3. Experimental Section
3.1. Materials and Methods
The starting materials and reagents, purchased from commercial suppliers, were used without further purification. All reactions were monitored by thin-layer chromatography (TLC), on aluminium sheets (Silica gel 60-F254, E. Merck). Compounds were visualized by UV light. Column chromatography was carried out using silica gel (200–300 mesh). All reaction solvents were dried prior to use according to standard procedures. All primary reagents were commercially available. Silica gel chromatography solvents were of analytical grade. Melting points were recorded on a micro melting point apparatus MP-500D and were uncorrected. NMR spectra were recorded on a Jeol JNM-ECP spectrometer at 600 MHz for 1H NMR and 150 MHz for 13C NMR with TMS as the internal standard. Chemical shifts are expressed in δ (ppm) and coupling constants (J) in Hz. Multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), dd (doublet of doublets), brs (broad singlet), etc. Mass spectra were recorded using a Q-TOF Ultima™ Global by chemical ionization.
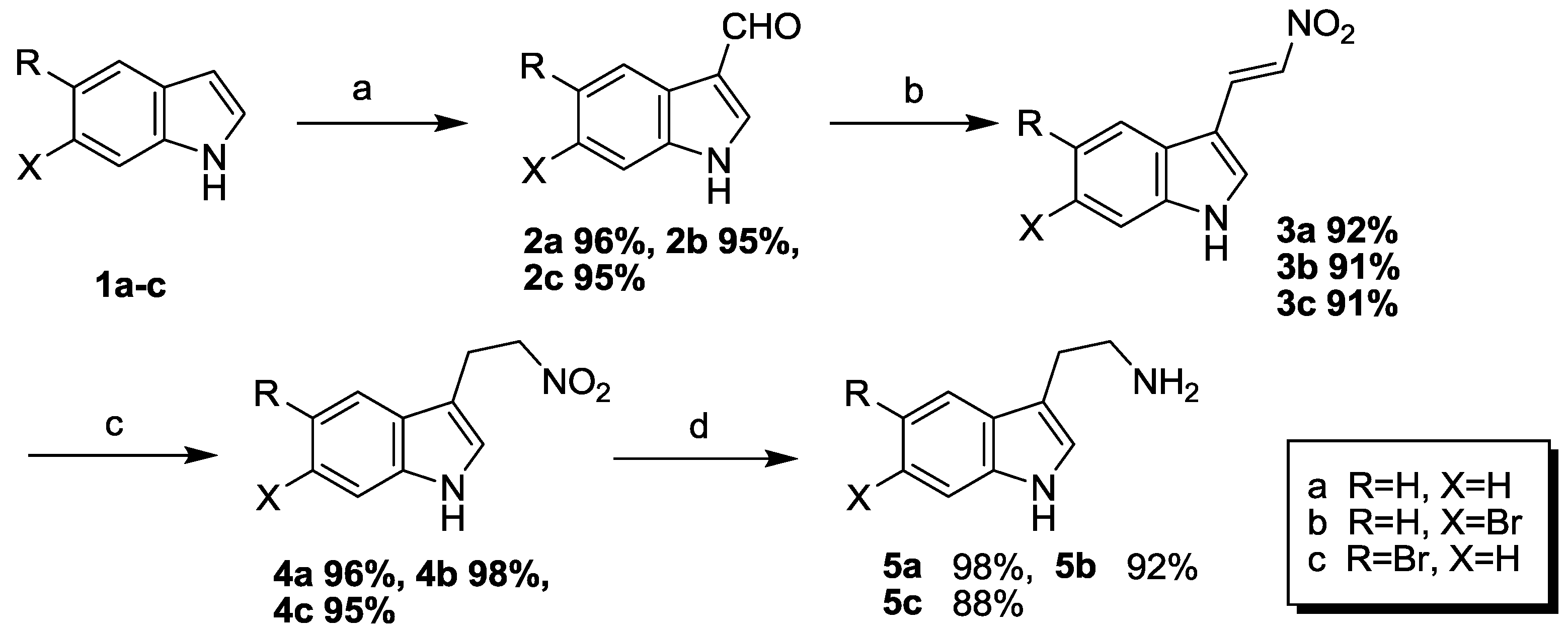
3.2. General Procedure for Compounds 2a–c
In a separate tear-shaped flask under a nitrogen atmosphere, 40 mL of DMF was cooled to 0 °C, and then POCl
3 (150 mmol) was added dropwise within 0.5 h under nitrogen. After the addition, stirring was continued for 1.5 h at 0 °C. It was followed by addition of a solution of indole
1a–
c (100 mmol) in DMF (20 mL) below 0 °C within 1.5 h. Then the reaction solution was heated to 40 °C for another 2 h and cooled to 0 °C. Subsequently, crushed ice (50 g) was added, and then the pH was adjusted to 8–9 by adding NaOH solution (20%, w/w). The resulting reaction mixture was then heated at reflux for 12 h. And then, the resulting suspension solution was cooled naturally to the room temperature and filtered through celite to afford a white crude product. The crude product was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide the compounds
2a–
c [
8].
1H-Indole-3-carbaldehyde (2a): White solid; yield 96%; 1H NMR (600 MHz, CDCl3) δ 11.21 (brs, 1H), 10.04 (s, 1H), 8.24 (d, J = 7.8 Hz, 1H), 8.21 (s, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.30–7.23 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 184.6, 137.3, 124.7, 123.7, 122.2, 121.4, 119.2, 112.2.
6-Bromo-1H-indole-3-carbaldehyde (2b): White solid; yield 95%; 1H NMR (600 MHz, CDCl3) δ 11.33 (brs, 1H), 10.03 (s, 1H), 8.24 (s, 1H), 8.15 (d, J = 8.3 Hz, 1H), 7.76 (d, J = 1.8 Hz, 1H), 7.39 (dd, J = 8.3, 1.8 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 184.7, 137.9, 125.3, 123.6, 122.9, 119.0, 116.5, 115.2.
5-Bromo-1H-indole-3-carbaldehyde (2c): White solid; yield 95%; 1H NMR (600 MHz, CDCl3) δ 11.36 (brs, 1H), 10.02 (s, 1H), 8.39 (d, J = 1.8 Hz, 1H), 8.26 (s, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.40 (dd, J = 8.7 Hz, 1.8 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 184.6, 138.2, 136.1, 126.5, 126.4, 123.8, 118.5, 115.3, 114.2.
3.3. General Procedure for Compounds 3a–c
To a solution of 1
H-indole-3-carbaldehyde
2a–
c (80 mmol) in CH
3NO
2 (80 mL), was added ammonium acetate (40 mmol) and benzene (1.1 mL, 12.4 mmol). The reaction mixture was heated at reflux for 12 h. After cooling to room temperature, some solvent was removed under reduced pressure and filtered through Celite to afford a yellow crude product. The crude product was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:2) as the mobile phase to provide the compounds
3a–
c [
9].
(E)-3-(2-Nitrovinyl)-1H-indole (3a): Deep yellow solid; yield 92%; 1H NMR (600 MHz, CDCl3) δ 11.33 (brs, 1H), 8.39 (d, J = 13.3 Hz, 1H), 8.16 (d, J = 3.2 Hz, 1H), 7.97 (dd, J = 5.9, 2.3 Hz, 1H), 7.92 (d, J = 13.3 Hz, 1H), 7.59 (dd, J = 5.9, 2.3 Hz, 1H), 7.32–7.29 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 138.2, 135.1, 133.9, 131.9, 125.1, 123.6, 122.1, 120.5, 112.8, 108.8.
(E)-6-Bromo-3-(2-nitrovinyl)-1H-indole (3b): Deep yellow solid; yield 91%; 1H NMR (600 MHz, CDCl3) δ 11.39 (brs, 1H), 8.36 (d, J = 13.3 Hz, 1H), 8.18 (s, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.91 (d, J = 13.3 Hz, 1H), 7.78 (d, J = 1.8 Hz, 1H), 7.41 (dd, J = 8.3, 1.8 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 138.9, 135.4, 133.2, 132.7, 124.9, 124.1, 122.0, 116.5, 115.7, 108.8.
(E)-5-Bromo-3-(2-nitrovinyl)-1H-indole (3c): Deep yellow solid; yield 91%; 1H NMR (600 MHz, CDCl3) δ 11.41 (brs, 1H), 8.35 (d, J = 13.7 Hz, 1H), 8.19 (s, 1H), 8.16 (d, J = 1.8 Hz, 1H), 7.97 (d, J = 13.7 Hz, 1H), 7.54 (d, J = 8.7 Hz, 1H), 7.42 (dd, J = 8.7, 1.8 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 136.7, 135.5, 133.0, 132.7, 126.8, 126.3, 122.9, 115.0, 114.5, 108.3.
3.4. General Procedure for Compounds 4a–c
To a solution of (
E)-3-(2-nitrovinyl)-1
H-indole
3a–
c (20mmol) in THF (60 mL) and CH
3OH (9 mL), was added NaBH
4 (40 mmol) in batch over 0.5 h. The above reaction solution was stirred at room temperature for about 1 h and the completion of the reaction was monitored by TLC. Then water (100 mL) and hydrochloric acid (100 mL, 10%, v/v) was added slowly. The resulting reaction mixture was extracted with CH
2Cl
2 (30 mL × 3). The combined organic phase was washed with H
2O (20 mL × 3) and brine (20 mL × 3), dried over anhydrous MgSO
4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide the compounds
4a–
c [
10].
3-(2-Nitroethyl)-1H-indole (4a): Brown solid; yield 96%; 1H NMR (600 MHz, CDCl3) δ 8.09 (brs, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.25 (t, J = 7.8, 7.3 Hz, 1H), 7.18 (t, J = 8.3, 7.3 Hz, 1H), 7.04 (s, 1H), 4.67 (t, J = 6.9 Hz, 2H), 3.49 (t, J = 6.9 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 136.3, 126.7, 122.7, 122.6, 119.9, 118.2, 111.6, 110.0, 75.8, 23.7.
6-Bromo-3-(2-nitroethyl)-1H-indole (4b): Brown solid; yield 98%; 1H NMR (600 MHz, CDCl3) δ 8.13 (brs, 1H), 7.49 (d, J = 1.8 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.24 (dd, J = 8.2, 1.8 Hz, 1H), 7.00 (d, J = 1.8 Hz, 1H), 4.64 (t, J = 6.9 Hz, 2H), 3.49 (t, J = 6.9 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 137.1, 125.7, 123.3, 123.2, 119.5, 116.1, 114.5, 110.3, 75.8, 23.5.
5-Bromo-3-(2-nitroethyl)-1H-indole (4c): Brown solid; yield 95%; 1H NMR (600 MHz, CDCl3) δ 8.18 (brs, 1H), 7.68 (d, J = 1.8 Hz, 1H), 7.29 (dd, J = 8.7, 1.8 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 7.03 (d, J = 1.8 Hz, 1H), 4.64 (t, J = 7.3 Hz, 2H), 3.41 (t, J = 7.3 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 134.9, 128.5, 125.4, 124.0, 120.8, 113.2, 113.1, 109.7, 75.7, 23.4.
2-(1H-indol-3-yl)ethanamine (5a): To a solution of 3-(2-nitroethyl)-1H-indole 4a (1.9 g, 10 mmol) in methanol (30 mL), was added 10% Pd/C (0.08 g). H2 was passed in at room temperature, then hydrogenated under normal pressure for 24 h. The completion of the reaction was monitored by TLC. The reaction mixture was then filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/methanol (10:1) as the mobile phase to provide 2-(1H-indol-3-yl)ethanamine 5a as an white solid in a yield of 98%. 1H NMR (600 MHz, CDCl3) δ 11.04 (brs, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.17 (s, 1H), 7.11 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H), 2.70 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 136.9, 127.9, 123.2, 121.4, 118.9, 113.0, 111.9, 43.1, 29.8.
3.5. General Procedure for Compounds 5b–c
To a solution of substituted 3-(2-nitroethyl)-1
H-indole
4b–
c (10 mmol) in THF (30 mL), was added LiAlH
4 (20 mmol) slowly. After the addition, it was heated at reflux for about 5 h. The completion of the reaction was monitored by TLC. The mixture was allowed to cool to room temperature and quenched by dropwise addition of saturated Na
2SO
4 solution. The resulting suspension mixture was filtered through Celite, and the filtrate was extracted with EtOAc (30 mL × 3). The combined organic phase was washed with H
2O (20 mL × 3) and brine (20 mL × 3), dried over anhydrous MgSO
4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/methanol (10:1) as the mobile phase to provide the compounds
5b–
c as a gray solid [
10].
2-(6-Bromo-1H-indol-3-yl)ethanamine (5b): White solid; yield 92%; 1H NMR (600 MHz, CDCl3) δ 11.08 (brs, 1H), 7.52 (d, J = 1.8 Hz, 1H), 7.47 (d, J = 8.7 Hz, 1H), 7.18 (s, 1H), 7.09 (dd, J = 8.7, 1.8 Hz, 1H), 3.53 (s, 2H), 2.83 (t, J = 6.4 Hz, 2H), 2.77 (t, J = 6.4 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 137.7, 126.9, 124.4, 121.5, 120.7, 114.5, 114.2, 113.1, 42.6, 28.7.
2-(5-Bromo-1H-indol-3-yl)ethanamine (5c): White solid; yield 88%; 1H NMR (600 MHz, CDCl3) δ 11.17 (brs, 1H), 7.75 (d, J = 1.8 Hz, 1H), 7.31–7.21 (m, 2H), 7.16 (s, 1H), 3.51 (s, 2H), 2.83 (t, J = 6.4 Hz, 2H), 2.74 (t, J = 6.4 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 135.4, 129.7, 124.9, 124.0, 123.7, 121.1, 113.9, 111.2, 42.4, 28.5.
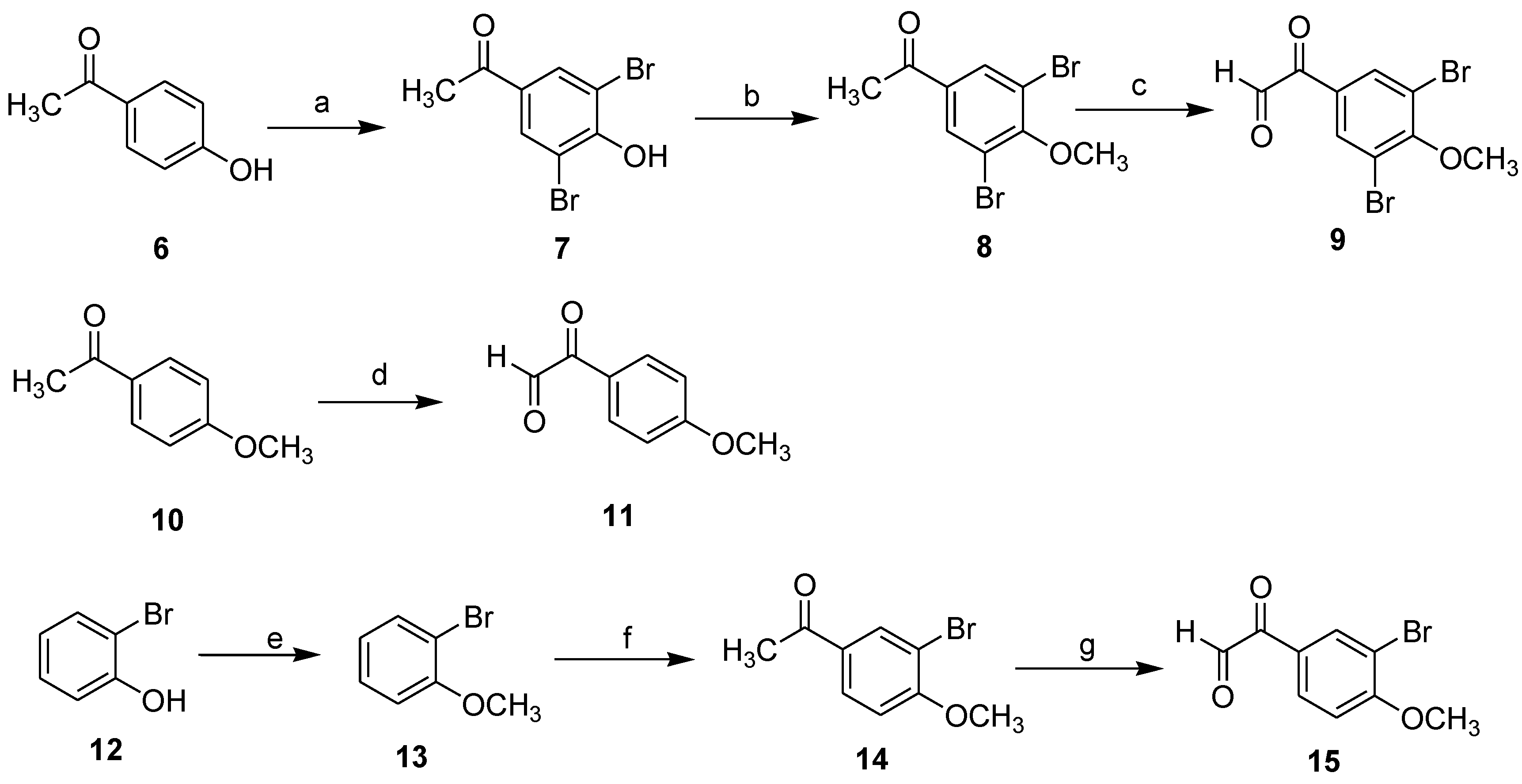
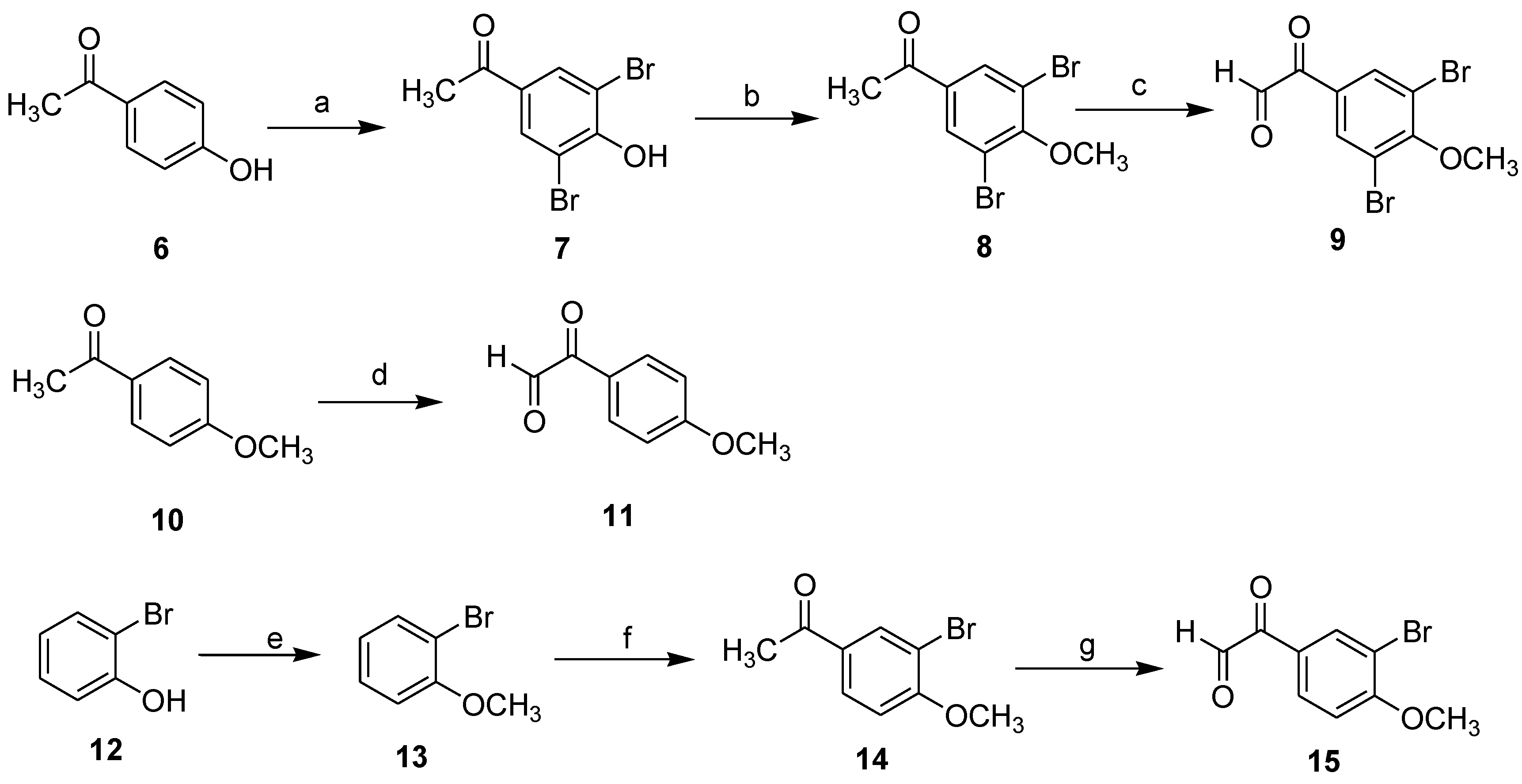
1-(3,5-Dibromo-4-hydroxyphenyl)ethanone (7): To a suspended mixture of 1-(4-hydroxyphenyl)ethanone (4.08 g, 30 mmol) and deionized water (200 mL), was added N-Bromosuccinimide (8.01 g, 45 mmol). The reaction mixture was heated to 60 °C, then 40% (v/v) H2SO4 (20 mL) was added. After stirred for about 10 h and the completion of the reaction was monitored by TLC, the reaction mixture was filtered through Celite to afford a white crude product. The crude product was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide a compound 1-(3,5-dibromo-4-hydroxyphenyl)ethanone (7) as a white solid in the yield of 92%.
1-(3,5-Dibromo-4-methoxyphenyl)ethanone (8): To a solution of 7 (20 mmol) in anhydrous acetone (50 mL) were added potassium carbonate (30.0 mmol) and dimethyl sulfate (2.84 mL, 30.0 mmol), and refluxed for 2 h. After cooled to room temperature, the reaction mixture was filtered through Celite, and the filtrate was concentrated under vacuum. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:2) as the mobile phase to provide the desired compound 8 as a white solid in the yield of 95%. 1H NMR (600 MHz, CDCl3) δ 8.08 (s, 2H), 3.93 (s, 3H), 2.56 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 194.5, 158.1, 135.1, 133.0, 118.7, 60.9, 26.6.
3.6. General Procedure for Compounds 9, 11, 15
To a solution of dioxane/deionized water (20:1) (30 mL) was added SeO
2 (12 mmol). The mixture was heated to 80 °C and stirred until the solid dissolved. It was followed by addition of substituted phenylethanone
8,
10,
14 (10 mmol), and was refluxed for about 12 h and the completion of the reaction was monitored by TLC. The hot solution was filtered through Celite, and the filtrate was concentrated under vacuum. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide the desired compound
9,
11,
15 [
11].
2-(3,5-Dibromo-4-methoxyphenyl)-2-oxoacetaldehyde (9): Light brown solid; yield 71%; 1H NMR (600 MHz, CDCl3) δ 9.42 (s, 1H), 8.28 (s, 1H), 8.22 (s, 1H), 3.85 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.6, 191.9, 157.9, 134.5, 132.3, 118.3, 61.2.
2-(4-Methoxyphenyl)-2-oxoacetaldehyde (11): Light brown solid; yield 75%; 1H NMR (600 MHz, CDCl3) δ 9.53 (s, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 3.83 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 195.2, 192.5, 164.0, 132.4, 126.7, 114.4, 56.1.
2-(3-Bromo-4-methoxyphenyl)-2-oxoacetaldehyde (15): Light brown solid; yield 74%; 1H NMR (600 MHz, CDCl3) δ 9.43 (s, 1H), 8.28 (s, 1H), 8.09 (d, J = 8.7 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H), 3.93 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 194.4, 192.6, 159.9, 134.8, 131.7, 127.8, 112.7, 110.9, 57.2.
1-(3-Bromo-4-methoxyphenyl)ethanone (14): To a suspension mixture of anhydrous AlCl3 (3.0 g, 22.5 mmol) in CS2 (50 mL) , was added 1-bromo-2-methoxybenzene (2.79 g, 15 mmol). The above mixture was maintained at room temperature and stirred for 0.5 h, and then acetyl chloride (1.6 mL, 22.5 mol) was added dropwise. After the addition, stirring was continued for another 0.5 h. And then, the reaction mixture was refluxed for 2 h. After cooled to room temperature, it was poured into 80 mL of ice-water containing 20 mL of concentrated hydrochloric acid and stirred to reach room temperature. The mixture was extracted with CH2Cl2 (20 mL × 3). The combined organic phase was washed with H2O (20 mL × 3), 10% aqueous NaOH (10 mL × 2), H2O (20 mL × 3) and brine (20 mL × 3), dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified by recrystallization from petroleum ether (20 mL) to afford 1-(3-bromo-4-methoxyphenyl)ethanone (14) as a brown solid in the yield of 76%.
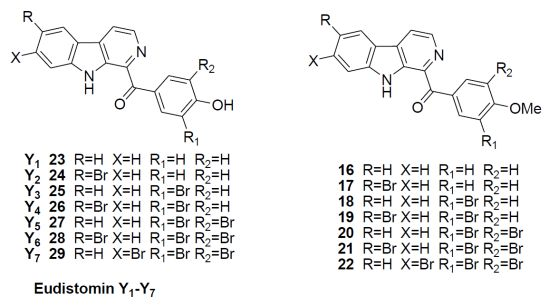
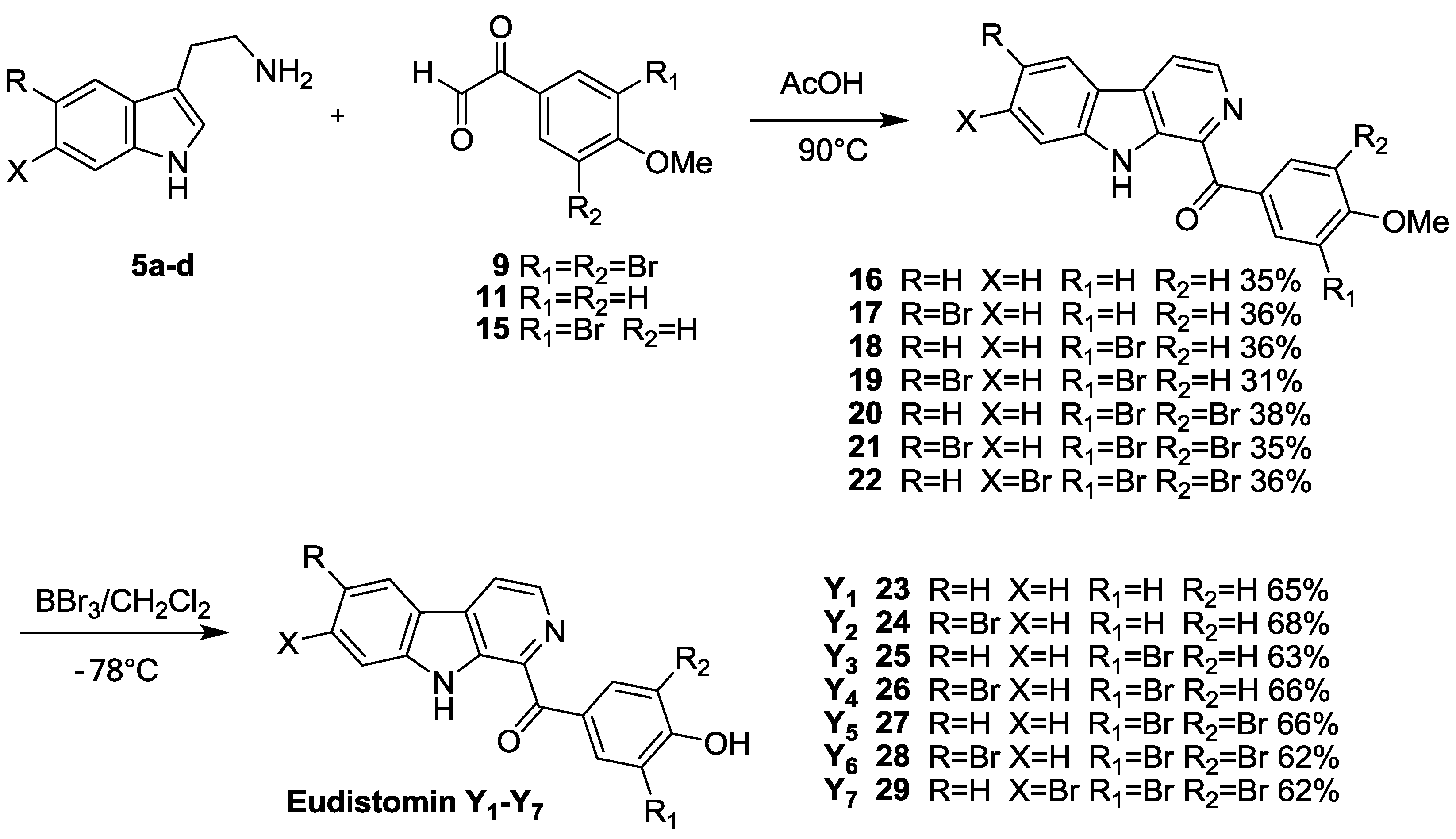
3.7. General Procedure for Compounds 16–22
To a solution of substituted phenylglyoxal 9, 11, 15 (3 mmol) in AcOH (30 mL), was added substituted tryptamine 5a–d (3 mmol). The above mixture was heated at 90 °C for 10 h, then cooled and adjusted pH to 5 by adding concentrated ammonium hydroxide. The resulting mixture was diluted with 100 mL of water and extracted with EtOAc (30 mL × 3). The combined organic phase was washed with H2O (20 mL × 3) and brine (20 mL × 3), dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide the desired compounds 16–22.
(4-Methoxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (16): Yellow solid; yield 35%; mp 182–183 °C; 1H NMR (600 MHz, CDCl3) δ 10.46 (brs, 1H), 8.60 (d, J = 5.0 Hz, 1H), 8.45 (d, J = 8.7 Hz, 2H), 8.17 (d, J = 7.8 Hz, 1H), 8.15 (d, J = 5.0 Hz, 1H), 7.62–7.58 (m, 2H), 7.34 (t, J = 7.8 Hz, 1H), 7.03 (d, J = 8.7 Hz, 2H), 3.91 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.7, 163.4, 141.1, 138.0, 137.4, 137.1, 134.0, 131.7, 130.4, 129.4, 122.0, 121.0, 120.8, 118.4, 113.6, 112.1, 55.7; HRMS: m/z calcd. for C19H15N2O2+, 303.1133; found: 303.1130.
(6-Bromo-9H-pyrido[3,4-b]indol-1-yl)(4-methoxyphenyl)methanone (17): Yellow solid; yield 36%; mp 194–195 °C; 1H NMR (600 MHz, CDCl3) δ 10.48 (brs, 1H), 8.62 (d, J = 5.0 Hz, 1H), 8.45 (d, J = 8.7 Hz, 2H), 8.29 (d, J = 1.8 Hz, 1H), 8.10 (d, J = 5.0 Hz, 1H), 7.68 (dd, J = 8.7, 1.8 Hz, 1H), 7.48 (d, J = 8.7 Hz, 1H), 7.04 (d, J = 8.7 Hz, 2H), 3.92 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 193.4, 163.5, 139.7, 138.3, 137.6, 137.4, 134.0, 132.1, 130.5, 130.1, 124.7, 122.8, 120.8, 118.5, 113.6, 113.5, 55.7; HRMS: m/z calcd. for C19H14N2O2Br+, 381.0239; found: 381.0237.
(3-Bromo-4-methoxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (18): Yellow solid; yield 36%; mp 192–193 °C; 1H NMR (600 MHz, CDCl3) δ 10.40 (brs, 1H), 8.69 (d, J = 1.8 Hz, 1H), 8.59 (d, J = 5.0 Hz, 1H), 8.49 (dd, J = 8.7, 2.2 Hz, 1H), 8.15 (m, 2H), 7.60 (m, 2H), 7.35–7.33 (m, 1H), 7.01 (d, J = 8.7 Hz, 1H), 3.99 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 192.2, 159.4, 141.1, 138.1, 137.5, 136.9, 136.5, 133.1, 131.8, 131.4, 129.5, 122.0, 120.9, 118.7, 112.2, 111.5, 110.9, 56.6; HRMS: m/z calcd. for C19H14N2O2Br+, 381.0239; found: 381.0220.
(3-Bromo-4-methoxyphenyl)(6-bromo-9H-pyrido[3,4-b]indol-1-yl)methanone (19): Yellow solid; yield 31%; mp 195–196 °C; 1H NMR (600 MHz, CDCl3) δ 10.50 (brs, 1H), 8.68 (d, J = 1.8 Hz, 1H), 8.59 (d, J = 5.0 Hz, 1H), 8.47 (dd, J = 8.7, 2.2 Hz, 1H), 8.26 (s, 1H), 8.08 (d, J = 5.0 Hz, 1H), 7.66 (dd, J = 8.7, 2.2 Hz, 1H), 7.46 (d, J = 8.7 Hz, 1H), 7.00 (d, J = 8.7 Hz, 1H), 3.99 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 191.9, 159.5, 139.6, 138.3, 137.5, 136.8, 133.2, 132.2, 131.1, 130.7, 124.7, 122.7, 118.7, 113.6, 111.5, 110.9, 56.6; HRMS: m/z calcd. for C19H13N2O2Br2+, 458.9344; found: 458.9327.
(3,5-Dibromo-4-methoxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (20): Yellow solid; yield 38%; mp 197–198 °C; 1H NMR (600 MHz, CDCl3) δ 10.38 (brs, 1H), 8.62 (d, J = 5.0 Hz, 1H), 8.59 (s, 1H), 8.20–8.18 (m, 1H), 7.65–7.62 (m, 2H), 7.38–7.36 (m, 1H), 3.98 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 191.5, 157.5, 141.2, 138.4, 137.5, 136.0, 135.6, 132.1, 129.7, 122.1, 121.2, 120.9, 119.2, 118.1, 112.2, 60.9; HRMS: m/z calcd. for C19H13N2O2Br2+, 458.9344; found: 458.9333.
(6-Bromo-9H-pyrido[3,4-b]indol-1-yl)(3,5-dibromo-4-methoxyphenyl)methanone (21): Yellow solid; yield 35%; mp 234–235 °C; 1H NMR (600 MHz, CDCl3) δ 10.41 (brs, 1H), 8.64 (d, J = 5.0 Hz, 1H), 8.60 (s, 2H), 8.32 (d, J = 1.8 Hz, 1H), 8,16 (d, J = 5.0 Hz, 1H), 7.72 (dd, J = 8.7, 1.8 Hz, 1H), 7.52 (d, J = 8.7 Hz, 1H), 3.99 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 191.3, 157.6, 139.7, 138.7, 137.6, 136.1, 135.4, 134.1, 132.5, 131.0, 124.9, 122.7, 119.3, 118.1, 114.0, 113.8, 60.9; HRMS: m/z calcd. for C19H12N2O2Br3+, 536.8449; found: 536.8442.
(7-Bromo-9H-pyrido[3,4-b]indol-1-yl)(3,5-dibromo-4-methoxyphenyl)methanone (22): Yellow solid; yield 36%; mp 216–217 °C; 1H NMR (600 MHz, CDCl3) δ 10.38 (brs, 1H), 8.63 (d, J = 5.0 Hz, 1H), 8.59 (s, 2H), 8,16 (d, J = 5.0 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.78 (d, J = 1.8 Hz, 1H), 7.48 (dd, J = 8.7, 1.8 Hz, 1H), 3.98 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 191.3, 157.7, 141.9, 138.9, 137.5, 136.1, 135.4, 131.5, 124.7, 123.5, 123.2, 119.9, 119.1, 118.1 115.4, 60.9; HRMS: m/z calcd. for C19H12N2O2Br3+, 536.8449; found: 536.8464.
3.8. General Procedure for Compounds 23–29
To a solution of the compounds 16–22 (0.5 mmol) in CH2Cl2 (10mL) at −78 °C under argon atmosphere, was slowly added dropwise BBr3 (5 mmol). The reaction mixture was stirred and warmed to r.t. and stirred for 24 h. NaOH solution (5 mL, 2 mol/L) was then slowly added dropwise. After addition, a short period of stirring was continued, and then the solution was acidified with hydrochloric acid (20 mL, 2 mol/L), followed by extraction with CH2Cl2 (30 mL × 3). The combined organic phase was washed with H2O (20 mL × 3) and brine (20 mL × 3), dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography using silica gel as the stationary phase and using ethyl acetate/hexane (1:3) as the mobile phase to provide the desired compounds eudistomins Y1–Y7 23–29.
(4-Hydroxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (Eudistomins Y1 23): Yellow solid; yield 65%; mp 217–218 °C; 1H NMR (600 MHz, acetone-d6) δ 11.30(s, 1H), 9.26 (s, 1H), 8.56 (d, J = 5.0 Hz, 1H), 8.47 (d, J = 8.8 Hz, 2H), 8.38 (d, J = 5.0 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7. 34 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 8.8 Hz, 2H); 13C NMR (151 MHz, acetone-d6) δ 192.0, 163.1, 142.5, 139.0, 138.4, 137.6, 135.0, 131.0, 130.2, 129.4, 122.7, 122.1, 121.3, 119.7, 115.6, 113.5; HRMS: m/z calcd. for C18H11N2O2−, 287.0821; found: 287.0830.
(6-Bromo-9H-pyrido[3,4-b]indol-1-yl)(4-hydroxyphenyl)methanone (Eudistomins Y2 24): Yellow solid; yield 68%; mp 247–248 °C; 1H NMR (600 MHz, acetone-d6) δ 11.46 (s, 1H), 9.30 (s, 1H), 8.60 (d, J = 5.0 Hz, 1H), 8.53 (d, J = 1.8 Hz, 1H), 8.47 (d, J =8.8 Hz, 2H), 8.43 (d, J = 5.0 Hz, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.76 (dd, J = 8.8, 1.8 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H); 13C NMR (151 MHz, acetone-d6) δ 192.2, 162.8, 141.1, 139.0, 138.5, 137.5, 135.1, 132.3, 131.1, 130.0, 125.4, 123.5, 119.1, 115.5, 113.5, 112.0; HRMS: m/z calcd. for C18H10N2O2Br−, 364.9926; found: 364.9925.
(3-Bromo-4-hydroxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (Eudistomins Y3 25): Yellow solid; yield 63%; mp 231–232 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.99 (s, 1H), 11.28 (s, 1H), 8.57 (d, J = 2.2 Hz, 1H), 8.54 (d, J = 4.4 Hz, 1H), 8.43 (d, J = 4.4 Hz, 1H), 8.32 (d, J = 7.7 Hz, 1H), 8.24 (dd, J = 8.8, 2.2 Hz, 1H), 7.79 (d, J = 8.8 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 190.1, 158.2, 141.6, 137.0, 136.7, 136.4, 135.8, 132.5, 131.0, 129.6, 128.9, 121.8, 120.1, 120.0, 118.7, 115.6, 112.9, 108.8; HRMS: m/z calcd. for C18H10N2O2Br−, 364.9926; found: 364.9932.
(3-Bromo-4-hydroxyphenyl)(6-bromo-9H-pyrido[3,4-b]indol-1-yl)methanone (Eudistomins Y4 26): Yellow solid; yield 66%; mp 265–266 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.13 (s, 1H), 11.30 (s, 1H), 8.61 (s, 1H), 8.57 (d, J = 5.5 Hz, 1H), 8.56 (s, 1H), 8.50 (d, J = 5.5 Hz, 1H), 8.23 (dd, J = 8.8, 2.2 Hz, 1H), 7.75–7.72 (m, 1H), 7.13 (d, J = 8.8 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 189.9, 158.3, 140.3, 137.3, 137.1, 136.4, 135.9, 132.5, 131.4, 129.9, 129.4, 124.5, 122.0, 119.1, 115.6, 114.9, 112.2, 108.8; HRMS: m/z calcd. for C18H9N2O2Br2−, 442.9031; found: 442.9012.
(3,5-Dibromo-4-hydroxyphenyl)(9H-pyrido[3,4-b]indol-1-yl)methanone (Eudistomins Y5 27): Yellow solid; yield 66%; mp 267–268 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.04 (s, 1H), 10.96 (s, 1H), 8.57 (d, J = 5.5 Hz, 1H), 8.54 (s, 2H), 8.47 (d, J = 5.5 Hz, 1H), 8.33 (d, J = 7.3 Hz, 1H), 7.81 (d, J = 8.2 Hz, 1H), 7.61 (t, J = 7.3 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 188.9, 154.5, 141.7, 137.1, 136.0, 135.9, 135.4, 131.2, 131.0, 129.0, 121.9, 120.3, 120.0, 119.1, 113.0, 110.9; HRMS: m/z calcd. for C18H9N2O2Br2−, 442.9031; found: 442.9030.
(6-Bromo-9H-pyrido[3,4-b]indol-1-yl)(3,5-dibromo-4-hydroxyphenyl)methanone (Eudistomins Y6 28): Yellow solid; yield 62%; mp 277–278 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.16 (s, 1H), 10.97 (brs, 1H), 8.61 (s, 1H), 8.59 (d, J = 4.4 Hz, 1H), 8.52 (s, 2H), 8.51 (d, J = 4.4 Hz, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.73 (dd, J = 8.8, 2.2 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 188.8, 154.6, 140.4, 137.4, 136.4, 136.0, 135.4, 131.5, 130.8, 130.1, 124.5, 122.0, 119.6, 115.0, 112.3, 110.9; HRMS: m/z calcd. for C18H8N2O2Br3−, 520.8136; found: 520.8134.
(7-Bromo-9H-pyrido[3,4-b]indol-1-yl)(3,5-dibromo-4-hydroxyphenyl)methanone (Eudistomins Y7 29): Yellow solid; yield 62%; mp 297–298 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.11 (s, 1H), 10.96 (brs, 1H), 8.58 (d, J = 4.6 Hz, 1H), 8.53 (s, 2H), 8.46 (d, J = 4.6 Hz, 1H), 8.27 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 1.8 Hz, 1H), 7.45 (dd, J = 8.2, 1.8 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 188.8, 154.6, 142.5, 137.7, 136.3, 136.0, 135.4, 130.8, 130.6, 123.7, 123.2, 121.8, 119.2, 115.6, 110.8; HRMS: m/z calcd. for C18H8N2O2Br3−, 520.8136; found: 520.8141.




{kind=link}
{kind=link}
{kind=link}
{kind=link}