Hippuristanol Reduces the Viability of Primary Effusion Lymphoma Cells both in Vitro and in Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
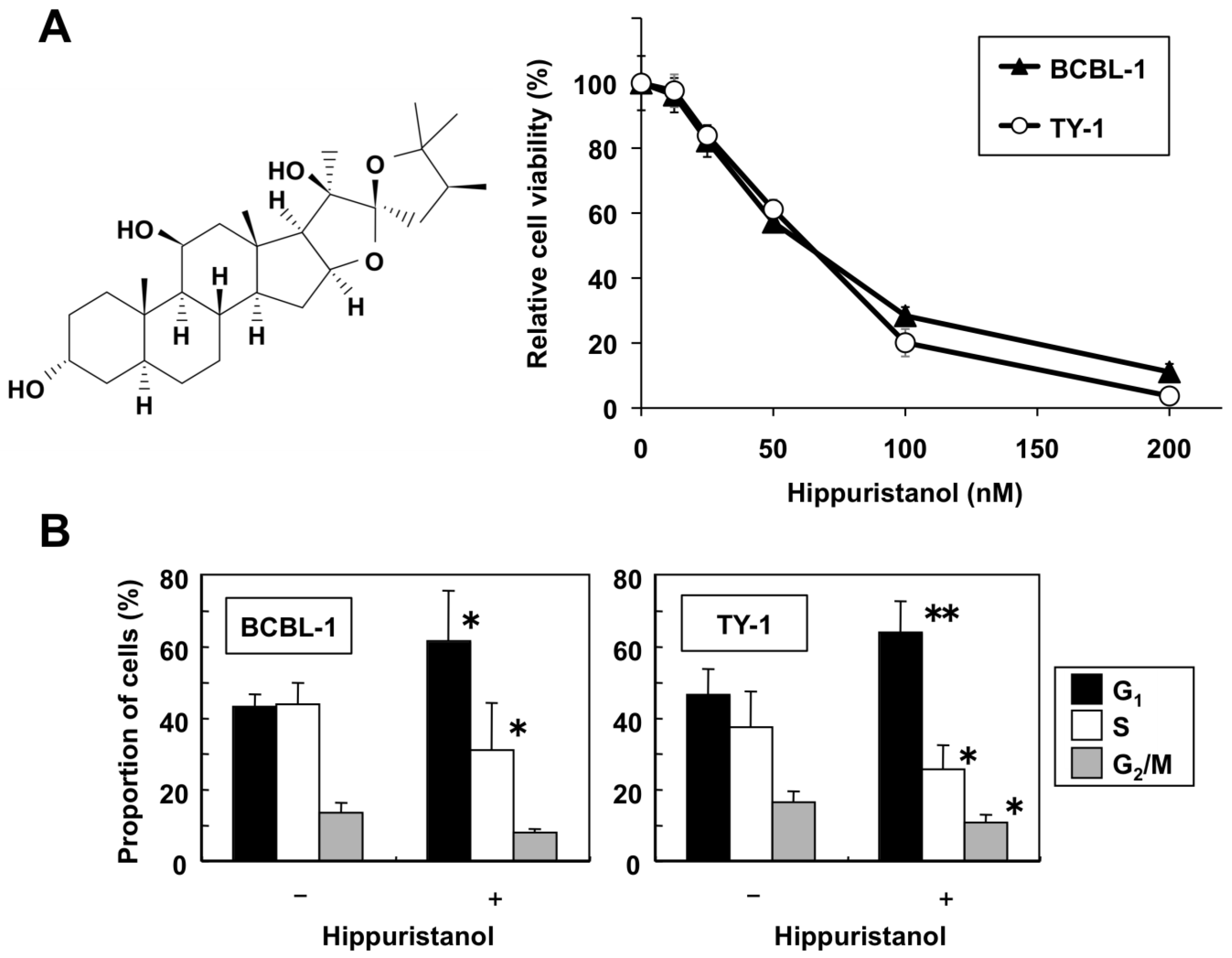
2.1. Hippuristanol Dose-Dependently Inhibits PEL Cell Viability
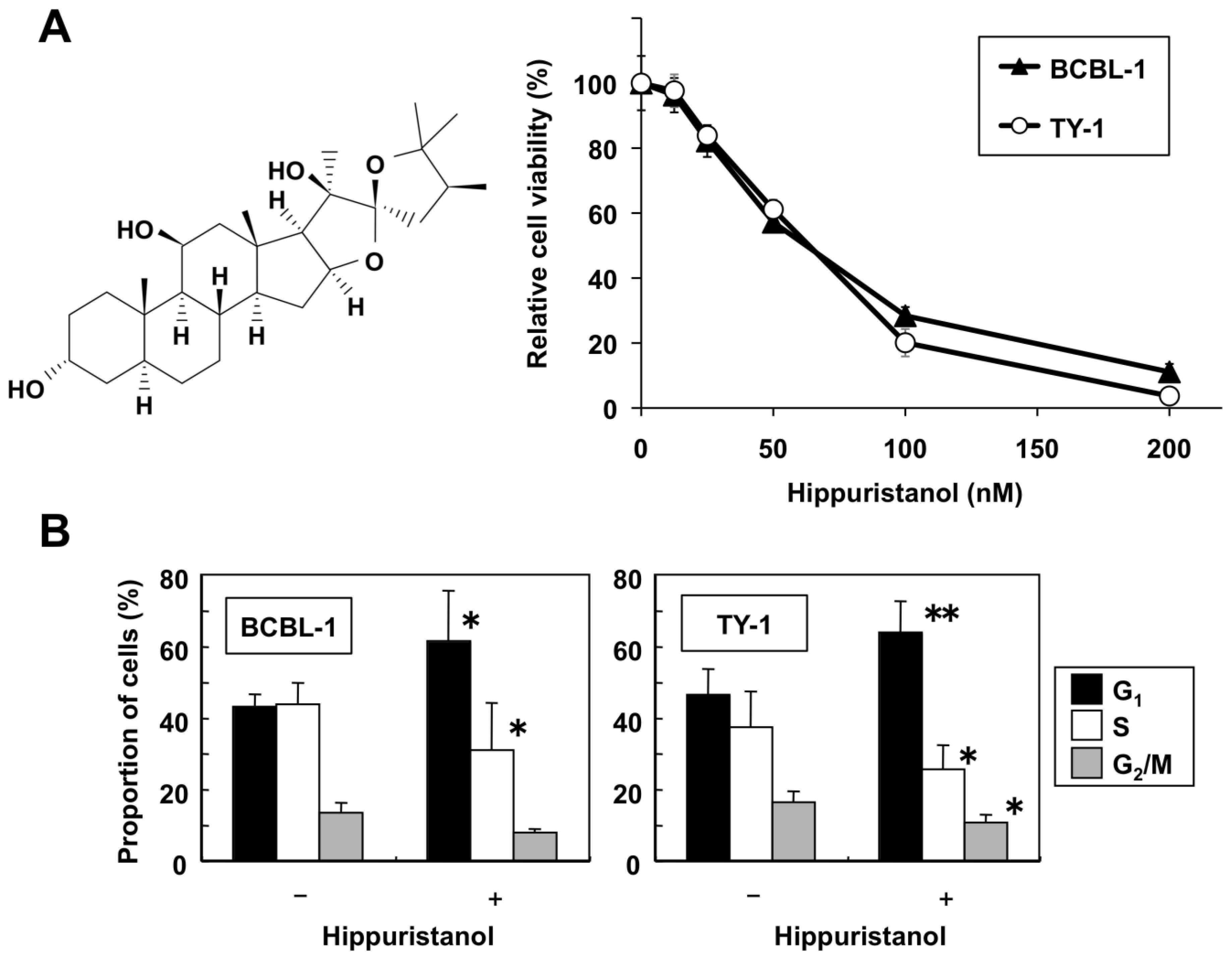
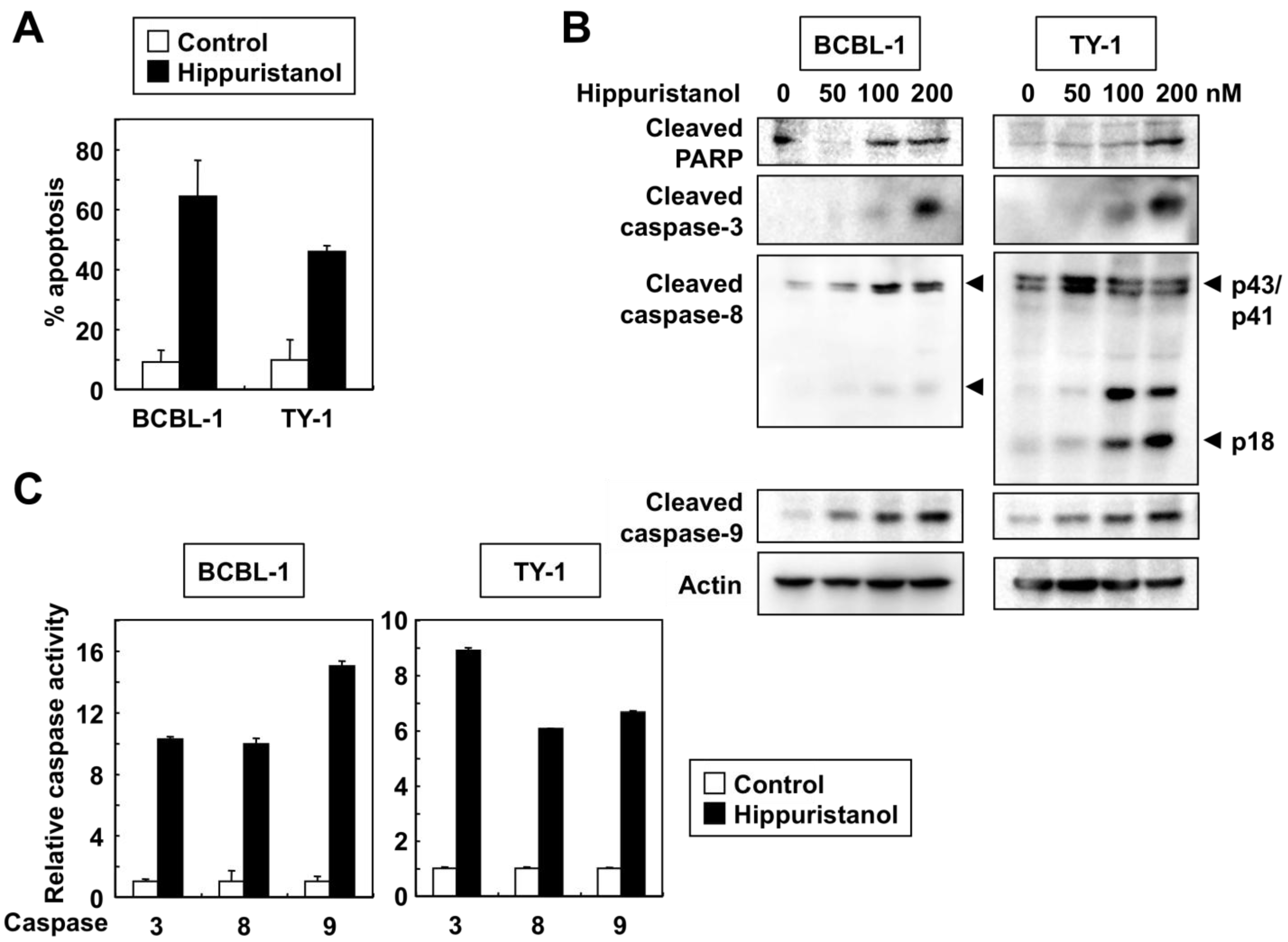
2.2. Effects of Hippuristanol on PEL Cell Cycle and Apoptosis
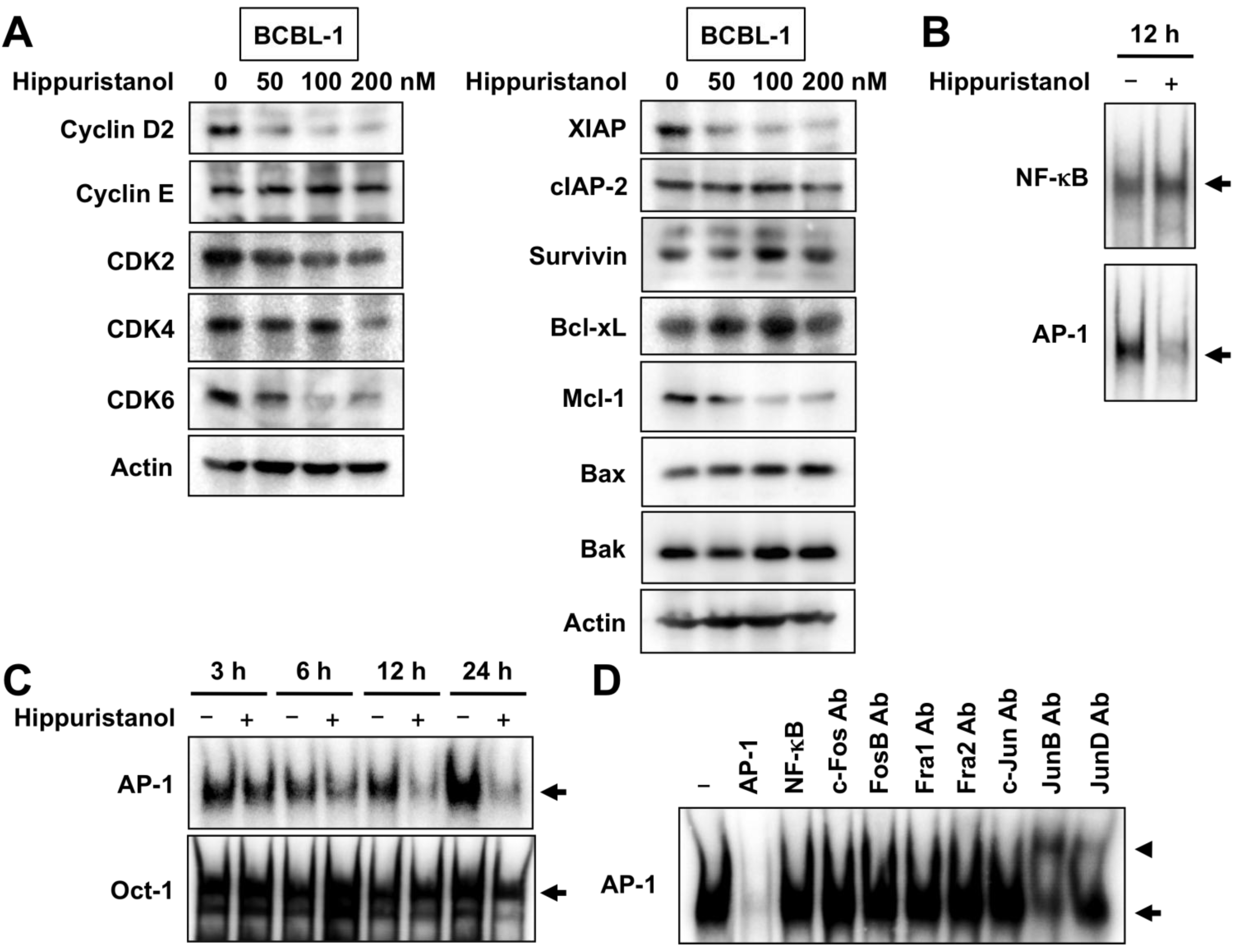
2.3. Effects of Hippuristanol on Expression of Cell Cycle and Apoptosis Regulatory Proteins in PEL Cells


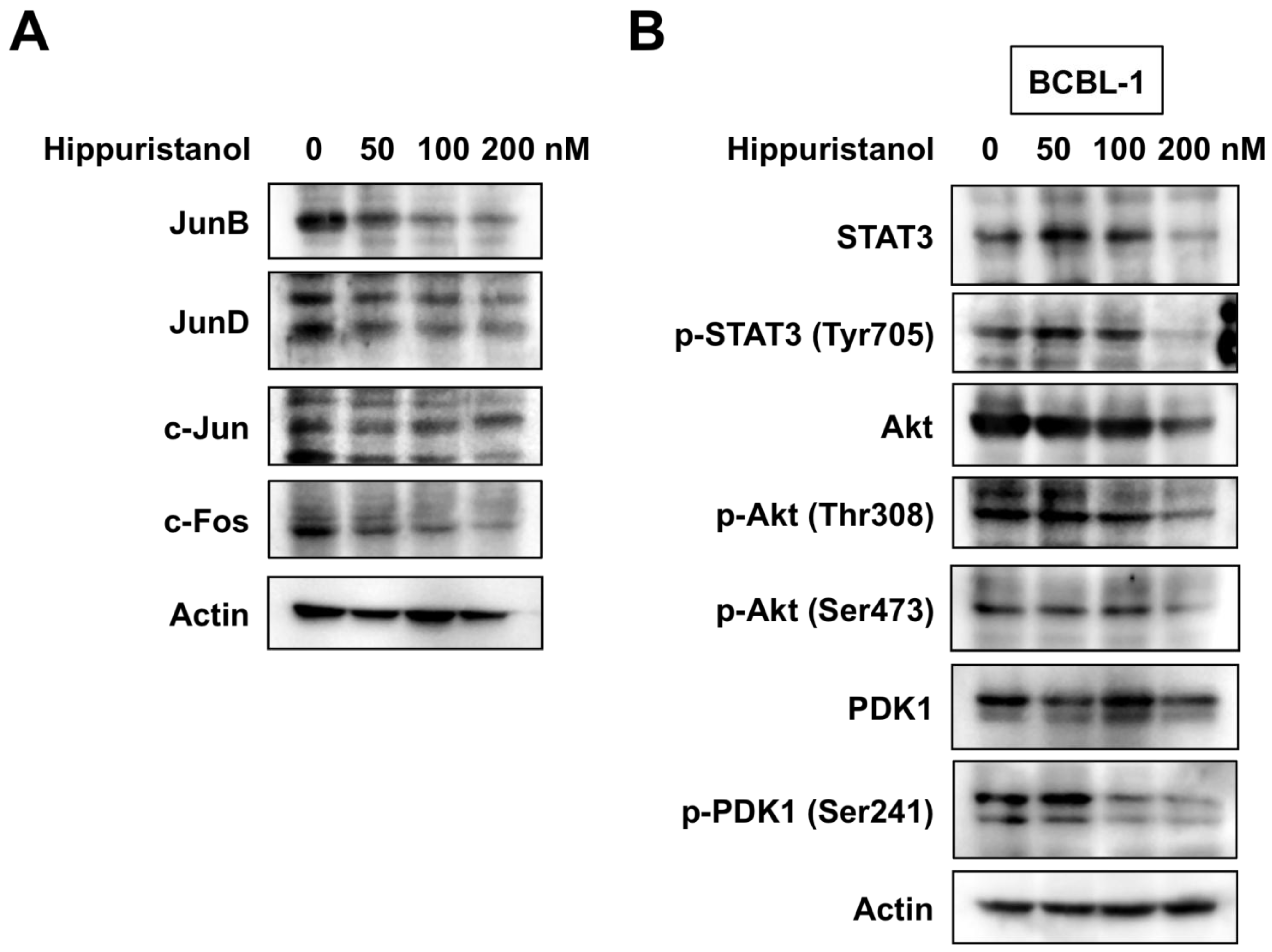
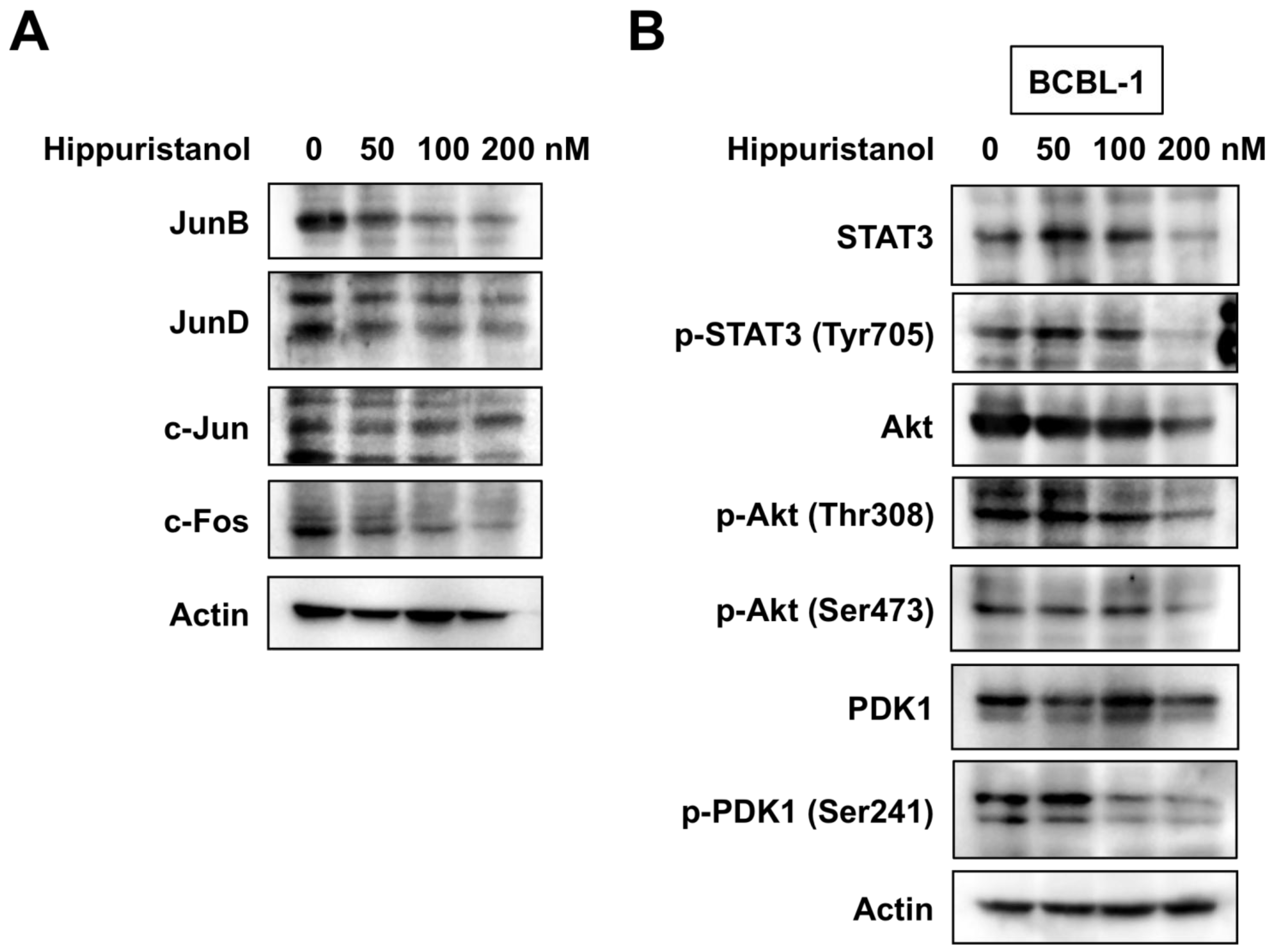
2.4. Hippuristanol Efficiently Blocks Constitutive Activation of AP-1 in PEL Cells
2.5. Hippuristanol Inhibits Activation of STAT3 and Akt in PEL Cells
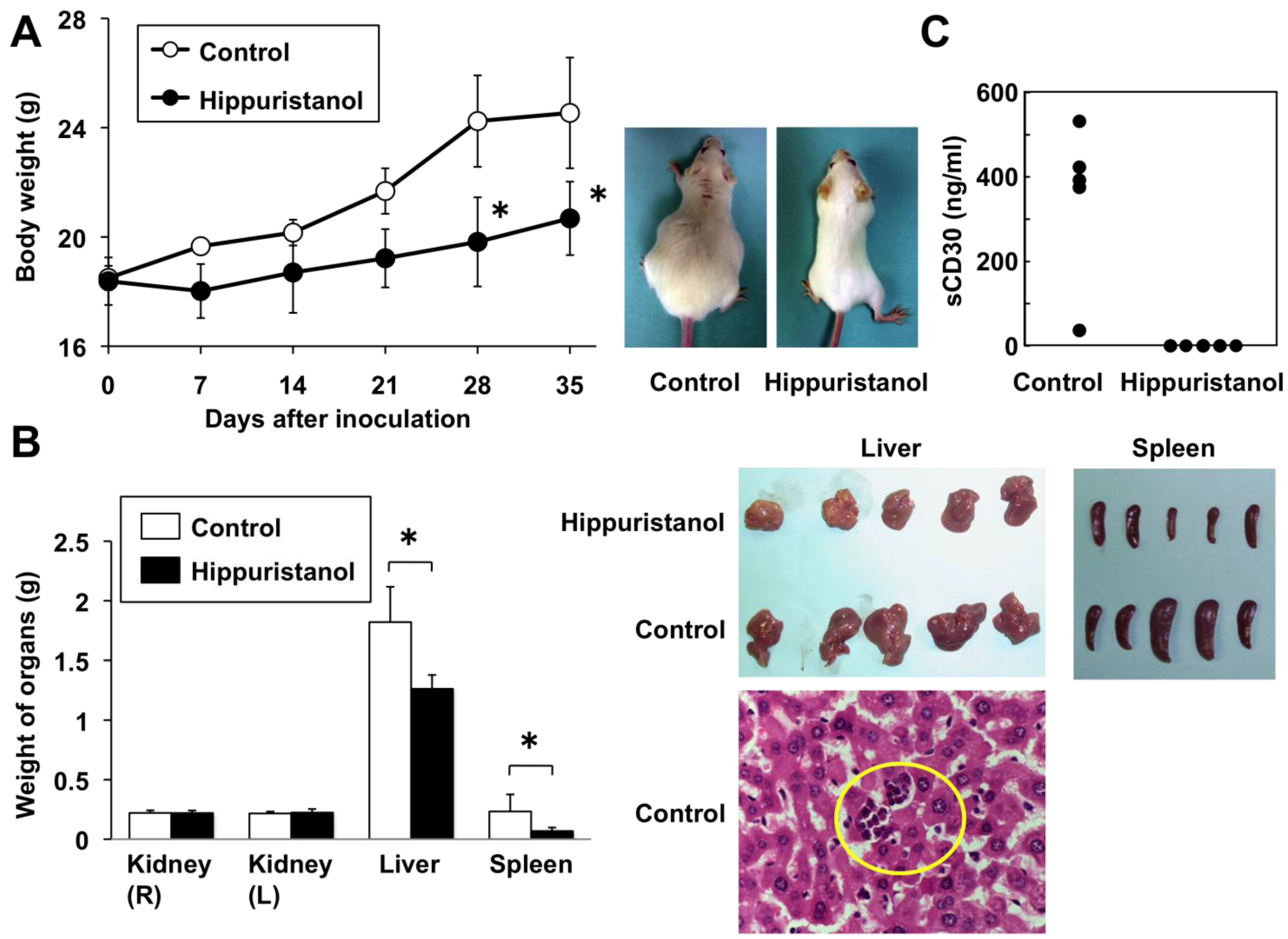
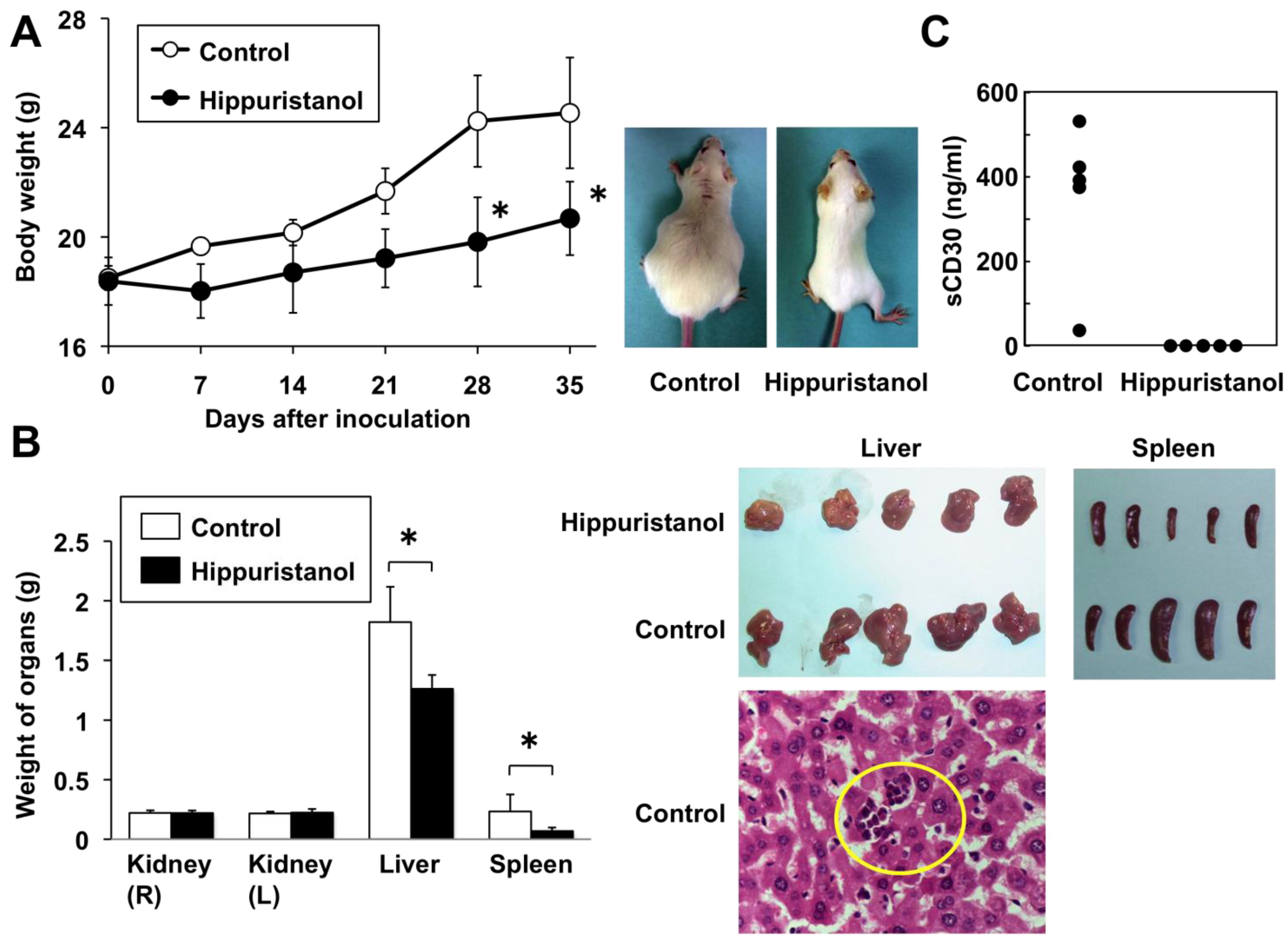
2.6. In Vivo Effects of Hippuristanol in Immunodeficient Mice Inoculated with PEL Cells
3. Discussion
4. Experimental Section
4.1. Cell Lines and Reagents
4.2. Assesement of Cell Viability and Apoptosis
4.3. Cell Cycle Analysis
4.4. In Vitro Measurement of Caspase Activity
4.5. Western Blotting
4.6. Electrophoretic Mobility Shift Assay (EMSA)
4.7. In Vivo Therapeutic Effect of Hippuristanol
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar]
- Green, I.; Espiritu, E.; Ladanyi, M.; Chaponda, R.; Wieczorek, R.; Gallo, L.; Feiner, H. Primary lymphomatous effusions in AIDS: A morphological, immunophenotypic, and molecular study. Mod. Pathol. 1995, 8, 39–45. [Google Scholar]
- Boulanger, E.; Gérard, L.; Gabarre, J.; Molina, J.-M.; Rapp, C.; Abino, J.-F.; Cadranel, J.; Chevret, S.; Oksenhendler, E. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J. Clin. Oncol. 2005, 23, 4372–4380. [Google Scholar] [CrossRef]
- Keller, S.A.; Schattner, E.J.; Cesarman, E. Inhibition of NF-κB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 2000, 96, 2537–2542. [Google Scholar]
- Aoki, Y.; Feldman, G.M.; Tosato, G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 2003, 101, 1535–1542. [Google Scholar] [CrossRef]
- Uddin, S.; Hussain, A.R.; Al-Hussein, K.A.; Manogaran, P.S.; Wickrema, A.; Gutierrez, M.I.; Bhatia, K.G. Inhibition of phosphatidylinositol 3′-kinase/AKT signaling promotes apoptosis of primary effusion lymphoma cells. Clin. Cancer Res. 2005, 11, 3102–3108. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ishikawa, C.; Katano, H.; Yasumoto, T.; Mori, N. Fucoxanthin and its deacetylated product, fucoxanthinol, induce apoptosis of primary effusion lymphomas. Cancer Lett. 2011, 300, 225–234. [Google Scholar] [CrossRef]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef]
- Bordeleau, M.-E.; Mori, A.; Oberer, M.; Lindqvist, L.; Chard, L.S.; Higa, T.; Belsham, G.J.; Wagner, G.; Tanaka, J.; Pelletier, J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–220. [Google Scholar] [CrossRef]
- Lindqvist, L.; Pelletier, J. Inhibitors of translation initiation as cancer therapeutics. Future Med. Chem. 2009, 1, 1709–1722. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Ishikawa, C.; Machijima, Y.; Nakachi, S.; Senba, M.; Tanaka, J.; Mori, N. Effects of hippuristanol, an inhibitor of eIF4A, on adult T-cell leukemia. Biochem. Pharmacol. 2011, 81, 713–722. [Google Scholar] [CrossRef]
- Zhang, C.; Ao, Z.; Seth, A.; Schlossman, S.F. A mitochondrial membrane protein defined by a novel monoclonal antibody is preferentially detected in apoptotic cells. J. Immunol. 1996, 157, 3980–3987. [Google Scholar]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef]
- An, J.; Sun, Y.; Rettig, M.B. Transcriptional coactivation of c-Jun by KSHV-encoded LANA. Blood 2004, 103, 222–228. [Google Scholar] [CrossRef]
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.T.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010, 143, 897–910. [Google Scholar] [CrossRef]
- Casamayor, A.; Morrice, N.A.; Alessi, D.R. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: Identification of five sites of phosphorylation in vivo. Biochem. J. 1999, 342, 287–292. [Google Scholar] [CrossRef]
- Michai, M.; Goto, H.; Hattori, S.; Vaeteewoottacharn, K.; Wongkham, C.; Wongkham, S.; Okada, S. Soluble CD30: A possible serum tumor marker for primary effusion lymphoma. Asian Pacific J. Cancer Prev. 2012, 13, 4939–4941. [Google Scholar] [CrossRef]
- Brooks, A.R.; Shiffman, D.; Chan, C.S.; Brooks, E.E.; Milner, P.G. Functional analysis of the human cyclin D2 and cyclin D3 promoters. J. Biol. Chem. 1996, 271, 9090–9099. [Google Scholar]
- Fukada, T.; Ohtani, T.; Yoshida, Y.; Shirogane, T.; Nishida, K.; Nakajima, K.; Hibi, M.; Hirano, T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998, 17, 6670–6677. [Google Scholar] [CrossRef]
- Wang, X.H.; Liu, B.R.; Qu, B.; Xing, H.; Gao, S.L.; Yin, J.M.; Wang, X.F.; Cheng, Y.Q. Silencing STAT3 may inhibit cell growth through regulating signaling pathway, telomerase, cell cycle, apoptosis and angiogenesis in hepatocellular carcinoma: Potential uses for gene therapy. Neoplasma 2011, 58, 158–171. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Kaneko, S.; Feldman, R.I.; Nicosia, S.V.; Wang, H.G.; Tsang, B.K.; Cheng, J.Q. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J. Biol. Chem. 2004, 279, 5405–5412. [Google Scholar]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.-M.; Yang-Yen, H.-F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Invest. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar]
- Alvarez, J.V.; Febbo, P.G.; Ramaswamy, S.; Loda, M.; Richardson, A.; Frank, D.A. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005, 65, 5054–5062. [Google Scholar] [CrossRef]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef]
- Katano, H.; Hoshino, Y.; Morishita, Y.; Nakamura, T.; Satoh, H.; Iwamoto, A.; Herndier, B.; Mori, S. Establishing and characterizing a CD30-positive cell line harboring HHV-8 from a primary effusion lymphoma. J. Med. Virol. 1999, 58, 394–401. [Google Scholar] [CrossRef]
- Antalis, T.M.; Godbolt, D. Isolation of intact nuclei from hematopoietic cell types. Nucl. Acids Res. 1991, 19, 4301. [Google Scholar] [CrossRef]
- Mori, N.; Prager, D. Transactivation of the interleukin-1α promoter by human T cell leukemia virus type I and type II Tax proteins. Blood 1996, 87, 3410–3417. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ishikawa, C.; Tanaka, J.; Katano, H.; Senba, M.; Mori, N. Hippuristanol Reduces the Viability of Primary Effusion Lymphoma Cells both in Vitro and in Vivo. Mar. Drugs 2013, 11, 3410-3424. https://doi.org/10.3390/md11093410
Ishikawa C, Tanaka J, Katano H, Senba M, Mori N. Hippuristanol Reduces the Viability of Primary Effusion Lymphoma Cells both in Vitro and in Vivo. Marine Drugs. 2013; 11(9):3410-3424. https://doi.org/10.3390/md11093410
Chicago/Turabian StyleIshikawa, Chie, Junichi Tanaka, Harutaka Katano, Masachika Senba, and Naoki Mori. 2013. "Hippuristanol Reduces the Viability of Primary Effusion Lymphoma Cells both in Vitro and in Vivo" Marine Drugs 11, no. 9: 3410-3424. https://doi.org/10.3390/md11093410