Bioproduction of Chitooligosaccharides: Present and Perspectives

Division of Applied Bioscience & Biotechnology, Institute of Environment-Friendly Agriculture (IEFA), College of Agricultural and Life Sciences, Chonnam National University, Gwangju 500-757, Korea
*
Author to whom correspondence should be addressed.
Mar. Drugs 2014, 12(11), 5328-5356; https://doi.org/10.3390/md12115328
Submission received: 19 August 2014
/
Revised: 20 October 2014
/
Accepted: 21 October 2014
/
Published: 28 October 2014
(This article belongs to the Special Issue Advances in Marine Chitin and Chitosan)
Abstract
:Chitin and chitosan oligosaccharides (COS) have been traditionally obtained by chemical digestion with strong acids. In light of the difficulties associated with these traditional production processes, environmentally compatible and reproducible production alternatives are desirable. Unlike chemical digestion, biodegradation of chitin and chitosan by enzymes or microorganisms does not require the use of toxic chemicals or excessive amounts of wastewater. Enzyme preparations with chitinase, chitosanase, and lysozymeare primarily used to hydrolyze chitin and chitosan. Commercial preparations of cellulase, protease, lipase, and pepsin provide another opportunity for oligosaccharide production. In addition to their hydrolytic activities, the transglycosylation activity of chitinolytic enzymes might be exploited for the synthesis of desired chitin oligomers and their derivatives. Chitin deacetylase is also potentially useful for the preparation of oligosaccharides. Recently, direct production of oligosaccharides from chitin and crab shells by a combination of mechanochemical grinding and enzymatic hydrolysis has been reported. Together with these, other emerging technologies such as direct degradation of chitin from crustacean shells and microbial cell walls, enzymatic synthesis of COS from small building blocks, and protein engineering technology for chitin-related enzymes have been discussed as the most significant challenge for industrial application.
1. Introduction
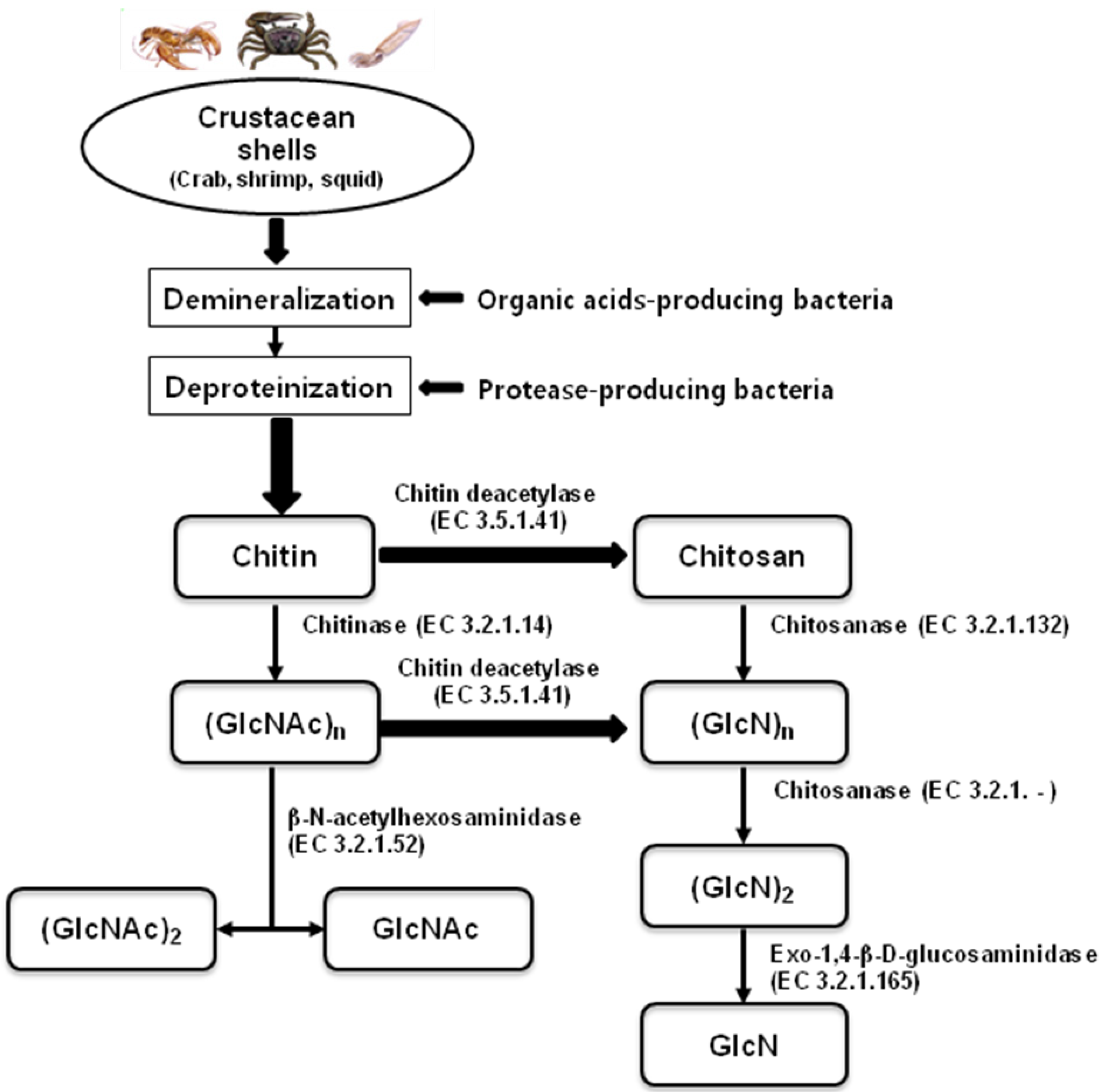
Chitin and chitosan have numerous applications as functional materials, as these natural polymers have excellent properties such as biocompatibility, biodegradability, non-toxicity, and adsorption. Chitin from crustacean shells is commonly obtained using inorganic acids for demineralization, with strong alkali for deproteinization [1,2]. These chemical processes have several drawbacks, including being a source of pollution [3] and reduction of depolymerization, and thus, chitin quality [4,5].
These chemical processes can be replaced by biotechnological processing with microbes and their metabolites, including organic acids and enzymes, as shown in Figure 1. The strains most frequently applied include Lactobacillus sp. and Serratia marcescens [6,7]. Biofermentation of crab shell wastes with 10% inoculums of S. marcescens FS-3 resulted in 84% deproteinization and 47% demineralization after 7 days of incubation [8]. Co-fermentation of the bacteria Lactobacillus paracasei KCTC-3074 and S. marcescens FS-3 [6] and the successive two-step fermentation with the two bacteria [7] provided more promising results for the production of chitin from crab shells.
Chitin and chitosan are high molecular-weight polymers with poor solubility at neutral pH values. This property limits their potential uses in the fields of food, health, and agriculture. However, these limitations may be overcome by the use of their oligomers or monomers. In humans, chitin monomers are precursors of the disaccharide units in glycosaminoglycans (such as hyaluronic acid, chondroitin sulfate, and keratin sulfate), which are necessary to repair and maintain healthy cartilage and joint function. Chitooligosaccharides (oligosaccharides derived mainly from chitin or chitosan, COS) have the potential ability to improve food quality [9,10,11] and human health [12,13].
COS mixtures can be prepared from chitosan by using different physical methods, like hydrothermal [14], microwave [15], ultra sonication [16] and gamma-rays [17]. Chemical methods using acid [18,19], H2O2 [20] or NaNO2 [21], can yield COS. Of chemical methods for hydrolysis of chitosan [18,19,20,21], acid hydrolysis is probably the best known. Early studies had shown that fully deacetylated chitosan is degradedto COS in concentrated hydrochloric acid [18]. In later studies [19], using a variety of chitosans, the acid-catalyzed degradation rates of chitosans were shown to depend on degree of deacetylation (DD). Acid hydrolysis was found to be highly specific to cleavage of GlcNAc-GlcNAc and GlcNAc-GlcN and A-D glycosidic linkages, with two to three orders of magnitude higher rates than the GlcN-GlcN and GlcN-GlcNAc linkages. In the same study, it was shown that the rate of deacetylation was less than one-tenth of the rate of depolymerization in concentrated acid, whereas the two rates were found to be equal in dilute acid [19]. Even though acid hydrolysis has been commonly used to prepare COS, these chemical and physical methods escape from the scope of this review and will not be dealt with more.
Enzymatic hydrolysis of chitin and chitosan has been proposed as an alternative method during the past few decades. Enzymes with hydrolytic activity on chitin and chitosan include chitinase, chitosanase, lysozyme, cellulase, pectinase, protease, lipase, and pepsin. These chitinolytic and chitosanolytic enzymes all have different modes of action and specificity for substrate size. A flow chart for the bioproduction of chitin, chitosan, and their oligosaccharides from natural resources by using enzymes and microorganisms is shown in Figure 1. Enzymatic hydrolysis seems to be generally preferable to chemical methods because the reaction is performed under more gentle conditions and the MW distribution of the product is more controllable [22,23]. However, production of well-defined COS in terms of length (degree of polymerization, DP), degree of de-N-acetylation (DD), and sequence (pattern of acetylation, PA) by enzymatic conversion processes is not straight forward [24]. The expensive cost of chitinases and chitosanases limits their wide application on an industrial scale, even using immobilized enzymes [22,25,26].
We are primarily concerned with the enzymatic hydrolysis of polysaccharides obtained from crustacean shells for the bioproduction of COS. In this review, we introduce the preparation and bioproduction of COS with enzymes from chitinase- and chitosanase-producing bacteria and fungi. Limitations and challenges in the bioproduction of COS are also discussed.
Figure 1.
Flow chart for the bioproduction of chitin, chitosan, and their oligosaccharides from biological resources.
Figure 1.
Flow chart for the bioproduction of chitin, chitosan, and their oligosaccharides from biological resources.

2. Bioproduction of Chitin Oligosaccharides and Its Monomer
Chitin oligosaccharides are produced from chitin by using chitinolytic enzymes. These enzymes hydrolyze the glycoside bonds between the sugars and are thus glycoside hydrolases (GH). GHs are classified in the Carbohydrate-Active enZYmes database (CAZy) [27,28,29,30]. The CAZy classification is purely based on the amino acid sequence similarity, which gives very useful information since sequence and structure, and hence mechanism, are related. In CAZy system, enzyme properties such as substrate and product activities, exo- versus endo-binding, processivity, and the presence of additional modules are not taken into account [27,28,29,30]. Chitinases occur in families GH18 and GH19 [31,32]. Chitinases have the unique ability to hydrolyze GlcNAc-GlcNAc bonds and this property discriminates these enzymes from chitosanases.
Chitinases can be classified into two major categories (endochitinases and exochitinases), according to their mode of action. First, endochitinases (EC 3.2.1.14; created 1961) cleave the linkage between GlcNAc-GlcNAc, GlcN-GlcNAc, and GlcNAc-GlcN in chitin chains to release smaller and more soluble chitin oligomers of variable size. Second, β-N-acetylhexosaminidase (EC 3.2.1.52; created 1972) includes subcategories of both chitobiase (EC 3.2.1.29; created 1961, deleted 1972) and β-d-acetylglucosaminidase (GlcNAcase, EC 3.2.1.30; created 1961, deleted 1992) [33]. Chitobiase catalyzes the progressive release of chitobiose starting at the nonreducing or reducing end of chitin. GlcNAcase progressively breaks down chitin polymer or chitin oligomers from the non-reducing or reducing end of the molecules, releasing β-d-GlcNAc or α-d-GlcNAc.These enzymes were prepared from bacteria and fungi, as shown in Table 1.
In COS production, it is practical to use crude enzyme preparations, such as microbial culture supernatants and their solid powders. Preparations of cellulase, protease, lipase, pepsin, chitinase, chitosanase, and lysozyme are mainly applied as commercial enzyme sources for the hydrolytic cleavage of chitin and chitosan (Table 1).
Pretreatment is necessary for enzymatic hydrolysis, because the strong crystallinity and insolubility of chitin in an aquatic environment. For example, chitin is normally treated with strong acids, such as hydrochloric acid (for colloidal chitin) [34] or phosphoric acid (for swollen chitin) [35] to break down crystal structure and increase the accessibility of the substrate to the enzyme. In contrast to chitin, chitosan is soluble in dilute mineral acids, thus forming salts with acids.
Enzymatic or chemical hydrolysis of chitin and chitosan results in a mixture of oligomers, not any specific oligomer, even with prolonged reaction time. Several techniques for separation and purification of COS have been reported, like gel filtration [36], ultrafiltration [37], and ion exchange [38] and metal affinity [39] chromatography. Preparative separation of COS is most commonly based on size, through size exclusion chromatography SEC). A SEC system using SuperdexTM 30 (GE Healthcare) columns, coupled in series, allowed separation of COS with similar DP values ranging from DP 2 to DP 20, independently of DD and PA [36]. Separation of COS can be achieved using cation-exchange chromatography, because protonated amino groups on the deacetylated sugars interact with the ion-exchange resin. With this method, COS of identical DP was separated based on the number of deacetylated units [38].
2.1. N-Acetyl-β-d-glucosamine
N-Acetyl-β-d-glucosamine (GlcNAc) was obtained with 85% yield from β- and α-chitin within 1 and 7 days, respectively, using chitinase from Burkholderia cepacia TU09 [40]. GlcNAc and N,N′-diacetylchitobiose [(GlcNAc)2] were obtained 75% and 20% yield from β-chitin within 6 days using chitinase from Bacillus licheniformis SK-1. In addition, chitinase from SK-1 produced GlcNAc with 41% yield from α-chitin. In most cases, β-chitin from squid pen is a better substrate than α-chitin from crab or shrimp shells.
GlcNAc and N-acetyl chitooligosaccharides were produced from colloidal α-chitin using a crude enzyme from Paenibacillus illinoisensis KJA-424 [7]. The production rate of monomer GlcNAc increased continuously during incubation, while that of GlcNAc oligomers declined. The maximum production of GlcNAc was 1.71 mg mL−1 (yield of 62.2%) after 24 h of incubation. At the same time (GlcNAc)2, tri-N-acetylchitotriose [(GlcNAc)3], hepta-N-acetylchitoheptaose [(GlcNAc)7] and octa-N-acetylchitooctaose [(GlcNAc)8] were 0.13 mg mL−1 (yield of 4.9%), 0.03 mg mL−1 (1.2%), 0.01 mg mL−1 (4.1%), and 0.24 mg mL−1 (9.6%), respectively.
Non-chitinase enzymes can be applied to the hydrolysis of chitin. GlcNAc was produced from α-chitin (from crab shell) and β-chitin (from squid pen) using crude cellulase preparations from Trichoderma viride (T) and Acremonium cellulolyticus (A) [41]. The yield of GlcNAc was enhanced by mixing cellulases T and A. Crystalline chitin (from bee) and α-chitin (from crab shell) were hydrolyzed with the enzymatic preparation Celloviridin G20x from strain Trichoderma reesei, which includes cellulases and β-glucanases [42]. After 10 days of incubation in these conditions, the yield of GlcNAc reached 86%.
The mixed enzymes (cellulase: lipase = 9:1) from Aspergillus niger, was used in various enzyme concentrations, to investigate the hydrolytic behavior on β-chitin [43]. Increasing substrate concentration while keeping the enzyme concentration constant resulted in higher GlcNAc yield. After 4 days of incubation, the yield of GlcNAc reached 61% from β-chitin (10 mg/mL).
Flake type of chitin together with swollen chitin, colloidal chitin, and powder chitin is also the substrate for the oligosaccharides production. GlcNAc was produced from crab shell α-chitin flake using crude enzyme extract from Aeromonas hydrophila H-2330, with 66%–77% yield after 10 days incubation at 17 °C [44].
Exo-type chitinases can use chitin oligosaccharides as a substrate. (GlcNAc)2 was gradually and completely degraded to GlcNAc by exo-type chitinase (ChiA71) from Bacillus thuringiensis subsp. pakistani with time [45] and by N-acetyl-β-hexosaminidase (StmHex) from Stenotrophomonas maltophilia [46]. GlcNAc was produced from (GlcNAc)4 by enzyme purified from Aeromonas hydrophila SUWA-9 [47].This kind of trial is not intended for the production of monomers, but for the elucidation of enzyme properties, as oligomers are much more expensive and valuable in application.
There are typical reports on the selective preparation of GlcNAc and (GlcNAc)2 [48]. It was found that α-chitin was effectively hydrolyzed to (GlcNAc) by Aeromonas sp. GJ-18 crude enzyme preparation below 45 °C. The enzyme preparation from Aeromonas sp. GJ-18 contained GlcNAcase and N,N′-diacetylchitobiohydrolase [49]. GlcNAcase was inactive above 50 °C, but N,N′-diacetylchitobiohydrolase was stable at this temperature. Therefore, GlcNAc and (GlcNAc)2 were selectively produced from α-chitin at two temperatures. At 45 °C, GlcNAc was the major hydrolytic product with a yield of 74% in 5 days’ incubation, while at 55 °C (GlcNAc)2 was the major product with a yield of 35% after 5 days’ incubation.

{kind=link}
| Chitinsource | Enzyme Source | Enzyme | Mol. Wt. | Condition | Product & Yield | Analysis | Reference |
|---|---|---|---|---|---|---|---|
| Swollen chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | 40 °C, 9 days | GlcNAc 94.9% | HPLC | [48] |
| NH2P-50 4E | |||||||
| Swollen chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | 45 °C, 5 days | GlcNAc 74% | HPLC | [49] |
| (GlcNAc)2 4.8% | |||||||
| 55 °C, 5 days | GlcNAc 3.9% | NH2P-50 4E | |||||
| (GlcNAc)2 34.7% | |||||||
| α-Chitin | Aeromonas sp. GJ-18 | Crude enzyme | - | Preincubation (50 °C, 60 min) 45 °C, 7 days | (GlcNAc)2 78.9% | HPLC | [50] |
| β-Chitin | (GlcNAc)2 56.6% | NH2P-50 4E | |||||
| α-Chitin | A. hydrophila H-2330 | Crude enzyme | - | 17 °C, 10 days | GlcNAc 64~77% | HPLC | [44] |
| (Flake & powder) | NH2P-50 | ||||||
| Chitin | A. hydrophila SUWA-9 | Chitinase | - | 37 °C, overnight | (GlcNAc)2~(GlcNAc)5 | TLC | [47] |
| GlcNAc | |||||||
| Chitosan (60% DD) | Bacillus cereus TKU027 | Chitinase | 65/63 kDa | 37 °C, 2 h 30 °C, 2 days | (GlcNAc/GlcN) DP 4~9 | MALDI-TOF MS | [44] |
| Culture supernatant | (GlcNAc/GlcN) DP 2~5 | ||||||
| β-Chitin | Bacillus cereus TKU022 | Chitosanase | 44 kDa | 37 °C, 2 days | (GlcNAc)2, (GlcNAc)4 | HPLC | [51] |
| (GlcNAc)5, (GlcNAc)6 | |||||||
| Colloidal chitin | Bacillus thuringiensis subsp. pakistani | Exochitinase | 66/60/47/32 kDa | 37 °C, 24 h | GlcNAc | TLC | [45] |
| α-Chitin | Burkholderia cepacia TU09 | Chitinase | 37 °C, 7 days 37 °C, 1 days | GlcNAc 85% | HPLCNH2P-50 | [40] | |
| β-Chitin | GlcNAc 85% | ||||||
| α-Chitin | Bacillus licheniformis SK-1 | Chitinase | 37 °C, 6 days | GlcNAc41% | HPLCNH2P-50 | ||
| β-Chitin | GlcNAc75% | ||||||
| Swollen chitin | Paenibacillus illinoisensis KJA-424 | Chitinase | 38/54/63 kDa | 37 °C, 24 h | GlcNAc 62.2% | HPLC | [7] |
| Chitin | Trichoderma reesei | Cellulases &β-glucanases | - | 37 °C, 10 days | GlcNAc 86% | TLC/HPLC Separon SGX NH2 | [42] |
| α-Chitin | Trichoderma viride | Cellulase | - | 37 °C, 3 days | GlcNAc 16% | HPLC/NMR | [41] |
| Acremonium cellulolyticus | GlcNAc 22% | ||||||
| β-Chitin | Aspergillus niger | Cellulose & lipase | - | 37 °C, 4 days | GlcNAc 61% | HPLC | [43] |
| NH2P-50 | |||||||
| Chitin | Thermococcus kodakaraensis | Chitinase | 90 kDa | 70 °C, 3 h | (GlcNAc)2 | TLC | [52] |
| KOD1 | |||||||
| Chitin | Vibrio anguillarum E-383a | Exochitinase | - | - | (GlcNAc)2 40.3% | HPLC | [47] |
| Chitin | Enterobacter sp. G1 | Chitosanase | 50 kDa | 35 °C, 5 min | (GlcNAc)2 | TLC/HPLC | [53] |
| Chitosan (80% DD) | |||||||
| Chitin | Corynebacterium sp. | Chitobiase | - | 40 °C, 24 h | GlcNAc | HPLC/NMR | [54] |
| Chitin, steam exploded | Lecanicillium lecanii | Chitinase | 40 °C, 6 days | GlcNAcDP 1~9 | HPLC/MALDI-TOF MS | [55] | |
| (GlcNAc)2, | Stenotrophomonas maltophilia | N-acetyl-β-hexosaminidase & Chitin synthase | 40 °C, 60 min | GlcNAc | HPLC | [46] | |
| (GlcNAc)6 |
2.2. N,N′-Diacetylchitobiose and Chitin Oligosaccharides
As mentioned above, (GlcNAc)2 can be obtained as a major hydrolytic product from enzymes by controlling the ratio of GlcNAcase to N,N′-diacetylchitobiohydrolase activities in the crude enzyme of Aeromonas sp. GJ-18 [50]. After 7 days of incubation, 78.9% and 56.6% of (GlcNAc)2 yields were obtained from swollen α-chitin and powdered β-chitin, respectively, using enzyme preparations that had been pretreated at 50 °C so as to inactivate GlcNAcase.
There are a few more reports on bioproduction of (GlcNAc)2. (GlcNAc)2 was produced from colloidal chitin by chitinase from Vibrio anguillarum E-383a, isolated from seawater [56]. The maximum yield of (GlcNAc)2 from chitin was 40.3%. (GlcNAc)2 was produced by shrimp α-chitin (100~200 mesh) from commercial bovine pepsin [57]. The yield of (GlcNAc)2 was 75%, while the yields of GlcNAc and (GlcNAc)3 were 19% and 9.5%, respectively. Tanaka et al. [52] also identified (GlcNAc)2 as an end product from colloidal chitin by using chitinase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1.
The hydrolytic products of chitinase and GlcNAcase are mixtures of hetero-COS (DP 1–15), depending on reaction conditions. Hetero-oligosaccharides [(GlcNAc/GlcN) DP 2~5] were produced from chitosan (60% DD) by culture supernatant obtained from of Bacillus cereus TKU027. Chitin oligosaccharides [(GlcNAc/GlcN) DP 4~9] were produced by chitinase (65 and 63 kDa), and oligomers were identified using MALDI-TOF MS [58]. To analysis the distribution of chitin oligosaccharides, chitin oligosaccharides were derivatized with 9-aminopyrene-1,4,6-trisulfonate (APTS) and separated by capillary electrophoresis (CE) with laser-induced fluorescence (LIF) detection [59].
3. Bioproduction of Chitosan Oligosaccharides and Its Monomer
The most important tool in the biodegradation of chitosan to its oligosaccharides is chitosanolytic enzymes. The chitosanases have been prepared from various bacteria and fungi, as shown in Table 2. Chitosanases in glycoside hydrolase (GH) families 5, 7, 8, 46, 75, and 80 [27,28,29,30], have been classified into subclasses I, II, and III based on their substrate specificities toward chitosan [19,60,61,62,63]. Class I chitosanases can hydrolyze both GlcNAc–GlcN and GlcN–GlcN linkages, and class II chitosanases can split only GlcN–GlcN linkages, whereas class III chitosanases can degrade both GlcN–GlcNAc and GlcN–GlcN linkages. Chitosanases can also be classified into two major categories (endochitosanases and exochitosanases), according to their cleavage sites. Endochitosanases (EC 3.2.1.132; created 1990, modified 2004) cleave a partly acetylated chitosan at random and produces COS. Exochitosanases are usually called exo-1,4-β-d-glucosaminidase (GlcNase, EC 3.2.1.165; created 2008), which cleaves β-d-glucosamine (GlcN) residues continuously from the non-reducing end of the substrate ([33].
| Chitosan Source | Enzyme Source | Enzyme | Mol. Wt. | Condition | Product & Yield | Analysis | Reference |
|---|---|---|---|---|---|---|---|
| Chitosan | Bacillus sp. KCTC 0377BP | Chitosanase | 45 kDa | 1700 (unit/mg) | (GlcN)3~(GlcN)7 | TLC/HPLC | [64] |
| Chitosan | Bacillus cereus S1 | Chitosanase | 45 kDa | 40 °C, 20 min | (GlcN)2 27.2% | HPLC | [65] |
| (GlcN)3 40.6% | |||||||
| (GlcN)4 32.2% | |||||||
| Chitosan | Bacillus sp. 16 | Chitosanase | 37 °C, 30 min | DP 2~9 (DP 5~6) | TLC/HPLC | [66] | |
| Chitosan | Bacillus sp. KFB-C108 | Chitosanase | 48 kDa | 55 °C, 12 h | (GlcN)3~(GlcN)5 | HPLC | [67] |
| Chitosan | Bacillus sp. HW-002 | Chitosanase | 46 kDa | 40 °C, 5 h | (GlcN)2 | HPLC | [68] |
| Chitosan | Bacillus pumilus BN-262 | Chitosanase | - | 45 °C, 1 h | (GlcN)4~(GlcN)6 | HPLC (NH2-60) Bio-Gel P-4gel | [69] |
| (GlcN)5~(GlcN)7 | |||||||
| Chitosan | Bacillus megaterium P1 | Chitosanase (A/B/C) | 43/39.5/22 kDa | 28 °C, 12 h/90 h | (GlcN)n oligomers | TLC | [70] |
| Chitosan | Acinetobacter sp. CHB101 | ChitosanaseI (endo) | 37 k Da | 37 °C, overnight | >(GlcN)5 | TLC | [71] |
| Chitosanase II (endo) | 30 kDa | ||||||
| Chitosan (60% DD) | Acinetobacter calcoaceticus TKU024 | Chitosanase (CHSA1) | 27 kDa | 37 °C, 30min | (GlcN)n oligomers | - | [72] |
| Chitosanase (CHSA2) | 66 kDa | ||||||
| Chitosan | Nocardia orientalis IFO 12806 | Chitosanase (exo) | 97 kDa (70 kDa) | 40 °C, 24 h | GlcN | TLC/HPLC | [73] |
| Chitosan | Matsuebacter chitosanotabidus 3001 | Chitosanase | 34 kDa | 30 °C, 10 min | (GlcN)2~(GlcN)6 | TLC | [74] |
| Chitosan | Aspergillus fumigatus KH-94 | Chitosanase I (endo) | 22.5 kDa | 50 °C, 30 min | DP 3~6 50% & >DP7 50% | TLC/HPLC | [75] |
| Chitosanase II (exo) | 108 kDa | 50 °C, 5 min | GlcN | ||||
| Chitosan | Aspergillus fumigates S-26 | Chitosanase | 104 kDa | 37 °C, 30 min | GlcN | TLC/HPLC | [76] |
| (GlcN)2~(GlcN)7 | |||||||
| Chitosan | Aspergillus oryzae IAM2660 | Chitosanase(endo) Chitosanase (exo) | 40 kDa | 37 °C, 20 min | >DP 5 | TLC | [77] |
| 135 kDa | 40 °C, overnight | GlcN | |||||
| Chitosan | Trichoderma reesei PC-3-7 | Chitosanase | 93 kDa | 37 °C, 15 h | GlcN | TLC | [78] |
| Chitosan | - | Immobilized papain | - | - | MW < 10,000 49.5% | MALDI-TOF MS | [79] |
| MW 600~2000 11.1% | |||||||
| Chitosan(82.8% DD) | Novozyme lipase | - | 37 °C, 24 h | DP 1–6 | TLC | [80] | |
| Chitosan (76% DD) | Commercial enzymes | Complex (cellulase, α-amylase, proteinase) | - | 40 °C, 40 min | DP 5~17 | MALDI-TOF-MS | [81] |
| Chitosan (95% DD) | Bacillus cereus TNU-FC-4 | Chitosanase | 46 kDa | 45 °C, 33 min | >DP 7 | HPLC | [25] |
| Rhizopus oligosporus cell wall | Streptomyces sp. N174 | Chitosanase | - | 40 °C, 24 h | (GlcN)2, GlcN-GlcNAc, (GlcN)2-GlcNAc | CP/MAS NMR/MALDI-TOF-MS | [ 82] |
| Chitosan | Rhodothermus obamensis | Branchzyme | 256 kDa | 50 °C, 24 h | DP 2–20 | GC-FID/SEC-HPLC | [83] |
| Chitosan (60% DD) | Penicillium janthinellum D4 | Chitosanase | 49 kDa | 50 °C, 60 h | DP 3–9 | MALDI-TOF-MS | [84] |
It is easier and more efficient to cleave the β-1,4-glycosidic linkages in chitosan than in chitin, because of its solubility in weakly acidic solutions. Chitosan, being a base, can be solubilized in both inorganic and organic acids, forming salts with acids. Acetic, lactic, and citric acids have all been used to facilitate enzyme hydrolysis. The solubility of chitosan depends on the molecular weight and degree of N-acetylation of the chitosan, concentration of the acid, and temperature. Homogeneous deacetylation of chitin to chitosan (approximately 50% DD) gives a water-soluble polymer.
The solubilized chitosan salt is a preferred substrate for chitosanases in homogeneous aqueous acids. Thus, chitosan is more rapidly hydrolyzed and gives a higher yield than chitin, which is not soluble and heterogeneous in aqueous environment. The products of the hydrolysis reaction of chitosan salt are accordingly the salt of its oligosaccharides. This means that a desalting process is necessary to obtain a pure oligosaccharide. Desalting is a rather complicated and costly process. In commercial products, the content has been expressed as being the salt of acetic, lactic, and citric acid of the oligosaccharides. Glucosamine is supplied in the marketplace as a dietary supplement in the form of a hydrochloric or sulfuric salt.
3.1. β-d-Glucosamine
The hydrolytic products of chitosan are mixtures of COS (DP 1–7), dependent on the reaction conditions, even though the monomer becomes a major end product after prolonged reaction time. Thus, chromatographic separation is necessary to purify each monomer, same as in chitin oligosaccharides. The enzymatic preparation of chitosan oligosaccharides is summarized in Table 2.
Koji mold Aspergillus oryzae is a strong producer of two different chitosanolytic enzymes, endochitosanase and exochitosanase (β-GlcNase). β-d-Glucosamine (GlcN) was produced as a final product from chitosan by a 135-kDa exochitosanase purified from A. oryzae IAM2660 [77]. In addition, chitosan oligosaccharides over DP 7 were produced from chitosan by a 40-kDa endochitosanase purified from A. oryzae. GlcN was produced from soluble chitosan by a 104-kDa exochitosanase purified from A. fumigates S-26 [76]. GlcN was produced from chitosan by a 108-kDa exochitosanase II purified from A. fumigatus KH-94 [75].
Chitosan oligosaccharides can be applied for chitosanase as a substrate, not for mass production of glucosamine but for elucidation of the reaction mechanism of chitosanase. GlcN resulted from the hydrolysis of chitobiose, chitopentose, and chitosan from the 79-kDa GlcNase of Nocardia orientalis IFO 12806 [73]. GlcN was also the final product from (GlcN)6 by the 93-kDa chitosanase purified from Trichoderma reesei PC-3-7 [78]. (GlcN)6 appeared to be hydrolyzed to GlcN5 and GlcN at the initial stage of the reaction.
3.2. Chitobiose and Chitosan Oligosaccharides
There are a few studies on the bioproduction of chitobiose (GlcN)2. Chitobiosewas produced from chitosan (75%~85% DD) in a mixture by chitosanase from Bacillus cereus S1 [65]. The composition of chitobiose, chitotriose, and chitotetraose was 27.2%, 40.6%, and 32.2%, respectively, after a 24-h reaction with a 45-kDa chitosanase. Chitobiose and chitotriose were produced from chitopentose and chitosan by crude proteins from Acinetobacter sp. CHB101 [71]. In the case of the 22.5-kDa endochitosanase purified from Aspergillus fumigatus KH-94, chitobiose, chitotriose, and chitotetraose were produced from chitohexaose [75]. In addition, chitosan oligomers of DP 3~6 (50% yield) and over DP 7 (50% yield) were produced from chitosan by the 22.5-kDa chitosanase.
These enzyme preparations contained endochitosanase activity. The endo-type activity is responsible for the production of chitosan oligosaccharides. Endo-type chitosanase is a major contributor to decreasing the viscosity of the reaction solution and fouling of the membrane reactor system [69]. The viscosity of high molecular weight material such as chitosan is involved in producing shearing forces in reactors, changes in enzyme-substrate affinity, and the fouling problem. When mixed with endochitosanase, the viscosity of chitosan decreased rapidly in accordance with production of the oligomers.
Thus, a great deal of interest has arisen regarding the reaction pattern of the endochitosanases. The chitosanase from Bacillus sp. KCTC 0377BP cleaved (GlcN)6 mainly into (GlcN)3 plus (GlcN)3 and to a lesser extent into (GlcN)2 plus (GlcN)4 [subsequently, (GlcN)4→(GlcN)2 + (GlcN)2] [64]. (GlcN)7 was cleaved into (GlcN)3 plus (GlcN)4 [subsequently, (GlcN)4→(GlcN)2 + (GlcN)2]. The purified endochitosanase (41-kDa) from Bacillus cereus D-11 hydrolyzed chito-oligomers (GlcN)5–7 into chitobiose, chitotriose and chitotetraose as the final products [85]. Minimal size of the oligosaccharides for enzymatic hydrolysis was pentamer. To further investigate the cleavage pattern of this enzyme, chitooligosaccharide alcohols (COS with the reducing unit converted into alditol) were prepared and used as substrates. The chitosanase split (GlcN)4 GlcNOH into (GlcN)3 + (GlcN)1GlcNOH, and (GlcN)5GlcNOH into (GlcN)4 + (GlcN)1GlcNOH and (GlcN)3 + (GlcN)2GlcNOH. The heptamer (GlcN)6GlcNOH was split into (GlcN)5 [subsequently,(GlcN)5→(GlcN)3 + (GlcN)2] + (GlcN)1GlcNOH, (GlcN)4 + (GlcN)2GlcNOH, and (GlcN)3 + (GlcN)3GlcNOH, whereas (GlcN)1–3GlcNOH were not hydrolyzed. The monomer GlcNor GlcNOH was never detected from the enzyme reaction. These results suggest that D-11 chitosanase recognizes three glucosamine residues in minus position and simultaneously two residues in plus position from the cleavage point [85].
Chitobiose as a main end product was produced from chitosan by a 46-kDa chitosanase purified from Bacillus sp. HW-002 [68]. A 34-kDa chitosanase purified from Matsuebacter chitosanotabidus 3001 was cleaved mainly (GlcN)5 and (GlcN)6 into (GlcN)2 plus (GlcN)3 [74]. Chitosan oligosaccharides [(GlcN)2–6] were produced from chitosan by the purified chitosanase.
Pantaleone et al. [86] reported the hydrolytic susceptibility of chitosan to a wide range of enzymes, including glycanases, proteases, and lipases derived from bacterial, fungal, mammalian, and plant sources. These nonspecific enzymes have proven hydrolytic ability on chitosan to produce various chitosan oligomers in immobilized papain [79], lysozyme [87], and pronase [88]. Xie et al. [89] reported that chitosan (80% DD) was depolymerized by the cellulase of Aspergillus niger to give COS with DP 3–11. Mixture of cellulase, α-amylase and proteinasewas effective in the production of COS (DP 5~17) [81]. This finding presents another opportunity in the industrial scale production of COS, because these enzymes are active enough, commercially available, cheaper than chitosanase, and easy to handle. A commercial lipase was also applied for enzymatic preparation of COS, where COS with DP 1–6 was produced with 93.8% yield for 24 h hydrolysis at 37 °C [80]. Recently, the commercial α-amylase was used to hydrolyze chitosan from Clanis bilineata larvae skin under the optimal pH and temperature, thereby affording COS with a DP 2–8 [90].
Most chitosanase-producing bacteria require chitosan as a carbon/nitrogen source and chitosanase-inducer. Thus, one can get COS from the culture supernatant of the microorganism. Reducing COS sugars from shellfish waste were produced in the culture supernatant of Acinetobacter calcoaceticus TKU024, including chitosanases (CHSA1 and CHSA2) after 5 days of incubation [72]. Even though the yield of this method is very low, this kind of trial could be considered as a stepping stone to the direct extraction of COS from bio resources.
Intact cells of Rhizopus oligosporus NRRL2710were digested with a GH-46 chitosanase from Streptomyces sp. N174 [82]. Valuable hetero- and homo-oligosaccharides GlcN–GlcNAc, (GlcN)2–GlcNAc, and (GlcN)2 were produced, functionally, by the enzymatic digestion of the intact cells. The chitosanase digestion of intact fungal cells should be an excellent system for bioconversion of abundant microbial biomass without any environmental impact.
4. Chitin Deacetylase and Chitooligosaccharides
The chemical conversion from chitin to chitosan, the most difficult and cost-demanding step, has been done with 50% NaOH and high temperature in industrial applications. In nature, this step occurs at comparatively lower temperatures and neutral pH, by chitin deacetylase (CDA, EC 3.5.1.41; created 1976) [91]. The enzyme hydrolyzes the linkage between the acetyl group and the amine group in N-acetyl-d-glucosamine residues of chitin. Thus, bioconversion of chitin oligosaccharides to chitosan oligosaccharides can be achieved by CDA and vice versa.
Chitin deacetylase from Absidia corymbifera DY-9 was active towards water-soluble chitin (WSCT-50), glycol chitin, chitosan (DD 71%–88%), and chitin oligosaccharides with DP 2~7 [92,93]. CDA displayed little activity on chitin flakes, chitin powder, swollen chitin, or β-chitin powder. Chitin oligosaccharides were a comparatively good substrate because their solubility increased availability and accessibility for the CDA. Deacetylation rate on (GlcNAc)2-6 was size-dependent; greater lengths produced a higher rate of activity. These results suggest that solubilization of chitin is a limiting factor for enzymatic bioconversion of chitin to chitosan by CDA. Extracellular CDA from Mortierella sp. DY-52 was active on WSCT (DD 50%), glycol chitin and crab chitosan (DD 71%–88%) and also on N-acetylglucosamine oligomers (GlcNAc)2–6 [94].
Deacetylation by CDA is apparently substrate size-specific. GlcNAc was converted into GlcN by CDA from Thermococcus kodakaraensis KOD1, but (GlcNAc)2 was converted into GlcN-GlcNAc, neither GlcNAc-GlcN or GlcN-GlcN [95]. Only the non-reducing residue of (GlcNAc)2 has been deacetylated. CDA from C. lindemuthianum ATCC 56676 converted (GlcNAc)2 not into (GlcN)2 but into hetero-disaccharide GlcN-GlcNAc, and transformed (GlcNAc)3 and (GlcNAc)4 into the deacetylated products (GlcN)3 and (GlcN)4, respectively [96]. Alfonso et al. [97] found that chitosan oligosaccharides, (GlcN)2-6, were produced from chitin by the joint action of endochitinase and CDA from Aspergillus nidulans, suggesting that deacetylation mainly occurs after chitin oligosaccharide production by the endochitinase.
A solid natural substrate shrimp chitin could be deacetylated with an 11% deacetylation by CDA from Saccharomyces cerevisia [98]. Pre-hydrolysis of crystalline shrimp chitin by grape chitinases increased the deacetylation triggered by CDA and produced COS with a degree of deacetylation of 67% [98]. It is well known that the high crystallinity of chitin microfibril, by the hydrogen bond-stabilized packaging of chitin polymer, greatly impedes the access of the enzyme to the deacetylation reaction site in the chitin molecule. Enzymatic deacetylation is profoundly affected by the physical properties of the substrate, such as crystallinity, degree of deacetylation, particle size, and origin [99]. Pretreatment to destroy the crystalline structure prior to addition of the enzyme seems to be desirable, in order to improve the deacetylation rate and produce novel chitosan polymers and oligomers. The preparation of COS including N-deacetylation and transglycosylation are summarized in Table 3.
Martinou et al. [100] investigated the mode of action of CDA from Mucor rouxiion fully water-soluble partially N-acetylated chitosans (DP 30) and found that the CDA hydrolyzed acetyl groups according to a multiple attack mechanism, that is, CDA does not preferentially attack any sequences in the chitosan chains. Together with this, a multiple chain mechanism has been suggested in CDA originating from Colletotrichum lindemuthianum ATCC 56676 [99].
| Product | Reaction Type | Reference |
|---|---|---|
| (GlcN)2~(GlcN)6 | N-Deacetylation with endochitinase and CDA from Aspergillus nidulans; substrates: chitin | [97] |
| GlcN-GlcNAc | N-Deacetylation with CDA from C. lindemuthianum ATCC 56676; substrates: (GlcNAc)2 | [96] |
| (GlcN)3 & (GlcN)4 | (GlcNAc)3 and (GlcNAc)4 | |
| Chitosan oligomers | N-Deacetylation with CDA from Mucor rouxii; substrates: partially N-acetylated chitosans (DPn, n = 30) | [100] |
| GlcN | N-Deacetylation with CDA from Thermococcus kodakaraensisKOD1; substrates: GlcNAc | [95] |
| (GlcN)2~(GlcN)7 | N-Deacetylation with CDA from Absidia corymbifera DY-9; substrates: (GlcNAc)2~(GlcNAc)7 and WSCT-50 | [94] |
| (GlcN)2~(GlcN)7 | N-Deacetylation with CDA from Mortierella sp. DY-52; substrates: (GlcNAc)2~(GlcNAc)7 | [91] |
| Chitosan (89% DD) & (GlcN)2~(GlcN)4 | N-Deacetylation with CDA from Saccharomyces cerevisiae; substrates: Chitin and (GlcNAc)2~(GlcNAc)4 | [98] |
| (GlcNAc)6~(GlcNAc)15 | Transglycosylation reaction on β-1,4-(GlcNAc)3with lysozyme containing (NH4)2SO4 (30% w/v) | [101] |
| β-1,4-(GlcNAc)2 & β-1,6-(GlcNAc)2 | Transglycosylation reaction on N-acetylchito-oligosaccharides [β-1,4-(GlcNAc)2 ~ β-1,4-(GlcNAc)6] with exo-β-d-GlcNase from Alteromonas sp. OK2607 | [102] |
| n-Butyl β-d-glucosaminide (C4GlcN) | Transglycosylation reaction on chitosan oligosaccharides & n-butanol with exo-β-d-GlcNase from Penicillium funiculosum KY616 | [103] |
5. Transglycosylation and Chitooligosaccharides
In addition to hydrolytic activity, some chitinolytic enzymes possess certain level of transglycosylation ability, that is, the ability to transfer the released oligosaccharide moiety to a suitable acceptor to form a new glycosidic bond. The transglycosylation activity of these chitinolytic enzymes suggests great potential for the synthesis of size- and stereo-specific chitin/chitosan oligomers, or even polymers and their derivatives.
Preparation of higher DP chitin oligosaccharides was achieved by the transglycosylation reaction of glycolytic enzymes including chitinase, chitosanase, and other glycosidases. Chitinase purified from Trichoderma reesei KDR-11 was shown to convert (GlcNAc)4 into (GlcNAc)2(55.7%) and (GlcNAc)6(39.6%) by a transglycosylation reaction [104]. Akiyama et al. [105] reported that COS with DP 4–12 were successfully synthesized by a lysozyme-catalyzed transglycosylation reaction using N,N′,N″-tri(monochloro)acetylchitotriose and N,N′,N″-triacetylchitotriose as substrates followed by a base-catalyzed removal of the N-monochloroacetyl groups. Recently, Hattori et al. [101] made progress in biopreparation of COS. They tried lysozyme-mediated transglycosylation using β-1,4-(GlcNAc)3 as starting substance and successfully produced chitin oligomers of (GlcNAc)6 to (GlcNAc)15 in an aqueous system containing 30% (NH4)2SO4.
When GlcNase purified from Penicillium funiculosum KY616 was incubated with a mixture of chitosan oligomers and n-butanol, n-butyl β-d-glucosaminide (C4GlcN) was synthesized as a product by transglycosylation [103]. Yields of C4GlcN from chitobiose, chitotriose, and chitotetraose were found to be 14%, 23%, and 30%, respectively. The unusual β-1,6-(GlcNAc)2 were synthesized from β-1,4-(GlcNAc)2–6 by transglycosylation in chitinase preparation of marine bacterium Alteromonas sp. OK2607 [102].
Another example of enzymatic transglycosylation shows its diversity and potential in commercial applications. Fujimoto et al. [106] have reported the synthesis of gentiooligosaccharides (DP 3–9) from gentiobiose using a crude enzyme preparation from P. multicolor. The transglycosylation was shown to take place in two stages by a combination of β-glucosidase and β-(1-6)-glucanase. In the beginning, β-glucosidase produced gentiotriose from gentiobiose, and then β-(1-6)-glucanase acted on the resulting gentiotriose to produce a series of gentiooligosaccharides (DP 3–9) by transglycosylation. The transglycosylation reaction has high potential in the small-scale preparation of high value glycoside products applicable in medical and industrial fields.
6. Chemoenzymatic Glycosylation and Chitooligosaccharides
Chemoenzymatic glycosylation of chitooligosaccharides was intensively reviewed by the Kobayashi group [31,107,108,109]. In vitro synthesis of chitooligosaccharides was first reported by utilizingchitinase (EC 3.2.1.14) from Bacillus sp. (classified into glycoside hydrolase family 18; GH18) as catalyst [31,32]. On the basis of the transition state analogue substrate (TSAS) concept [31], the N,N′-diacetylchitobiose (GlcNAc-β-(1→4)-GlcNAc) oxazoline monomer was introduced. The enzymatic polymerization of this monomer proceeded via ringopening poly addition under weak alkaline conditions (pH 9.0~11.0), giving a synthetic COS with perfectly controlled stereo- and region selectivity. The DPs were evaluated as 10–20 depending on the reaction conditions [107].
The chitinase-catalyzed glycosylation using the sugar oxazoline substrate was applied to the stepwise elongation of GlcNAc unit by utilizing two enzymes, chitinase for ring-opening polyaddition of N-acetyllactosamine(Gal-β-(1→4)-GlcNAc) oxazoline (Gal = galactose) and β-galactosidase for the removal of the galactose unit from the produced oligosaccharide. [110,111,112,113,114]. The hydrolytic removal affords a new glycosyl acceptor with the GlcNAc unit at the non-reducing end. Repetition of these procedures using the two enzymes enabled the synthesis of chitooligosaccharides with desired chain lengths.
Interestingly, N-acetyllactosamine oxazoline was found to bepolymerized by chitinase A1 from Bacillus circulans WL-12 catalysis under basic conditions, giving rise to a novel oligosaccharide having the β-(1→4)-β-(1→6)-linked repeating unit in the main chain [115]. The DP of the resulting oligosaccharides was up to 5 based on the disaccharide. This was the first example of enzymatic glycosylation forming β-(1→6)-glycosidic linkage by chitinase catalysis.
The main disadvantage using the chemo-enzymatic approach is poor yield, because the product becomes necessarily a substrate for the enzyme (Table 4). Suppression of the chitinase-catalyzed hydrolysis of the product during the enzymatic polymerization is a challenge for chemoenzymatic glycosylation of COS. Wild-type chitinaseA1 from Bacillus circulans WL-12 has D202 and E204 residues as a DXE (D = Asp, X = any amino acid, E = Glu), a general sequence at the catalytic domain of chitinase [116,117]. A mutant chitinase E204Q (Q = Gln) exhibited less hydrolysis activity of the produced oligosaccharides by the enzymatic glycosylation, probably less protonation ability toward the oxygen of the glycosidic linkage by replacement of COOH in Glu with CONH2 in Gln [118].
Table 4.
Chemoenzymatic preparation of chitin and chitosan oligosaccharides (COS) and its derivatives.
| Product | Reaction Type | Reference |
|---|---|---|
| N-Acetylchitooligosaccharides | Chemoenzymatic elongation of N-GlcNAc unit by combined use of chitinase and β-galactosidase | [115] |
| Chitin derivatives with the deacetylated extents ranging from 0% to 50% | Chitinase-catalyzed copolymerization of N-acetylchitobiose oxazoline with the N,N′-diacetylchitobiose oxazoline | [54] |
| 6- O-Carboxymethylated chitotetraose alternatingly | Chitinase-catalyzed chemoenzymatic glycosylation of 6- O- and 6′-O-carboxymethyled chitobioseoxazolines | [119] |
| 3- O-Methylated chitotetraose | Chitinase-catalyzed chemoenzymatic glycosylation with 3- O-methyl- and 3′-O-methylchitobiose oxazolines | [118] |
| Oligo- N-acetyllactosamine derivatives with β-(1→4)-β-(1→6)-linked repeating unit | Chemoenzymatic polymerization by using transition state analogue substrate with chitinase A1 | [116] |
| Alternatingly d-Glcβ-(1→4) N-GlcNAc repeating units, a cellulose-chitin hybrid polysaccharide | Chemoenzymatic glycosylation of Glcβ (1→4) GlcN Acoxazoline and GlcNAcβ (1→4) Glc fluoride by chitinase and cellulose, respectively | [103] |
| Alternatingly d-GlcNβ-(1→4) N-GlcNAc repeating units, a chitin-chitosan hybrid polysaccharide | Chitinase-catalyzed chemoenzymatic glycosylation of C-2′ position modified N-acetylchitobiose oxazolines | [55] |
| Fluorinated chitins | Chitinase-catalyzed polymerization C-6, C-6′, or both modified N-acetylchitobiose oxazolines | [120,121] |
7. Perspectives
7.1. Direct Degradation and Separation of Chitin from Crustacean Shells or Squid Pens
The direct degradation and separation of α-chitin from crab and shrimp shells, and microbial cell walls poses a significant challenge. In light of the difficulties associated with traditional chitin oligosaccharides’ production processes, environmentally compatible and reproducible degradation alternatives are desirable. However, crustacean shells are not soluble in standard aqueous media and their crystallinity is potentially too high to be degraded enzymatically. To solve these problems, pretreatment to break the chitin crystal structure is widely considered.
In this regard, chitin substrates were pretreated with steam explosion prior to enzymatic reaction [122]. A 11.28% reduction of the crystallinity index was observed with steam explosion and a 23.6% yield of chitin oligosaccharides with DP up to 5 was achieved. Interestingly, Nakagawa et al. [123] reported a breakthrough in the direct production of oligosaccharides from chitin and crab shells. They introduced a combination protocol of mechanochemical grinding and enzymatic hydrolysis, and produced GlcNAc and (GlcNAc)2 directly from crab shells and chitin. The direct degradation ratio of the chitin in crab shell was close to 100%. For this purpose, they developed a novel “converge” mill [124], a derivative of the medium ball mill. Mechanochemical grinding, with the converge mill, was found to be extremely effective for pretreating chitin and crab shell before enzymatic digestion [125].
Breaking down the chitin crystal structure of biomaterials improves enzymatic degradation, allowing the enzymes to easily access and exert catalytic action. Wu and Miao [126] showed that mechanochemical treatment markedly increases the glucose yield from enzymatic corn flour hydrolysis. Similar results were observed by Fujimoto et al. [127] for lignocellulosic biomass. Van Craeyveld et al. [128] also showed improvement of extractability and molecular properties of Psyllium seed husk arabinoxylan by ball milling.
Mechanochemical grinding also provides an additional advantage. In the case of chitosan oligosaccharide production, salts are inevitably formed from aquatic reaction mixtures. Sometimes the bound salts limit application of COS. Salt production can be circumvented by mechanochemical grinding of the substrate, followed by enzymatic hydrolysis.
7.2. New Enzymes with Better Properties
Solubilization has become the first choice for breaking down the chitin crystal structure in chitin and crab shells to improve enzymatic degradation. Solvents are necessary to solvate the substrate, and these are still being studied along with continuing research seeking enzymes that are stable in organic solvents or heterogeneous solvent systems. Enzymes that function in an extreme environment are important for this purpose and their mode of action differ fundamentally from of glycoside hydrolases.
During researches on enzymes capable of efficiently degrading recalcitrant polysaccharides such as cellulose and chitin for biofuels, it has been speculated about the existence of a substrate-disrupting factor that could make the crystalline substrate more accessible to hydrolytic enzymes [129]. Vaaje-Kolstad et al. [130] showed that CBP21 (CBP for chitin-binding protein), produced by the chitinolytic bacterium Serratia marcescens, is an enzyme that catalyzes cleavage of glycosidic bonds in crystalline chitin, thus opening up the in accessible polysaccharide material for hydrolysis by normal glycoside hydrolases. The products were identified as chitin oligosaccharides with a normal sugar at the non-reducing end and an oxidized sugar, 2-(acetylamino)-2-deoxy-d-gluconic acid (GlcNAcA), at the other end. This is an oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. This finding helps understand the factors involved in degradation of crystallinechitin and the chitin cycle in nature and provides a stake to more precisely handle the process for COS production from crystalline chitin and even raw materials such as crab and shrimp shells.
Recently, several commercially available hydrolytic enzymes including lysozyme, cellulase, papain, pectinases, and hemicellulase were found to catalyze the cleavage of the glycoside bond in chitin and chitosan [83]. Use of these nonspecific enzymes together with specific chitinase, chotosanase, glycosyltransferase, and CDA certainly opens a possible route to optimize the hydrolysis reactions controlling the production of chitooligosaccharides. Although different nonspecific enzymes have been used to obtain COS from chitin and chitosan, due to the limited capacity of most hydrolytic enzymes there is still interest in finding new enzymes with better properties.
Branchzyme is a relatively inexpensive commercial preparation that contains a branching glycosyltransferase from Rhodothermus obamensis expressed in Bacillus subtilis. This enzyme catalyzes the transfer of a segment of a 1,4-α-d-glucan chain to a primary hydroxyl group in a similar glucan chain to create 1,6-α-linkages, thereby increasing the number of branch points [131]. Interestingly, Branchzyme was active on chitosan and produced COS with DP 2-20, with a higher concentration having COS with DP 3–8 [132]. Recently, a family 46 chitosanase from S. coelicolor A3(2) was employed to degrade both a fully deacetylated chitosan and a 68% deacetylated chitosan for the production of a series of COS and to study in-depth the enzyme's mode of action [133].
7.3. Manipulation of the Size-Distribution of Oligomers Produced by Enzymatic Bioconversion
The DD, DP, the molecular weight distribution, and N-acetylation pattern (PA) of the resulting COS mixture depend on the starting material (mostly DD of chitin and chitosan) and the specificity of the enzyme used. Product mixtures can be enriched for certain compounds by optimizing the substrate-enzyme combination. This is illustrated by several studies on enzymatic degradation of chitosans [134,135,136,137,138,139,140]. The degradation of chitosan by the family 18 chitinases, ChiA, ChiB and ChiC, from Serratia marcescens has been studied in detail [36,134,141,142]. The biphasic kinetics of the reaction with ChiB was illustrated, as in the initial rapid phase and the second much slower phase of the reaction [36]. Sorbotten et al. [36] showed that the size-distribution of oligomers could be manipulated by varying the DD of the chitosans (DD 87%, 68%, 50%, and 35%) with ChiB; the products get longer as the DD goes up. Very interestingly, Sikorski et al. [137,142] have produced a model for the degradation of different chitosans with ChiB, which is capable of predicting the length distributions and yield of the products after extended hydrolysis. These profiles allow for selection of optimal reaction and substrate parameters for efficient production of oligomers with desired lengths. Altogether, this suggests that production of oligomers with desired lengths can be achieved by manipulating the choice of the starting materials with specific DD, and PA, the choice of the enzyme, and the choice of the processing time [130].
7.4. Genetic Engineering Technology
Genetic engineering technology is applicable and promising for mass production of recombinant enzymes and understanding mode of action, which is useful for bioproduction of COS and synthesis of structure-specific oligosaccharides. Martinez et al. [51] declared that enzymatic synthesis of COS has been a matter of research by exploiting the transglycosylation activity of retaining family 18 chitinases. The mutagenesis of two GH-18 glycoside hydrolase, B. circulans WL-12 chitinase A1 (Bc ChiA1) and Trichoderma harzanium chitinase 42 (Th Chit42) abolished the hydrolytic activity of chitin [51]. The mutants D200A and D202A of Bc ChiA1, together with D170N of Th Chit42 proved to be active for chitinbiose oxazoline polymerization, and also for coupling reaction between Gal (β1→4) chitinbiose oxazoline and chitinpentaose at neutral pH. These mutants have retained the ability to catalyze transglycosylation reaction on natural COS. Such mutants could be considered as chitin transglycosylases.
Chitin synthase, a processive inverting enzyme of glycosyltransferase family 2, transfers GlcNAc from UDP-GlcNAc to preexisting chitin chains in reactions that are typically stimulated by free GlcNAc. This enzyme can be applied for synthesis of chitin oligosaccharides. By using a recombinant Saccharomyces cerevisiae chitin synthase with UDP-GlcNAc, GlcNAc2 was obtained as a major reaction product [143]. Formation of both COS and insoluble chitin was stimulated by GlcNAc2 and by N-propanoyl-, N-butanoyl-, and N-glycolyl-glucosamine.
7.5. Direct Fermentation of Raw Materials such as Crab and Shrimp Shells
The direct fermentation of raw materials such as crab and shrimp shells presents another opportunity in the production of chitin and COS. Chitin oligosaccharides were directly obtained from 1.5% shrimp head powder after 2 days of fermentation with protease- and chitosanase-producing Bacillus cereusTKU022 [144]. (GlcNAc)2, (GlcNAc)4, (GlcNAc)5, and (GlcNAc)6 were all identified in the culture supernatant. Even though the concentration of the products is still very low (0.3–201.5 μg/mL), these findings are meaningful in terms of direct production from raw materials.
7.6. Reactor Systems
Finally, the reactor system used should be considered to be a critical factor for efficiency and high yield in bioproduction. There are three types of reactors normally used in the preparation of chitosan oligosaccharides, including batch reactors, column reactors and ultrafiltration reactors [145]. Batch reactors represent the simplest method used for the enzymatic production of COS. This method has a few drawbacks, including limited reuse of reacting enzyme and continuous production of COS, difficulty in controlling molecular weight of COS, low yield and high cost.
Continuous preparation of COS is possible by packing immobilized enzymes into a column reactor that the substrate passes through. Though this method has several advantages over batch reactors, the poorer affinity of immobilized enzymes to chitosan substrate than that of free enzyme limits the activity of enzyme and the usage of column reactors in the commercial preparation of COS.
The ultrafiltration membrane reactor has been applied for overcoming the problems of the reusability of enzymes in batch reactors and the poor affinity of the substrate toward immobilized enzymes in column reactor systems, but suffers from the problem of membrane fouling [25,69]. A dual ultrafiltration membrane reactor system composed of a column reactor packed with immobilized enzymes and an ultrafiltration membrane reactor, has been invented and applied in COS production [146]. Using this reactor, chitosanase from Bacillus pumilus BN-262 continuously produced chitosan oligomers of DP 3~6 from chitosan (89% DD), free from any fouling problem.
8. Conclusions
Bioproduction of COS with enzymes and microorganismshas been studied for decades. However, the yield of bioproduction is still lower and the cost is higher than traditional chemical methods. Crude rather than pure enzyme preparations of chitinase, chitosanase, lysozyme, cellulase, protease, lipase, and pepsin were preferred for this purpose for the practical production of the oligosaccharides. The transglycosylation activity of chitinolytic enzymes was successfully exploited for the synthesis of desired chitin oligomers and their derivatives. Chitin deacetylase is also applicable to the preparation of oligosaccharides.
The direct degradation and separation of chitin from crab and shrimp shells and microbial cell walls presents a significant challenge in bioproduction of COS. The direct production of oligosaccharides from chitin and crab shells was achieved by using a combination of mechanochemical grinding and enzymatic hydrolysis. In case of chitosan oligosaccharides, salt forms are inevitably formed from aquatic reaction mixtures, which limit the application. However, the mechanochemical grinding of the substrate followed by enzymatic hydrolysis can circumvent this.
Breaking down the crystal structure of chitin and crab shells is necessary to improve enzymatic degradation of the substrate. Solubilization is the first choice for destruction of chitin crystallinity. Once solubilized in organic or heterogeneous solvent systems, the substrate must receive hydrolytic enzymes with special properties. Thus, screening high-potential enzymes that function in extreme environments is an important factor for the optimization of bioproduction.
The direct fermentation of raw biomaterials like crab and shrimp shells presents another opportunity in the production of chitin and COS. Even though the efficiency of this method is still very low, it is significant in terms of the direct production of COS from the row materials.
To overcome these inefficiencies, the optimization of a reactor system appropriate to enzymatic bioproduction is called for. A dual ultrafiltration membrane reactor system has been described that is capable of overcoming the problems of enzyme reusability in batch reactors as well as the poor affinity of the substrate toward immobilized enzymes in standard column reactor systems.
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) through the National Research Lab. Program funded by the Ministry of Science and Technology (No. R0A-2003-000-10322-0), and by Bio-industry Technology Development Program, Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- No, H.K.; Meyers, S.P.; Lee, K.S. Isolation and characterization of chitin from crawfish shell waste. J. Agric. Food Chem. 1989, 37, 575–579. [Google Scholar]
- Aye, K.N.; Stevens, W.F. Technical note: Improved chitin production by pre-treatment of shrimp shells. J. Chem. Technol. Biotechnol. 2004, 79, 421–425. [Google Scholar] [CrossRef]
- Allan, G.G.; Fox, J.R.; Kong, N. Marine polymers, part 8. A critical evaluation of the potential sources of chitin and chitosan. In Proceeding of the 1st International Conference on Chitin/Chitosan; Muzzarelli, R.A.A., Pariser, E.R., Eds.; MIT Sea Grant: Cambridge, MA, USA, 1978; pp. 64–78. [Google Scholar]
- Simpson, B.K.; Gagné, N.; Simpson, M.V. Bioprocessing of chitin and chitosan. In Fisheries Processing: Biotechnological Applications; Martin, A.M., Ed.; Chapman & Hall: London, UK, 1994; pp. 155–173. [Google Scholar]
- Healy, M.G.; Romo, C.R.; Bustos, R. Bioconversion of marine crustacean shell waste. Res. Conserv. Recycl. 1994, 11, 139–147. [Google Scholar] [CrossRef]
- Jung, W.J.; Jo, G.Y.; Kuk, J.H.; Kim, K.Y.; Park, R.D. Extraction of chitin from red crab shell waste by cofermentation with Lactobacillus paracasei subsp. tolerans KCTC-3074 and Serratia marcescens FS-3. Appl. Microbiol. Biot. 2006, 71, 234–237. [Google Scholar]
- Jung, W.J.; Jo, G.Y.; Kuk, J.H.; Kim, Y.J.; Oh, K.T.; Park, R.D. Production of chitin from red crab shell waste by successive fermentation with Lactobacillus paracasei KCTC-3074 and Serratia marcescens FS-3. Carbohydr. Polym. 2007, 68, 746–750. [Google Scholar] [CrossRef]
- Jo, G.H.; Jung, W.J.; Kuk, J.H.; Oh, K.T.; Kim, Y.J.; Park, R.D. Screening of protease-producing Serratia marcescens FS-3 and its application to deproteinization of crab shell waste for chitin extraction. Carbohydr. Polym. 2008, 74, 504–508. [Google Scholar] [CrossRef]
- Seyfarth, F.; Schliemann, S.; Elsner, P.; Hipler, U.C. Antifungal effect of high- and low-molecular-weight chitosan hydrochloride, carboxymethyl chitosan, chitosan oligosaccharide and N-acetyl-d-glucosamine against Candida albicans, Candida krusei and Candida glabrata. Int. J. Pharm. 2008, 353, 139–148. [Google Scholar] [PubMed]
- Oliveira, E.N., Jr.; El Gueddari, N.E.; Moerschbacher, B.M.; Peter, M.G.; Franco, T.T. Growth of phytopathogenic fungi in the presence of partially acetylated chitooligosaccharides. Mycopathologia 2008, 166, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.; Fukamizo, T. Production of food oligosaccharides by chitinases and chitosanases: Structural biological perspective. New Food Ind. 2001, 43, 7–14. [Google Scholar]
- Lee, H.W.; Park, Y.S.; Choi, J.W.; Yi, S.Y.; Shin, W.S. Antidiabetic effects of chitosan oligosaccharides in neonatal streptozotocin-induced noninsulin-dependent diabetes mellitus in rats. Biol. Pharm. Bull. 2003, 26, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Sato, K.; Saimoto, H.; Morimoto, M.; Shigemasa, Y. Depolymerization of chitin and chitosan under hydrothermal conditions. Sen-I Gakkaishi 2003, 59, 104–109. [Google Scholar] [CrossRef]
- Xing, R.E.; Liu, S.; Yu, H.H.; Guo, Z.Y.; Wang, P.B.; Li, C.P.; Li, Z.; Li, P.C. Salt-assisted acid hydrolysis of chitosan to oligomers under microwave irradiation. Carbohydr. Res. 2005, 340, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zivanovic, S.; Hayes, D.G.; Weiss, J. Efficient reduction of chitosan molecular weight by high-intensity ultrasound: Underlying mechanism and effect of process parameters. J. Agric. Food Chem. 2008, 56, 5112–5119. [Google Scholar] [CrossRef] [PubMed]
- Yoksan, R.; Akashi, M.; Miyata, M.; Chirachanchai, S. Optimal gamma-ray dose and irradiation conditions for producing low-molecular-weight chitosan that retains its chemical structure. Radiat. Res. 2004, 161, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Domard, A.; Cartier, N. Glucosamine oligomers: 4. Solid state-crystallization and sustained dissolution. Int. J. Biol. Macromol. 1992, 14, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Einbu, A.; Vårum, K.M. Depolymerization and de-N-acetylation of chitin oligomers in hydrochloric acid. Biomacromolecules 2007, 8, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Jia, X.G.; Lei, W.X.; Li, Z.J.; Zhang, T.Y. Spectra analyses of chitosans degraded by hydrogen peroxide under optimal conditions. Spectrosc. Spectr. Anal. 2009, 29, 43–47. [Google Scholar]
- Morris, V.B.; Neethu, S.; Abraham, T.E.; Pillai, C.K.S.; Sharma, C.P. Studies on the condensation of depolymerized chitosans with DNA for preparing chitosan-DNA nanoparticles for gene delivery applications. J. Biomed. Mater. Res. Part B 2009, 89B, 282–292. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Park, P.J.; Kim, S.K. Antimicrobial effect of chitooligosaccharides produced by bioreactor. Carbohydr. Polym. 2001, 44, 71–76. [Google Scholar]
- Qin, C.; Zhou, B.; Zeng, L.; Zhang, Z.; Liu, Y.; Du, Y.; Xiao, L. The physicochemical properties and antitumor activity of cellulase-treated chitosan. Food Chem. 2004, 84, 107–115. [Google Scholar]
- Aam, B.B.; Heggset, E.B.; Norberg, A.L.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Production of chitooligosaccharides and their potential applications in medicine. Mar. Drugs 2010, 8, 1482–1517. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Hsiao, Y.C.; Chiang, B.H. Production of high degree polymerized chitooligosaccharides in a membrane reactor using purified chitosanase from Bacillus cereus. Food Res. Int. 2009, 42, 1355–1361. [Google Scholar] [CrossRef]
- Zeng, H.; Zheng, L.Y. Studies on Penicillium sp. ZDZ1 chitosanase immobilized on chitin by cross-linking reaction. Process Biochem. 2002, 38, 531–535. [Google Scholar]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active enZYmes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- CAZy. The Carbohydrate-Active enZYmes database. Available online: http://www.cazy.org (accessed on 27 April 2010).
- Davies, G.; Henrissat, B. Structures and mechanisms of glycosyl hydrolases. Structure 1995, 3, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 1996, 316, 695–696. [Google Scholar] [PubMed]
- Kobayashi, S.; Kiyosada, T.; Shoda, S. Synthesis of artificial chitin: Irreversible catalytic behavior of a glycosyl hydrolase through a transition state analogue substrate. J. Am. Chem. Soc. 1996, 118, 13113–13114. [Google Scholar]
- Sato, H.; Mizutani, S.; Tsuge, S.; Ohtani, H.; Aoi, K.; Takasu, A.; Okada, M.; Kobayashi, S.; Kiyosada, T.; Shoda, S. Determination of the degree of acetylation of chitin/chitosan by pyrolysis-gas chromatography in the presence of oxalic acid. Anal. Chem. 1998, 70, 7–12. [Google Scholar] [CrossRef]
- ExplorEnz—The Enzyme Database. Available online: http://www.enzyme-explorer.org/ (accessed on 27 April 2010).
- Berger, L.R.; Reynolds, D.M. The chitinase system of a strain of Streptomyces griseus. Biochem. Biophys. Acta 1958, 29, 522–534. [Google Scholar]
- Monreal, J.; Reese, E.T. The chitinase of Serratia marcescens. J. Microbiol. 1969, 15, 689–696. [Google Scholar]
- Sørbotten, A.; Horn, S.J.; Eijsink, V.G.; Vårum, K.M. Degradation of chitosans with chitinase B from Serratia marcescens. Production of chito-oligosaccharides and insight into enzyme processivity. FEBS J. 2005, 272, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Lopatin, S.A.; Derbeneva, M.S.; Kulikov, S.N.; Varlamov, V.P.; Shpigun, O.A. Fractionation of chitosan by ultrafiltration. J. Anal. Chem. 2009, 64, 648–651. [Google Scholar] [CrossRef]
- Haebel, S.; Bahrke, S.; Peter, M.G. Quantitative sequencing of complex mixtures of heterochitooligosaccharides by MALDI-linear ion trap mass spectrometry. Anal. Chem. 2007, 79, 5557–5566. [Google Scholar]
- Le Dévédec, F.; Bazinet, L.; Furtos, A.; Venne, K.; Brunet, S.; Mateescu, M.A. Separation of chitosan oligomers by immobilized metal affinity chromatography. J. Chromatogr. A 2008, 1194, 165–171. [Google Scholar] [CrossRef]
- Pichyangkura, R.; Kudan, S.; Kuttiyawong, K.; Sukwattanasinitt, M.; Aiba, S. Quantitative production of 2-acetamido-2-deoxy-d-glucose from crystalline chitin by bacterial chitinase. Carbohydr. Res. 2002, 337, 557–559. [Google Scholar] [CrossRef]
- Sashiwa, H.; Fujishima, S.; Yamano, N.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Sukwattanasinitt, M.; Pichyangkurac, R.; Aiba, S. Enzymatic production of N-acetyl-d-glucosamine from chitin. Degradation study of N-acetylchitooligosaccharide and the effect of mixing of crude enzymes. Carbohydr. Polym. 2003, 51, 391–395. [Google Scholar] [CrossRef]
- Il’ina, A.V.; Zueva, O.Y.; Lopatin, S.A.; Varlamov, V.P. Enzymatic hydrolysis of α-chitin. Appl. Biochem. Microbiol. 2004, 40, 35–38. [Google Scholar]
- Sukwattanasinitt, M.; Zhu, H.; Sashiwa, H.; Aiba, S. Utilization of commercial non-chitinase enzymes from fungi for preparation of 2-acetamido-2-deox-d-glucose from β-chitin. Carbohydr. Res. 2002, 337, 133–137. [Google Scholar] [CrossRef]
- Sashiwa, H.; Fujishima, S.; Yamano, N.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Hiraga, K.; Oda, K.; Aiba, S. Production of N-acetyl-d-glucosamine from α-chitin by crude enzymes from Aeromonas hydrophila H-2330. Carbohydr. Res. 2002, 337, 761–763. [Google Scholar] [CrossRef]
- Thamthiankul, S.; Suan-Ngay, S.; Tantimavanich, S.; Panbangred, W. Chitinase from Bacillus thuringiensis subsp pakistani. Appl. Microbiol. Biotechnol. 2001, 56, 395–401. [Google Scholar]
- Katta, S.; Ankati, S.; Podile, A.R. Chitooligosaccharides are converted to N-acetylglucosamine by N-acetyl-β-hexosaminidase from Stenotrophomonas maltophilia. FEMS Microbiol. Lett. 2013, 348, 19–25. [Google Scholar] [CrossRef]
- Lan, X.; Ozawa, N.; Nishiwaki, N.; Kodaira, R.; Okazaki, M.; Shimosaka, M. Purification, cloning, and sequence analysis of β-N-acetylglucosaminidase from the chitinolytic bacterium Aeromonas hydrophila strain SUWA-9. Biosci. Biotechnol. Biochem. 2004, 68, 1082–1090. [Google Scholar]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Ahn, J.S.; Kim, K.Y.; Park, R.D. Selective preparation of N-acetyl-d-glucosamine and N,N′-diacetylchitobiose from chitin using a crude enzyme preparation from Aeromonas sp. Biotechnol. Lett. 2005, 27, 7–11. [Google Scholar] [CrossRef]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Kim, Y.C.; Kim, K.Y.; Park, R.D. Production of N-acetyl-β-d-glucosamine from chitin by Aeromonas sp. GJ-18 crude enzyme. Appl. Microbiol. Biotechnol. 2005, 68, 384–389. [Google Scholar] [CrossRef]
- Kuk, J.H.; Jung, W.J.; Jo, G.H.; Kim, K.Y.; Park, R.D. Production of N,N′-diacetylchitobiose from chitin using temperature-sensitive chitinolytic enzyme preparations of Aeromonas sp. GJ-18. World J. Microbiol. Biotechnol. 2006, 22, 135–139. [Google Scholar] [CrossRef]
- Martinez, E.A.; Boer, H.; Koivula, A.; Samain, E.; Driguez, H.; Armand, S.; Cottaz, S. Engineering chitinases for the synthesis of chitin oligosaccharides: Catalytic amino acid mutations convert the GH-18 family glycoside hydrolases into transglycosylases. J. Mol. Catalys. B Enzym. 2012, 74, 89–96. [Google Scholar]
- Tanaka, T.; Fukui, T.; Atomi, H.; Imanaka, T. Characterization of an exo-β-d-glucosaminidase involved in a novel chitinolytic pathway from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bact. 2003, 185, 5175–5181. [Google Scholar]
- Yamasaki, Y.; Hayashi, I.; Ohta, Y.; Nakagawa, T.; Kawamukai, M.; Matsuda, H. Purification and mode of action of chitosanolytic enzyme from Enterobacter sp. G-1. Biosci. Biotechnol. Biochem. 1993, 57, 444–449. [Google Scholar]
- Makino, A.; Sakamoto, J.; Ohmae, M.; Kobayashi, S. Effect of fluorine substituent on the chitinase-catalyzed polymerization of sugar oxazoline derivatives. Chem. Lett. 2006, 35, 160–161. [Google Scholar] [CrossRef]
- Makino, A.; Ohmae, M.; Kobayashi, S. Chitinase-catalyzed copolymerization to a chitin derivative having glucosamine unit in controlled proportion. Polym. J. 2006, 38, 1182–1188. [Google Scholar] [CrossRef]
- Takiguchi, Y.; Shimahara, K. N,N-Diacetylchitobiose production from chitin by Vibrio anguillarum strain E-383a. Lett. Appl. Microbiol. 1988, 6, 129–131. [Google Scholar] [CrossRef]
- Ilankovan, P.; Hein, S.; Ng, C.H.; Trung, T.S.; Stevens, W.F. Production of N-acetyl chitobiose from various chitin substrates using commercial enzymes. Carbohydr. Polym. 2006, 63, 245–250. [Google Scholar] [CrossRef]
- Wang, S.L.; Liu, C.P.; Liang, T.W. Fermented and enzymatic production of chitin/chitosan oligosaccharides by extracellular chitinases from Bacillus cereus TKU027. Carbohydr. Polym. 2012, 90, 1305–1313. [Google Scholar]
- Wang, C.Y.; Hsieh, Y.Z. Analysis of chitin oligosaccharides by capillary electrophoresis with laser-inducedfluorescence. J. Chromatogr. A 2002, 979, 431–438. [Google Scholar]
- Saito, J.; Kita, A.; Higuchi, Y.; Nagata, Y.; Ando, A.; Miki, K. Crystal structure of chitosanase from Bacillus circulans MH-K1 at 1.6-angstrom resolution and its substrate recognition mechanism. J. Biol. Chem. 1999, 274, 30818–30825. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Ohkawa, T.; Ikeda, Y.; Goto, S. Specificity of chitosanase from Bacillus pumilus. Biochim. Biophys. Acta 1994, 1205, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Chang, C.H.; Wu, Y.J.; Li, Y.K. Exploration of glycosyl hydrolase family 75, a chitosanase from Aspergillus fumigatus. J. Biol. Chem. 2006, 281, 3137–3144. [Google Scholar] [CrossRef]
- Fukamizo, T.; Brzezinski, R. Chitosanase from Streptomyces sp. strain N174: A comparative review of its structure and function. Biochem. Cell Biol. 1997, 75, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, E.J.; Piao, Z.; Yun, Y.C.; Shin, Y.C. Purification and characterization of chitosanase from Bacillus sp. strain KCTC 0377BP and its application for the production of chitosan oligosaccharides. Appl. Environ. Microbiol. 2004, 70, 4522–4531. [Google Scholar]
- Kurakake, M.; You, S.; Nakagawa, K.; Sugihara, M.; Komaki, T. Properties of chitosanase from Bacillus cereus S1. Curr. Microbiol. 2000, 40, 6–9. [Google Scholar] [CrossRef]
- Park, R.D.; Rhee, C.O.; Lee, H.C.; Cho, C.S.; Jo, D.H. A comparative study of chitooligosaccharide production from chitosan. J. Chitin Chitosan 1999, 4, 96–101. [Google Scholar]
- Yoon, H.G.; Ha, S.C.; Lim, Y.H.; Cho, H.Y. New thermostable chitosanase from Bacillus sp.: Purification and characterization. J. Microbiol. Biotechnol. 1998, 8, 449–454. [Google Scholar]
- Lee, H.W.; Choi, J.W.; Han, D.P.; Park, M.J.; Lee, N.W.; Yi, D.H. Purification and characteristics of chitosanase from Bacillus sp. HW-002. J. Microbiol. Biotechnol. 1996, 6, 19–25. [Google Scholar]
- Jeon, Y.J.; Kim, S.K. Production of chitooligosaccharides using ultrafilteration membrane reactor and their antibacterial activity. Carbohydr. Polym. 2000, 41, 133–144. [Google Scholar] [CrossRef]
- Pelletier, A.; Sygusch, J. Purification and characterization of three chitosanase activities from Bacillus megaterium P1. Appl. Environ. Microbiol. 1990, 56, 844–848. [Google Scholar]
- Shimosaka, M.; Nogawa, M.; Wang, X.Y.; Kumehara, M.; Okazaki, M. Production of two chitosanases from a chitosan-assimilating bacterium, Acinetobacter sp. strain CHB101. Appl. Environ. Microbiol. 1995, 61, 438–442. [Google Scholar]
- Wang, S.L.; Tseng, W.N.; Liang, T.W. Biodegradation of shellfish wastes and production of chitosanases by a squid pen-assimilating bacterium, Acinetobacter calcoaceticus TKU024. Biodegradation 2011, 22, 939–948. [Google Scholar] [CrossRef]
- Nanjo, F.; Katsumi, R.; Sakai, K. Purification and characterization of an exo-β-d-glucosaminidase, a novel type of enzyme, from Nocardia orientalis. J. Biol. Chem. 1990, 265, 10088–10094. [Google Scholar]
- Park, J.K.; Shimono, K.; Ochiai, N.; Shigeru, K.; Kurita, M.; Ohta, Y.; Tanaka, K.; Matsuda, H.; Kawamukai, M. Purification, characterization, and gene analysis of a chitosanase (ChoA) from Matsuebacter chitosanotabidus 3001. J. Bact. 1999, 181, 6642–6649. [Google Scholar]
- Kim, S.Y.; Shon, D.H.; Lee, K.H. Purification and characteristics of two types of chitosanases from Aspergillus fumigatus. J. Microbiol. Biotechnol. 1998, 8, 568–574. [Google Scholar]
- Jung, W.J.; Kuk, J.H.; Kim, K.Y.; Jung, K.C.; Park, R.D. Purification and characterization of exo-β-d-glucosaminidase from Aspergillus fumigatus S-26. Protein Expr. Purif. 2006, 45, 125–131. [Google Scholar]
- Zhang, X.Y.; Dae, A.L.; Zhang, X.K.; Kuroiwa, K. Purification and characterization of chitosanase and exo-β-d-glucosaminidase from Koji Mold, Aspergillus oryzae IAM2660. Biosci. Biotechnol. Biochem. 2000, 64, 1896–1902. [Google Scholar]
- Nogawa, M.; Takahashi, H.; Kasiwagi, A.; Ohshima, K.; Okada, H.; Morikawa, Y. Purification and characterization of exo-β-d-glucosaminidase from a cellulolytic fungus, Trichoderma reesei PC-3-7. Appl. Envoron. Microbiol. 1998, 64, 890–895. [Google Scholar]
- Lin, H.; Wang, H.; Xue, C.; Ye, M. Preparation of chitosan oligomers by immobilized papain. Enzyme Microb. Technol. 2002, 31, 588–592. [Google Scholar] [CrossRef]
- Lee, D.X.; Xia, W.S.; Zhang, J.L. Enzymatic preparation of chitooligosaccharides by commercial lipase. Food Chem. 2008, 111, 291–295. [Google Scholar] [CrossRef]
- Zhang, H.; Du, Y.; Yu, X.; Mitsutomi, M.; Aiba, S. Preparation of chitooligosaccharides from chitosan by acomplex enzyme. Carbohydr. Res. 1999, 320, 257–260. [Google Scholar] [CrossRef]
- Mahata, M.; Shinya, S.; Masaki, E.; Yamamoto, T.; Ohnuma, T.; Brzezinski, R.; Mazumder, T.K.; Yamashita, K.; Narihiro, K.; Fukamizo, T. Production of chitooligosaccharides from Rhizopus oligosporus NRRL2710 cells by chitosanase digestion. Carbohydr. Res. 2014, 383, 27–33. [Google Scholar] [CrossRef]
- Yalpani, M.; Pantaleone, D. An examination of the unusual susceptibility of aminoglycans to enzymatic hydrolysis. Carbohydr. Res. 1994, 256, 159–175. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Huang, C.C.; Liang, T.W.; Nguyen, V.B.; Pan, P.S.; Wang, S.L. Production and purification of a fungal chitosanase and chitooligomers from Penicillium janthinellum D4 and discovery of the enzyme activators. Carbohydr. Polym. 2014, 108, 331–337. [Google Scholar] [CrossRef]
- Gao, X.A.; Jung, W.J.; Kuk, J.H.; Park, R.D. Reaction pattern of Bacillus cereus D-11 chitosanase on chito-oligosaccharide alcohols. J. Microbiol. Biotechnol. 2009, 19, 358–361. [Google Scholar]
- Pantaleone, D.; Yalpani, M.; Scollar, M. Unusual susceptibility of chitosan to enzymic hydrolysis. Carbohydr. Res.1992, 1992, 237, 325–332. [Google Scholar]
- Aiba, S.I. Preparation of N-acetylchitooligosaccharides from lysozymatic hydroxylate of partially N-acetylated chitosans. Carbohydr. Res. 1994, 261, 297–306. [Google Scholar] [CrossRef]
- Kumar, A.B.V.; Gowda, L.R.; Tharanathan, R.N. Non-specific depolymerization of chitosan by pronase and characterization of the resultant products. Eur. J. Biochem. 2004, 271, 713–723. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, J.; Wei, Y.; Hong, X. Preparation of chitooligosaccharides by the enzymatic hydrolysis of chitosan. Polym. Degrad. Stab. 2009, 94, 1895–1899. [Google Scholar] [CrossRef]
- Wu, S. Preparation of chitooligosaccharides from Clanis bilineata larvae skin and their antibacterial activity. Int. J. Biol. Macromol. 2012, 51, 1147–1150. [Google Scholar]
- Zhao, Y.; Park, R.D.; Muzzarelli, R.A.A. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef]
- Zhao, Y.; Kim, Y.J.; Oh, K.T.; Nguyen, V.N.; Park, R.D. Production and characterization of extracellular chitin deacetylase from Absidia corymbifera DY-9. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 119–126. [Google Scholar] [CrossRef]
- Zhao, Y.; Ju, W.T.; Jo, G.H.; Jung, W.J.; Park, R.D. Perspectives of chitin deacetylase research. In Biotechnology of Biopolymers; Elnashar, M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 131–144. [Google Scholar]
- Kim, Y.J.; Zhao, Y.; Oh, K.T.; Nguyen, V.N.; Park, R.D. Enzymatic deacetylation of chitin by extracellular chitin deacetylase from newly screened Mortierella sp. DY-52. J. Microbiol. Biotechnol. 2008, 18, 759–766. [Google Scholar]
- Tanaka, T.; Fukui, T.; Fujiwara, S.; Atomi, H.; Imanaka, T. Concerted action of diacetylchitobiose deacetylase and exo-beta-d-glucosaminidase in a novel Biotechnology of Biopolymers chitinolytic pathway in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Biol. Chem. 2004, 279, 30021–30027. [Google Scholar]
- Tokuyasu, K.; Ono, H.; Ohnishi-Kameyama, M.; Hayashi, K.; Mori, Y. Deacetylation of chitin oligosaccharides of dp 2-4 by chitin deacetylase from Colletotrichum lindemuthianum. Carbohydr. Res. 1997, 303, 353–358. [Google Scholar] [CrossRef]
- Alfonso, C.; Nuero, O.M.; Santamarla, F.; Reyes, F. Purification of a heat-stable chitin deacetylase from Aspergillus nidulans and its role in cell wall degradation. Curr. Microbiol. 1995, 30, 49–54. [Google Scholar] [CrossRef]
- Aguila, E.M.D.; Gomes, L.P.; Andrade, C.T.; Silva, J.T.; Paschoalin, V.M.F. Biocatalytic production of chitosan polymers from shrimp shells, using a recombinant enzyme produced by Pichia pastoris. Am. J. Mol. Biol. 2012, 2, 341–350. [Google Scholar] [CrossRef]
- Blair, D.E.; Hekmat, O.; Schuttelkopf, A.W.; Shrestha, B.; Tokuyasu, K.; Withers, S.G.; van Aalten, D.M. Structure and mechanism of chitin deacetylase from the fungal pathogen Colletotrichum lindemuthianum. Biochemistry 2006, 45, 9416–9426. [Google Scholar] [CrossRef] [PubMed]
- Martinou, A.; Bouriotis, V.; Stokke, B.T.; Vårum, K.M. Mode of action of chitin deacetylase from Mucor rouxii on partially N-acetylated chitosans. Carbohydr. Res. 1998, 311, 71–78. [Google Scholar] [CrossRef]
- Hattori, T.; Sakabe, Y.; Ogata, M.; Michishita, K.; Dohra, H.; Kawagishi, H.; Totani, K.; Nikaido, M.; Nakamura, T.; Koshino, H.; Usui, T. Enzymatic synthesis of an α-chitin-like substance via lysozyme-mediated transglycosylation. Carbohydr. Res. 2012, 347, 16–22. [Google Scholar] [CrossRef]
- Shimoda, K.; Nakajima, K.; Hiratsuka, Y.; Nishimura, S.I.; Kurita, K. Efficient preparation of β-(1→6)-(GlcNAc)2 by enzymatic conversion of chitin and chito-oligosaccharides. Carbohydr. Polym. 1996, 29, 149–154. [Google Scholar] [CrossRef]
- Matsumura, S.; Yao, E.; Toshima, K. One-step preparation of alkyl β-d-glucosaminide by the transglycosylation of chitosan and alcohol using purified exo-β-d-glucosaminidase. Biotechnol. Lett. 1999, 21, 451–456. [Google Scholar]
- Usui, T.; Matsu, H.; Isobe, K. Enzymic synthesis of useful chito-oligosaccharides utilizing transglycosylation by chitinolytic enzymes in a buffer containing ammonium sulfate. Carbohydr. Res. 1990, 203, 65–77. [Google Scholar] [CrossRef]
- Akiyama, K.; Kawazu, K.; Kobayashi, A. A novel method for chemo-enzymatic synthesis of elicitor-active chitosan oligomers and partially N-acetylated chitin oligomers using N-acetylated chitotriose as substrates in a lysozyme-catalyzed transglycosylation reaction system. Carbohydr. Res. 1995, 279, 151–160. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Hattori, T.; Uno, S.; Murata, T.; Usui, T. Enzymatic synthesis of gentiooligosaccharides by transglycosylation with β-glycosidases from Penicillium multicolor. Carbohydr. Res. 2009, 344, 972–978. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A.; Matsumoto, H.; Kunii, S.; Ohmae, M.; Kiyosada, T.; Makiguchi, K.; Matsumoto, A.; Horie, M.; Shoda, S.I. Enzymatic polymerization to novel polysaccharides having a glucose-N-acetylglucosamine repeating unit, a cellulose-chitin hybrid polysaccharide. Biomacromolecules 2006, 7, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Kadokawa, J.I. Precision polysaccharide synthesis catalyzed by enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar]
- Shoda, S.; Izumi, R.; Fujita, M. Green process in glycotechnology. Bull. Chem. Soc. Jpn. 2003, 76, 1–13. [Google Scholar]
- Kadokawa, J.; Shoda, S.J. New methods for architectures of glyco-materials. Synthetic Org. Chem. Jpn. 2003, 61, 1207. [Google Scholar] [CrossRef]
- Misawa, Y.; Lohavisavapanichi, C.; Shoda, S. Chemo-enzymatic synthesis of oligosaccharides having a polymerizable group at the reducing end. Glycoconj. J. 1999, 16, S122. [Google Scholar]
- Shoda, S.; Fujita, M.; Lohavisavapanichi, C.; Misawa, Y.; Ushizaki, K.; Tawata, Y.; Kuriyama, M.; Kohri, M.; Kuwata, H.; Watanabe, T. Efficient method for the elongation of the N-acetylglucosamine unit by combined use of chitinase and β-galactosidase. Helv. Chim. Acta 2002, 85, 3919. [Google Scholar] [CrossRef]
- Kadokawa, J.; Shoda, S. Enzymatic synthesis of glyco-macromonomers. Sen’i Gakkaishi 2003, 59, 74–78. [Google Scholar] [CrossRef]
- Shoda, S.; Misawa, Y.; Nishijima, Y.; Tawata, Y.; Kotake, T.; Noguchi, M.; Kobayashi, A.; Watanabe, T. Chemo-enzymatic synthesis of novel oligo-N-acetyllactosamine derivatives having a β(1-4)–β(1-6) repeating unit by using transition state analogue substrate. Cellulose 2006, 13, 477–484. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobori, K.; Miyashita, K.; Fujii, T.; Sakai, H.; Uchida, M.; Tanaka, H. Identification of glutamic acid 204 and aspartic acid 200 in chitinase A1 of Bacillus circulans WL-12 as essential residues for chitinase activity. J. Biol. Chem. 1993, 268, 18567–18572. [Google Scholar]
- Watanabe, T.; Suzuki, K.; Oyanagi, W.; Ohnishi, K.; Tanaka, H. Gene cloning of chitinase A1 from Bacillus circulans WL-12 revealed its evolutionary relationship to Serratia chitinase and to the type III homology units of fibronectin. J. Biol. Chem. 1990, 265, 15659–15665. [Google Scholar]
- Sakamoto, J.; Watanabe, T.; Ariga, Y.; Kobayashi, S. Ring-opening glycosylation of a chitobiose oxazoline catalyzed by a non-chitinolytic mutant of chitinase. Chem. Lett. 2001, 30, 1180–1181. [Google Scholar]
- Ochiai, H.; Ohmae, M.; Kobayashi, S. Enzymatic synthesis of alternatingly 6-O-carboxymethylated chitotetraose by selective glycosidation with chitinase catalysis. Chem. Lett. 2004, 33, 694–695. [Google Scholar] [CrossRef]
- Ochiai, H.; Ohmae, M.; Kobayashi, S. Enzymatic glycosidation of sugar oxazolines having a carboxylate group catalyzed by chitinase. Carbohydr. Res. 2004, 339, 2769–2788. [Google Scholar] [CrossRef]
- Makino, A.; Kurosaki, K.; Ohmae, M.; Kobayashi, S. Chitinase-catalyzed synthesis of alternatingly N-Deacetylated chitin: A chitin–chitosan hybrid polysaccharide. Biomacromolecules 2006, 7, 950–957. [Google Scholar] [CrossRef]
- Villa-Lerma, G.; Gonzalez-Marquez, H.; Gimeno, M.; Lopez-Luna, A.; Barzana, E.; Shirai, K. Ultrasonication and steam-explosion as chitin pretreatments for chitin oligosaccharide production by chitinases of Lecanicillium lecanii. Biores. Technol. 2013, 146, 794–798. [Google Scholar]
- Nakagawa, Y.S.; Oyama, Y.; Kon, N.; Nikaido, M.; Tanno, K.; Kogawa, J.; Inomata, S.; Masui, A.; Yamamura, A.; Kawaguchi, M.; et al. Development of innovative technologies to decrease the environmental burdens associated with using chitin as a biomass resource: Mechanochemical grinding and enzymatic degradation. Carbohydr. Polym. 2011, 83, 1843–1849. [Google Scholar] [CrossRef]
- Sato, T.; Asada, K.; Takeda, M.; Tanno, K. Mechano-chemical synthesis of compound powders in ZnO–TiO2 system by a new high intensive ball mill. J. Jpn. Soc. Powder Metall. 2006, 53, 62–67. [Google Scholar]
- Takeda, T.; Nikaido, M.; Totani, T.; Obara, M.; Nakano, Y.; Uchimiya, H. Evaluation of high intensive ball mill method for effective saccharification of plant cell wall materials. J. Appl. Glycosci. 2009, 56, 71–76. [Google Scholar]
- Wu, Q.; Miao, Y. Mechanochemical effects of micronization on enzymatic hydrolysis of corn flour. Carbohydr. Polym. 2008, 72, 398–402. [Google Scholar]
- Fujimoto, S.; Inoue, H.; Yano, S.; Sakaki, T.; Minowa, T.; Endo, T. Bioethanol production from lignocellulosic biomass requiring no sulfuric acid: Mechanochemical pretreatment and enzymic saccharification. J. Jpn. Petrol. Inst. 2008, 51, 264–273. [Google Scholar] [CrossRef]
- Van Craeyveld, V.; Delcour, J.A.; Courtin, C.M. Ball milling improves extractability and affects molecular properties of Psyllium (Plantago ovate Forsk) seed husk arabinoxylan. J. Agric. Food Chem. 2008, 56, 11306–11311. [Google Scholar] [CrossRef]
- Reese, E.T.; Siu, R.G.H.; Levinson, H.S. The biological degradation of soluble cellulose derivatives and its relationship to the mechanism of cellulose hydrolysis. J. Bacteriol. 1950, 59, 485–497. [Google Scholar]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Shinohara, M.L.; Ihara, M.; Abo, M.; Hashida, M.; Takagi, S.; Beck, T.C. A novel thermostable branching enzyme from an extremely thermophilic bacterial species, Rhodothermus obamensis. Appl. Microbiol. Biotechnol. 2001, 57, 653–659. [Google Scholar]
- Montilla, A.; Ruiz-Matute, A.I.; Corzo, N.; Cecilia Giacomini, C.; Irazoqui, G. Enzymatic generation of chitooligosaccharides from chitosan using soluble and immobilized glycosyltransferase (Branchzyme). J. Agric. Food Chem. 2013, 61, 10360–10367. [Google Scholar]
- Heggset, E.B.; Dybvik, A.I.; Hoell, I.A.; Norberg, A.L.; Sørlie, M.; Eijsink, V.G.H.; Vårum, K.M. Degradation of chitosans with a family 46 chitosanase from Streptomyces coelicolor A3(2). Biomacromolecules 2010, 11, 2487–2497. [Google Scholar] [CrossRef]
- Horn, S.J.; Sørbotten, A.; Synstad, B.; Sikorski, P.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G. Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J. 2006, 273, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Fukamizo, T.; Honda, Y.; Goto, S.; Boucher, I.; Brzezinski, R. Reaction mechanism of chitosanase from Streptomyces sp. N174. Biochem. J. 1995, 311, 377–383. [Google Scholar]
- Amano, K.; Ito, E. The action of lysozyme on partially deacetylated chitin. Eur. J. Biochem. 1978, 85, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.; Stokke, B.T.; Sørbotten, A.; Vårum, K.M.; Horn, S.J.; Eijsink, V.G. Development and application of a model for chitosan hydrolysis by a family 18 chitinase. Biopolymers 2005, 77, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Heggset, E.B.; Hoell, I.A.; Kristoffersen, M.; Eijsink, V.G.; Vårum, K.M. Degradation of chitosans with chitinase G from Streptomyces coelicolor A3(2): Production of chito-oligosaccharides and insight into subsite specificities. Biomacromolecules 2009, 10, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Mitsutomi, M.; Hata, T.; Kuwahara, T. Purification and characterization of novel chitinases from Streptomyces griseus Hut 6037. J. Ferment. Bioeng. 1995, 80, 153–158. [Google Scholar] [CrossRef]
- Sasaki, C.; Vårum, K.M.; Itoh, Y.; Tamoi, M.; Fukamizo, T. Rice chitinases: Sugar recognition specificities of the individual subsites. Glycobiology 2006, 16, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.J.; Sikorski, P.; Cederkvist, J.B.; Vaaje-Kolstad, G.; Sørlie, M.; Synstad, B.; Vriend, G.; Vårum, K.M.; Eijsink, V.G.H. Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. USA 2006, 103, 18089–18094. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, P.; Sørbotten, A.; Horn, S.J.; Eijsink, V.G.; Vårum, K.M. Serratia marcescens chitinases with tunnel-shaped substrate-binding grooves show endo activity and different degrees of processivity during enzymatic hydrolysis of chitosan. Biochemistry 2006, 45, 9566–9574. [Google Scholar] [CrossRef] [PubMed]
- Gyore, J.; Parameswar, A.R.; Hebbard, C.F.F.; Oh, Y.; Bi, E.; Demchenko, A.V.; Price, N.P.; Orlean, P. 2-Acylamido analogues of N-acetylglucosamine prime formation of chitin oligosaccharides by yeast chitin synthase 2. J. Biol. Chem. 2014, 289, 12835–12841. [Google Scholar] [CrossRef]
- Liang, T.W.; Hsieh, J.L.; Wang, S.L. Production and purification of a protease, a chitosanase, and chitin oligosaccharides by Bacillus cereus TKU022 fermentation. Carbohydr. Res. 2012, 362, 38–46. [Google Scholar] [CrossRef]
- Vandanarachchi, J.K.; Kurukulasuriya, M.S.; Kim, S.K. Chitin, chitosan, and their oligosaccharides in food industry. In Chitin, Chitosan, Oligosaccharides and Their Derivatives; Kim, S.K., Ed.; CRC Press: Boca Raton, FL, USA, 2011; pp. 543–560. [Google Scholar]
- Jeon, Y.J.; Kim, S.K. Continuous production of chitooligosaccharides using a dual reactor system. Process Biochem. 2000, 35, 623–632. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jung, W.-J.; Park, R.-D. Bioproduction of Chitooligosaccharides: Present and Perspectives. Mar. Drugs 2014, 12, 5328-5356. https://doi.org/10.3390/md12115328
AMA Style
Jung W-J, Park R-D. Bioproduction of Chitooligosaccharides: Present and Perspectives. Marine Drugs. 2014; 12(11):5328-5356. https://doi.org/10.3390/md12115328
Chicago/Turabian StyleJung, Woo-Jin, and Ro-Dong Park. 2014. "Bioproduction of Chitooligosaccharides: Present and Perspectives" Marine Drugs 12, no. 11: 5328-5356. https://doi.org/10.3390/md12115328