic half-life (t½ = 1.29 h) in patients [1,9]. If the aqueous lifetime were too short, the prodrug would be inactivated long before it were able to reach its sub-cellular target; too long it could be metabolised and excreted without taking effect. The pH dependence of TMZ and MTIC hydrolysis are complementary, so that it is only around neutral pH that the whole reaction process from prodrug activation to methyl group transfer occurs effectively, indeed MTIC and similar triazenes may be prepared by alkaline hydrolysis of their imidazotetrazines [20].
Superficially, imidazotetrazine activation is hydrolysis of a carbonyl compound with water or hydroxide ion attacking at C4, so that both acid and base catalysed reactions would be expected, in common with other carbonyl group reactions. The reason for the absence of the acid catalysed variant is the presence of the weakly basic imidazole N7. 15N-NMR experiments showed that the resonance of N7 was shifted and inverted in the presence of TFA, indicating that it carried a proton and was also, formally, the site of the positive charge (Figure 6) [26]. Since protonation occurs at a distant site, it is not able to activate the 4-carbonyl group to nucleophilic attack. The site of protonation at N7 was independently determined in the X-ray crystal structure of the salt TMZ·HCl·2H2O (vide infra, Figure 9) [27].
Figure 5.
pH Dependence of the pseudo-first order rate constants for hydrolysis of TMZ and MTIC. Adapted from reference [16].
Figure 5.
pH Dependence of the pseudo-first order rate constants for hydrolysis of TMZ and MTIC. Adapted from reference [16].

Figure 6.
15N-NMR spectra of TMZ. (A) 15N{1H} in DMSO-d6; (B) with full 1H nOe in TFA. Both experiments were acquired in the presence of Cr(acac)3 and referenced to external CH3NO2 [26].
Figure 6.
15N-NMR spectra of TMZ. (A) 15N{1H} in DMSO-d6; (B) with full 1H nOe in TFA. Both experiments were acquired in the presence of Cr(acac)3 and referenced to external CH3NO2 [26].

In addition to pH, the aqueous reactivity of imidazotetrazines generally is also dependent on the nature of the 3-substituent, such that the rate of hydrolysis is increased by electron withdrawing groups [20], notwithstanding the ability of powerfully electron withdrawing groups to switch the mechanism away from hydrolysis to the retro-cycloaddition reaction.
4.2. Ultimate Electrophile Lifetime
The other significant kinetic parameter is the lifetime of the ultimate electrophile released from the prodrug. Hydrolysis of methyldiazonium in a purely chemical system has t½ = 0.39 s [28]. Again, a sub-optimal value detracts from clinical effectiveness: ethyldiazonium eliminates or reacts with water before it has time to encounter a reactive nucleic acid target site [25], while longer-lived intermediary electrophiles such as chloronium achieve clinical efficacy [29]. For TMZ, these clinically-useful, if not formally optimized, pharmacokinetic properties were achieved serendipitously.
5. Temozolomide Co-Crystals
Drug stability, solubility and formulation are important factors when considering activity in vivo, as they can be barriers to bioavailability and efficacy. These factors are influenced by the solid form in which the compound of interest resides, whether this be as polymorphs, salts, solvates, co-crystals or even incorporated into microspheres [30].
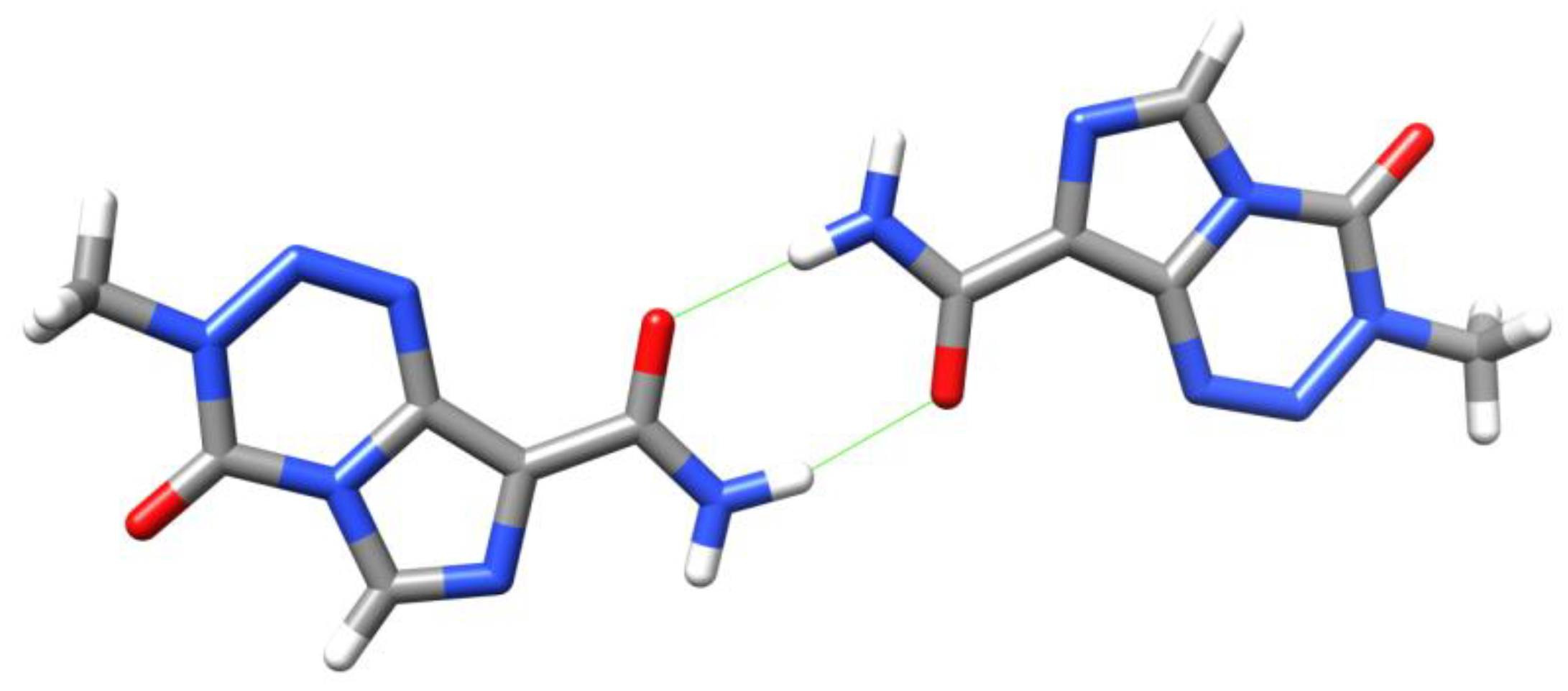
Drug compounds can frequently exist in a number of different polymorphs. Ten polymorphs of TMZ are covered in the patent literature [31,32], of which four have had X-ray structures solved [33,34]. TMZ can easily form a dimer via intermolecular hydrogen bonds between the carboxamide groups (Figure 7). It has also been proposed that the energetic preference for the observed conformer can be attributed to an intramolecular hydrogen bonding interaction between amide NH and imidazole N7, although this shows significant (74°) deviation from linearity [27,33].
Figure 7.
Dimers seen in Lowe’s crystal structure of TMZ [33]. Strong H-bonds are shown in green.
Figure 7.
Dimers seen in Lowe’s crystal structure of TMZ [33]. Strong H-bonds are shown in green.

Stored under the correct conditions, commercial TMZ is relatively stable. However, prolonged storage or exposure to humid conditions can accelerate degradation of the prodrug and reduce efficacy. Salt formation and crystallisation are widely accepted methods of altering physicochemical properties of drug molecules.
In recent years, a great deal of work has been done, primarily by Babu and co-workers, to investigate the co-crystalisation of TMZ with different molecules as a means of enhancing stability and solubility. Given the higher stability of TMZ under acidic conditions, Babu et al. reasoned that co-crystallisation with compounds bearing generally-recognised-as-safe (GRAS) carboxylic acid groups could act as pH adjusters to inhibit hydrolytic degradation and thus improve aqueous stability. Recognising that complexation must be carefully controlled to ensure that the active species is still released, they made a number of co-crystals with organic acids which were characterised by powder X-ray diffraction and IR before testing (for an example, see Figure 8). As expected, these co-crystals all exhibited a stable hydrogen bonding interaction between the acidic group of the co-former and the amide group of TMZ. Several co-crystals showed dissolution rates comparable to that of TMZ and they all showed a decrease in decomposition rate in pH 7.0 buffer, with a moderate correlation between drug half-life and acidity of the co-former. Whereas TMZ would exhibit discolouration after just one week at ambient conditions, co-crystals with succinic acid, oxalic acid or salicylic acid showed no sign of discolouration or degradation after one year under the same conditions [35].
Figure 8.
Temozolomide–salicylic acid co-crystal [35]. Strong H-bonds are shown in green.
Figure 8.
Temozolomide–salicylic acid co-crystal [35]. Strong H-bonds are shown in green.

In a similar vein, Sanphiu, Babu and Nangia have also created a number of co-crystals using GRAS carboxamides such as saccharin and caffeine as co-formers [36]. These two co-crystals were more stable than other carboxamides used, and indeed temozolomide; in the case of saccharin this can be attributed to its acidic nature. However, the stability improvement was less than that seen with carboxylic acid co-formers.
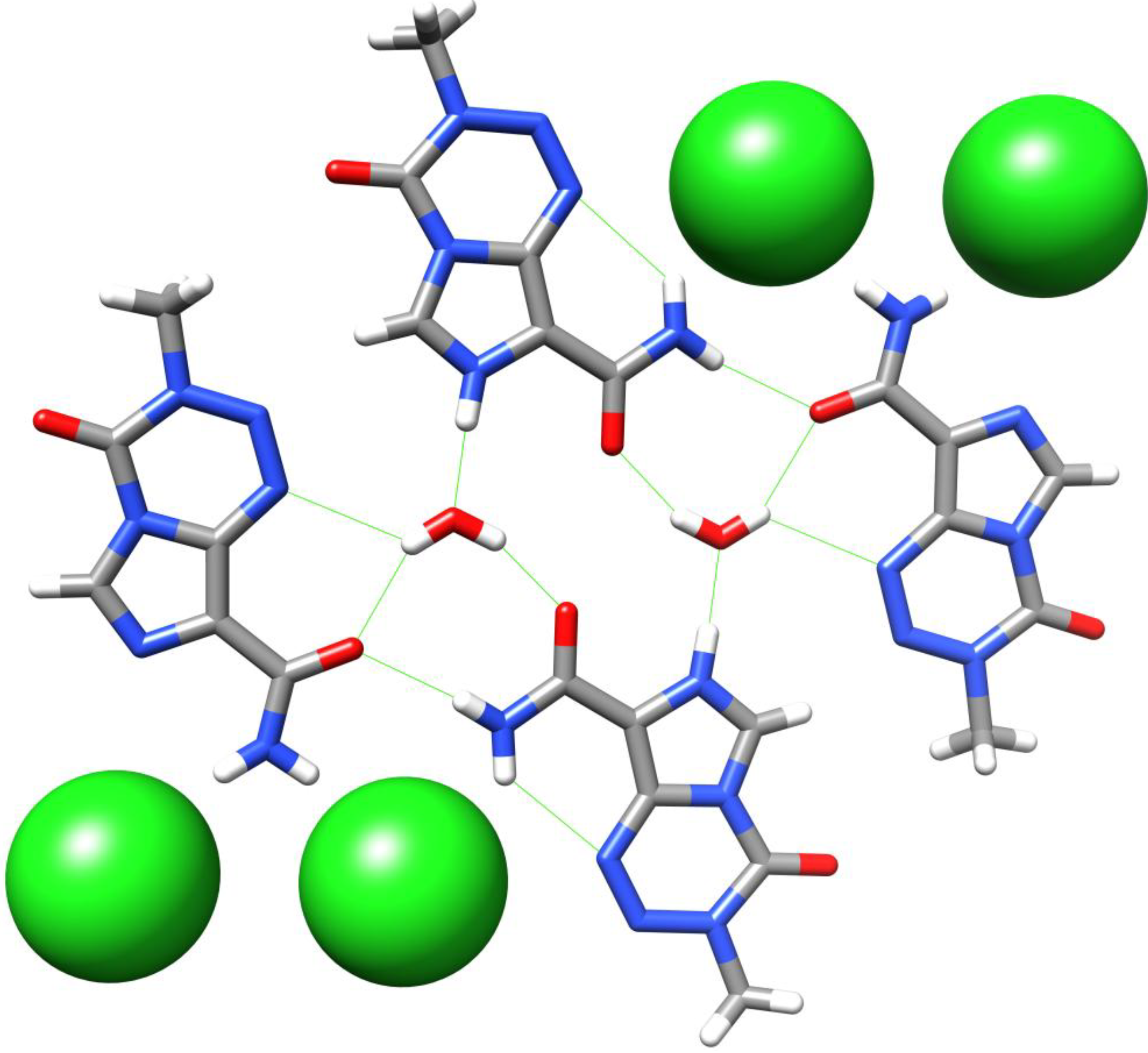
Babu et al. have recently obtained a crystal structure of TMZ hydrochloride in its dihydrate form. It is shown that the lattice structure for this crystallisation form contains one neutral TMZ, one TMZ·HCl, one H3OCl and three water molecules, with protonation, as expected from previous NMR studies [26], occurring at the imidazole N7 of TMZ (Figure 9). The network of hydrogen bonds forms a tetrameric TMZ unit which extends into parallel sets of ribbons, connected by strong hydrogen bonds to water and chloride ions [27]. Under accelerated humidity conditions, after one week the PXRD plot of the hydrochloride salt had reverted to that of neutral TMZ.
Figure 9.
A tetrameric unit of TMZ in the crystal structure of TMZ·HCl·2H2O [27]. Strong H-bonds are shown in green.
Figure 9.
A tetrameric unit of TMZ in the crystal structure of TMZ·HCl·2H2O [27]. Strong H-bonds are shown in green.

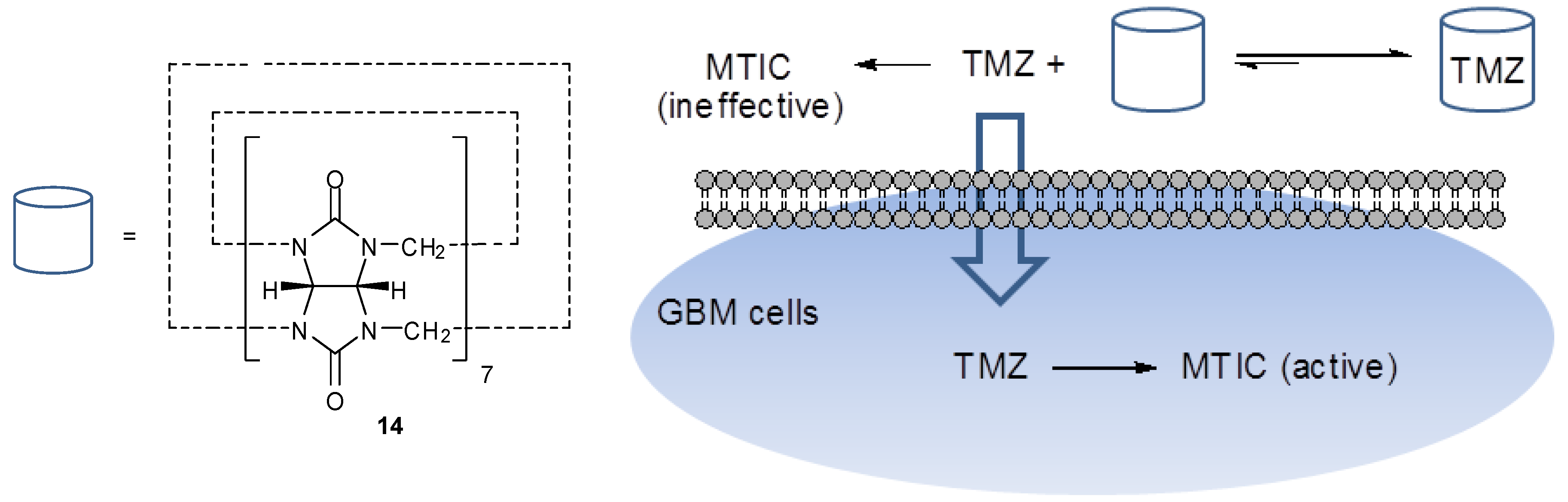
Other TMZ complexes include that with cadmium chloride, synthesised by Liu [37]. Appel et al. exploited the use cucurbit[n]uril (14), which has previously been shown to improve the solubilty, stability and cellular uptake of drug molecules. They formed a 1:1 complex with TMZ, encapsulating the drug within the natural cavity of the macrocycle (Figure 10). In doing so, the stability of the imidazotetrazine was increased, resulting in a longer half-life (5 h in pH 7.0 buffer, compared with 3 h for the control solution of TMZ alone). It was observed that the activity of the drug against glioblastoma multiforme (GBM) primary cells was increased, presumably due to the prolonged lifetime of TMZ in solution. This may allow increased absorption into cells prior to hydrolysis and thus increased exposure to the active drug since MTIC itself does not cross the blood-brain barrier or effectively penetrate cells [38].
Other TMZ complexes include that with cadmium chloride, synthesised by Liu [37]. Appelet al. exploited the use cucurbit[n]uril (14), which has previously been shown to improve the solubilty, stability and cellular uptake of drug molecules. They formed a 1:1 complex with TMZ, encapsulating the drug within the natural cavity of the macrocycle (Figure 10). In doing so, the stability of the imidazotetrazine was increased, resulting in a longer half-life (5 h in pH 7.0 buffer, compared with 3 h for the control solution of TMZ alone). It was observed that the activity of the drug against glioblastoma multiforme (GBM) primary cells was increased, presumably due to the prolonged lifetime of TMZ in solution. This may allow increased absorption into cells prior to hydrolysis and thus increased exposure to the active drug since MTIC itself does not cross the blood-brain barrier or effectively penetrate cells [38].
Figure 10.
Cucurbit[n]uril (14) and uptake of TMZ into GBM cells [38].
Figure 10.
Cucurbit[n]uril (14) and uptake of TMZ into GBM cells [38].

6. Synthesis of Temozolomide and the Imidazotetrazine Core
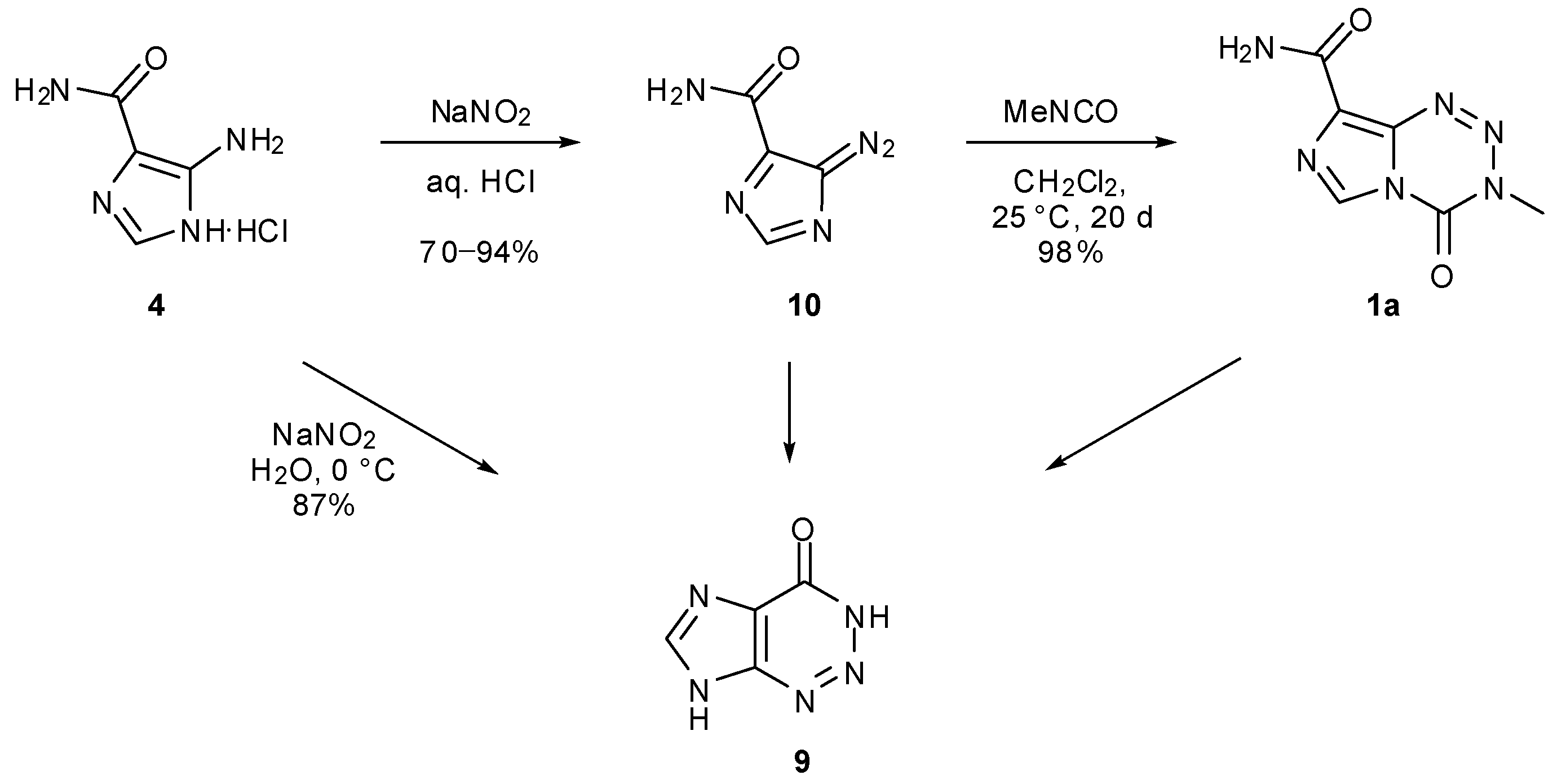
The original synthetic strategy towards TMZ and other imidazotetrazines is based on a general method for azolotetrazines [39], and involves the coupling of 5-diazoimidazole-4-carboxamide (diazo-IC, 10) or its analogues with the relevant isocyanate (Scheme 5). Evidence suggests this occurs in a stepwise rather than a concerted mechanism [20,25]. Diazo-IC can be readily prepared on a multigram scale from commercially available AIC hydrochloride 4 by treatment under diazotisation conditions (NaNO2/HCl), which produces the desired product in yields of 70%–94% [26,40]. Incidentally, if acidic conditions are not used in this reaction, AHX is instead furnished as the sole product [41]. Cyclisation of diazo-IC to AHX occurs rapidly under basic conditions but also takes place more slowly in neutral and acidic media, doing so more readily when light is present [40,42]. In addition, AHX has been observed as result of imidazotetrazine decomposition [20,25].
Early examples of the coupling of diazo-IC with isocyanates were performed in dichloromethane or ethyl acetate, keeping the reaction in the dark to avoid UV-promoted decomposition of diazo-IC. Although use of these reaction solvents results in clinical grade material, reaction times are very slow due to the insolubility of diazo-IC, typically taking several weeks to reach completion [20]. More recent synthetic protocols generally employ DMSO as the reaction solvent, giving rise to much faster reaction times [26,43]. Attempts using a range of imidazolonium and phosphonium ionic liquids failed to improve on the rate, yield or purity of the DMSO reaction [44]. A mixed DMSO–EtOAc solvent has also been used to give large amounts of clinical grade TMZ in three days [45]. Isolation of the imidazotetrazine products can often be achieved by addition of water or ethyl acetate and collection of the resultant precipitate.

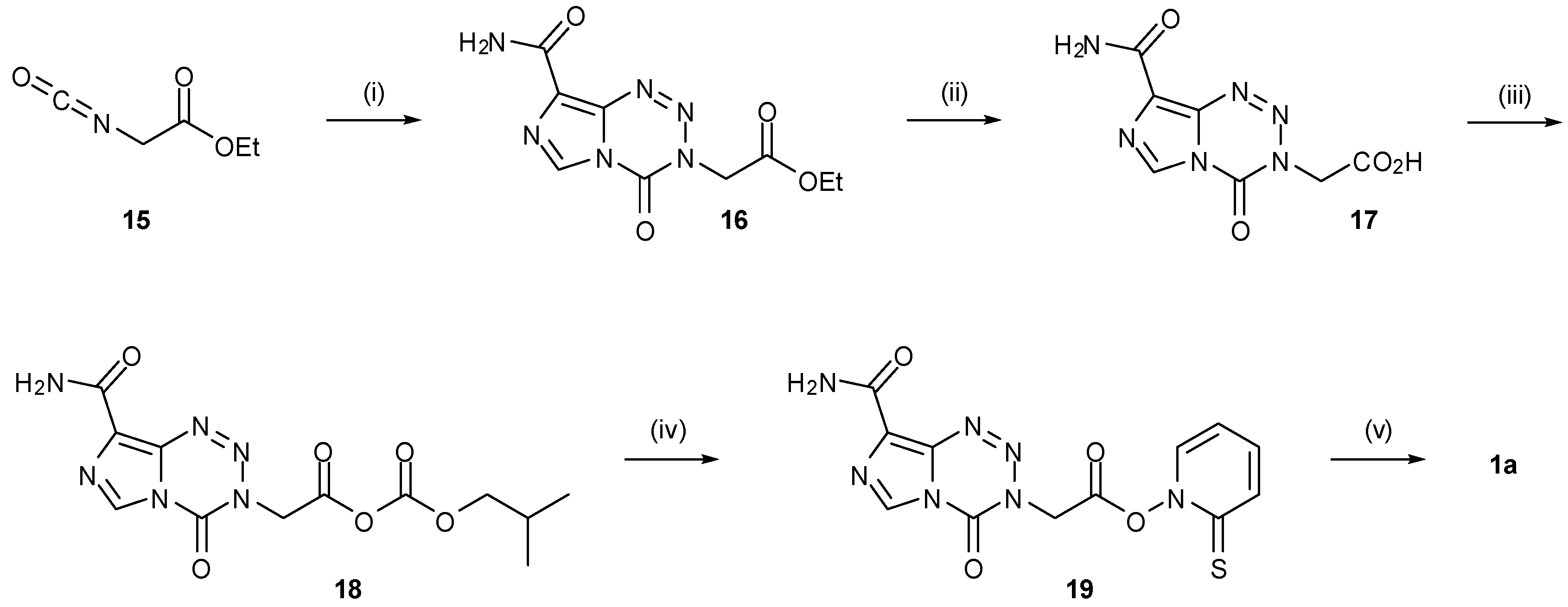
The stigma associated with the use of methyl isocyanate, following the 1984 Bhopal disaster and subsequent near-disappearance of the compound from the market, prompted the development of a number of alternative routes to TMZ and thus the imidazotetrazine core structure [47]. The first of these approaches was to use a masked methyl isocyanate equivalent (Scheme 6). Wang used commercially available isocyanoacetate 15 to synthesise an ester analogue of TMZ (16) by reacting with either Diazo-IC or AIC∙HCl. This compound was subsequently functionalized via acid 17 to carbonic anhydride 18, converted to activated ester 19 and then subjected to Barton decarboxylation conditions to reveal the desired methyl group of TMZ [46].
Scheme 6.
Masked methyl isocyanate approach to TMZ [46].
Scheme 6.
Masked methyl isocyanate approach to TMZ [46].

Reagents and conditions: (i) Diazo-IC, DMSO, 25 °C (80%) or AIC∙HCl, pyridine, DMSO, 20 °C followed by NaNO2, 2 M HCl, 0 °C (71%); (ii) 5 M HCl, 45 °C; (iii) Me2CHCH2CO2Cl, N‑methylmorpholine; (iv) 2-mercaptopyridine-N-oxide, Et3N, −15 °C; (v) Bu3SnH, AIBN (cat.), DMF, hν (26%) [46].
Alternative masking of the methyl group can be achieved using silicon. TMS protected imidazotetrazine derivative 20 was synthesised in 33% yield from the appropriate isocyanate and diazo-IC; treatment with TBAF then furnished TMZ (Scheme 7) [48].
Scheme 7.
TMS deprotection to give TMZ [48].
Scheme 7.
TMS deprotection to give TMZ [48].

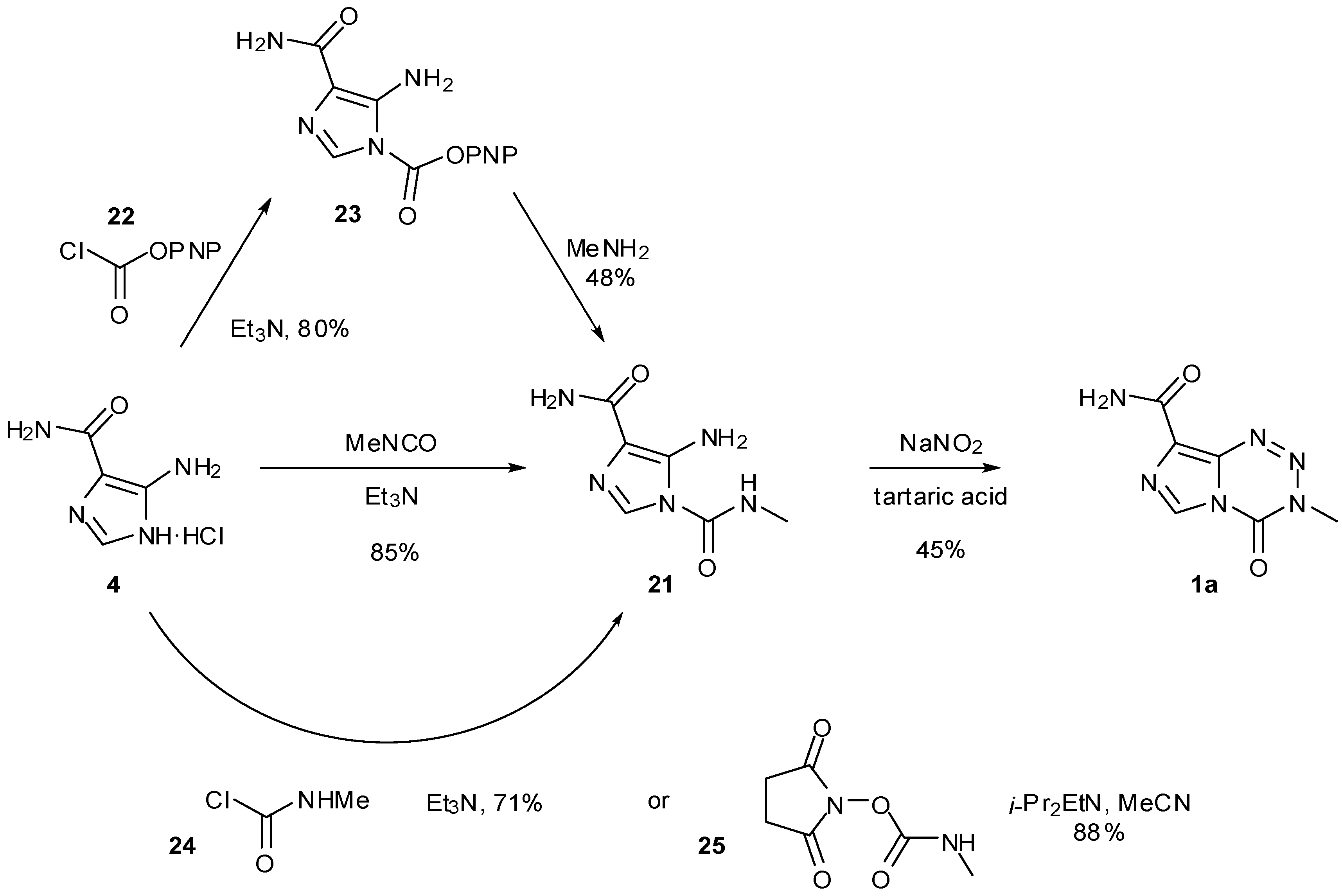
In addition to the use of alternative isocyanates, routes to imidazotetrazines avoiding the use of potentially-explosive diazo-IC were also explored (Scheme 8). The first of these started from AIC∙HCl but reversed the two steps of the original synthesis to proceed via urea 21. Formation of ureas gives high yields however the subsequent imidazotetrazine cyclisation was found to be generally low yielding (30%–35% in the case of TMZ under standard conditions), with a significant amount of AHX seen as a by-product [47,49]. Alternative routes to urea 21 also obviated methyl isocyanate from this process. Reaction of AIC with p-nitrophenyl chloroformate (22) furnished carbamate 23 which was treated with methylamine to give the desired urea. A second modification allowed direct formation of the urea by carbamoylation of AIC with methylcarbamoyl chloride (24), whereas a related route by Turchetta et al. involved the use of N-succinimidyl-N'-methyl carbamate (25) as the carbamoylating agent [50]. Exploration of a variety of solvents, nitrosating agents and acids gave the optimum imidazotetrazine-forming conditions to TMZ as outlined in Scheme 8. A more recent patent gives alternative conditions for diazotisation using LiCl to give TMZ in up to 65% yield [51].

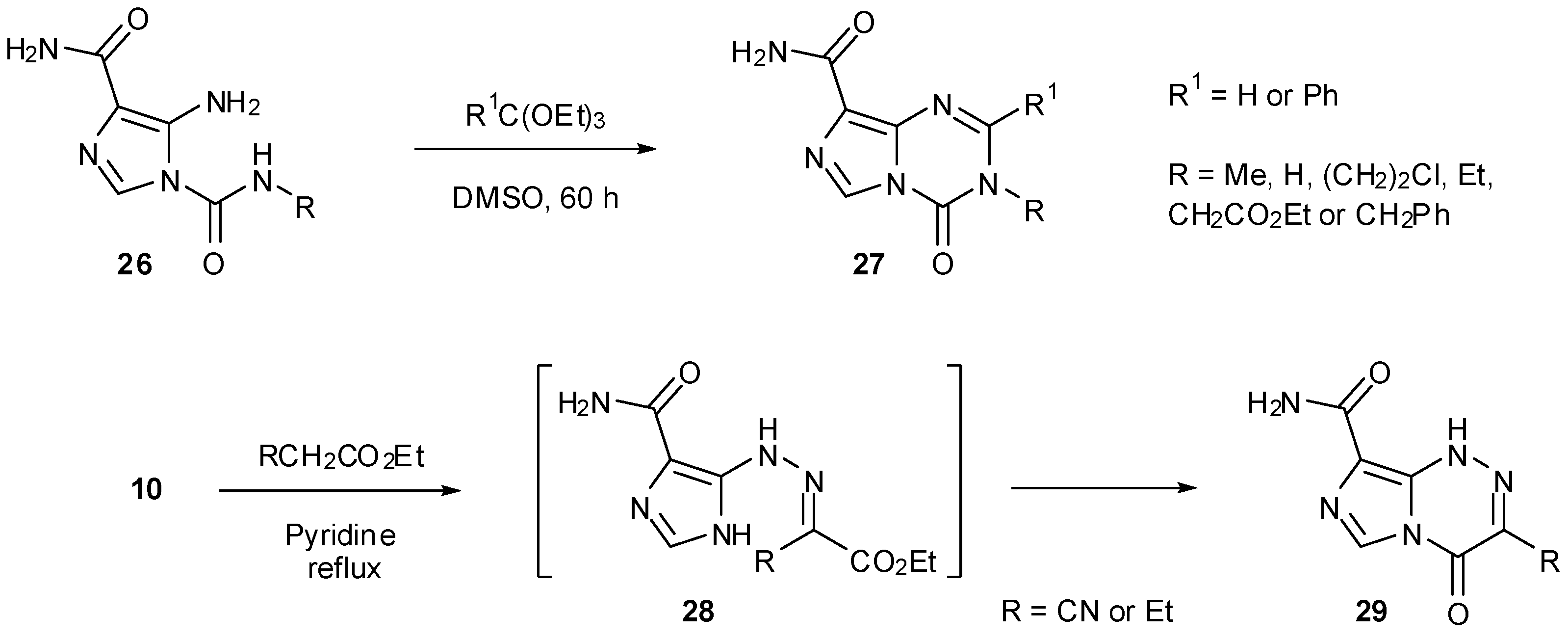
Treatment of ureas 26 with orthoformates rather than nitration gave rise to imidazotriazinones 27 (Scheme 9). Alternatively, reaction of diazo-IC with ethyl cyanoacetate or ethyl acetoacetate afforded regioisomeric imidazotriazinones 29 via unstable hydrazones 28 [25]. These types of heterocyclic structures, however, cannot give rise to biological activity in the same manner as the imidazotetrazines.
Scheme 9.
Synthesis of imidazotriazinones [25].
Scheme 9.
Synthesis of imidazotriazinones [25].

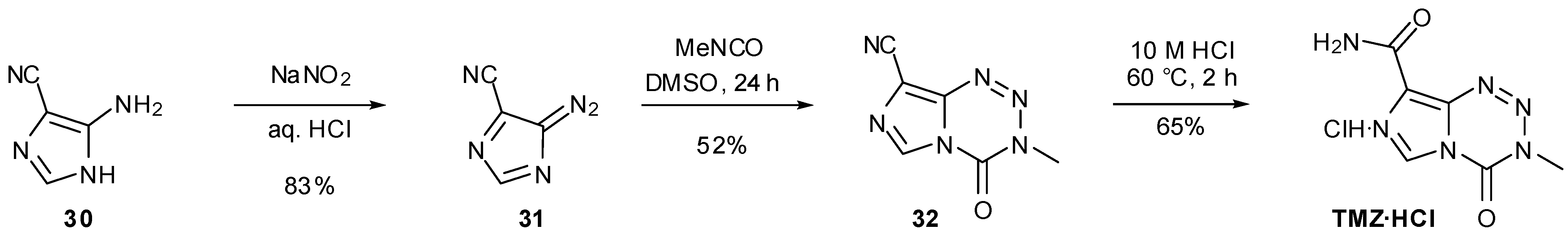
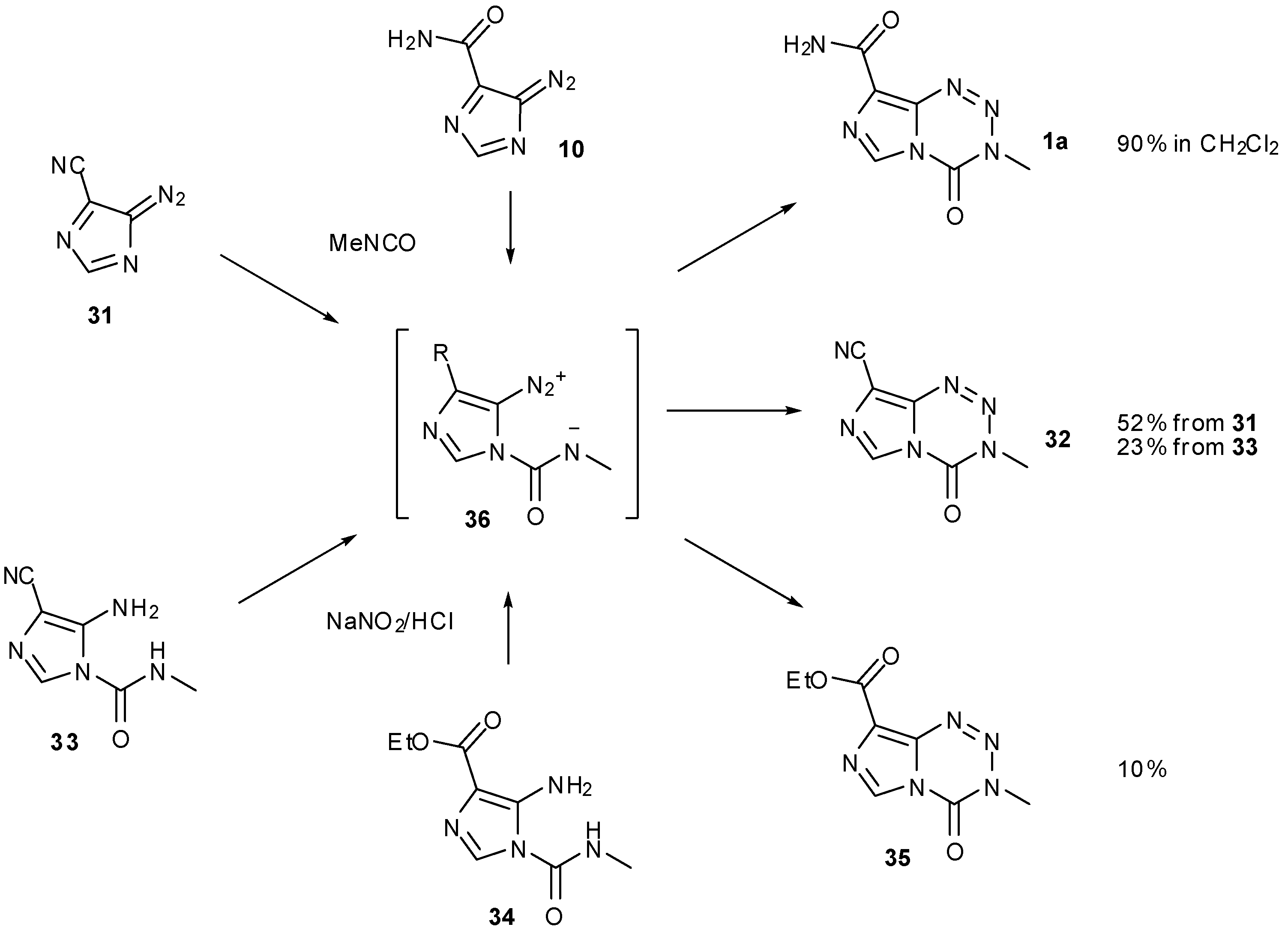
A further alternative to bypass diazo-IC in imidazotetrazine synthesis involved switching the amide to a nitrile group (Scheme 10). It was hypothesised that removal of the amide functionality would suppress competing formation of unwanted side-product AHX. In fact, the yield from cyclisation of this nitrile compound 31 was lower than that obtained using diazo-IC. Following dehydration of AIC with phosphorus oxychloride, nitrile 30 was subjected to the standard diazotisation/cycloaddition conditions to give TMZ analogue 32 in 44% yield. Reversing these steps offered no improvement as formation of urea 33 was achieved in only 5% yield, resulting in a 1% yield of imidazotetrazine 32 over the two steps (Scheme 11).
Scheme 10.
Synthesis of TMZ∙HCl from diazo-ICN (31) [47].
Scheme 10.
Synthesis of TMZ∙HCl from diazo-ICN (31) [47].

The acid stability of TMZ allowed hydrolysis of the nitrile group with HCl, restoring the amide group and giving the desired product TMZ∙HCl in 65% yield for the final step [47]. The yield for this hydrolysis has since been improved by Etlin et al. (89%) by lowering the reaction temperature, along with a method for subsequent neutralisation of the hydrochloride salt [52].
No improvement to the cyclisation was achieved when the ethyl ester urea 34 was subjected to nitration conditions; in this case only a 10% yield of tetrazine 35 was observed (Scheme 11).
Scheme 11.
Summary of routes to amide alternatives [47].
Scheme 11.
Summary of routes to amide alternatives [47].

Neither nitrile nor ester intermediates 36 are able to undergo competing intramolecular cyclisation to give AHX-type compounds. The most likely explanation for the poor yields is therefore that such intermediates are sensitive to inductive effects; these side chains may be too electron withdrawing compared with the amide of TMZ, thus impeding cyclisation and allowing other degradation pathways to manifest. Wang et al. point out subtle reactivity patterns amongst these derivatized intermediates, highlighting the complexity in constructing and handling such ring systems [47].
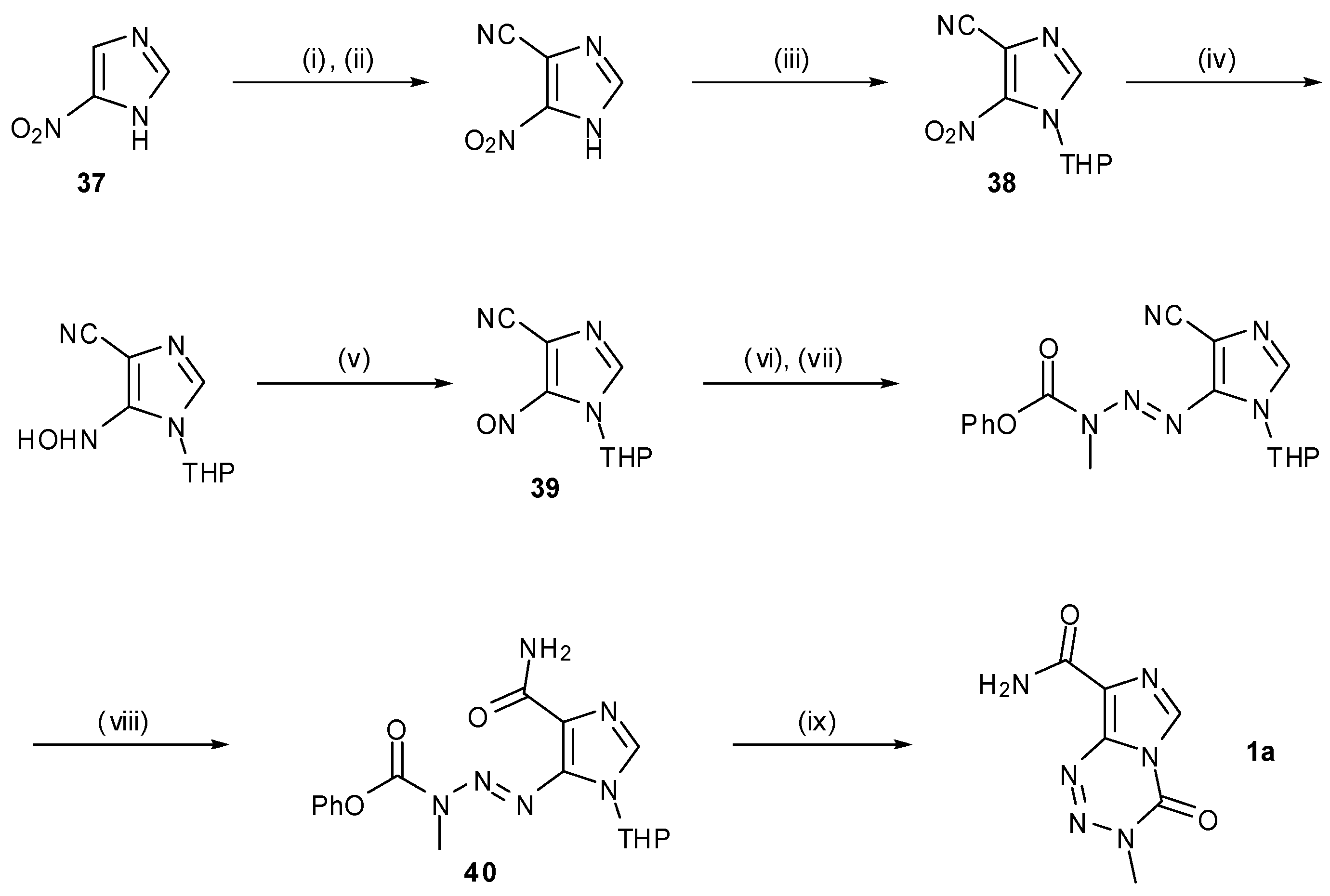
A somewhat different approach to TMZ synthesis using an MTIC analogue was taken by Wanner and Koomen (Scheme 12) [53]. This route exploits a UV-activated double bond isomerisation of triazene 40, which triggers spontaneous formation of TMZ. Beginning with 5-nitroimidazole (37), functionality is built up stepwise, culminating in condensation of 39 with phenylmethylcarbazate to provide the triazene moiety, followed by nitrile hydrolysis to furnish 40 prior to isomerisation. Whilst an elegant final step, the synthetic route to this key cyclisation precursor is comparatively lengthy vis-à-vis most other syntheses, with TMZ being made in an overall yield of 23% from imidazole 37.
Scheme 12.
Wanner and Koomen’s route to TMZ.

Reagents and conditions: (i) HNO3, Ac2O, 85%; (ii) KCN, NaHCO3, MeOH, H2O, rt, 62%; (iii) dihydropyran, p-TsOH, EtOAc, 96%; (iv) H2, 10% Pt/C, EtOAc, rt, 1 h; (v) NaIO4 in H2O, EtOAc, 0 °C, 30 min; (vi) H2N-N(Me)COOPh, CH2Cl2–AcOH (5:2), rt, 2 h; (vii) TFA–AcOH–MeOH (3:6:10), rt, 5 h, 76% from 38; (viii) c. HCl–AcOH (2:1), 60–65 °C, 25 min, 74%; (ix) acetone–MeOH (2:1), 366 nm, rt, 1 h, 79% [53].
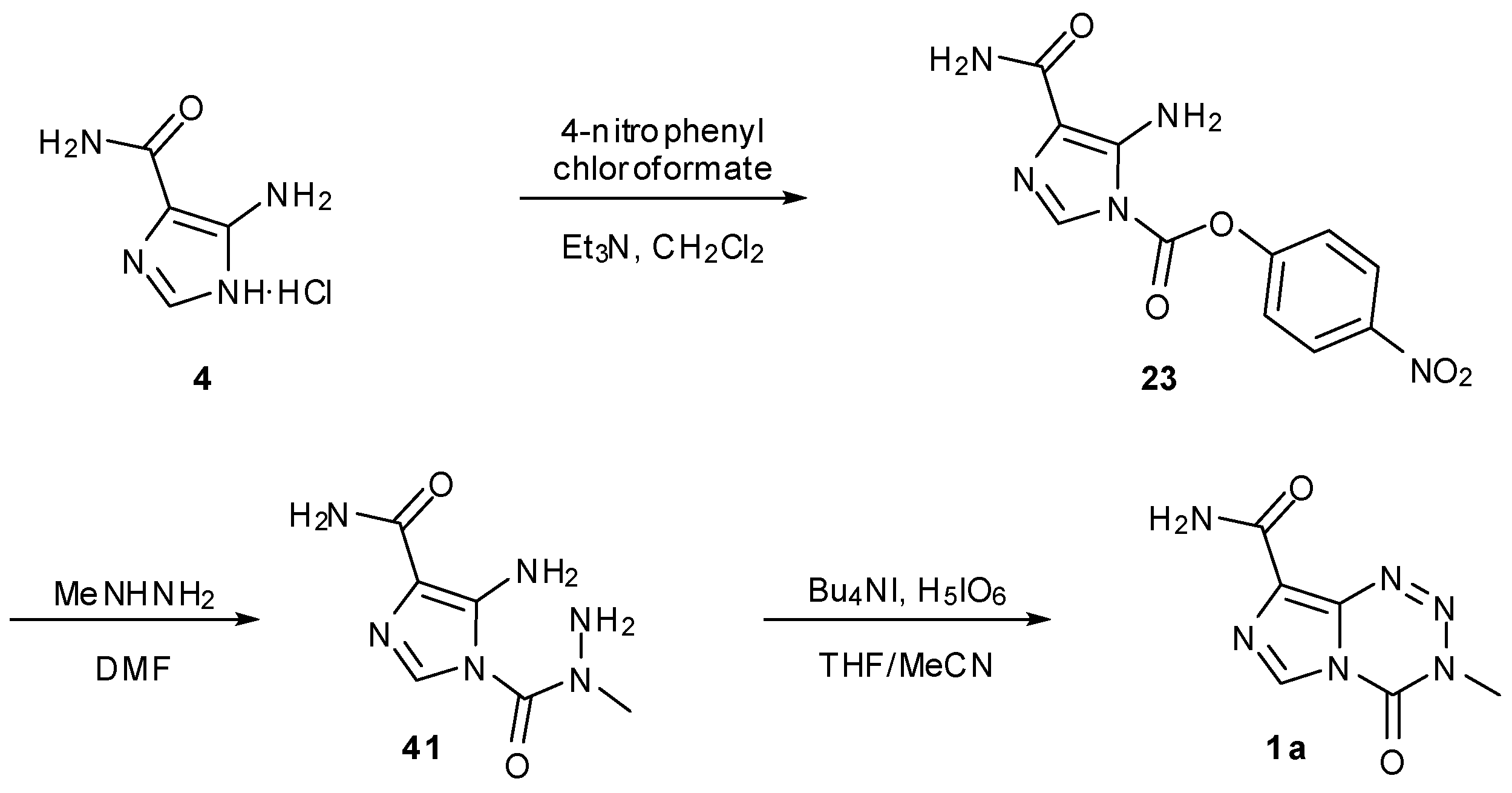
Schering-Plough Corporation has two patented procedures for the potential industrial scale synthesis of TMZ. The first of these avoids the handling of both diazo-IC and isocyanates but nevertheless has a familiar starting material. Following formation of carbamate 23 from AIC∙HCl; this activated intermediate was then treated with methyl hydrazine to give hydrazide 41. Finally, periodic acid effected an unusual oxidative cyclisation to furnish TMZ (Scheme 13) [54].
Scheme 13.
Schering-Plough industrial route to TMZ [54].
Scheme 13.
Schering-Plough industrial route to TMZ [54].

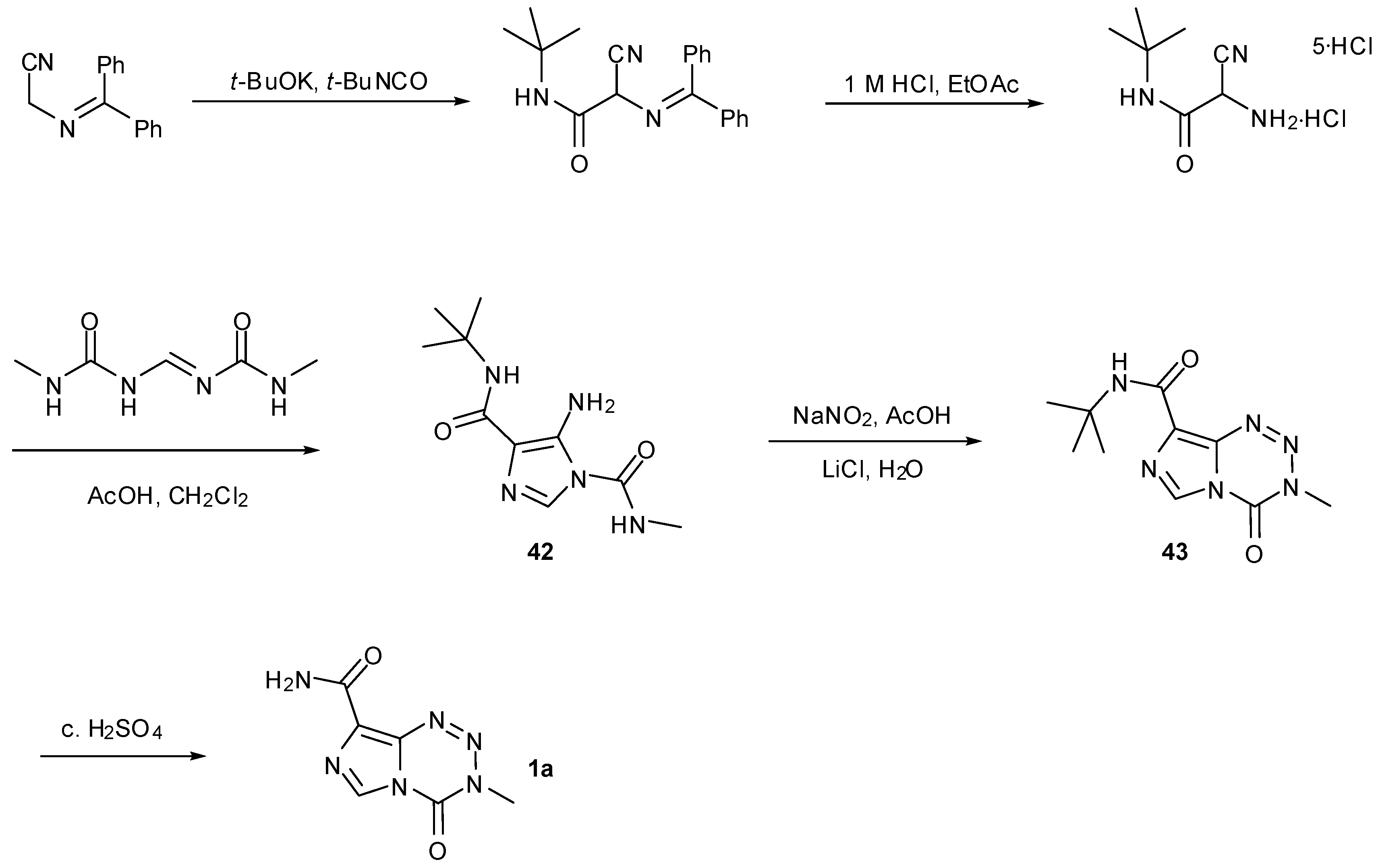
Their second and more cumbersome route requires construction of the imidazole ring during a lengthy route to urea 42. Improved conditions for cyclisation afford imidazotetrazine 43 and acidic cleavage of the tert-butyl protecting group finally furnished TMZ (Scheme 14) [55,56].

For small scale research purposes, coupling of diazo-IC with isocyanates in DMSO remains the method of choice for imidazotetrazine construction and offers unrivalled efficiency in terms of atom economy. Isocyanates 7 are most commonly formed via Curtius rearrangement of the corresponding azide 44. The azides themselves can be constructed by subjecting the hydrazide 45 to diazotisation conditions, or by treatment of an activated carbonyl 46 with sodium azide or a carboxylate with dppa (diphenylphosphoryl azide) (Scheme 15). Isocyanates are notoriously difficult to handle and purify; Curtius rearrangements, as well as urea thermolysis, give access to isocyanates with high purity, a factor which has proved to be important in the synthesis of more elaborate imidazotetrazine compounds.
Scheme 15.
Common routes to isocyanates.

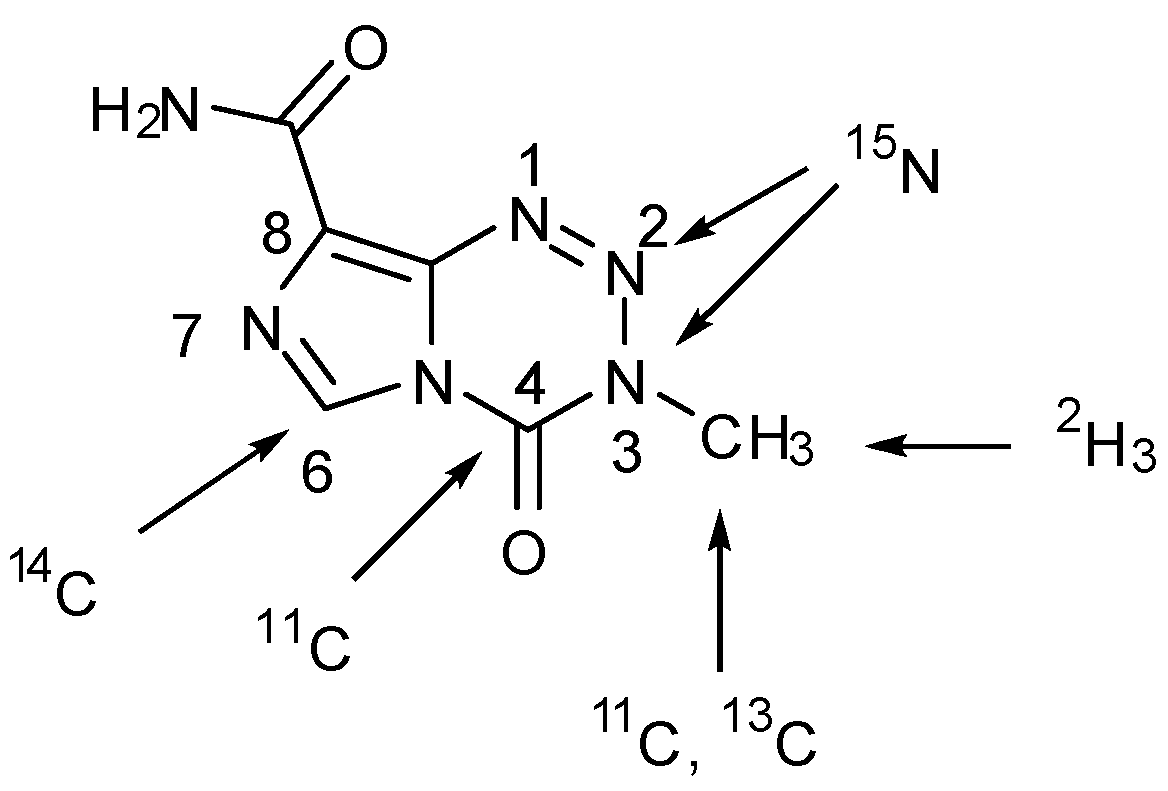
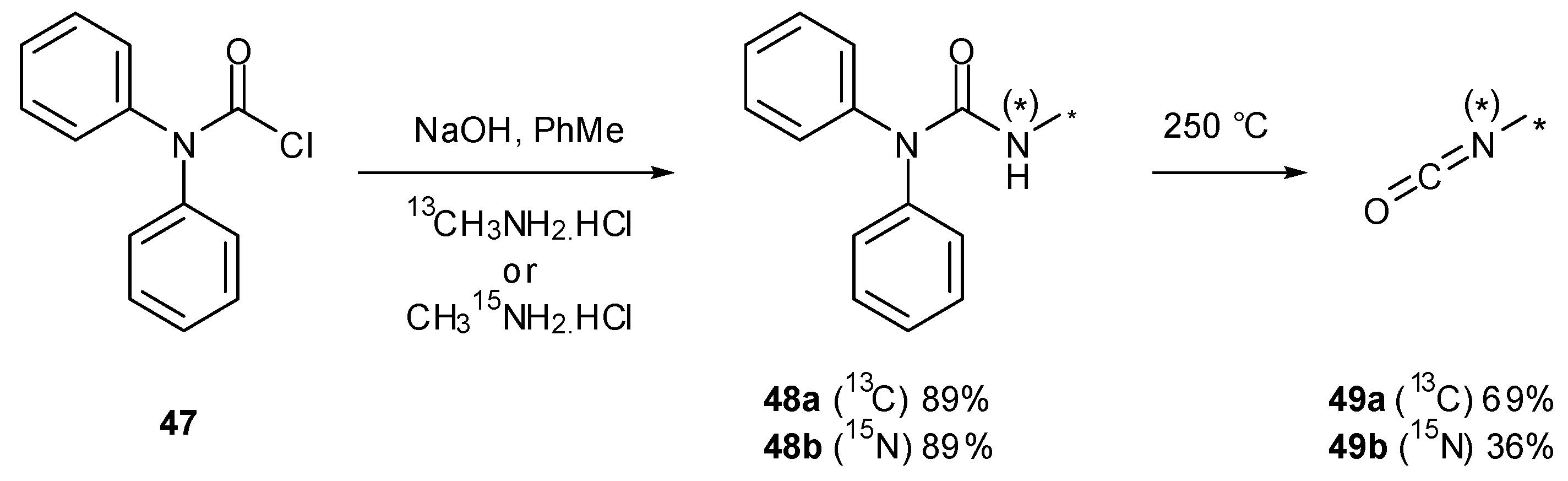
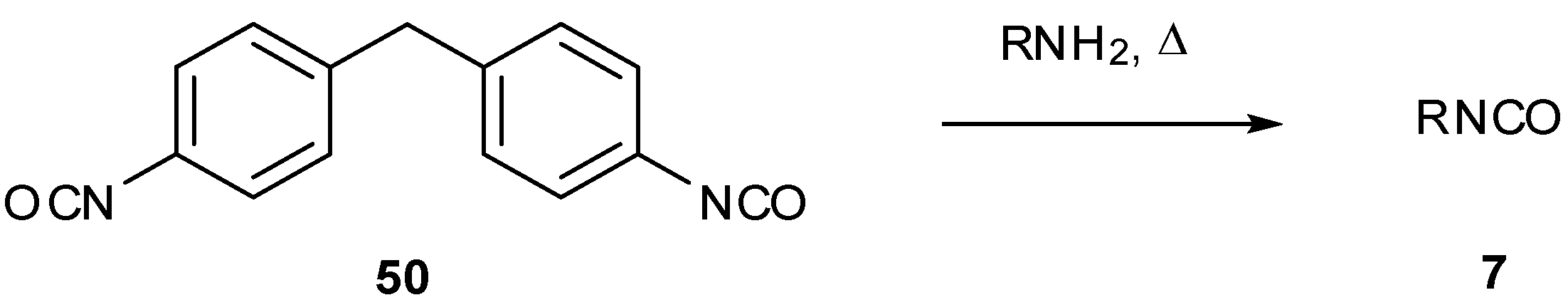
Various studies have required the synthesis of isotopically labelled TMZ. The pyrolysis of ureas is an attractive method for the preparation of volatile isocyanates since pure products are collected via distillation [57]; this was exploited in the synthesis of isotopically labelled isocyanates in order to assemble 13C/15N-labelled TMZ. Reaction of diphenylcarbamoyl chloride (47) with either 13C- or 15N-methylamine gave ureas 48a and 48b, which under thermolysis created 13C- and 15N-methyl isocyanate (49a and 49b) respectively (Scheme 16) [26]. This strategy has also been used for a number of other isocyanates during the formation of imidazotetrazines [25], including a process scale production of TMZ [58]. Access to isocyanates can also be achieved by the pyrolysis of 4,4'-methylenebis(phenylisocyanate) (50) in the presence of amines (Scheme 17) [59].
Scheme 16.
Urea pyrolysis to give labelled methylisocyanates [26].
Scheme 16.
Urea pyrolysis to give labelled methylisocyanates [26].

Scheme 17.
Thermolysis of 4,4'-methylenebis(phenylisocyanate) (50) [59].
Scheme 17.
Thermolysis of 4,4'-methylenebis(phenylisocyanate) (50) [59].

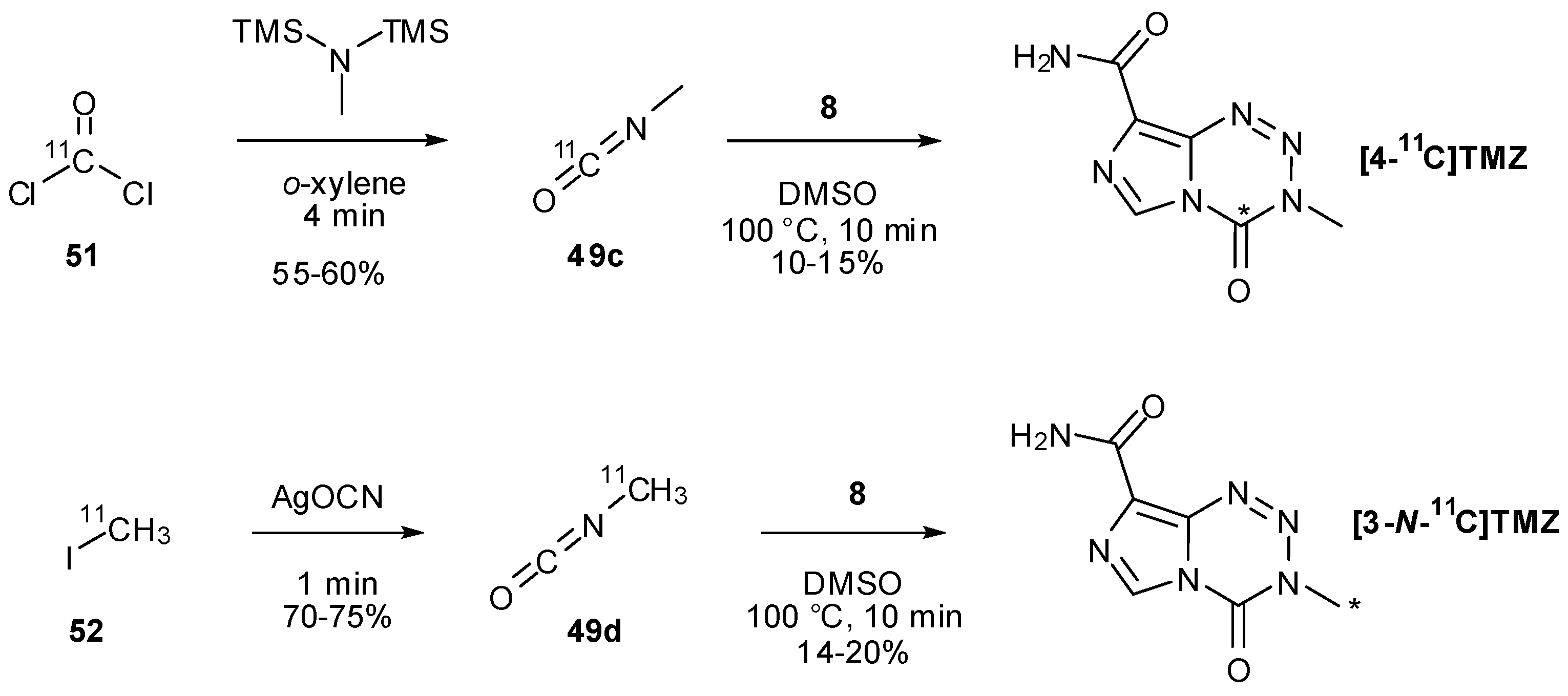
Two other isocyanate-forming methods are demonstrated by Brown et al., who initially attempted to synthesise 11C-labelled TMZ exploiting the known intermediate in the TMZ activation pathway, MTIC (2), and coupling this with isotopically labelled phosgene [8]. As with previous efforts to effect this reaction [47], this was without success. They were able instead to utilise the radiolabelled phosgene (51) to rapidly obtain [11C-carbonyl]methyl isocyanate (49c) which was then used in a standard coupling reaction, gaining access to [4-11C-carbonyl]TMZ (Scheme 18).
Scheme 18.
Alternative routes to isocyanates used in labelling studies [8].
Scheme 18.
Alternative routes to isocyanates used in labelling studies [8].

An alternative synthetic route to the isocyanate employing [11C]iodomethane (52) and silver cyanate also allowed access to [3-N-11C-methyl]TMZ. Due to the short half-life (t½ = 20.33 min) of 11C, expeditious reactions are necessary in such radiosyntheses; Brown et al. found they were able to use elevated temperatures (100 °C for 10 min rather than room temperature for several hours) to significantly reduce reaction time in the coupling of isocyanates 49c/d with diazo-IC (Scheme 18) [8].
2-15N-TMZ and [2H3-methyl]TMZ have also been synthesised by the standard method using [15N]diazo-IC (from Na15NO2) [26], and [2H3]methyl isocyanate respectively [25], and [6-14C-imidazole]TMZ has been used in several pharmacokinetic studies [7,60,61,62]. A summary of positions which have been labelled is shown in Figure 11.

7. Synthesis of Structural Analogues
Various TMZ analogues have been synthesized in efforts to find new, improved anti-cancer agents, using a range of methods to alter different parts of its structure.
7.1. Alternative Cores
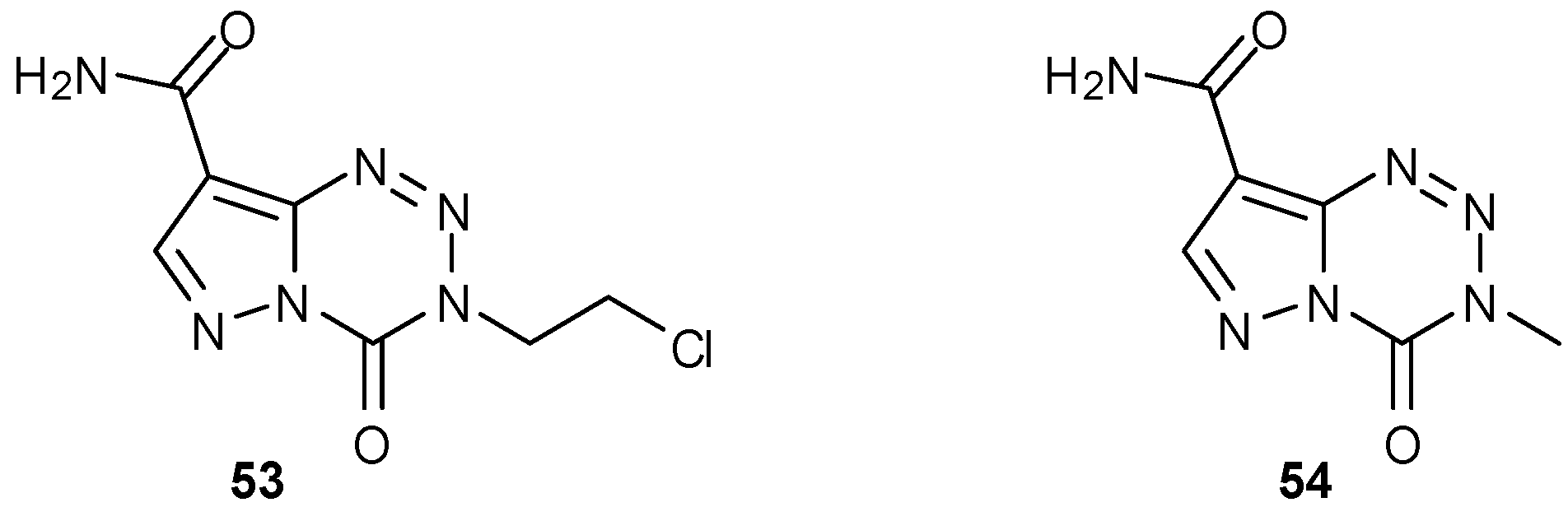
It is clear from the mechanism of action that the tetrazinone ring is crucial for the activity of imidazotetrazine anticancer agents, which function via DNA alkylation; the importance of the imidazole on the other hand was initially less well understood. Pyrazolo analogues of MTZ and TMZ were initially made by Cheng et al., in order to generate agents with lower light sensitivity [64]. Whilst MTZ analogue 53 showed excellent antineoplastic activity against leukemia cell lines P388 and L1210, the corresponding TMZ analogue 54 was totally inactive [64]. It has, however, shown potency in the lymphoblastoid GM892A (MGMT−) cell line [24].
Figure 12.
Pyrazolo analogues of MTZ and TMZ [64].
Figure 12.
Pyrazolo analogues of MTZ and TMZ [64].

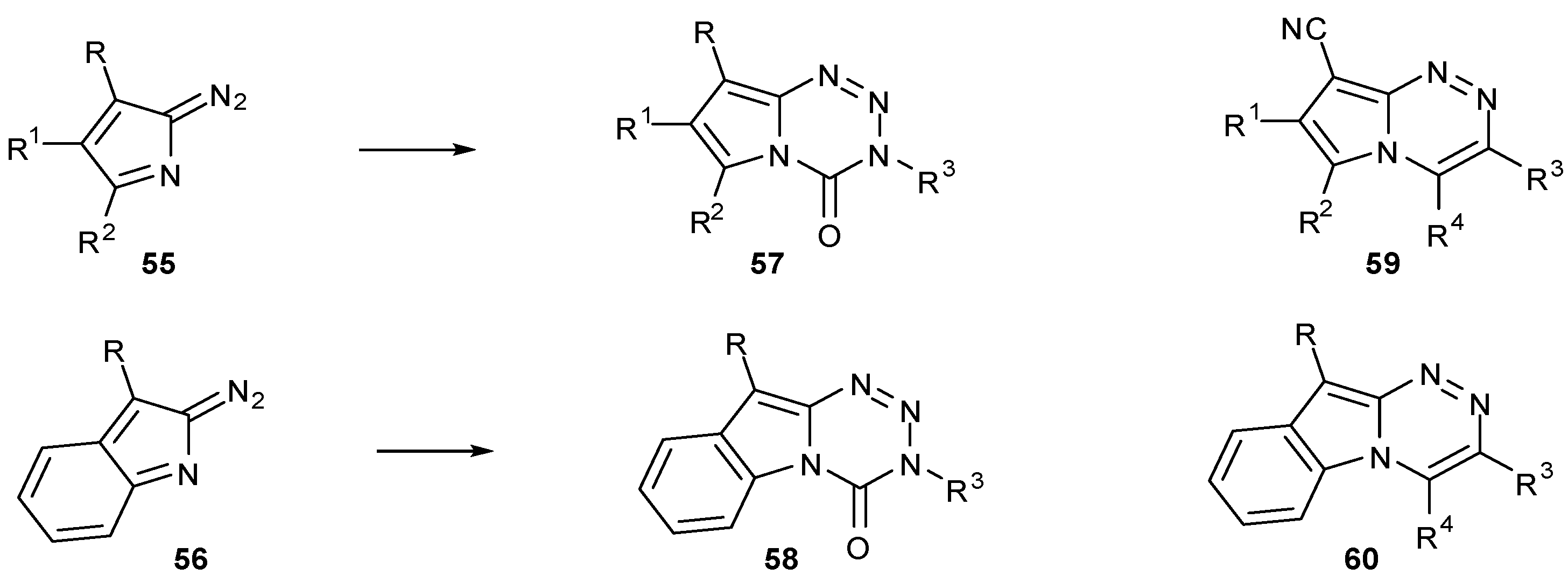
A more thorough investigation into fused heterocycle effects has been made by the Cirrincione group, who have synthesised a range of alternative bicyclic ring structures related to the imidazotetrazines. Coupling isocyanates with diazopyrroles 55 and 2-diazoindoles 56 afforded pyrrolo[2,1-d][1,2,3,5]tetrazinones 57 and indolo[2,1-d][1,2,3,5]-tetrazinones 58 respectively (Scheme 19) [65,66]. Whereas the latter could be easily made using the standard procedure, the pyrrole series required more forcing conditions owing to reduced electrophilicity of the diazo group [65].
Some compounds showed modest activity in a range of cell lines in the NCI60 panel; of those in which growth inhibition was observed, GI50 values were generally in the 10–100 μM range. Indolo compounds 58 as a class were inactive (GI50 > 100 μM) [67], whereas pyrrolo compounds 57 showed a range of activity (GI50 > 100 μM to < 10 nM); the most potent compound in the series had a GI50 of <30 nM in a number of cell lines. QSAR trends and COMPARE analysis against the standard agents database indicated that the mode of action of pyrrolotetrazinones 57 differs from that of the imidazotetrazines [68,69]. Interestingly, some analogous triazines 59 and 60 also showed antiproliferative activity although as expected, COMPARE data highlighted that these too function by an alternative mechanism of action from members of the standard agents database [70,71].

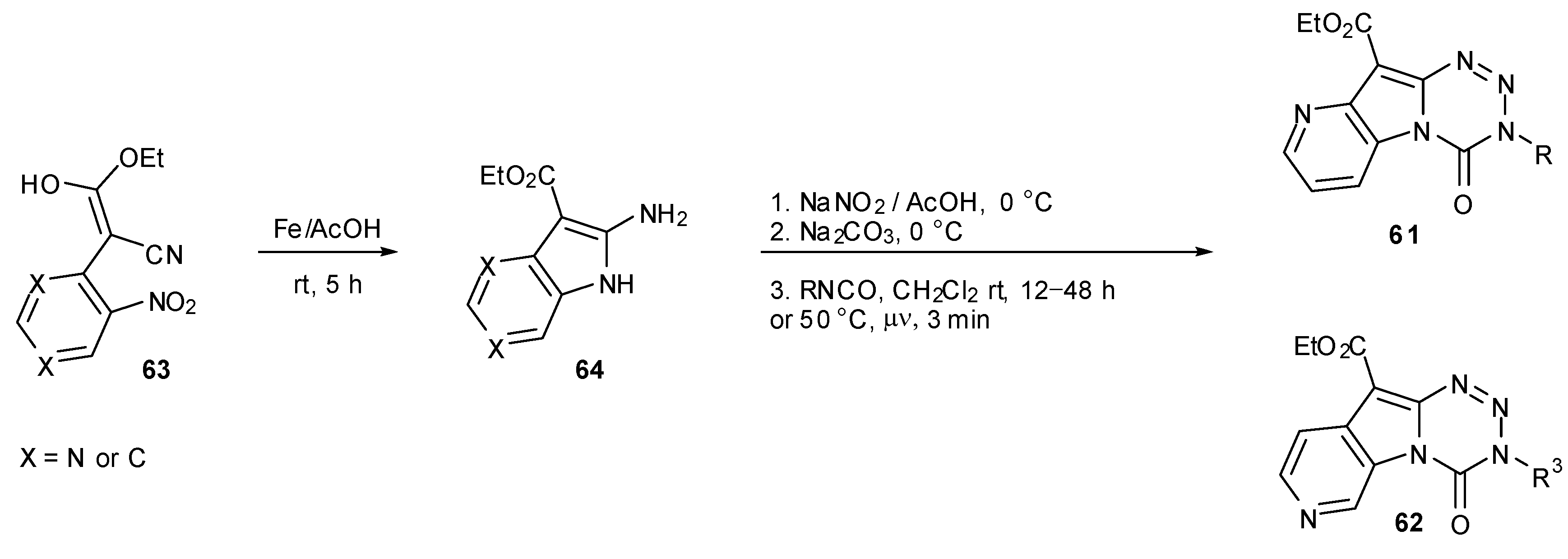
Diana and co-workers prepared two series of analogous pyridopyrrolotetrazinones 61 and 62. Following construction of the pyrrolopyridine fragments 63 and 64, standard diazotisation and coupling protocols gave the desired products. It is worthy of note that coupling with isocyanates was found to elicit higher yields when performed under microwave irradiation with greatly reduced reaction times (Scheme 20). Three of the compounds 62 showed growth inhibitory activity against a limited number of cell lines [72], but none of the compounds 61 tested in the NCI60 panel showed any significant activity [73].

Two dihydropyrazolotetrazepines 65 were synthesised by Maggio and co-workers (Figure 13); however, these compounds were inherently unstable and thus their biological profiles could not be evaluated [74]. A number of pyrazolotetrazinones 66 has also been synthesized for use as herbicidal agents [75,76].

7.2. 6- and 8-Analogues
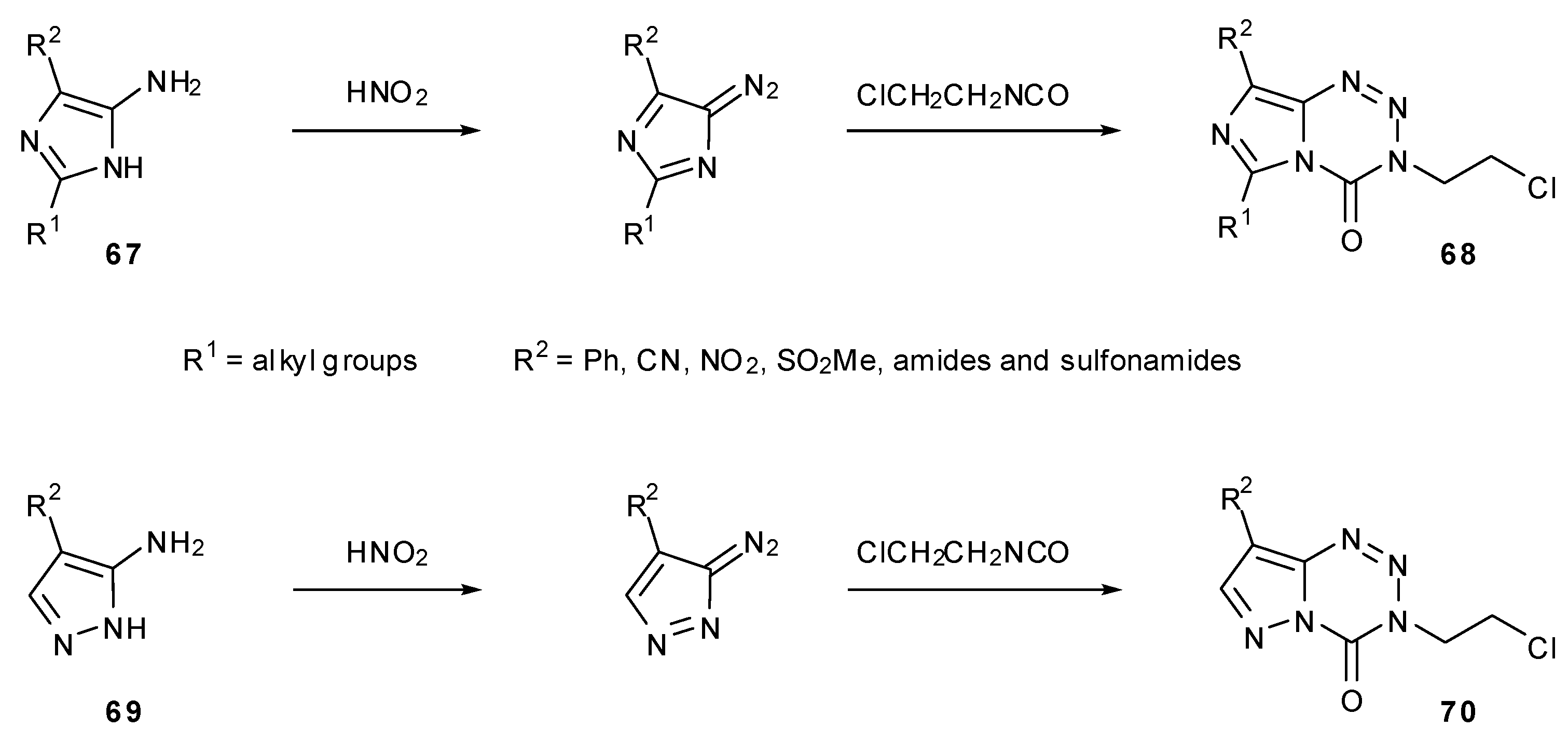
Lunt and co-workers used a variety of alternatively substituted imidazoles 67 and pyrazoles 69 to create MTZ analogues 68 and 70 (Scheme 21) [19]. An assessment of the structure–activity relationship was first made by alteration of groups in the 6-position (R1). Several compounds were made with straight chain and branched alkyl groups of increasing size. Smaller substituents showed similar activity to MTZ but more bulky groups appeared to be less cytotoxic which is likely to be due to a steric barrier to hydrolysis.
Scheme 21.
Synthesis of 6- and 8-analogues via substituted imidazoles [19].
Scheme 21.
Synthesis of 6- and 8-analogues via substituted imidazoles [19].

A large range of analogues with different groups in the 8-position (R2), these mostly being amides and sulfonamides, exhibited a more complex structure–activity relationship. Increasing bulk of the amide substituent appeared to reduce cytotoxicity; the same trend was seen with sulfamoyl compounds but these as a class, along with 8-methylsulfonyl, showed enhanced potency compared with the amides. The 8-phenyl, 8-cyano and 8-nitro compounds tested showed no useful in vivo activity. Pyrazolotetrazines showed similar trends, but as well as being more complicated to synthesise, showed lower potency than the corresponding imidazotetrazines [19].
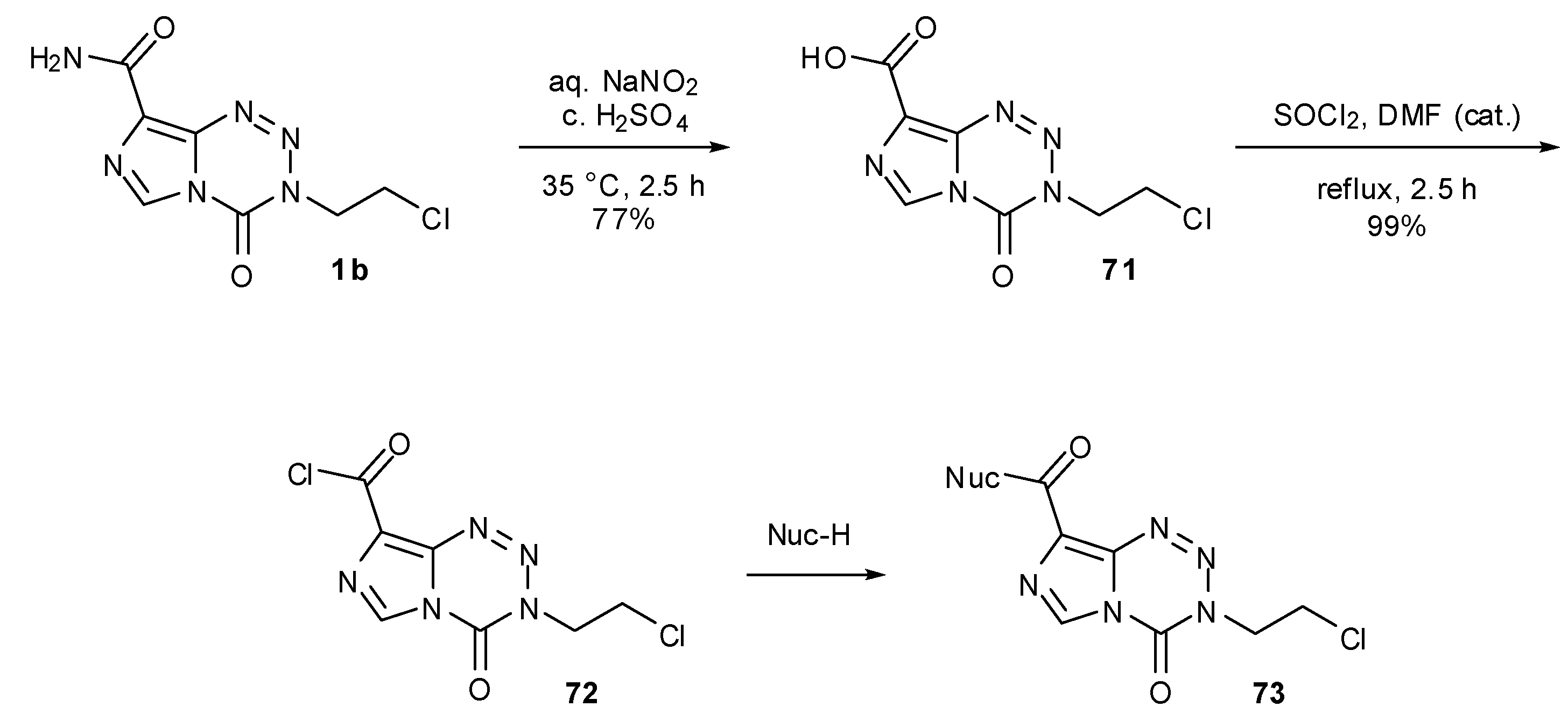
Derivatisation of the 8-position of MTZ and TMZ can be achieved via treatment with nitrosylsulfuric acid (formed in situ) to hydrolse the amide moiety to carboxylic acid 71 (Scheme 22). Conversion to acid chloride 72 using refluxing thionyl chloride provides the necessary activation for selective reaction with nucleophiles, without destructive nucleophilic attack at the crucial endocyclic carbonyl. In this manner, large numbers of ester and amide analogues of MTZ 73 have been synthesised [77]. These compounds showed a broad range of activity in the TLX5 lymphoma cell line but no clear structure–activity relationship could be determined. This acid chloride approach was also utilised by Liu in the construction of a range of TMZ-based amides and esters. Esters were shown to have at least 60-fold higher water solubility than TMZ as well as more potent antitumour activity in HL-60 leukemia cell line [78].
Scheme 22.
General synthesis of esters and amides via MTZ acid chloride [78].
Scheme 22.
General synthesis of esters and amides via MTZ acid chloride [78].

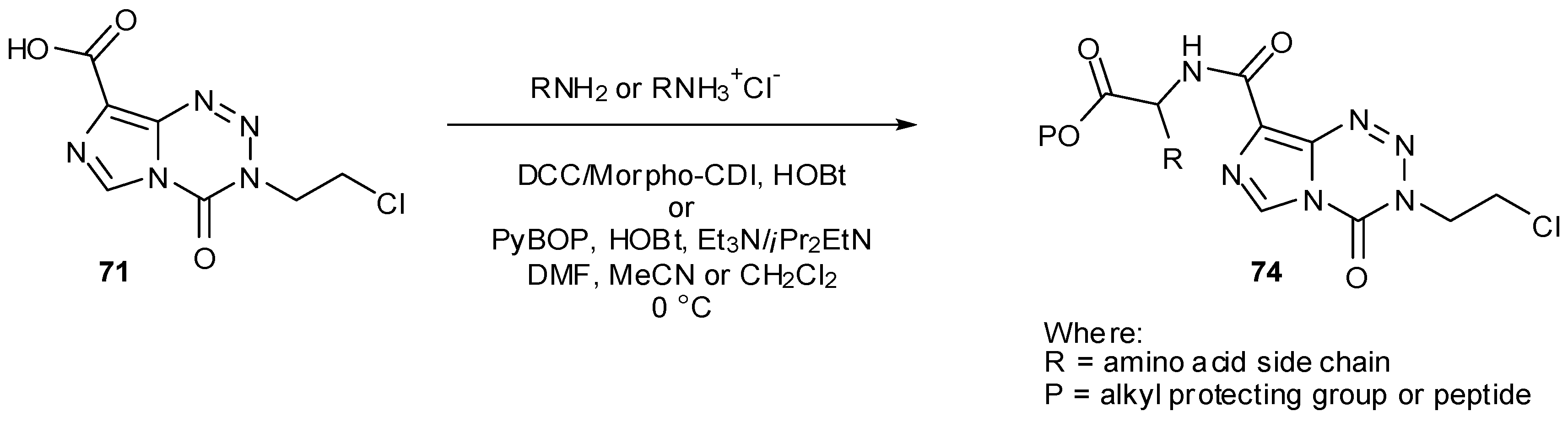
Esters and amides can also be achieved directly from carboxylic acid 71 by the use of alkyl halides or amines with coupling reagents such as DCC or phosphonium salts [79]. A number of conjugates 74 with amino acids on MTZ were prepared using this method, employing solid phase synthesis to generate longer chain DNA major-groove targeting peptidic analogues (Scheme 23) [45]. Care must of course be taken to avoid basic conditions in the presence of imidazotetrazines and with this in mind only acid labile groups were utilised for protection of amino acid side chain residues.
Scheme 23.
Synthesis of amides from MTZ carboxylic acid [45].
Scheme 23.
Synthesis of amides from MTZ carboxylic acid [45].

No significant change in aqueous stability was observed in this set of amide compounds and in vitro activity was not sufficiently improved to warrant further investigation. Imidazotetrazines conjugated to lexitropsins, a group of known distamycin-derived minor groove DNA binders were also synthesised by amide coupling. These conjugates were surprisingly water soluble but showed no enhanced activity. Regardless of the nature of the conjugate (major or minor groove targeting), alkylation was always seen at guanine-rich sequences in the major groove of DNA [45].
This coupling method was used in the synthesis of an assortment of temozolomide alkyl esters, thioesters and amides as prodrugs for topical application. Hexyl ester 75 was tested both in vitro and in vivo and showed improved dermal permeability compared with that of TMZ as well as demonstrating effective anti-melanoma properties (Figure 14) [80].
Wang also synthesized a number of amide/ester linked imidazotetrazines using PyBOP coupling in order to explore the dual action of MTZ/TMZ with cephalosporin antibiotic agents, with an aim to increase the spectrum of activity of the parent antibiotics (Figure 14) [81]. However, dual action β-lactam agents 76 were not as effective as ampicillin against most bacterial strains and little synergistic activity was seen.

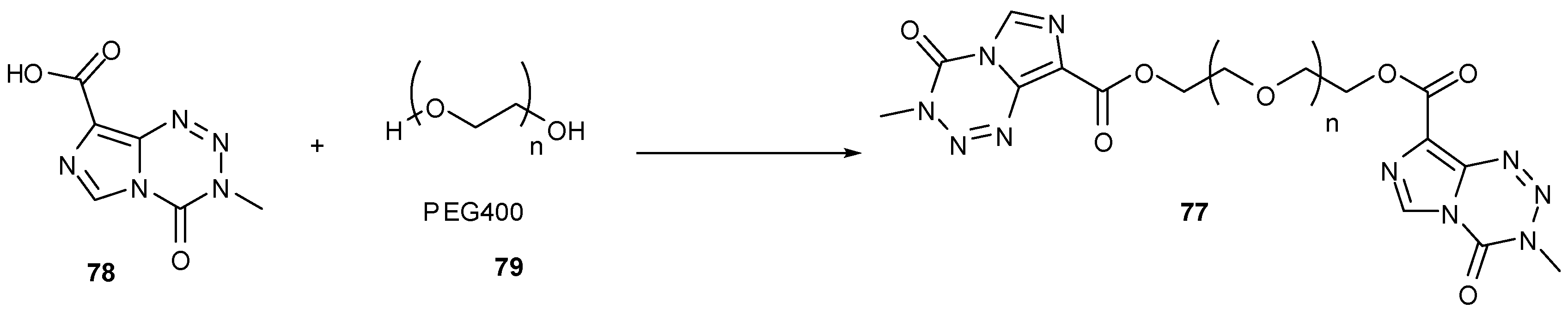
An ethylene glycol linked dimer of TMZ 77 (Scheme 24), synthesised from acid 78 and PEG400 (79), when administered intravenously showed equal efficacy against glioma cells in vivo compared with oral dosing of TMZ. In addition, a mouse xenograft model showed tumour growth inhibition comparable to that exhibited by TMZ [82].
Scheme 24.
Glycol linker dimer [82].
Scheme 24.
Glycol linker dimer [82].

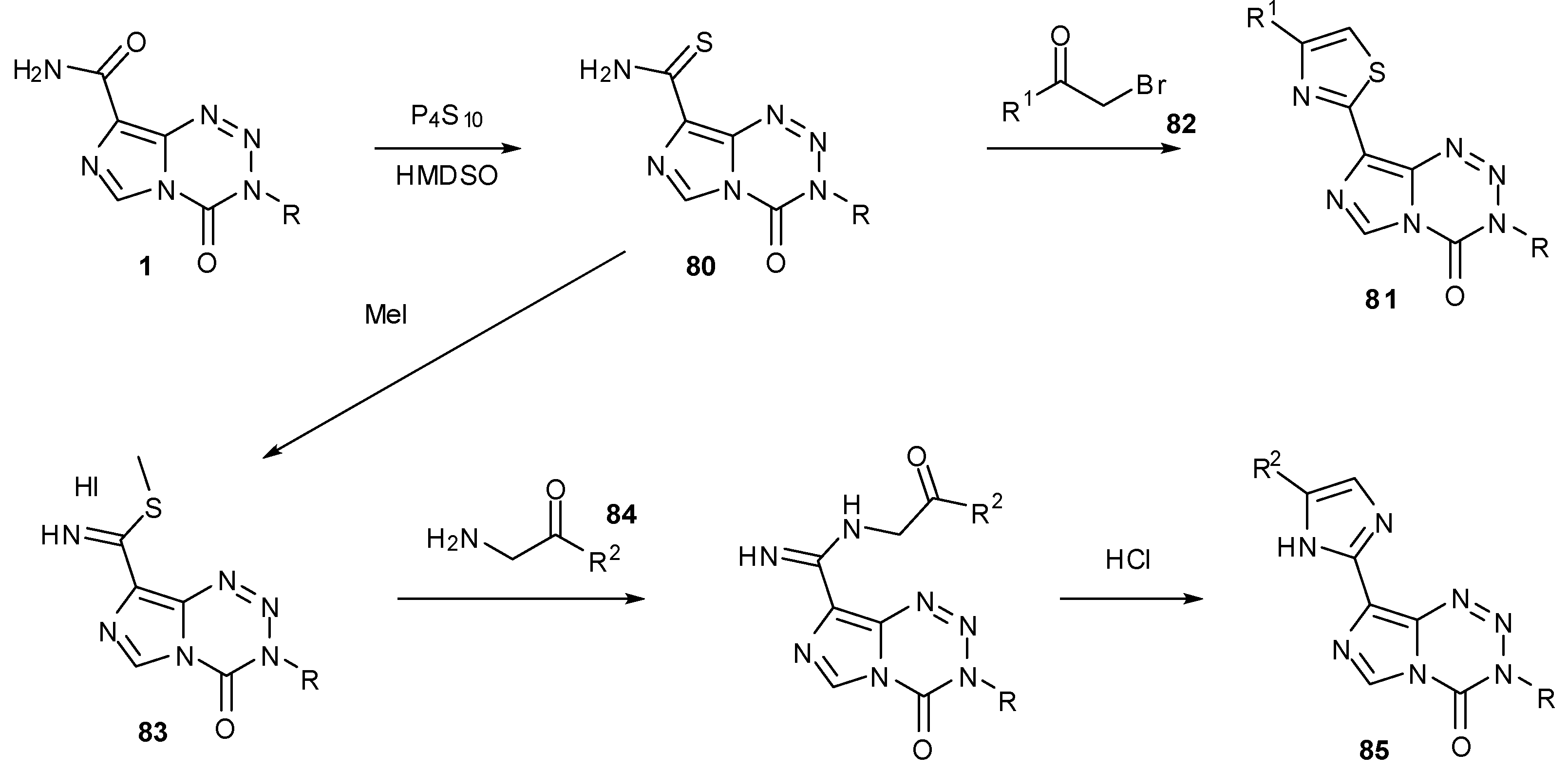
An alternative way to derivatise the 8-position is to convert the carboxamide to a thioamide. Hummersone and co-workers used phosphorus pentasulfide and hexamethyldisiloxane to elaborate on the 8-position. These thioamides 80 can then be further derivatised to thiazoles 81 by condensation with an appropriate α-bromo ketone 82. Alternatively, conversion of thioamides to carbimidothioates 83 then reaction with aminoketones 84 gives access to imidazoles 85 (Scheme 25) [83].
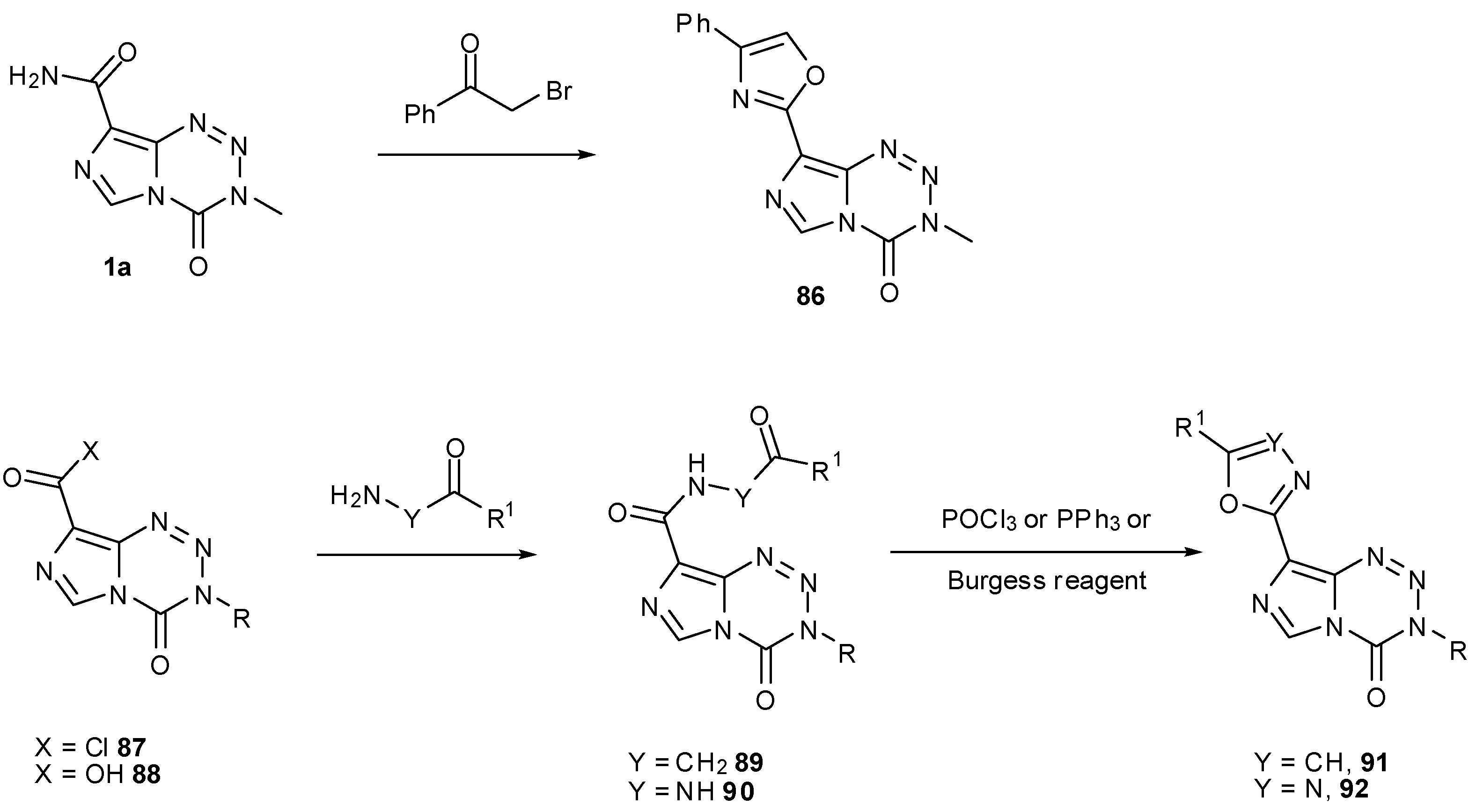
Oxazoles 86 can similarly be synthesised by treatment of carboxamides, e.g., 1a with α-bromo ketones. Alternatively, reaction of acid chloride 87 with an appropriate amino ketone to form keto amides of the form 89 followed by ring closure and dehydration gives regioisomeric oxazoles 91. In a similar manner, reaction of carboxylic acids 88 with hydrazides allows construction of oxadiazoles 92 via 90 (Scheme 26). A large number of these 8-substituted compounds was found to have effective biological activity, even in cell lines that have active MGMT; many compounds showed GI50 values <45 μM (compared to 45.6 μM for TMZ) in SNB19V (MGMT−) and <70 μM (compared to 526.3 μM for TMZ) in SNB19M (MGMT+) [83].
Scheme 25.
Formation of thiazoles and imidazoles [83].
Scheme 25.
Formation of thiazoles and imidazoles [83].

Scheme 26.
Construction of oxazoles and oxadiazoles [83].
Scheme 26.
Construction of oxazoles and oxadiazoles [83].

7.3. 3-Analogues
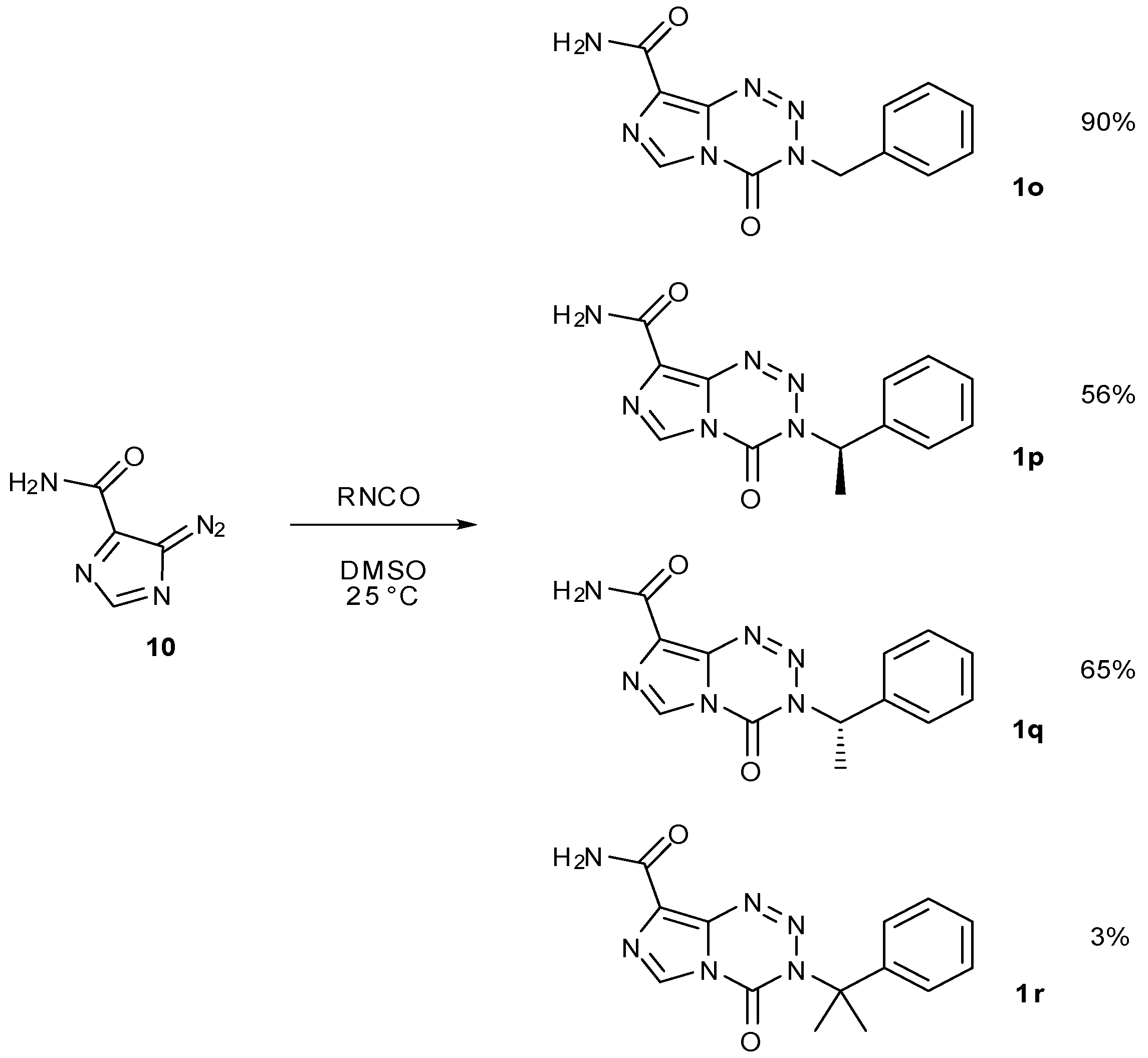
The 3-substituent in imidazotetrazine compounds is crucial as this is the group that controls reactivity of the imidazotetrazine and will also be delivered and subsequently bonded to DNA. A large number of 3-N-substituted imidazotetrazines has been synthesised, bearing a variety of functionalised alkyl groups as well as alkyl-linked aromatics and heterocycles [25,84,85]. These have been constructed mostly using isocyanate methods. The effects of steric hindrance on this coupling reaction with diazo-IC (8) are evident; benzyl isocyanate gives 90% yield of 1o [43], whereas (R)- and (S)-1-phenylethyl isocyanate after 16 h afford imidazotetrazines 1p and 1q in 56% and 65% yield respectively. Somewhat surprisingly, reaction with, α-dimethylbenzyl isocyanate does furnish the gem-dimethyl analogue 1r, albeit it in a yield of only 3% after 16 h reaction time (Scheme 27) [84,85].
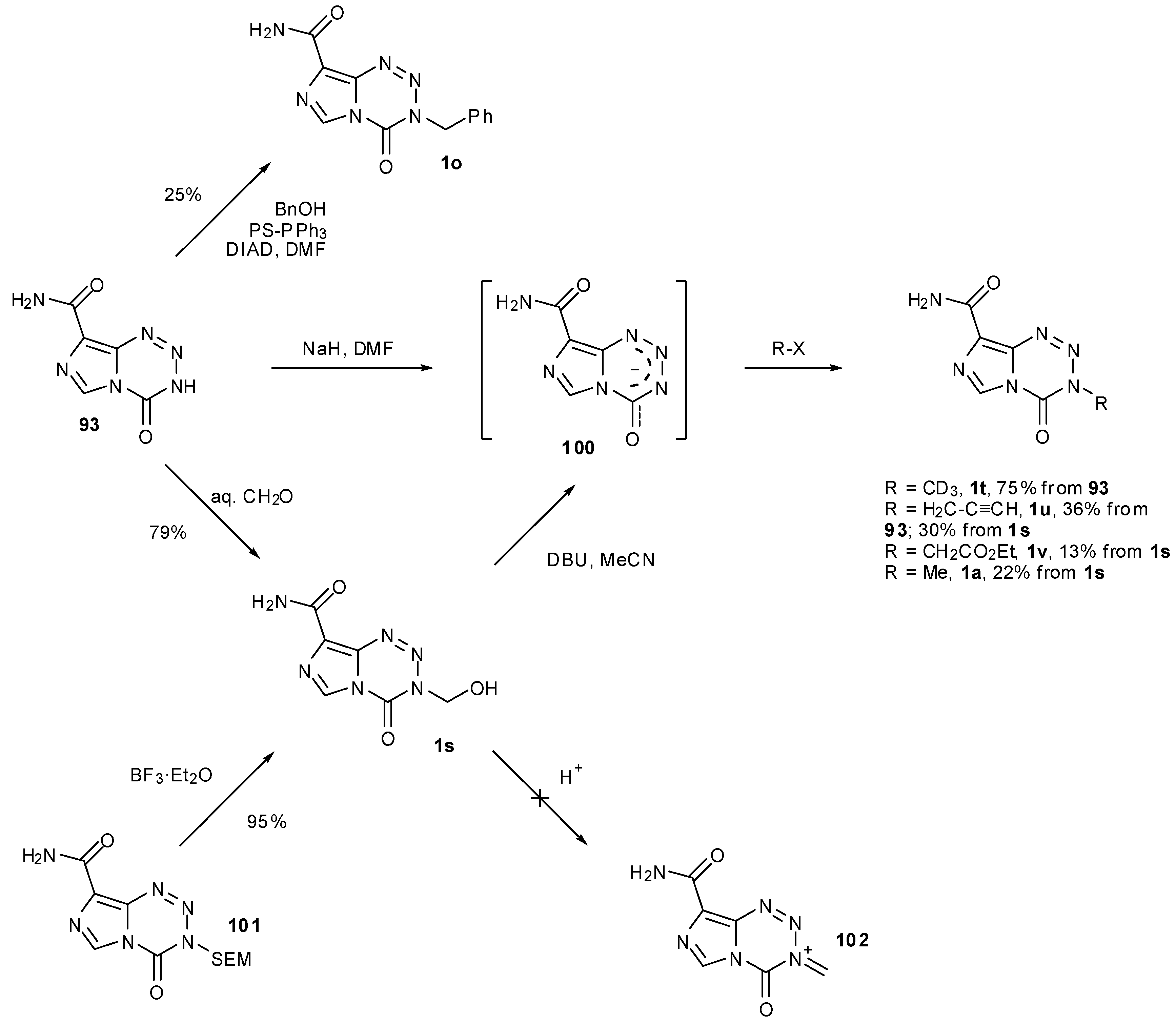
A more recent development in imidazotetrazine synthesis employs the intermediate, nortemozolomide (93). The route to 93 uses diazo-IC (10) and glycine-derived isocyanate 94, itself formed via azide displacement of the anhydride resulting from reaction with Boc-glycine (95) and ethyl chloroformate (96), and subsequent Curtius rearrangement. Following imidazotetrazine formation, Boc-deprotection of 96 leads to spontaneous elimination of formaldehyde imine, thus resulting in formation of nortemozolomide (Scheme 28) [86,87]. This imidazotetrazine compound opens up scope for not only the synthesis of TMZ but also creates opportunities for alternative routes to 3-N-substituted analogues.


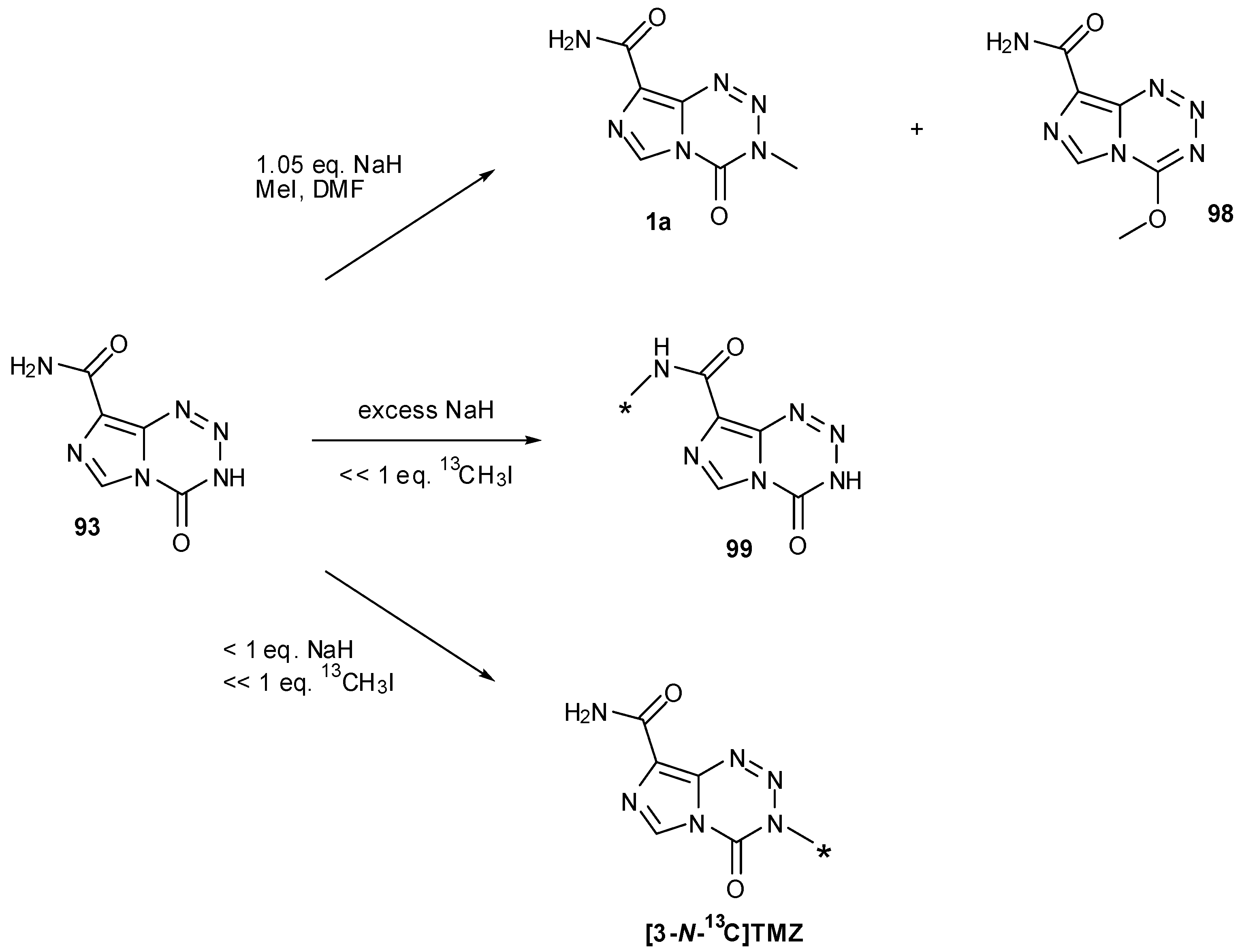
Methylation of 93 to give TMZ was achieved by Cousin et al., in 61% yield by deprotonation with sodium hydride and subsequent quenching of the anion with an excess of methyl iodide in DMF. An O-methylated side-product was also detected, suspected to be 98; alteration of base had little effect on the outcome of the reaction but less side-product was obtained when the reaction was performed at lower temperatures (−20 °C) [87]. During the course of their [13C]-and [11C]-labelled TMZ syntheses, Moseley and co-workers determined that sodium hydride was the optimum base for methylation of nortemozolomide in order to reduce 8-N-methylation. However, they observed that the reaction is sensitive to the amount of base and found that with [13C]-methyl iodide, excess NaH resulted in exclusive 8-N-methylation to give 99 and that substoichiometric quantities favoured 3-N-methylation (Scheme 29) [63].

The derivatization of 93 using a number of alkylating agents was also investigated. The scope was shown to be limited, as alkyl halides with bulky or electron withdrawing substituents failed to yield the desired products. The benzyl derivative 1o was obtained under Mitsunobu conditions whereas hydroxymethyl compound 1s was formed from simple treatment of 93 with formaldehyde (Scheme 30). This alcohol itself was next probed as a gateway to diversification. Exposure to acid gave only ring decomposition products rather than the anticipated iminium species 102 which it was envisioned could have provided access to ethers and thioethers. On the other hand, treatment of 1s with DBU and trapping with electrophiles provided alternative, if less efficient conditions for access to simple alkylated systems, presumably via anion 100. Alcohol 1s is also accessible in high yield from SEM protected nortemozolomide 101 via BF3 etherate deprotection, albeit with some BF3 contamination [84,87].

8. Design of MGMT/MMR-Independent Anti-Cancer Agents
The principal constraints on TMZ efficacy are the dependence on MMR and resistance caused by MGMT, the result of which is that the number of tumours able to respond to TMZ therapy is limited; current efforts in the design of new analogues seek to bypass these limitations.
Although much attention has been paid to the modification of the 8-substituent and exploring the effects of targeting ligands attached at this position, it is actually the 3-subsituent that generates the pharmacological activity of the imidazotetrazines. Recent efforts to modify tumour response to TMZ-type drugs have focused on this position. Key to rationally altering the 3-substituent and the biological activity is a full appreciation of the role of the 3-substituent in the imidazotetrazine mechanism of activation and reactivity.
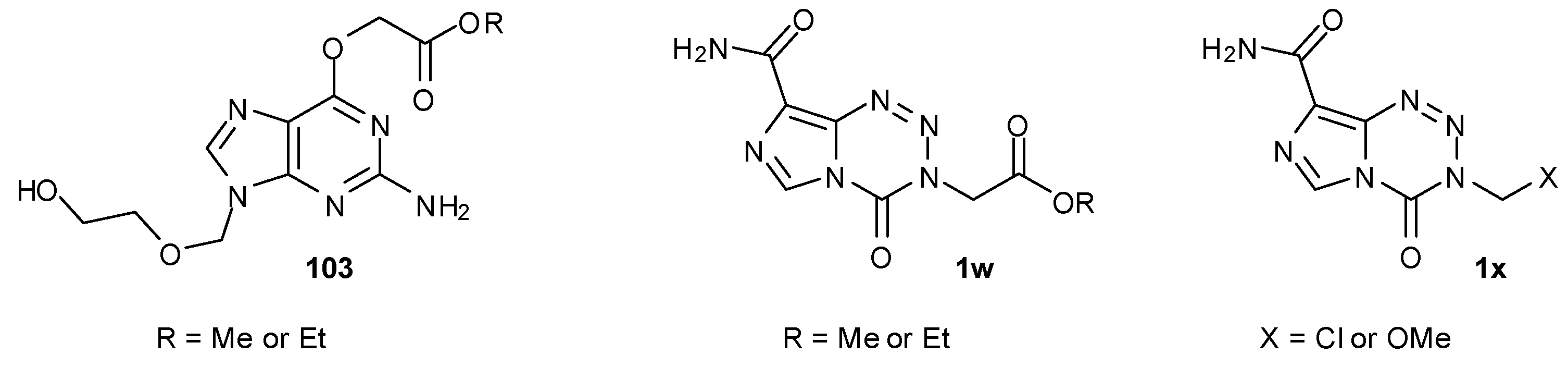
One approach towards achieving this is to modify the 3-substituent of the imidazotetrazine such that the group transferred to DNA GO6 sites is a poor substrate for MGMT and thus is not susceptible to recognition or DNA repair by the protein. A family of alkylated guanine molecules 103 with polar or ionisable O6 substituents has been synthesised and demonstrated resistance to MGMT cleavage (Figure 15) [88]. A number of imidazotetrazine 3-N-derivatives bearing such types of substituents (e.g., 1w and 1x) has demonstrated MGMT- and MMR-independent activity; several cell lines that are resistant to TMZ, whether due to high levels of MGMT or MMR mutation/deficiency, have shown sensitivity to these new compounds [85,89].

Another approach is to subtly shift the mode of action, which has been achieved by embracing a neighbouring group participation (NGP) mechanism to alter the behaviour of the active species resulting from prodrug release.
8.1. Introduction to NGP Analogues
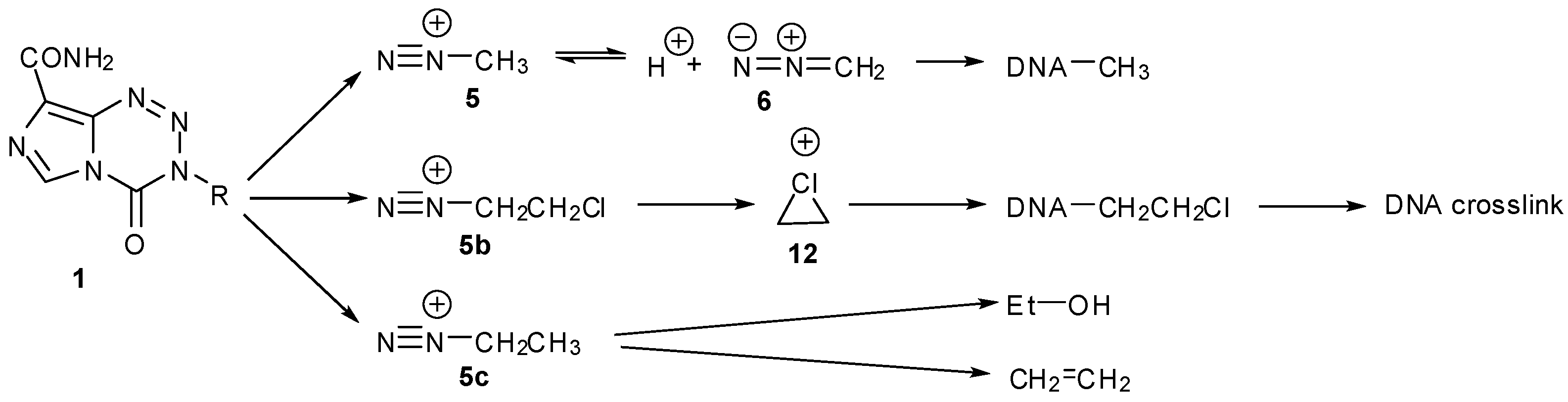
Earlier, the aqueous chemistry of the early imidazotetrazines was discussed (vide supra, Section 3), summarised in Scheme 31. The important lesson here is that for a drug to elicit a biological response, fine tuning of the latent electrophile is required. Firstly so that unproductive side-reactions, such as inactivation by elimination, hydrolysis or rearrangement, are disfavoured. More subtly, it is also important that the ultimate electrophile has sufficient aqueous lifetime to locate an appropriate reaction site on DNA. Stabilisation of diazonium ions 5, 5b, by either equilibration with diazomethane (6) or involvement of the neighbouring chlorine atom to form a chloronium intermediate 12, appears adequate to ensure useful pharmacological activity. In contrast, ethyl diazonium (5c) is not stabilised by equilibration with the diazo form but is susceptible to elimination which provides a “dead end” to the sequence of reactive intermediates.
Scheme 31.
Summary of alkyldiazonium ions derived from early imidazotetrazines.

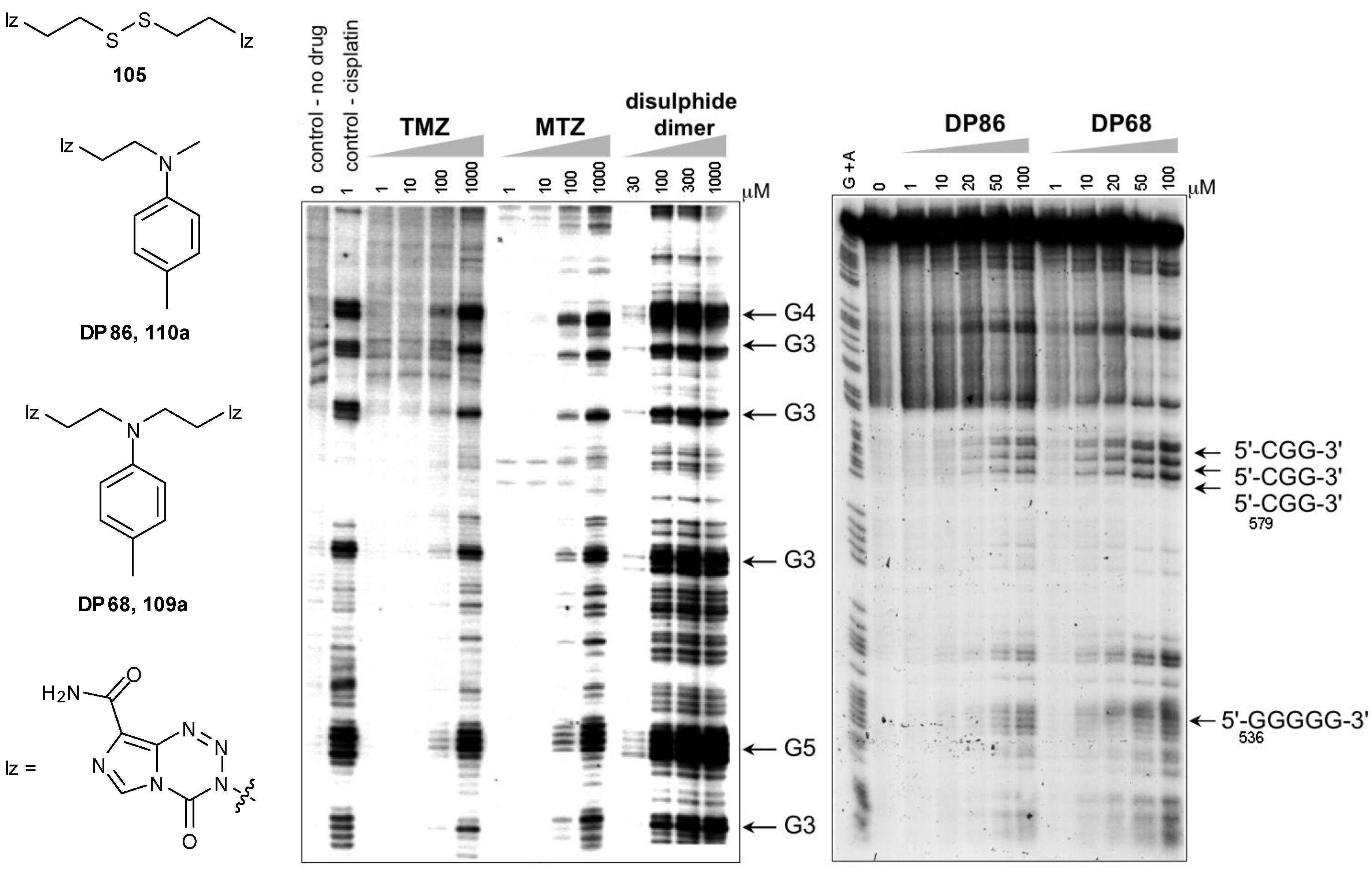
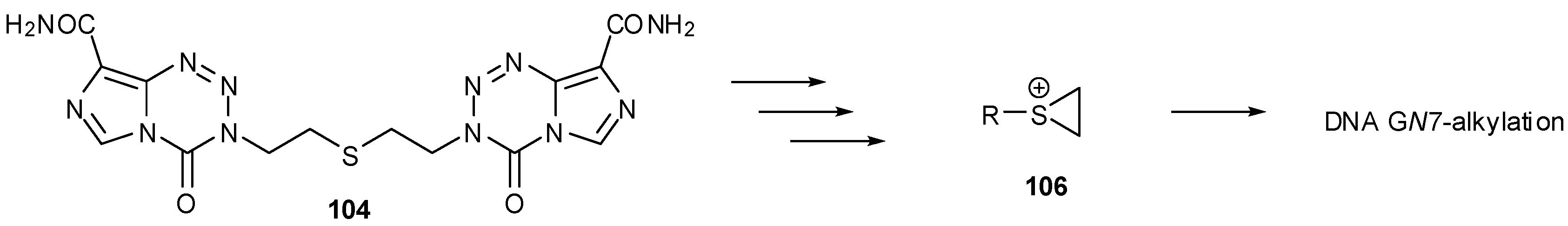
Control of the reactive intermediates has been achieved using neighbouring group participation mechanisms, inspired in part by the chloronium ion of MTZ and the aziridinium intermediates generated by nitrogen mustard prodrugs. Moreover, bisimidazotetrazines were designed that combined two imidazotetrazines to form potential DNA crosslinking agents. The first examples used sulfur as the heteroatom, e.g., 104, 105 and led to both effective DNA alkylation (Figure 16), and activity in the NCI60 panel [90]. The good reactivity with DNA was consistent with the generation of episulfonium ions 106 which are relatively long-lived intermediates in aqueous systems (Scheme 32) [91]. Whilst demonstrating the validity of the design principles, further development of the thioether and disulphide linked compounds was disfavoured as NCI COMPARE analysis indicated close relationships with existing agents such as thiotepa, chlorambucil and uracil mustard (p > 0.8) [92].
Figure 16.
(Left) Comparison of reaction of TMZ, MTZ and a disulphide-linked dimer 105 with DNA showing the enhanced reactivity of the episulphonium prodrug, sites of DNA alkylation detected by the Taq DNA polymerase stop method (adapted from reference [90]); (Right) Reaction of Aziridinium Prodrugs (DP86 and DP68, 107a and 108a) showing the increased reactivity of the bisimidazotetrazine. Sites of GN7 alkylation on the upper strand of pBR322 DNA detected by the piperidine cleavage method. Arrows indicate the position and sequence context of the alkylated guanines. Adapted from reference [93].
Figure 16.
(Left) Comparison of reaction of TMZ, MTZ and a disulphide-linked dimer 105 with DNA showing the enhanced reactivity of the episulphonium prodrug, sites of DNA alkylation detected by the Taq DNA polymerase stop method (adapted from reference [90]); (Right) Reaction of Aziridinium Prodrugs (DP86 and DP68, 107a and 108a) showing the increased reactivity of the bisimidazotetrazine. Sites of GN7 alkylation on the upper strand of pBR322 DNA detected by the piperidine cleavage method. Arrows indicate the position and sequence context of the alkylated guanines. Adapted from reference [93].

Scheme 32.
Generation of episulfonium ions from a sulfur-linker dimer [90].
Scheme 32.
Generation of episulfonium ions from a sulfur-linker dimer [90].

Potential aziridinium ion precursors were attractive as these are reactive intermediates of proven clinical utility, being found widely in, or generated by, synthetic and natural product anti-tumour drugs and prodrugs so may be expected empirically to display suitable pharmacokinetics.
8.2. Synthesis of Novel N-Linked Imidazotetrazine Dimers
This new class of compounds employed nitrogen as the heteroatom controlling the cascade of reactive intermediates in a manner analogous to the nitrogen mustard drugs 107 (Figure 17). These anticancer agents are DNA cross-linkers which react via aziridinium ions 108; these are ring opened during nucleophilic substitution reactions with groups on DNA, principally N7 of guanine residues.
Figure 17.
Nitrogen mustards, resulting aziridinium ions and novel imidazotetrazine dimers.

These novel N-linked dimers 109 were rationally designed as acid-stable precursors to aziridinium ions, to combine the useful pharmacokinetic properties of the imidazotetrazines with the proven clinical utility of DNA cross-linkers. By exploiting preferential GN7 alkylation (70% for TMZ), as opposed to the mere 5% alkylation at GO6, an efficiency gain was sought; moreover, the requirements for MMR and absence of MGMT may be negated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
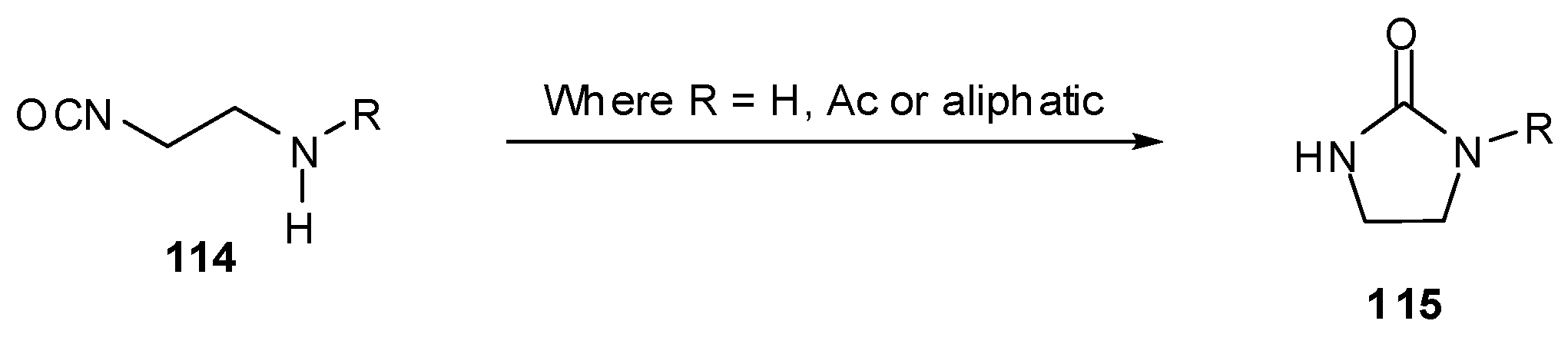{kind=link}
{kind=link}
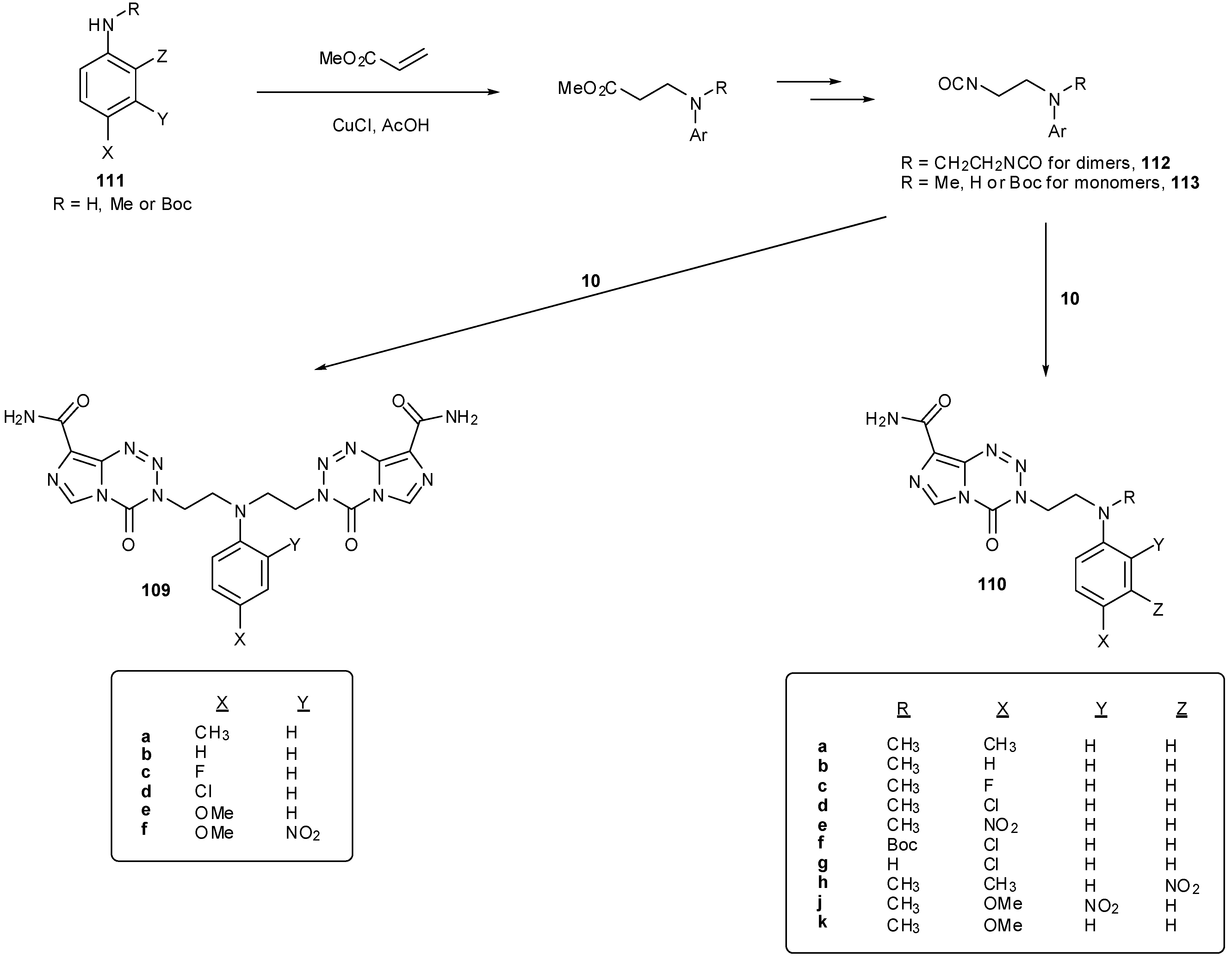
Synthesis of these dimer compounds 109 was via diisocyanates, an approach that had already proved successful in the construction of dimeric imidazotetrazines [25,45]. A family of monomeric imidazotetrazines 110 with aminoethyl side chains has been similarly prepared. The required substrates were obtained from conjugate addition of primary and secondary anilines 111 with methyl acrylate followed by functional group interconversion to give diisocyanates 112 and isocyanates 113 respectively (Scheme 33) [93,94,95].
Imidazotetrazine rings cannot be closed in the presence of tertiary amines unless the nucleophilic centre is deactivated. This can be achieved through the use of anilines or by N-Boc protection; these techniques also serve the purpose of preventing undesired cyclisation of β-aminoisocyanates 114 to cyclic ureas 115 (Scheme 34) [95]. It is worthy of note that an acetate group is insufficiently electron withdrawing for N-deactivation. Little difference in reaction yield was seen between secondary and tertiary anilines and Boc-aniline isocyanates 113 to give 110.
Scheme 34.
Cyclisation of β-aminoisocyanates.

8.3. Properties and Activity of N-Linked Compounds
Both classes of new compound show similar hydrolysis kinetics to TMZ, with almost identical half-lives at pH 7.4 and pH 8.0; acid stability is still robust although not as pronounced as for TMZ [93].
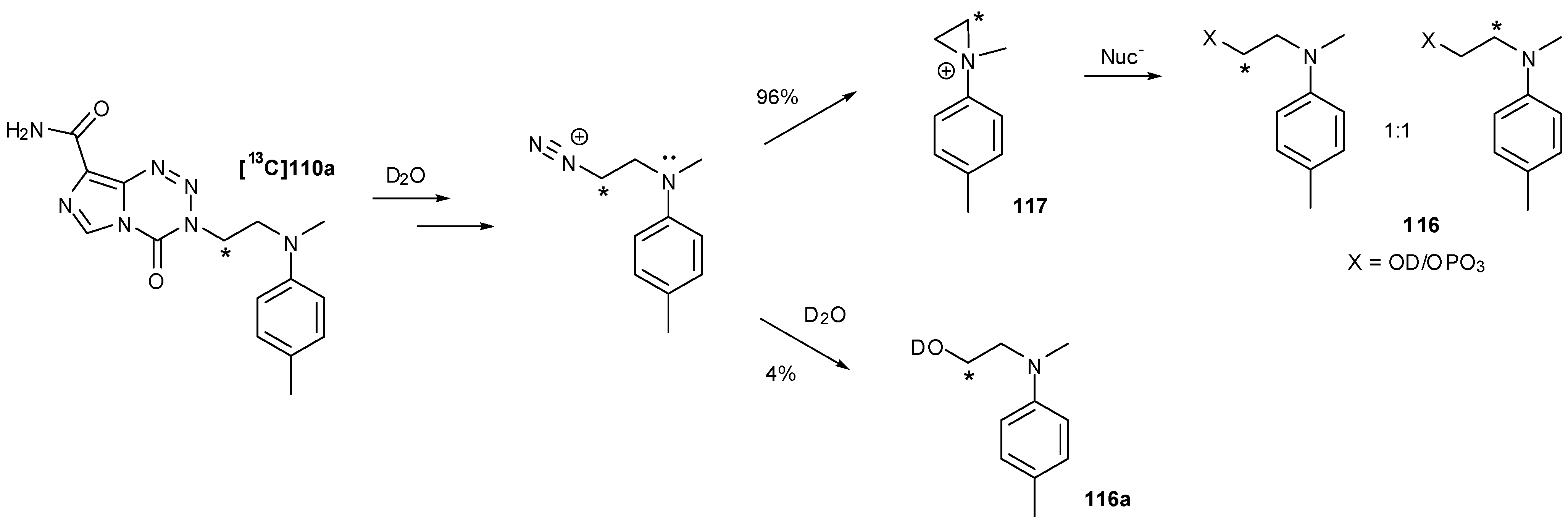
An isotopically labelled version of monomer 110a was prepared for mechanism studies using enriched 13C(2)-acrylate. On exposure to D2O phosphate buffer (pD 7.8), a mixture of phosphate and alcohol (hydrolysis) products 116 was observed. From 13C-NMR studies it could be seen that there was a 4% excess of the alcohol 116a resulting from direct hydrolysis, yet amongst the remaining 96% there was a complete scrambling of the labelled centre, thus proving the formation of an aziridine intermediate 117 (Scheme 35) [93].
Scheme 35.
Aziridinium release [93].
Scheme 35.
Aziridinium release [93].

Reaction of 109a and 110a with DNA in a piperidine cleavage assay showed that alkylation sites were identical to those obtained with melphalan. In this assay, dimeric compound 109a was more reactive than monomer 110a and showed reactivity with DNA equivalent to melphalan (Figure 16) [93]; in whole cells, dimer 109a showed DNA crosslinking activity equivalent to melphalan [96].
Both dimers 109 and monomers 110 showed promising in vitro anti-cancer activity. Preliminary screening was undertaken against an MGMT and MMR-proficient cell line (A2780) and the associated MMR-deficient A2780-cp70; MGMT was inactivated with PaTrin2 in order to evaluate both MMR and MGMT dependence. Pleasingly, members of both classes of compound showed anti-proliferative activity, regardless of MGMT and MMR status of the cell lines tested. The monofunctional agents showed activity equivalent to MTZ, whereas the bifunctional agents showed significantly greater potency than MTZ [95].
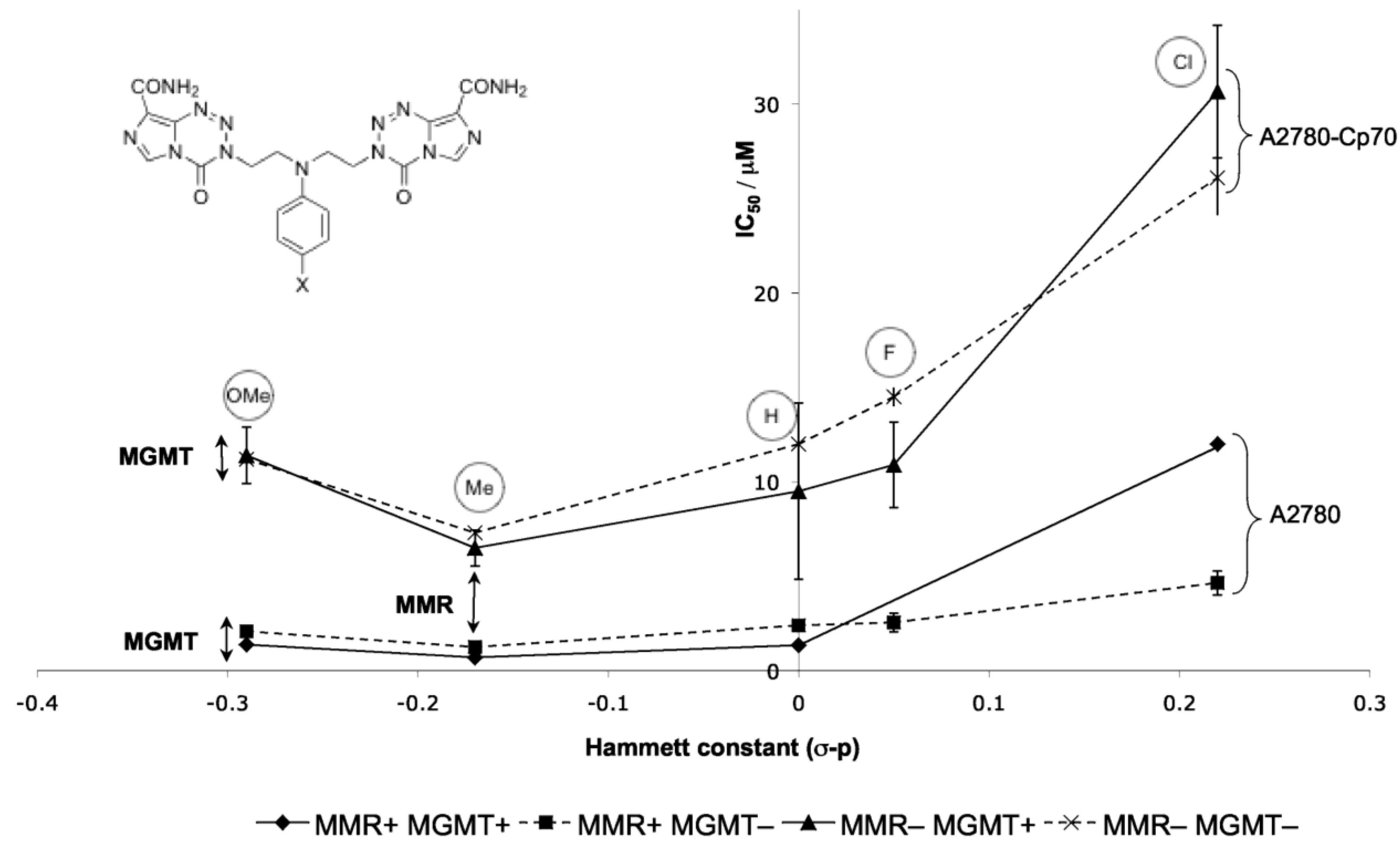
Figure 18.
Effect of electron-withdrawing capabilities of substituent X on the activity of aniline dimers 109 [95]. The differences between sets of lines indicate the limited MMR/MGMT effects.
Figure 18.
Effect of electron-withdrawing capabilities of substituent X on the activity of aniline dimers 109 [95]. The differences between sets of lines indicate the limited MMR/MGMT effects.

Probing more deeply into the in vitro SAR of the aniline dimers 109 showed a trend in Hammett constant vs. IC50; with the exception of the 4-methoxy derivative, the more electron-donating the aniline substituent, the better the activity (Figure 18). This shows a clear link between electronics of the 4-subtituent and pharmacological activity. This can be rationalised by electron donation providing enhanced formation and stability of the aziridinium alkylating agent; the anomalous methoxy group on the other hand potentially increases basicity too much so that appreciable protonation of the aniline occurs, thus reducing nucleophilicity.
Table 2.
Results of Matrix COMPARE (P values) for NCEs 109c–e, 110d, with selected standard agents. DTIC, 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide (3); MEL, melphalan; CHB, chlorambucil; BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; CCNU, N-(2-chloroethyl)-N'-cyclohexyl-N-nitrosourea; CP, cisplatin; Data from reference [95].
| 109c | 109d | 109e | 110d | MTZ | DTIC | MEL | CHB | BCNU | CCNU | CP | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 109c | 1 | 0.92 | 0.92 | 0.83 | 0.35 | 0.72 | 0.4 | 0.33 | 0.32 | 0.06 | 0.32 |
| 109d | 0.92 | 1 | 0.81 | 0.79 | 0.38 | 0.64 | 0.36 | 0.29 | 0.27 | 0.05 | 0.35 |
| 109e | 0.92 | 0.81 | 1 | 0.77 | 0.46 | 0.71 | 0.59 | 0.53 | 0.45 | 0.10 | 0.42 |
| 110d | 0.83 | 0.79 | 0.77 | 1 | 0.42 | 0.62 | 0.35 | 0.33 | 0.25 | 0.08 | 0.27 |
Moreover, NCI screening data confirmed the lack of relationship between MGMT, MMR and efficacy. Matrix COMPARE analysis showed the distinctiveness of these compounds against other aziridinium/diazonium-releasing or crosslinking agents such as MTZ, the nitrosoureas, nitrogen mustards and cisplatin (Table 2).
9. Conclusions
The imidazotetrazine prodrugs are an interesting class of pharmaceutically active compounds. The original synthetic strategy towards the bicyclic imidazotetrazine core employed coupling of diazo-IC with isocyanates; the driving forces for alternative syntheses were based around the stigma and lack of availability of methyl isocyanate as well as avoidance of potentially explosive diazo-IC. Ultimately, however, none of the procedures have bettered the initial route and this remains the preferred method for laboratory scale synthesis.
Ring opening follows two mechanisms, the retro-cycloaddition and hydrolytic prodrug activation mechanism. The latter occurs in two steps: hydrolysis of the imidazotetrazine ring followed by acid-mediated release of alkyl diazonium ions. The mechanism and kinetics of this prodrug activation and thus the circulating half-life are determined by the 3-substituent of the imidazotetrazine. The nature of this group plays a huge role as it also ultimately determines the biological response. In addition to controlling prodrug activation, controlling the reactivity of the released electrophile is of vital importance to allow reaction with DNA.
Response of a tumour to TMZ treatment is dependent on two factors: absence of MGMT, the protein which results in inherent resistance, and presence of MMR, the actions of which ultimately lead to cell death. In addition to these constraints, it is only a minor proportion of DNA alkylation (5% on GO6) which actually results in the anti-tumour response. Recent efforts to combat MGMT resistance have focused on compounds which create adducts unrecognisable by MGMT or those which instead are able to elicit a pharmacological response from GN7 alkylation. Moreover, the need for a non-mutagentic TMZ derivative is urgent, now that the molecular details of the relationship between TMZ-induced mutation and the evolution of aggressive, drug resistant tumour regrowth, following initial good response to therapy, has been established [97].
Acknowledgments
Dedicated to Malcolm Stevens (“With the possible exception of Taxol, anticancer drugs do not grow on trees”) who started it all and to the other azolotetrazinone chemists whose work we have cited. The authors thank Yorkshire Cancer Research for support (grant B210).
Conflict of Interest
The authors declare no conflict of interest
References
- Newlands, E.S.; Stevens, M.F.G.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Stevens, M.F.G. Temozolomide: From cytotoxic to molecularly-targeted agent. In Cancer Drug Design and Discovery; Neidle, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 157–172. [Google Scholar]
- Darkes, M.J.M.; Plosker, G.L.; Jarvis, B. Temozolomide a review of its use in the treatment of malignant gliomas, malignant melanoma and other advanced cancers. Am. J. Cancer 2002, 1, 55–80. [Google Scholar]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar]
- Threadgill, M.D. The Chemistry of azolotetrazinones. In The Chemistry of Antitumour Agents; Wilman, D.E.V., Ed.; Blackie & Son Ltd.: Bel Air, CA, USA, 1990; pp. 187–201. [Google Scholar]
- Gaudilliere, B.; Berna, P. Chapter 30. To market, to market. Annu. Rep. Med. Chem. 2000, 35, 331–356. [Google Scholar] [CrossRef]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Brown, G.D.; Luthra, S.K.; Brock, C.S.; Stevens, M.F.G.; Price, P.M.; Brady, F. Antitumor imidazotetrazines. 40. Radiosyntheses of [4-11C-carbonyl]- and [3-N-11C-methyl]-8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (temozolomide) for positron emission tomography (PET) Studies. J. Med. Chem. 2002, 45, 5448–5457. [Google Scholar]
- Stevens, M.F.G.; Hickman, J.A.; Langdon, S.P.; Chubb, D.; Vickers, L.; Stone, R.; Baig, G.; Goddard, C.; Gibson, N.W.; Slack, J.A.; et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987, 47, 5846–5852. [Google Scholar]
- Tsang, L.L.H.; Quarterman, C.P.; Gescher, A.; Slack, J.A. Comparison of the cytotoxicity in vitro of temozolomide and dacarbazine, prodrugs of 3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother. Pharmacol. 1991, 27, 342–346. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar]
- Happold, C.; Roth, P.; Wick, W.; Schmidt, N.; Florea, A.-M.; Silginer, M.; Reifenberger, G.; Weller, M. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 2012, 122, 444–455. [Google Scholar] [Green Version]
- Barvaux, V.A.; Ranson, M.; Brown, R.; McElhinney, R.S.; McMurry, T.B.H.; Margison, G.P. Dual repair modulation reverses temozolomide resistance in vitro. Mol. Cancer Ther. 2004, 3, 123–127. [Google Scholar]
- Wheelhouse, R.T.; Stevens, M.F.G. Decomposition of the antitumour drug temozolomide in deuteriated phosphate buffer: methyl group transfer is accompanied by deuterium exchange. Chem. Commun. 1993, 1177–1178. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.G.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Wheelhouse, R.T.; Denny, B.J.; Stevens, M.F.G. Novel approaches in anticancer drug design. In Proceedings of the International Symposium on Novel Approaches in Cancer Therapy, Heidelberg, Germany, December 1993; Zeller, W.J., D’Incalci, M., Newell, D.R., Eds.; Contrib. to Oncol. 1995; 49, pp. 40–49. [Google Scholar]
- Campbell, J.R. Diazomethane-d2 (CD2N2). Chem. Ind. 1972, 540. [Google Scholar]
- Lunt, E.; Newton, C.G.; Smith, C.; Stevens, G.P.; Stevens, M.F.G.; Straw, C.G.; Walsh, R.J.A.; Warren, P.J.; Fizames, C.; Lavelle, F.; et al. Antitumor imidazotetrazines. 14. Synthesis and antitumor activity of 6- and 8-substituted imidazo[5,1-d]-1,2,3,5-tetrazinones and 8-substituted pyrazolo[5,1-d]-1,2,3,5-tetrazinones. J. Med. Chem. 1987, 30, 357–366. [Google Scholar] [CrossRef]
- Stevens, M.F.G.; Hickman, J.A.; Stone, R.; Gibson, N.W.; Baig, G.U.; Lunt, E.; Newton, C.G. Antitumor imidazotetrazines. 1. Synthesis and chemistry of 8-carbamoyl-3-(2-chloroethyl)imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, a novel broad-spectrum antitumor agent. J. Med. Chem. 1984, 27, 196–201. [Google Scholar]
- Lown, J.W.; Chauhan, S.M.S. Synthesis of specifically 15N- and 13C-labeled antitumor (2-haloethyl)nitrosoureas. The study of their conformations in solution by nitrogen-15 and carbon-13 nuclear magnetic resonance and evidence for stereoelectronic control in their aqueous decomposition. J. Org. Chem. 1981, 46, 5309–5321. [Google Scholar] [CrossRef]
- Hartley, J.A.; Gibson, N.W.; Kohn, K.W.; Mattes, W.B. DNA sequence selectivity of guanine-N7 alkylation by three antitumor chloroethylating agents. Cancer Res. 1986, 46, 1943–1947. [Google Scholar]
- Horgan, C.M.T.; Tisdale, M.J. Antitumour imidazotetrazines—IV An investigation into the mechanism of antitumour activity of a novel and potent antitumour agent, mitozolomide (CCRG 81010, M & B 39565; NSC 353451). Biochem. Pharmacol. 1984, 33, 2185–2192. [Google Scholar] [CrossRef]
- Clark, A.S.; Deans, B.; Stevens, M.F.G.; Tisdale, M.J.; Wheelhouse, R.T.; Denny, B.J.; Hartley, J.A. Antitumor imidazotetrazines. 32. Synthesis of novel imidazotetrazinones and related bicyclic heterocycles to probe the mode of action of the antitumor drug temozolomide. J. Med. Chem. 1995, 38, 1493–1504. [Google Scholar]
- Wang, Y.; Wheelhouse, R.T.; Zhao, L.; Langnel, D.A.F.; Stevens, M.F.G. Antitumour imidazotetrazines. Part 36. Conversion of 5-amino-imidazole-4-carboxamide to imidazo[5,1-d][1,2,3,5]tetrazin-4(3H)-ones and imidazo[1,5-a][1,3,5]triazin-4(3H)-ones related in structure to the antitumour agents temozolomide and mitozolomide. J. Chem. Soc. Perkin Trans. 1998, 1, 1669–1675. [Google Scholar]
- Wheelhouse, R.T.; Wilman, D.E.V; Thomson, W.; Stevens, M.F.G. Antitumour imidazotetrazines. Part 31. The synthesis of isotopically labelled temozolomide and a multinuclear (1H, 13C, 15N) magnetic resonance investigation of temozolomide and mitozolomide. J. Chem. Soc. Perkin Trans. 1995, 1, 249–252. [Google Scholar]
- Babu, N.J.; Sanphui, P.; Nath, N.K.; Khandavilli, U.B.R.; Nangia, A. Temozolomide hydrochloride dihydrate. CrystEngComm 2013, 15, 666–671. [Google Scholar] [CrossRef]
- McGarrity, J.F.; Smyth, T. Hydrolysis of diazomethane—Kinetics and mechanism. J. Am. Chem. Soc. 1980, 102, 7303–7308. [Google Scholar] [CrossRef]
- Newlands, E.S.; Blackledge, G.; Slack, J.A.; Goddard, C.; Brindley, C.J.; Holden, L.; Stevens, M.F.G. Phase-I clinical trial of mitozolomide. Cancer Treat. Rep. 1985, 69, 801–805. [Google Scholar]
- Zhang, D.; Tian, A.; Xue, X.; Wang, M.; Qiu, B.; Wu, A. The effect of temozolomide/poly(lactide-co-glycolide) (PLGA)/nano-hydroxyapatite microspheres on glioma U87 cells behavior. Int. J. Mol. Sci. 2012, 13, 1109–1125. [Google Scholar]
- Panda, B.; Maikap, G.C.; Agarwal, S.K.; Singh, M.K.; Jaggi, M. Crystalline temozolomide monohydrate and process for preparation thereof. Int. Pat. WO2008111092A1, 18 September 2008. [Google Scholar]
- Adin, I.; Iustain, C. Novel crystalline forms of temozolomide. US Pat. US20100022609A1, 28 January 2010. [Google Scholar]
- Lowe, P.R.; Sansom, C.E.; Schwalbe, C.H.; Stevens, M.F.G.; Clark, A.S. Antitumor imidazotetrazines. 25. Crystal structure of 8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (temozolomide) and structural comparisons with the related drugs mitozolomide and DTIC. J. Med. Chem. 1992, 35, 3377–3382. [Google Scholar] [CrossRef]
- Babu, N.J.; Reddy, L.S.; Aitipamula, S.; Nangia, A. Polymorphs and polymorphic cocrystals of temozolomide. Chem. Asian J. 2008, 3, 1122–1133. [Google Scholar] [CrossRef]
- Babu, N.J.; Sanphui, P.; Nangia, A. Crystal engineering of stable temozolomide cocrystals. Chem. Asian J. 2012, 7, 2274–2285. [Google Scholar]
- Sanphui, P.; Babu, N.J.; Nangia, A. Temozolomide cocrystals with carboxamide coformers. Cryst. Growth Des. 2013, 13, 2208–2219. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Wei, J.; Zhou, R.; Chen, Z.; Liang, H. Synthesis and crystal structure of a cadmium complex of temozolomide. J. Chem. Res. 2012, 36, 520–522. [Google Scholar] [CrossRef]
- Appel, E.A.; Rowland, M.J.; Loh, X.J.; Heywood, R.M.; Watts, C.; Scherman, O.A. Enhanced stability and activity of temozolomide in primary glioblastoma multiforme cells with cucurbit[n]uril. Chem. Commun. 2012, 48, 9843–9845. [Google Scholar]
- Ege, G.; Gilbert, K. [7+2]- and [11+2]-Cycloaddition reactions of diazo-azoles with isocyanates to azolo[5,1-d][1,2,3,5]tetrazine-4-ones. Tetrahedron Lett. 1979, 20, 4253–4256. [Google Scholar]
- Shealy, Y.F.; Struck, R.F.; Holum, L.B.; Montgomery, J.A. Synthesis of potential anticancer agents. XXIX. 5-Diazoimidazole-4-carboxamide and 5-diazo-ν-triazole-4-carboxamide. J. Org. Chem. 1961, 26, 2396–2401. [Google Scholar] [CrossRef]
- Woolley, D.W.; Shaw, E. Some imidazo-1,2,3-triazenes and their biological relationship to the purines. J. Biol. Chem. 1951, 189, 401–410. [Google Scholar]
- Horton, K.; Stevens, M.F.G. Triazenes and related products. Part 23. New photo-products from 5-diazoimidazole-4-carboxamide (diazo-IC). J. Chem. Soc. Perkin Trans. 1981, 1, 1433–1436. [Google Scholar] [CrossRef]
- Wang, Y.; Stevens, M.F.G. Synthetic studies of 8-carbamoylimidazo-[5,1-d]-1,2,3,5-tetrazin-4(3H)-one: A key derivative of antitumour drug temozolomide. Bioorganic Med. Chem. Lett. 1996, 3, 185–188. [Google Scholar] [CrossRef]
- Pletsas, D.; Karodia, N.; Wheelhouse, R.T.; University of Bradford, Bradford, UK. Unpublished data.
- Arrowsmith, J.; Jennings, S.A.; Clark, A.S.; Stevens, M.F.G. Antitumor imidazotetrazines. 41. Conjugation of the antitumor agents mitozolomide and temozolomide to peptides and lexitropsins bearing DNA major and minor groove-binding structural motifs. J. Med. Chem. 2002, 45, 5458–5470. [Google Scholar]
- Wang, Y.; Stevens, M.F.G.; Thomson, W. Alternative syntheses of the antitumour drug temozolomide avoiding the use of methyl isocyanate. J. Chem. Soc. Chem. Commun. 1994, 1687–1688. [Google Scholar]
- Wang, Y.; Stevens, M.F.G.; Chan, T.; DiBenedetto, D.; Ding, Z.; Gala, D.; Hou, D.; Kugelman, M.; Leong, W.; Kuo, S.; et al. Antitumor imidazotetrazines. 35. New synthetic routes to the antitumor drug temozolomide. J. Org. Chem. 1997, 62, 7288–7294. [Google Scholar] [CrossRef]
- Wang, Y.; Stevens, M.F.G.; Thomson, W.T.; Shutts, B.P. Antitumour imidazotetrazines. Part 33. New syntheses of the antitumour drug temozolomide using “masked” methyl isocyanates. J. Chem. Soc. Perkin Trans. 1995, 1, 2783–2787. [Google Scholar]
- Wang, Y.; Lowe, P.R.; Thomson, W.T.; Clark, J.; Stevens, M.F.G. A new route to the antitumour drug temozolomide, but not thiotemozolomide. Chem. Commun. 1997, 363–364. [Google Scholar]
- Turchetta, S.; De Ferra, L.; Zenoni, M.; Anibaldi, M. Process for preparing temozolomide. Eur. Pat. EP2151442A2, 10 February 2010. [Google Scholar]
- Pathi, S.L.; Rao, D.R.; Kankan, R.N. Process for the preparation of temozolomide and analogs. US Pat. 2009. [Google Scholar]
- Etlin, O.; Alnabari, M.; Sery, Y.; Danon, E.; Arad, O.; Kaspi, J. Process for preparing temozolomide. US Pat. US20060183898A1, 17 August 2006. [Google Scholar]
- Wanner, M.J.; Koomen, G. A new synthesis of temozolomide. J. Chem. Soc. Perkin Trans. 2002, 1, 1877–1880. [Google Scholar] [CrossRef]
- Kuo, S.-C. Synthesis of temozolomide and analogs. US Pat. US6844434B2, 18 Janaury 2005. [Google Scholar]
- Kuo, S.-C.; Mas, J.L.; Hou, D. Synthesis of temozolomide and analogs. US Pat. US7087751B2, 8 August 2006. [Google Scholar]
- Łaszcz, M.; Kubiszewski, M.; Jedynak, Ł.; Kaczmarska, M.; Kaczmarek, L.; Łuniewski, W.; Gabarski, K.; Witkowska, A.; Kuziak, K.; Malińska, M. Identification and physicochemical characteristics of temozolomide process-related impurities. Molecules 2013, 18, 15344–15356. [Google Scholar] [CrossRef]
- Schwartzman, S.; Lima, D.A. Method of generating lower alkyl and cycloalkyl isocyanates. US Pat. US4141913A, 27 February 1979. [Google Scholar]
- Palle, R.V.; Marathe, A.M.; Manda, A. Process for preparing temozolomide. US Pat. US2007225496A1, 27 September 2007. [Google Scholar]
- Mormann, W.; Leukel, G. A simple and versatile synthesis of trimethylsiloxy-substituted isocyanates. Synthesis-Stuttgart 1988, 12, 990–992. [Google Scholar] [CrossRef]
- Tsang, L.L.H.; Farmer, P.B.; Gescher, A.; Slack, J.A. Characterisation of urinary metabolites of temozolomide in humans and mice and evaluation of their cytotoxicity. Cancer Chemother. Pharmacol. 1990, 26, 429–436. [Google Scholar] [CrossRef]
- Reyderman, L.; Statkevich, P.; Thonoor, C.M.; Patrick, J.; Batra, V.K.; Wirth, M. Disposition and pharmacokinetics of temozolomide in rat. Xenobiotica 2004, 34, 487–500. [Google Scholar]
- Ningaraj, N.S.; Sankpal, U.T.; Khaitan, D.; Meister, E.A.; Vats, T. Activation of KATP channels increases anticancer drug delivery to brain tumors and survival. Eur. J. Pharmacol. 2009, 602, 188–193. [Google Scholar]
- Moseley, C.K.; Carlin, S.M.; Neelamegam, R.; Hooker, J.M. An efficient and practical radiosynthesis of [11C]temozolomide. Org. Lett. 2012, 14, 5872–8575. [Google Scholar] [CrossRef]
- Cheng, C.C.; Elslager, E.F.; Werbel, L.M.; Priebe, S.R.; Leopold, W.R., III. Pyrazole derivatives. 5. Synthesis and antineoplastic activity of 3-(2-chloroethyl)-3,4-dihydro-4-oxopyrazolo[5,1-d]-1,2,3,5-tetrazine-8-carboxamide and related compounds. J. Med. Chem. 1986, 29, 1544–1547. [Google Scholar] [CrossRef]
- Diana, P.; Barraja, P.; Lauria, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Pyrrolo[2,1-d][1,2,3,5]tetrazines, a new class of azolotetrazines related to the antitumor drug temozolomide. Synthesis-Stuttgart 1999, 12, 2082–2086. [Google Scholar]
- Barraja, P.; Diana, P.; Lauria, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. 2-Diazoindoles: building blocks for the synthesis of antineoplastic agents. Farmaco 2002, 57, 97–100. [Google Scholar] [CrossRef]
- Barraja, P.; Diana, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Synthesis and antiproliferative activity of [1,2,3,5]tetrazino[5,4-a]indoles, a new class of azolo-tetrazinones. Bioorg. Med. Chem. 2005, 13, 295–300. [Google Scholar] [CrossRef]
- Diana, P.; Barraja, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Pyrrolo[2,1-d][1,2,3,5]tetrazine-4(3H)-ones, a new class of azolotetrazines with potent antitumor activity. Bioorg. Med. Chem. 2003, 11, 2371–2380. [Google Scholar] [CrossRef]
- Tripathi, G.; Mistra, J.P. QSAR studies on pyrrolo[2,1-d][1,2,3,5]tetrazinones, a new class of azolotetrazines. Indian J. Chem. 2005, 44, 1398–1400. [Google Scholar]
- Diana, P.; Barraja, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Pyrrolo[2,1-c][1,2,4]triazines from 2-diazopyrroles: Synthesis and antiproliferative activity. Eur. J. Med. Chem. 2002, 37, 267–272. [Google Scholar] [CrossRef]
- Barraja, P.; Diana, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Synthesis and antiproliferative activity of [1,2,4]triazino[4,3-a]indoles. Anticancer Res. 2004, 24, 3775–3779. [Google Scholar]
- Diana, P.; Stagno, A.; Barraja, P.; Carbone, A.; Montalbano, A.; Martorana, A.; Dattolo, G.; Cirrincione, G. Pyrido[4',3':4,5]pyrrolo[2,1-d][1,2,3,5]tetrazine a new class of temozolomide heteroanalogues. Arkivoc 2009, 1–11. [Google Scholar]
- Diana, P.; Stagno, A.; Barraja, P.; Montalbano, A.; Carbone, A.; Dattolo, G.; Cirrincione, G. Pyrido[2',3':4,5]pyrrolo[2,1-d][1,2,3,5]tetrazine-4(3H)-ones, a new class of temozolomide heteroanalogues. Arkivoc 2009, 177–186. [Google Scholar]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Plescia, F.; Cascioferro, S.; Daidone, G. Synthesis of alkyl-5,8-dimethyl-6-phenyl-5,6-dihydropyrazolo[3,4-f][1,2,3,5]tetrazepin-4(3H)-ones of pharmaceutical interest. Arkivoc 2006, 120–126. [Google Scholar]
- Zhu, Y.-Q.; Wu, C.; Li, H.-B.; Zou, X.-M.; Si, X.-K.; Hu, F.-Z.; Yang, H.-Z. Design, synthesis, and quantitative structure–activity relationship study of herbicidal analogues of pyrazolo[5,1-d][1,2,3,5]tetrazin-4(3H)ones. J. Agric. Food Chem. 2007, 55, 1364–1369. [Google Scholar] [CrossRef]
- Zhu, Y.-Q.; Cheng, J.; Zou, X.-M.; Hu, F.-Z.; Xiao, T.-H.; Yang, H.-Z. Design, synthesis and quantitative structure–activity relationship study of herbicidal analogues of pyrazolotetrazinones. Chin. J. Chem. 2008, 28, 1044–1049. [Google Scholar]
- Horspool, K.R.; Stevens, M.F.; Newton, C.G.; Lunt, E.; Walsh, R.J.; Pedgrift, B.L.; Baig, G.U.; Lavelle, F.; Fizames, C. Antitumor imidazotetrazines. 20. Preparation of the 8-acid derivative of mitozolomide and its utility in the preparation of active antitumor agents. J. Med. Chem. 1990, 33, 1393–1399. [Google Scholar]
- Liu, D.; Yang, J.-G.; Cheng, J.; Zhao, L.-X. Synthesis and antitumor activity of 3-methyl-4-oxo-3,4-dihydroimidazo [5,1-d][1,2,3,5]tetrazine-8-carboxylates and -carboxamides. Molecules 2010, 15, 9427–9437. [Google Scholar] [CrossRef]
- Suppasansatorn, P.; Wang, G.; Conway, B.R.; Wang, W.; Wang, Y. Skin delivery potency and antitumor activities of temozolomide ester prodrugs. Cancer Lett. 2006, 244, 42–52. [Google Scholar] [CrossRef]
- Wang, Y. Synthesis of temozolomide esters as potent anticancer pro-drugs for topical and transdermal applications in treatments of cancers. US Pat. US2006047117A1, 2 March 2006. [Google Scholar]
- Wang, Y.; Lambert, P.; Zhao, L.; Wang, D. Synthesis and antibacterial activity of dual-action agents of a β-lactam antibiotic with cytotoxic agent mitozolomide or temozolomide. Eur. J. Med. Chem. 2002, 37, 323–332. [Google Scholar] [CrossRef]
- Li, R.; Tang, D.; Zhang, J.; Wu, J.; Wang, L.; Dong, J. The temozolomide derivative 2T-P400 inhibits glioma growth via administration route of intravenous injection. J. Neurooncol. 2014, 116, 25–30. [Google Scholar]
- Hummersone, M.G.; Cousin, D. 3-Substituted-8-substituted-3H-imidazo[5,1-d][1,2,3,5]tetrazin-4-one compounds and their use. US Pat. US20120083513A1, 5 April 2012. [Google Scholar]
- Stevens, M.F.G.; Cousin, D.; Jennings, S.; McCarroll, A.J.; Williams, J.G.; Hummersone, M.G.; Zhang, J. 3-Substituted-4-oxo-3,4-dihydro-imidazo-[5,1-d][1,2,3,5]tetrazine-8-carboxylic acid amides and their use. Int. Pat. WO2009077741A2, 25 June 2009. [Google Scholar]
- Stevens, M.G.F.; Cousin, D.; Jennings, S.; McCarroll, A.J.; Williams, J.G.; Hummersone, M.G.; Zhang, J. 3-Substituted-4-oxo-3,4-dihydro-imidazo[5,1-d]1,2,3,5-tetrazine-8-carboxylic acid amides and their Use. US Pat. US20130338104A1, 19 December 2013. [Google Scholar]
- Hummersone, M.G.; Cousin, D. Methods and intermediates for the synthesis of 4-oxo-3,4-dihydro-imidazo[5,1-d][1,2,3,5]tetrazines. Int. Pat. WO2011107726A1, 9 September 2011. [Google Scholar]
- Cousin, D.; Stevens, M.F.G.; Hummersone, M.G. Antitumour imidazotetrazines. Synthesis and chemistry of 4-oxo-3,4-dihydroimidazo[5,1-d][1,2,3,5]tetrazine-8-carboxamide (nor-temozolomide): An intermediate for the preparation of the antitumour drug temozolomide and analogues, avoiding the use of isocyan. Med. Chem. Comm. 2012, 3, 1419–1422. [Google Scholar]
- Pletsas, D.; Wheelhouse, R.T.; Pletsa, V.; Nicolaou, A.; Jenkins, T.C.; Bibby, M.C.; Kyrtopoulos, S.A. Polar, functionalized guanine-O6 derivatives resistant to repair by O6-alkylguanine-DNA alkyltransferase: implications for the design of DNA-modifying drugs. Eur. J. Med. Chem. 2006, 41, 330–339. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Hummersone, M.; Madhusudan, S.; Laughton, C.A.; Bradshaw, T.D. Certain imidazotetrazines escape O6-methylguanine-DNA methyltransferase and mismatch repair. Oncology 2011, 80, 195–207. [Google Scholar] [CrossRef]
- Arrowsmith, J.; Jennings, S.A.; Langnel, D.A.F.; Wheelhouse, R.T.; Stevens, M.F.G. Antitumour imidazotetrazines. Part 39. Synthesis of bis(imidazotetrazine)s with saturated spacer groups. J. Chem. Soc. Perkin Trans. 2000, 1, 4432–4438. [Google Scholar]
- Henkel, J.G.; Amato, G.S. Methyl mercapturate episulfonium ion: a model reactive metabolite of dihaloethanes. J. Med. Chem. 1988, 31, 1279–1282. [Google Scholar] [CrossRef]
- Arrowsmith, J. Antitumour imidazotetrazines major and minor groove interactions. Ph.D. Thesis, The University of Nottingham, Nottingham, UK, 1997. [Google Scholar]
- Garelnabi, E.A.E.; Pletsas, D.; Li, L.; Kiakos, K.; Karodia, N.; Hartley, J.A.; Phillips, R.M.; Wheelhouse, R.T. A strategy for imidazotetrazine prodrugs with anti-cancer activity independent of MGMT and MMR. ACS Med. Chem. Lett. 2012, 3, 965–968. [Google Scholar]
- Wheelhouse, R.T.; Pletsas, D. Aminoalkyl-imidazotetrazines for treatment of cancer. Int. Pat. WO2009127815A1, 22 October 2009. [Google Scholar]
- Pletsas, D.; Garelnabi, E.A.E.; Li, L.; Phillips, R.M.; Wheelhouse, R.T. Synthesis and quantitative structure–activity relationship of imidazotetrazine prodrugs with activity independent of O6-methylguanine-DNA-methyltransferase, DNA mismatch repair, and p53. J. Med. Chem. 2013, 56, 7120–7132.96. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, Y.P.; Mladek, A.C.; Phillips, R.M.; Gyntherd, M.; Rautio, J.; Ross, A.H.; Wheelhouse, R.T.; Sakaria, J.N. Evaluation of novel imidazotetrazine analogues designed to overcome temozolomide resistance and glioblastoma regrowth. Submitted for publication.
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Moody, C.L.; Wheelhouse, R.T. The Medicinal Chemistry of Imidazotetrazine Prodrugs. Pharmaceuticals 2014, 7, 797-838. https://doi.org/10.3390/ph7070797
AMA Style
Moody CL, Wheelhouse RT. The Medicinal Chemistry of Imidazotetrazine Prodrugs. Pharmaceuticals. 2014; 7(7):797-838. https://doi.org/10.3390/ph7070797
Chicago/Turabian StyleMoody, Catherine L., and Richard T. Wheelhouse. 2014. "The Medicinal Chemistry of Imidazotetrazine Prodrugs" Pharmaceuticals 7, no. 7: 797-838. https://doi.org/10.3390/ph7070797