Synthetic Approaches to the Lamellarins—A Comprehensive Review


Abstract
:
1. Introduction

2. Syntheses of type-II Lamellarins
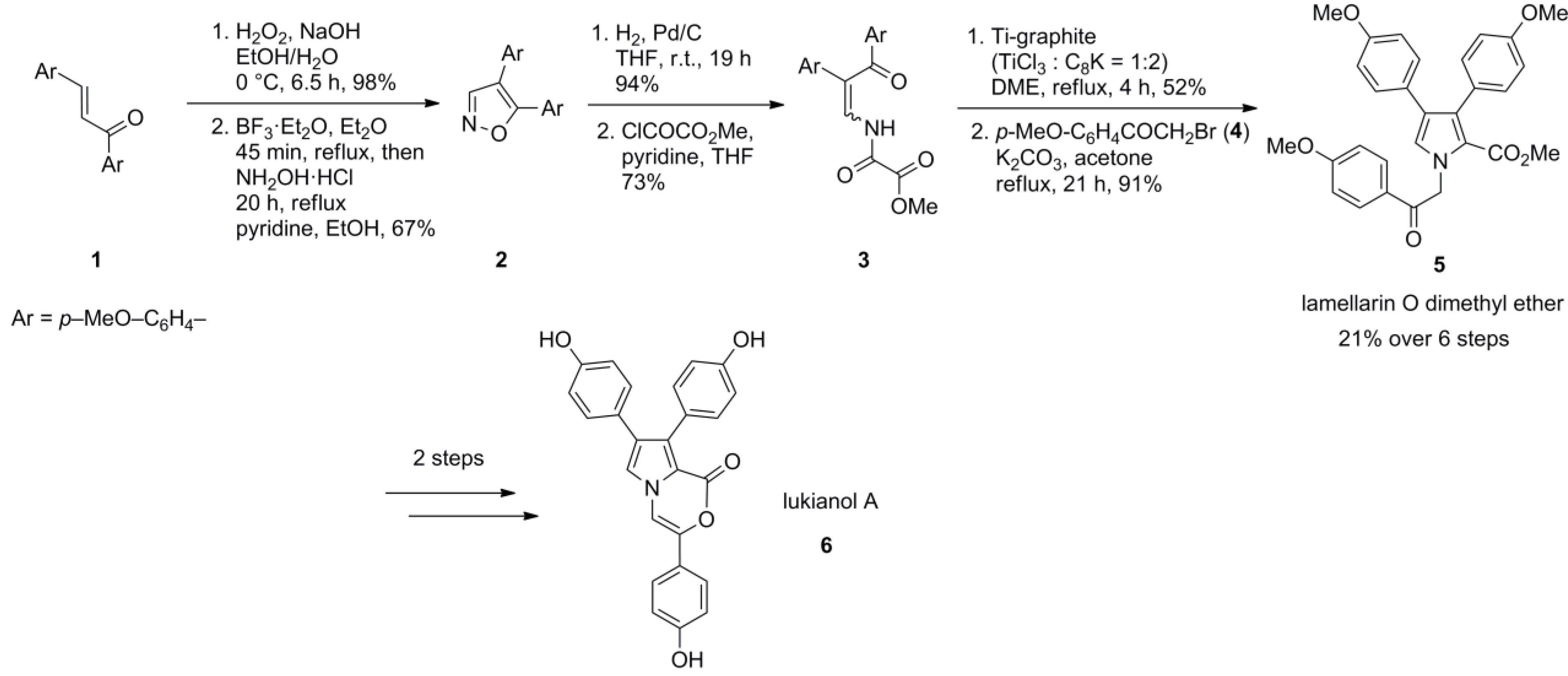
2.1. Fürstner 1995 [65]

2.2. Banwell 1997 [66]
2.3. Boger 1999 [67]


2.4. Álvarez 2004 [68]

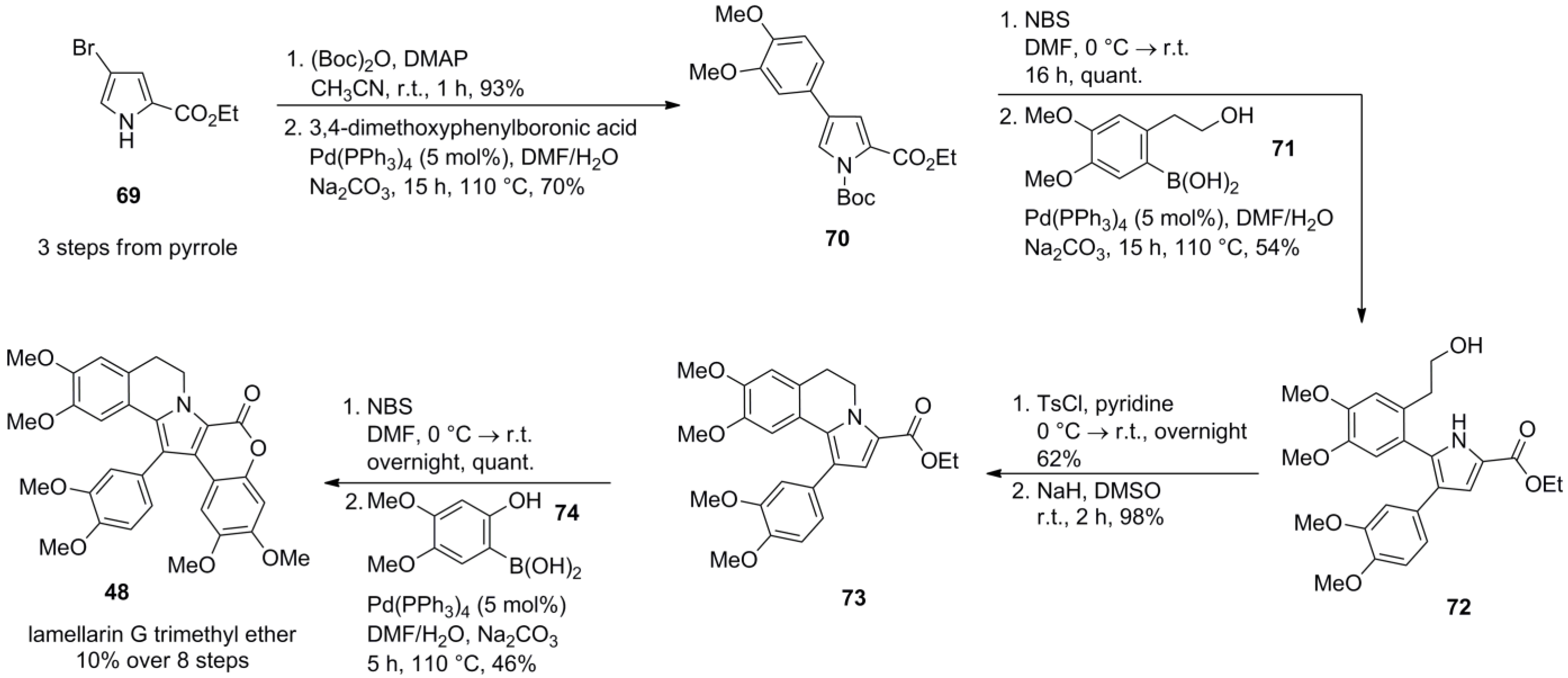
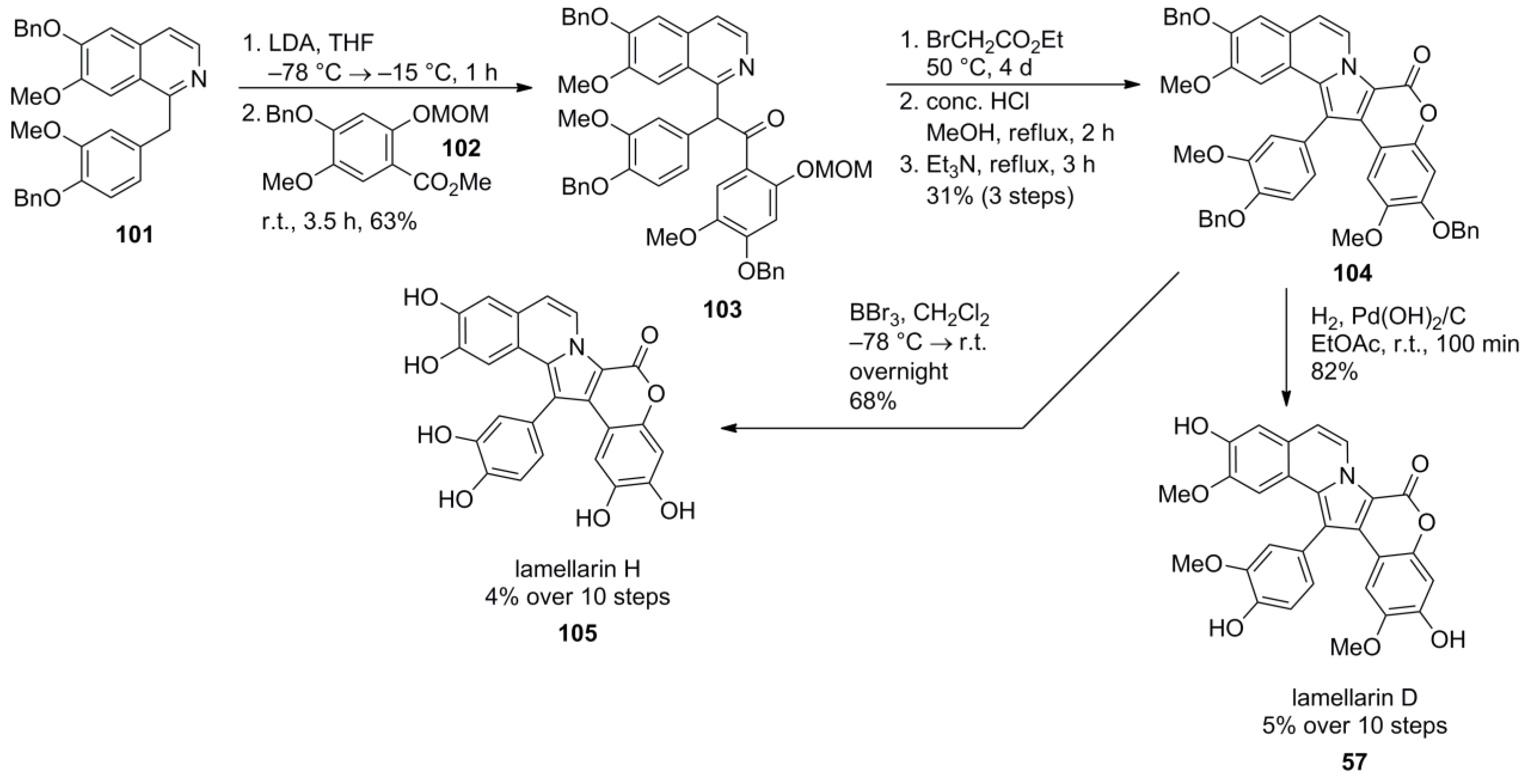
2.5. Iwao 2008 [69]
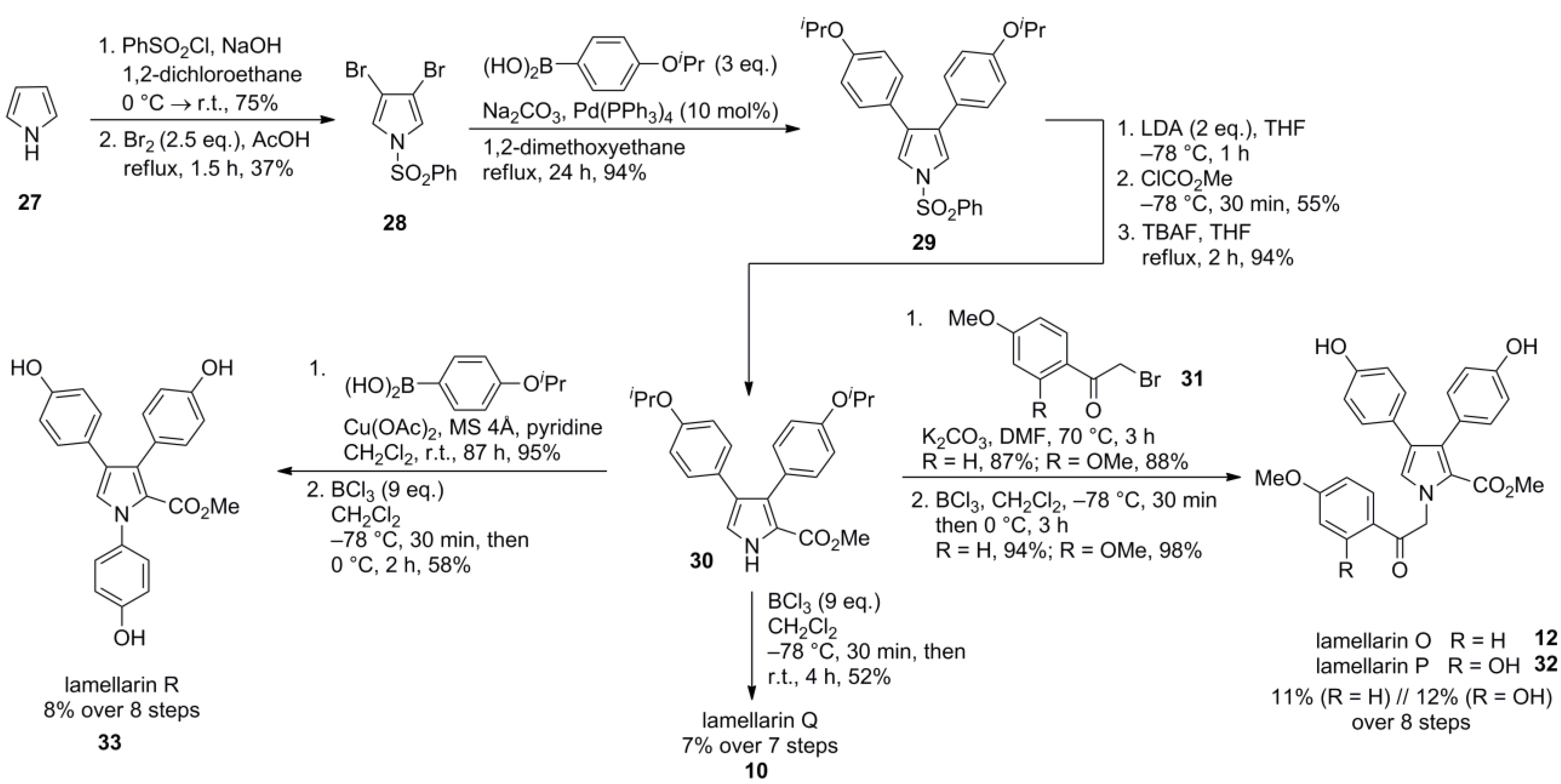
2.6. Jia 2011 [70]

2.7. Vazquez 2012 [71]

3. Syntheses of Type I-Lamellarins—Approaches via Halogenation and Cross-Coupling Reactions
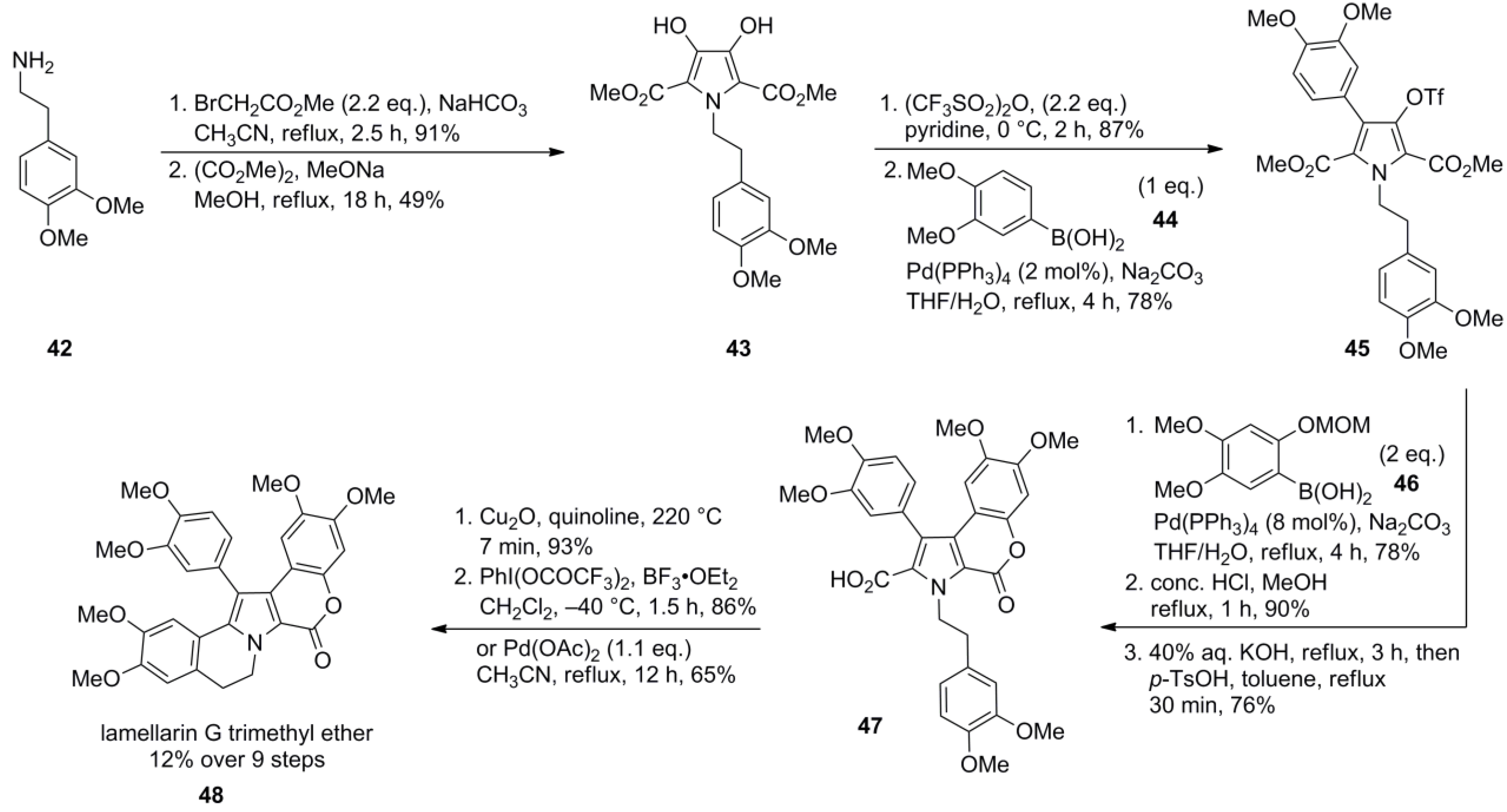
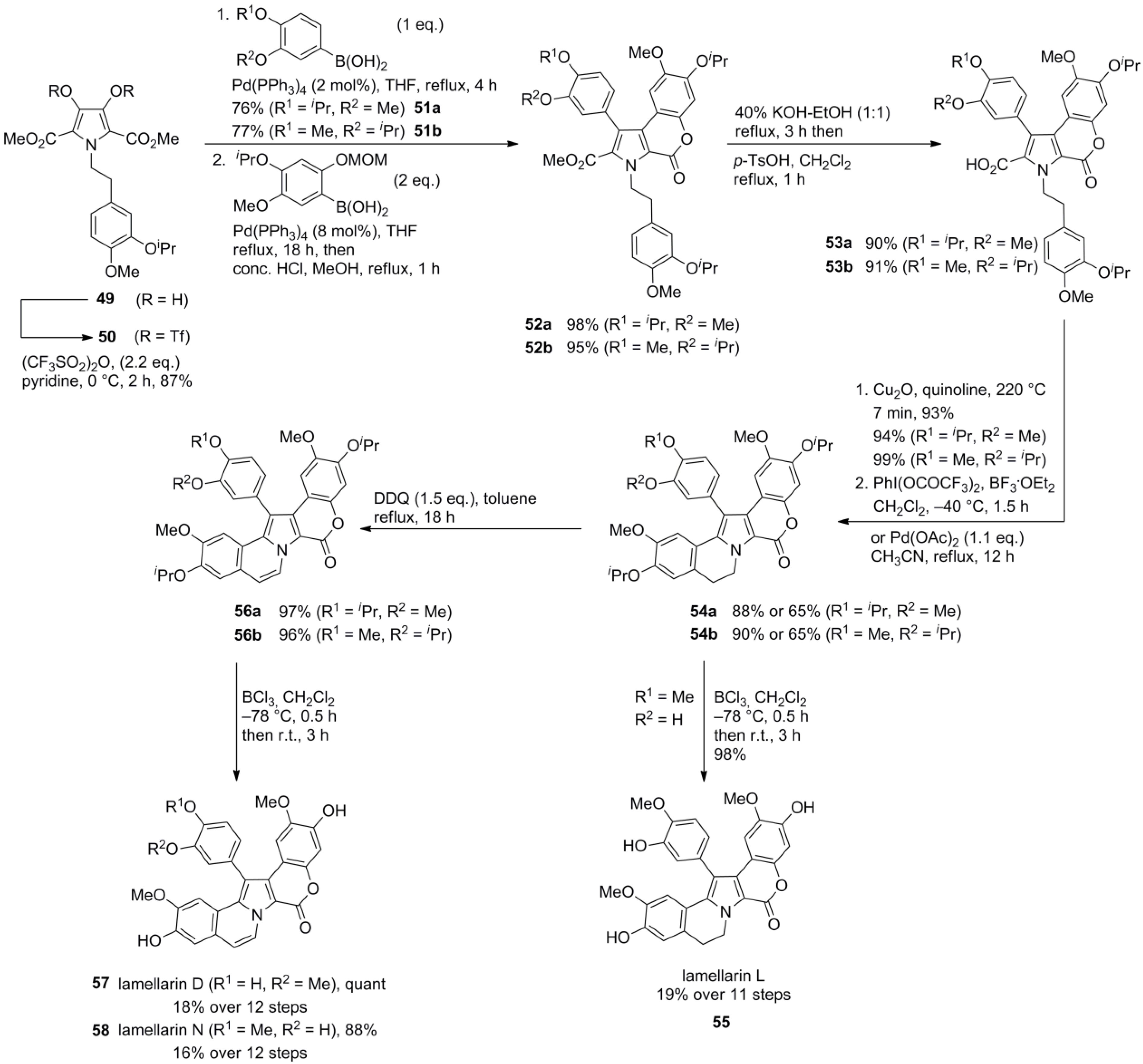
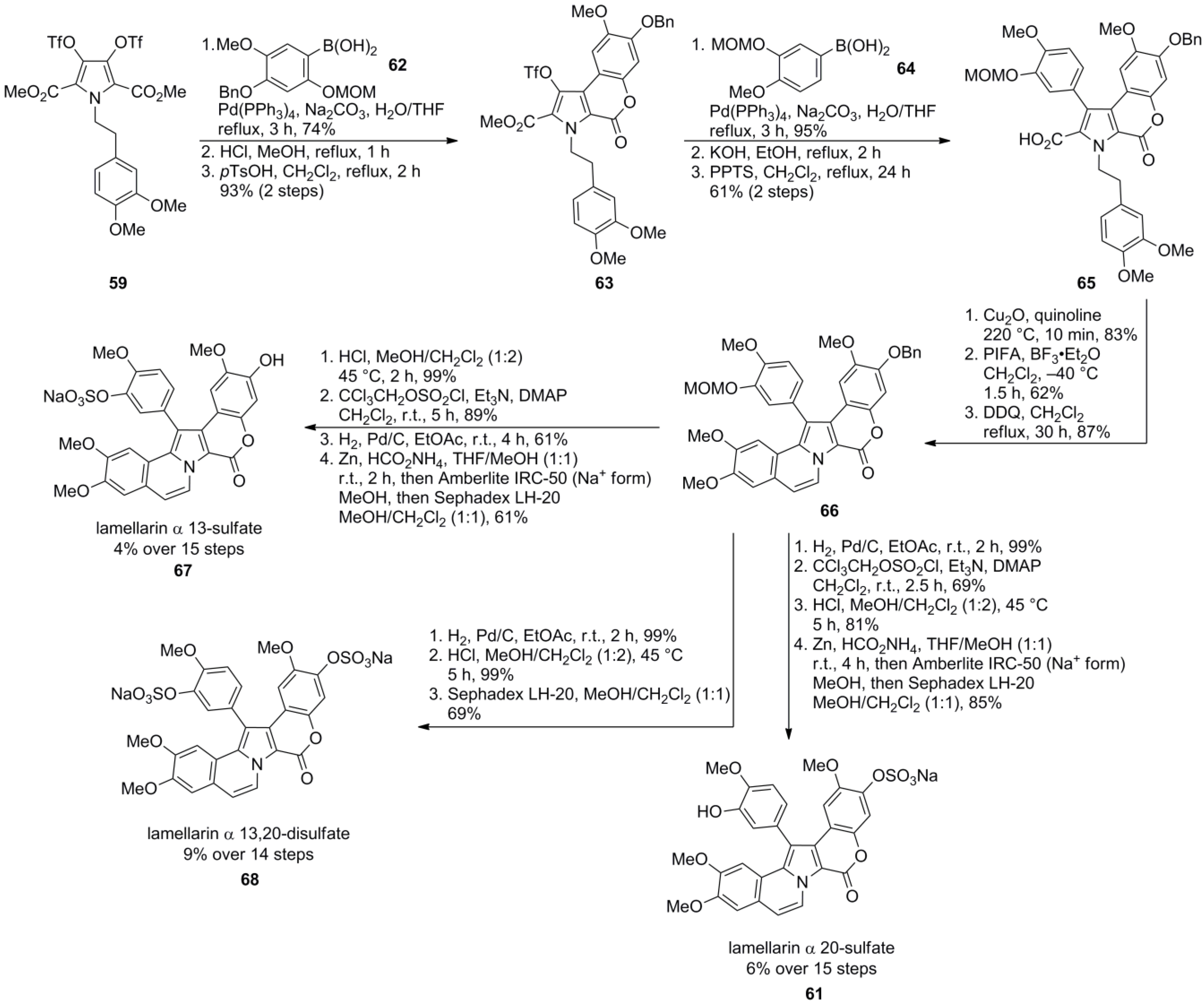
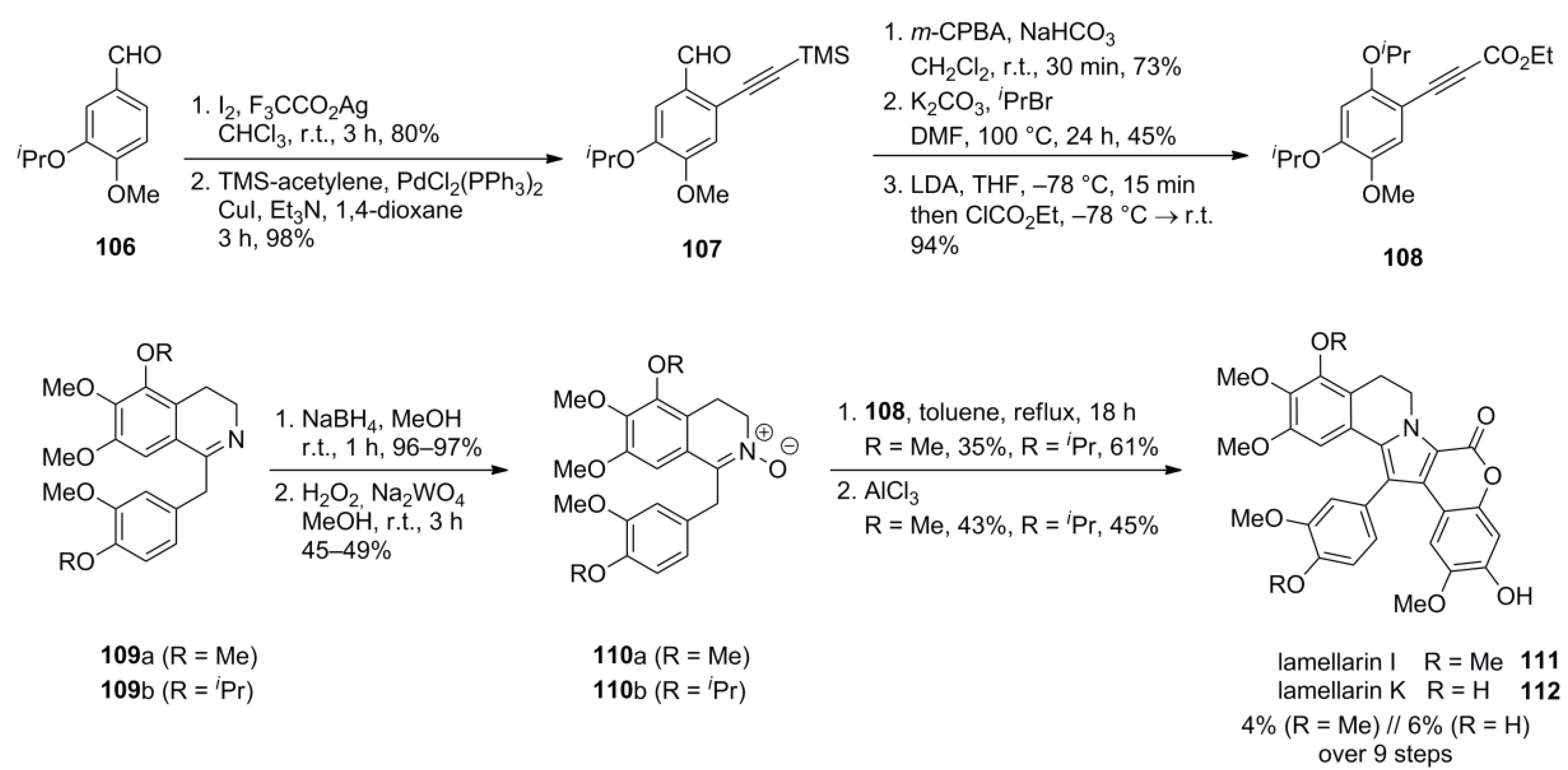
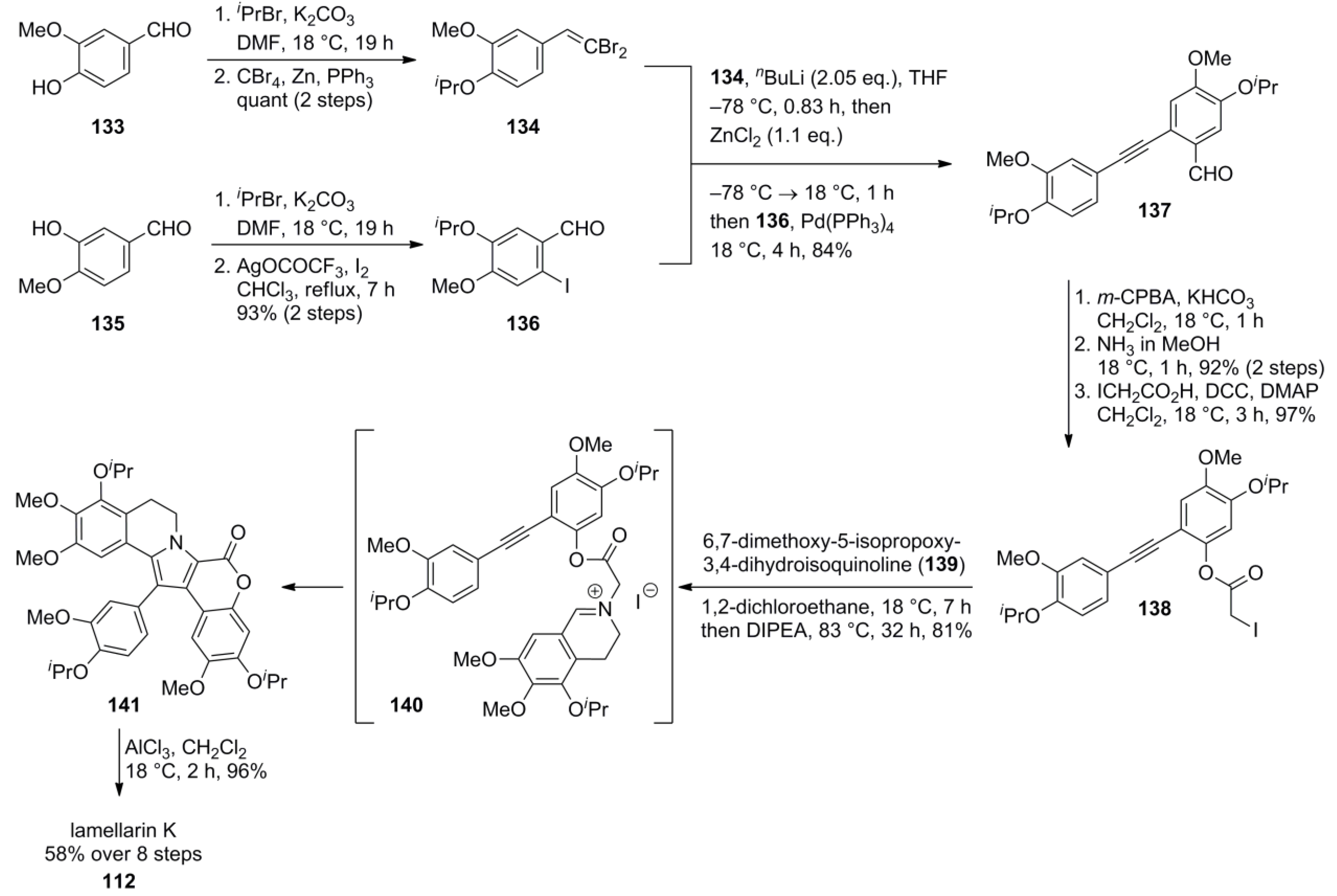
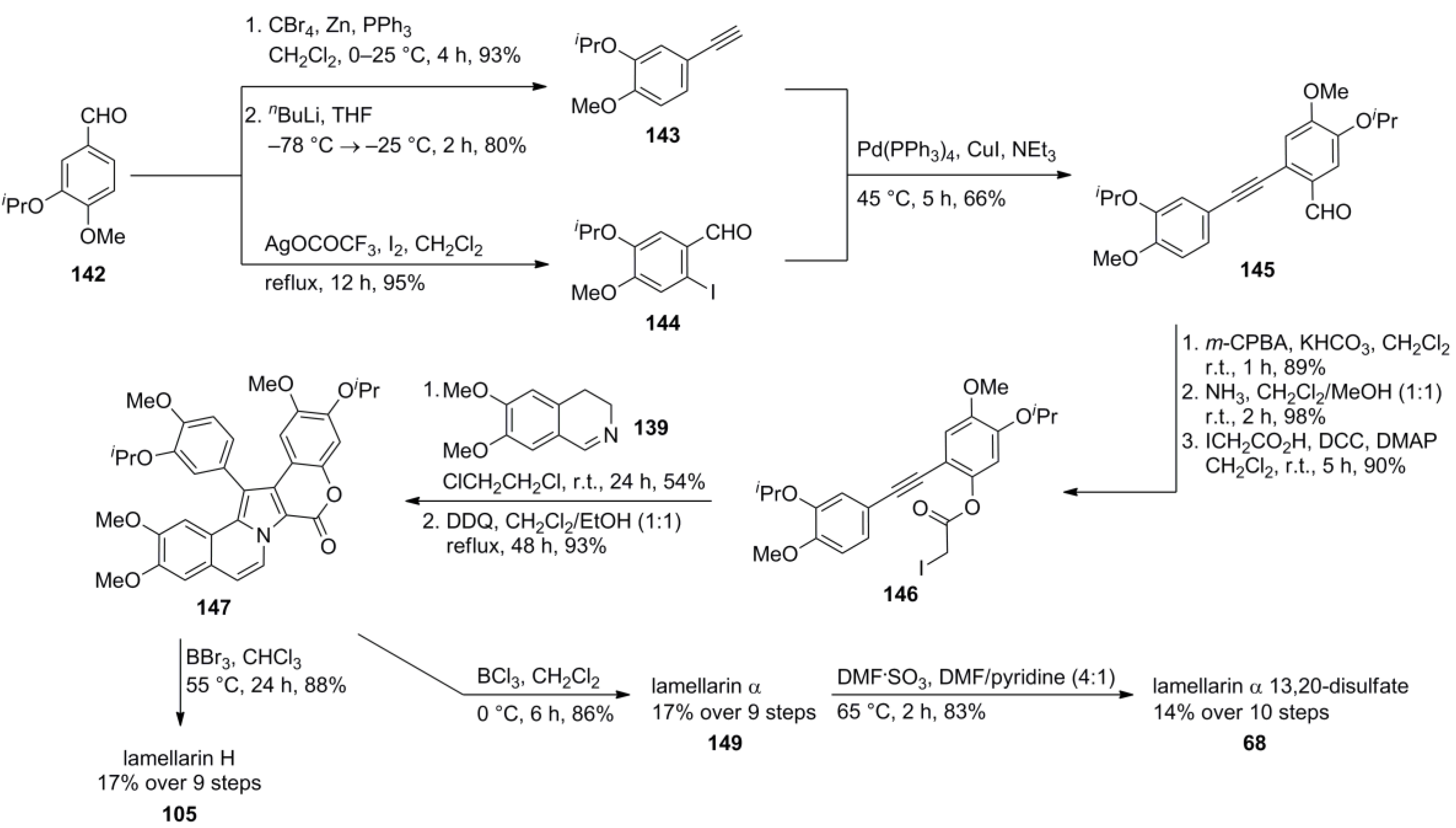
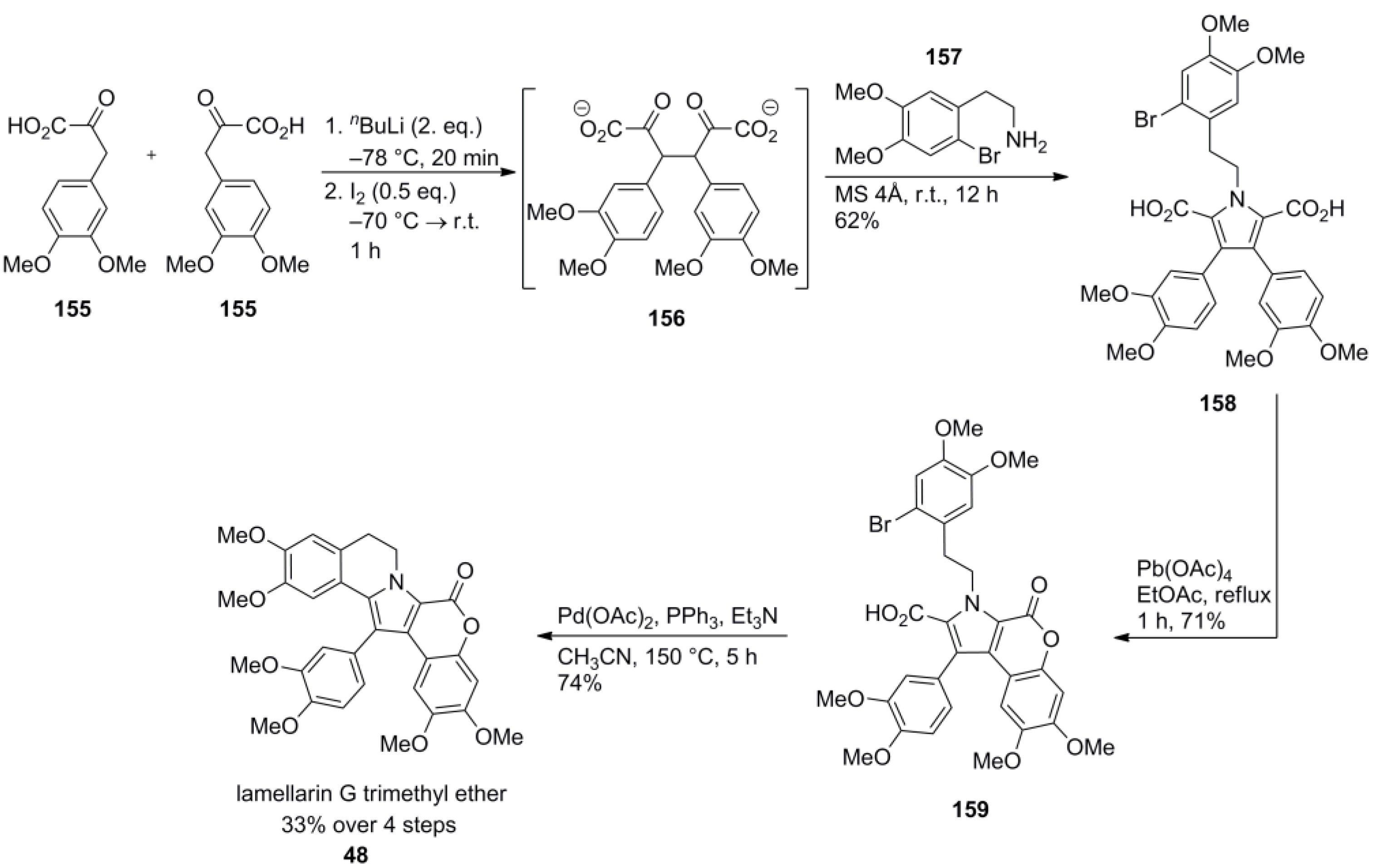
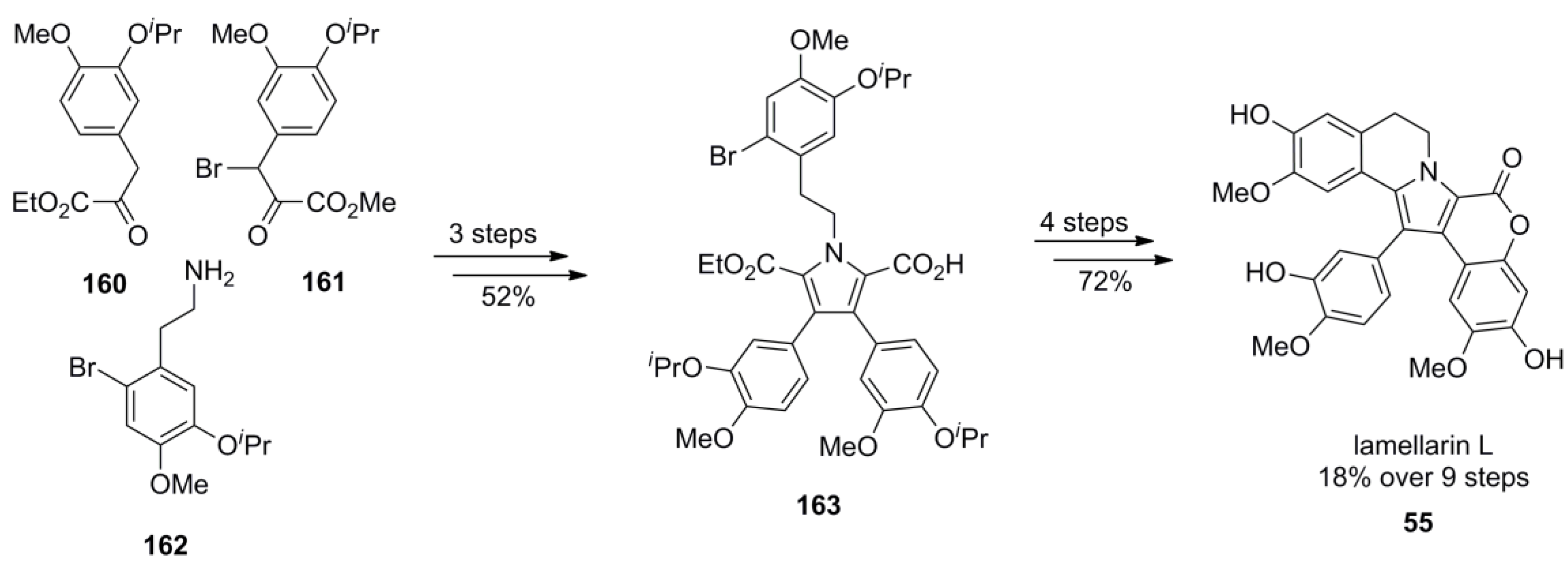
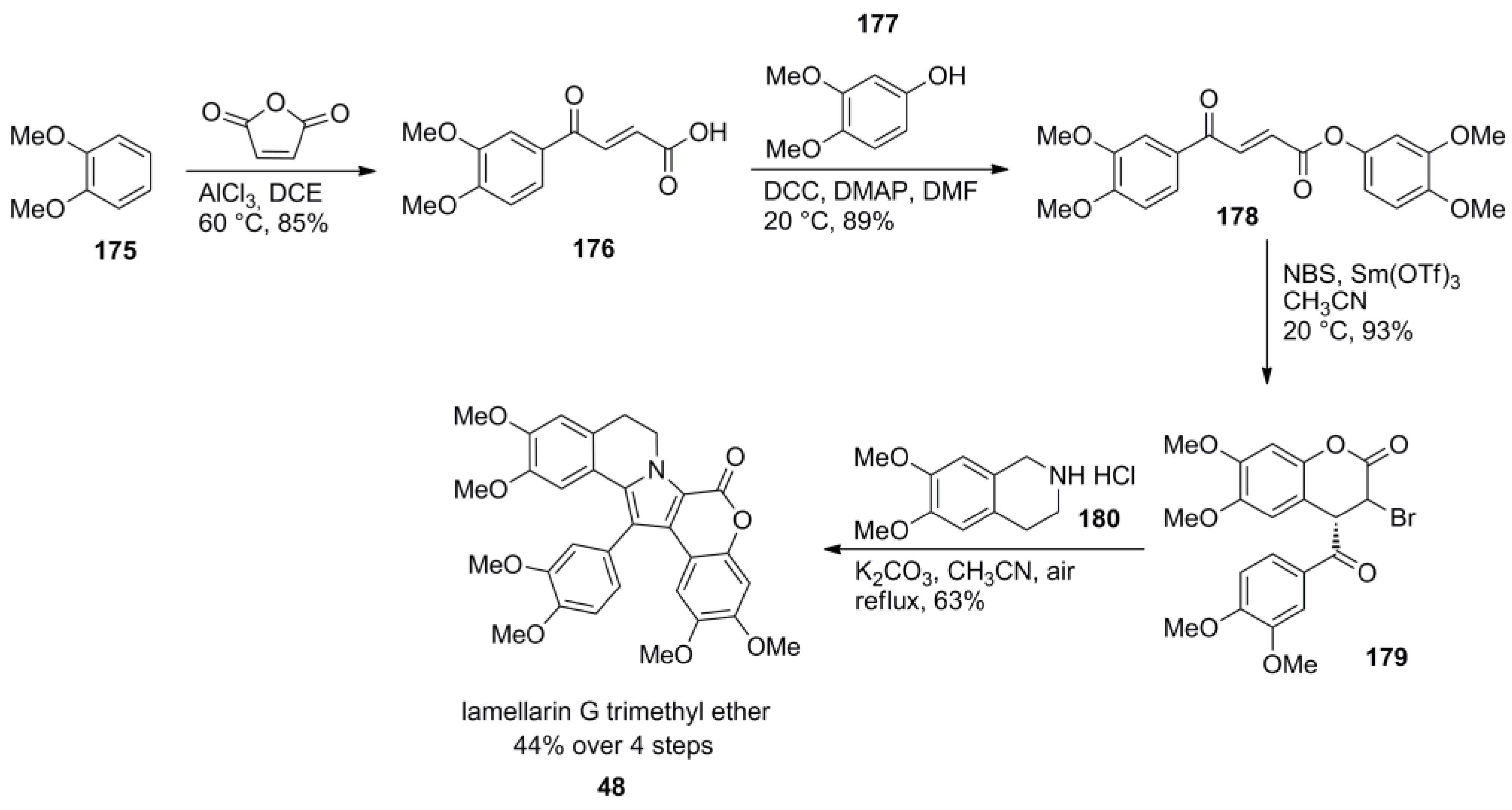
3.1. Iwao 2003 (lamellarin G trimethyl ether) [72]-2006 (lamellarin D, L and N) [73]-2006 (lamellarin α 20‑sulfate) [74]-2010 (lamellarin α 20-sulfate, α 13-sulfate and α 13,20-disulfate) [75]




3.2. Handy 2004 [78]

3.3. Álvarez 2005 [79]

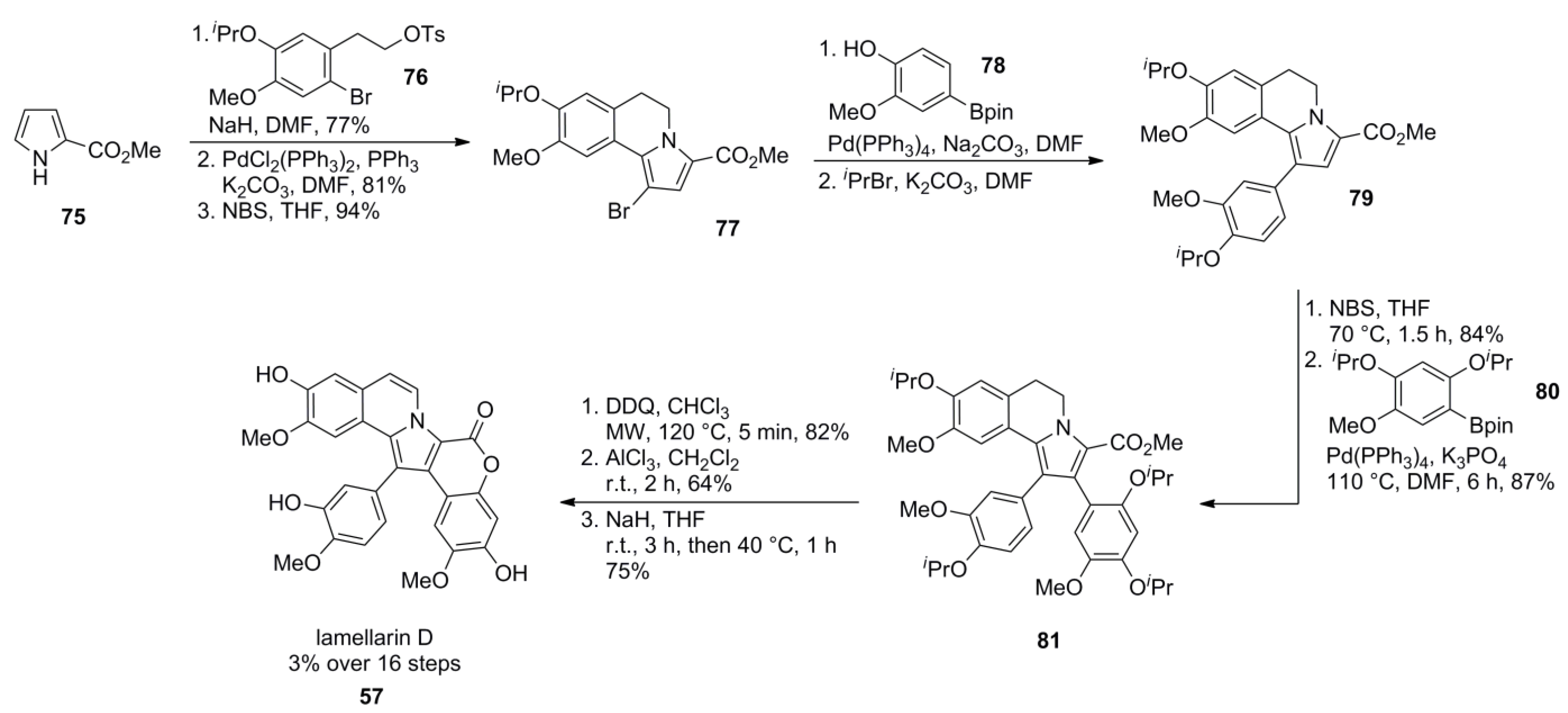
3.4. Banwell 2011 [80]

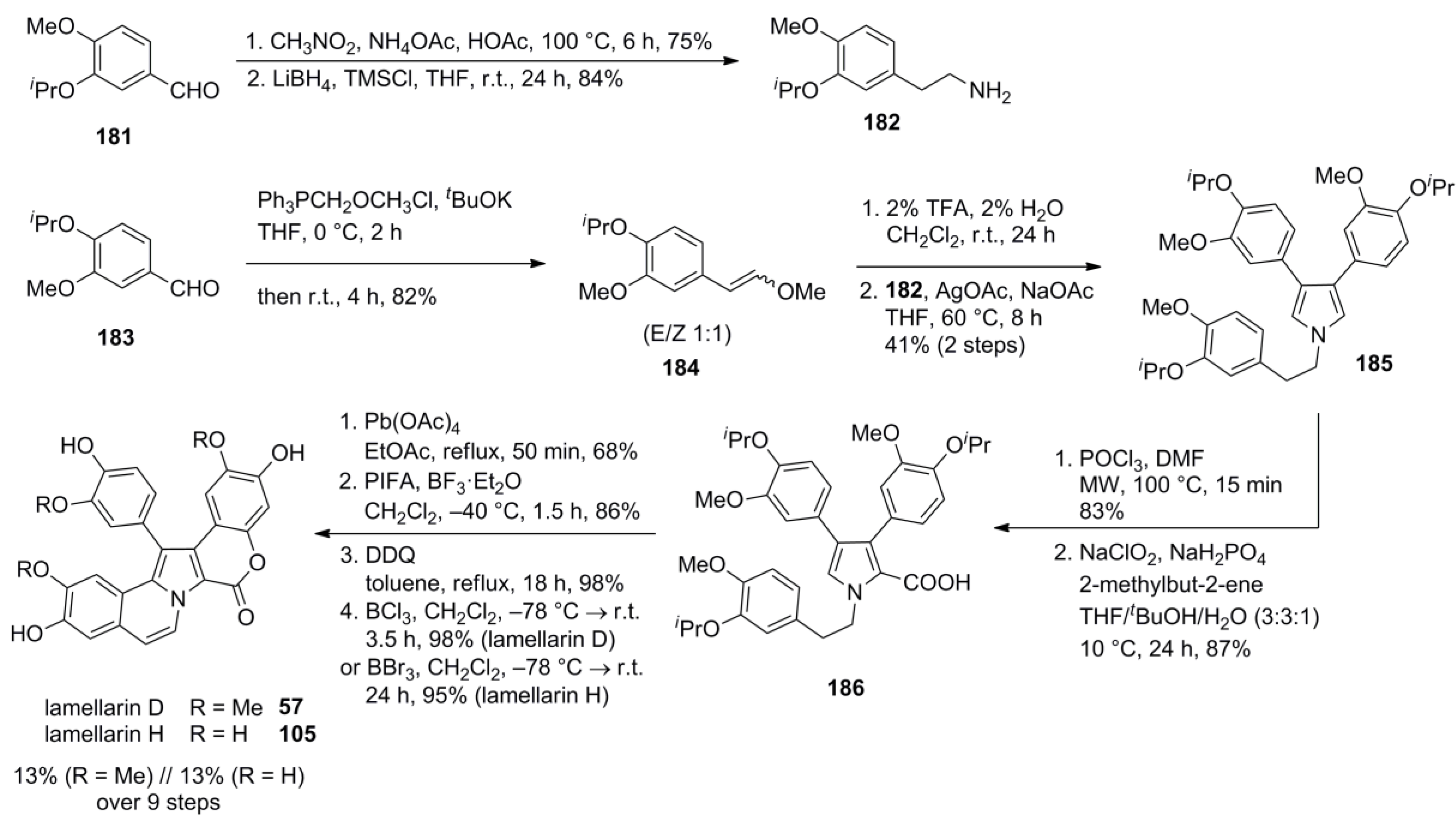
3.5. Iwao 2014 [81]

4. Syntheses of Type I-lamellarins—Approaches via N-Ylide-Mediated Pyrrole Ring Formation
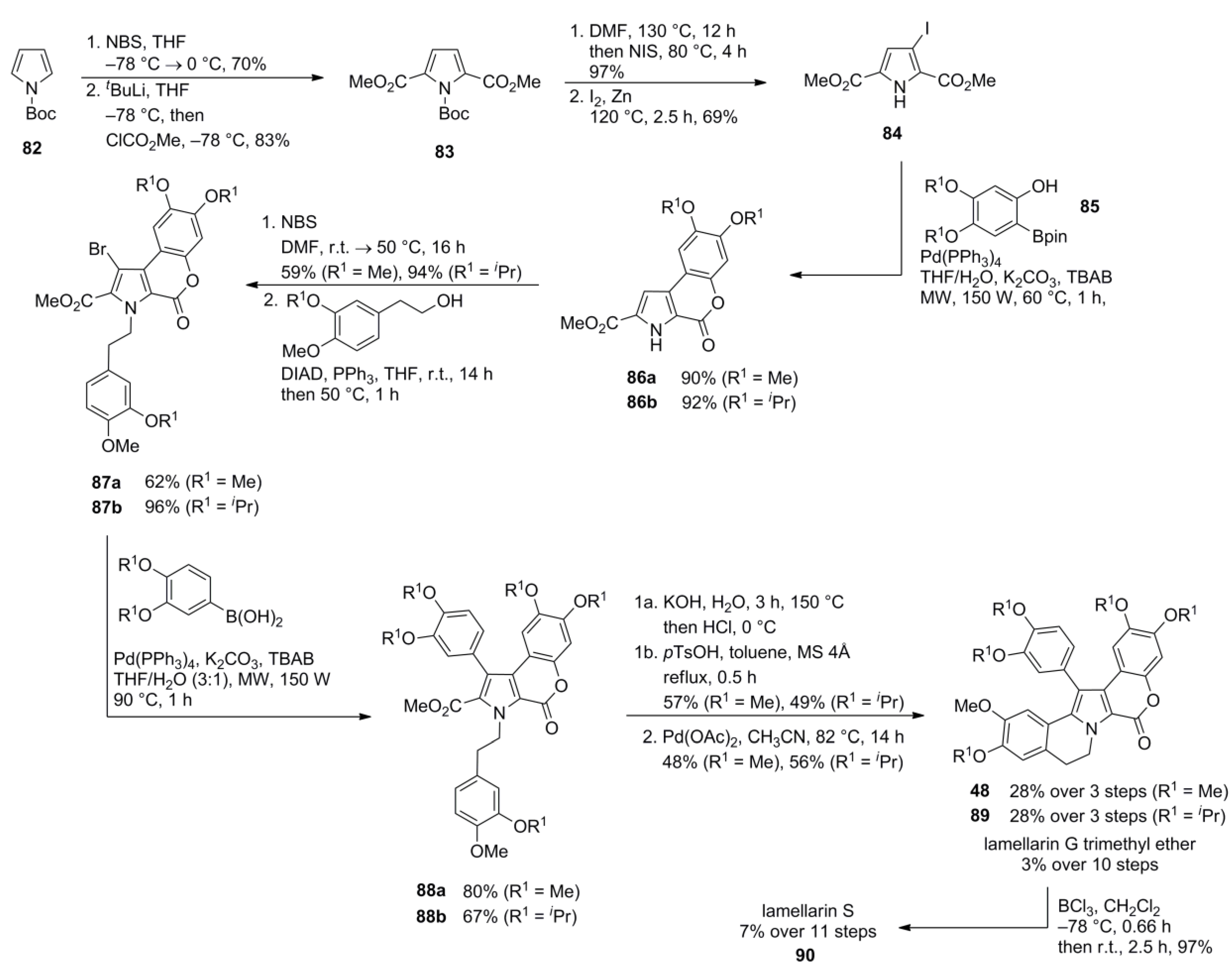
4.1. Iwao 1997 [82]

4.2. Guitián 2001 [83]

4.3. Ruchirawat 2001 [84]

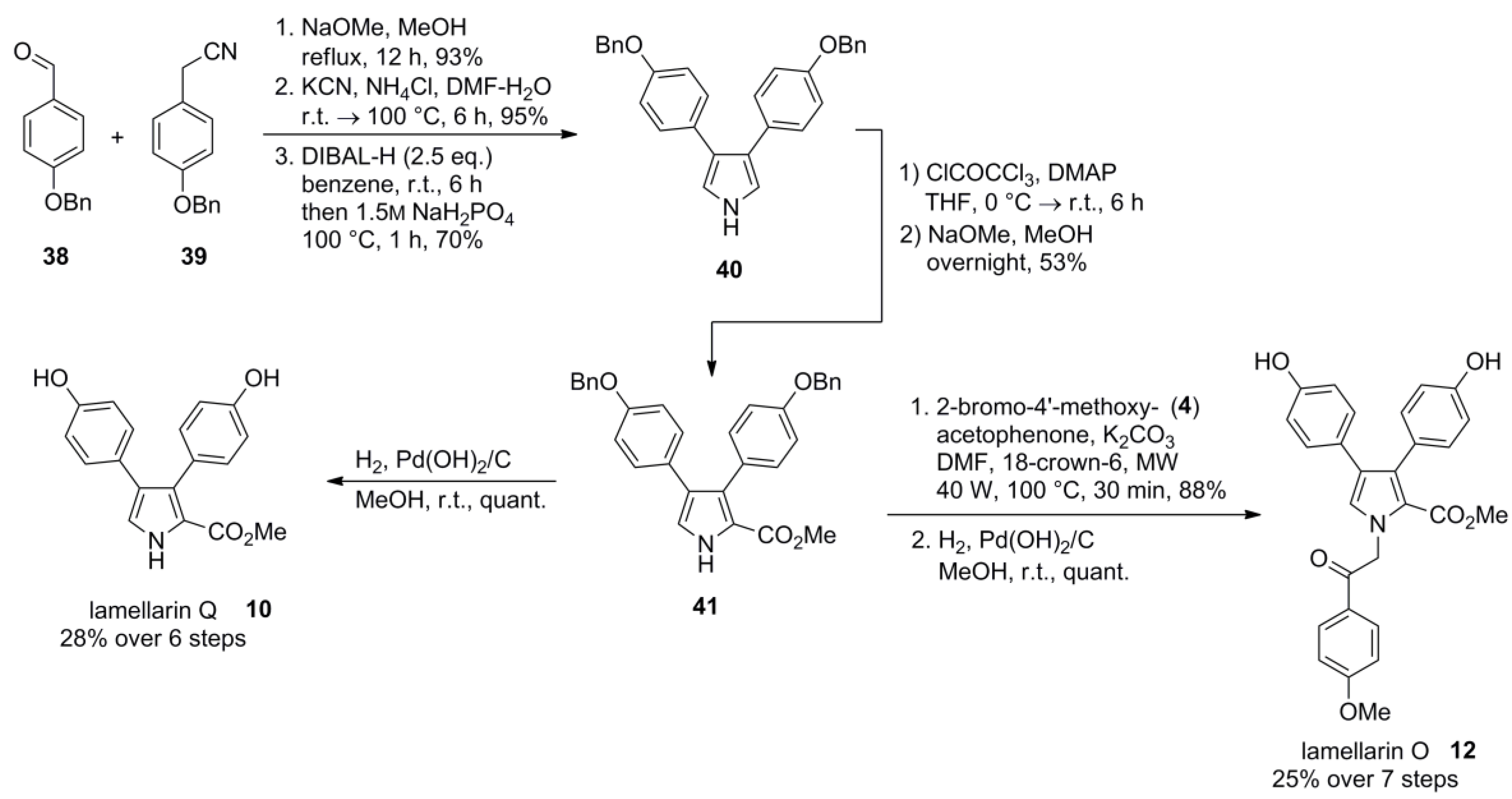
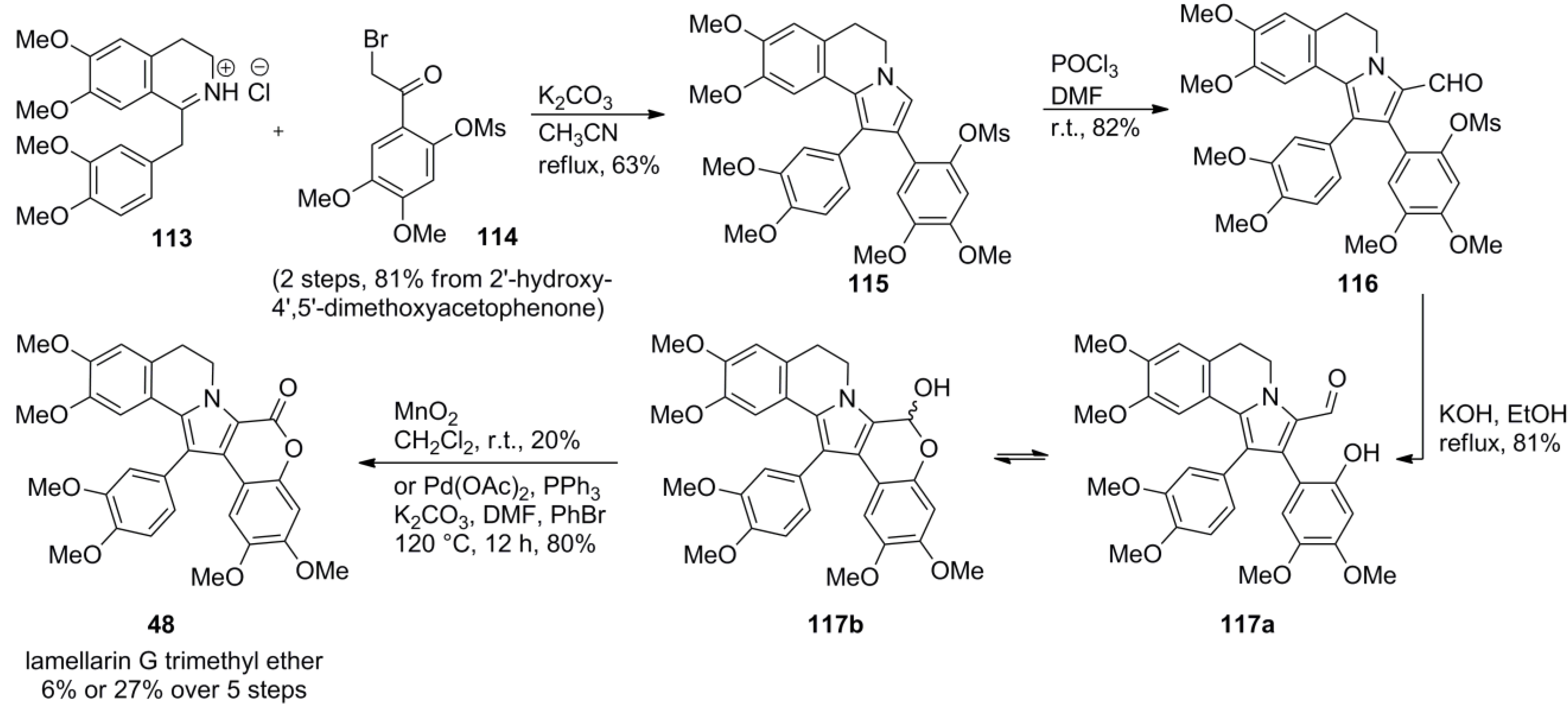
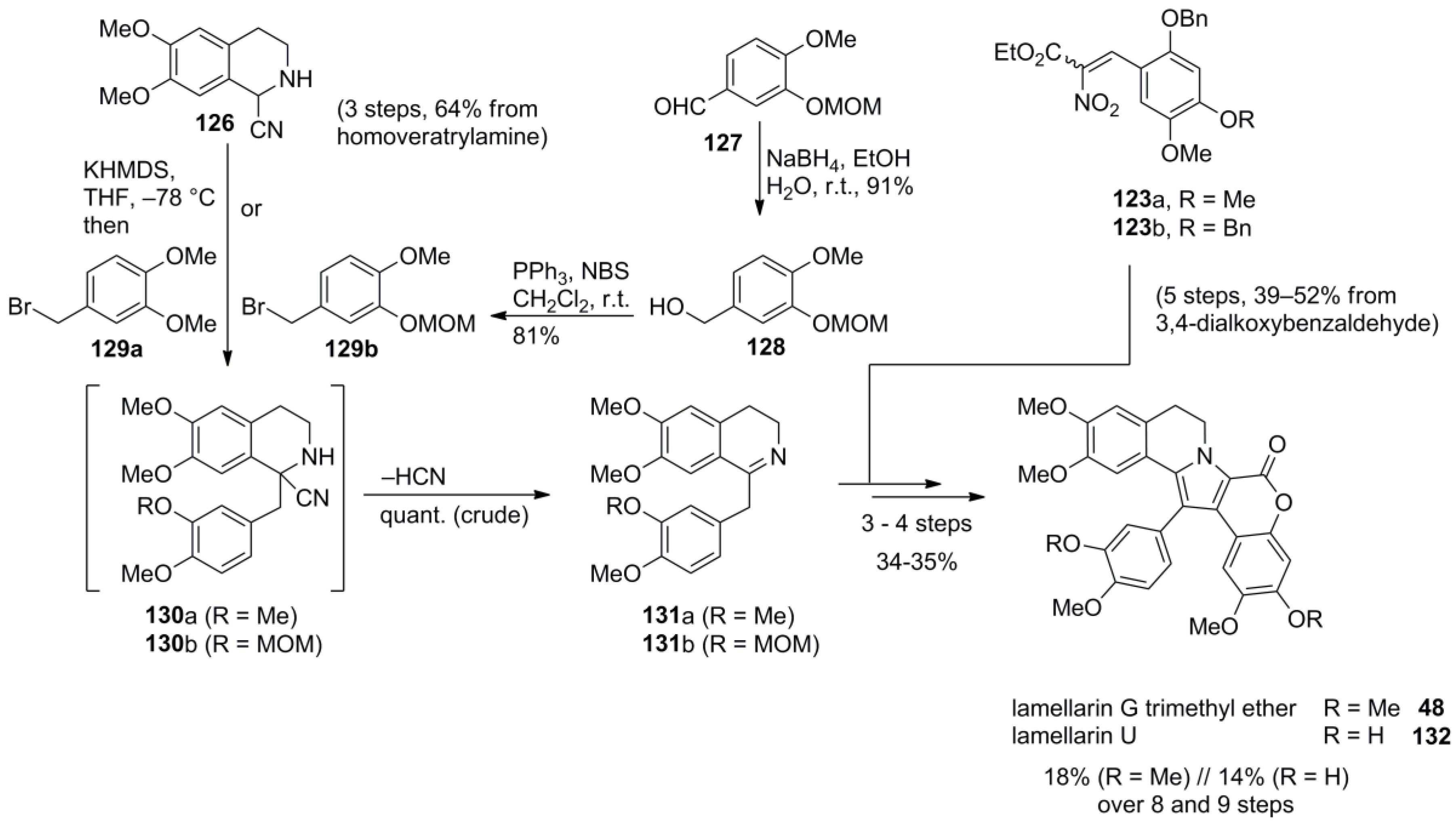
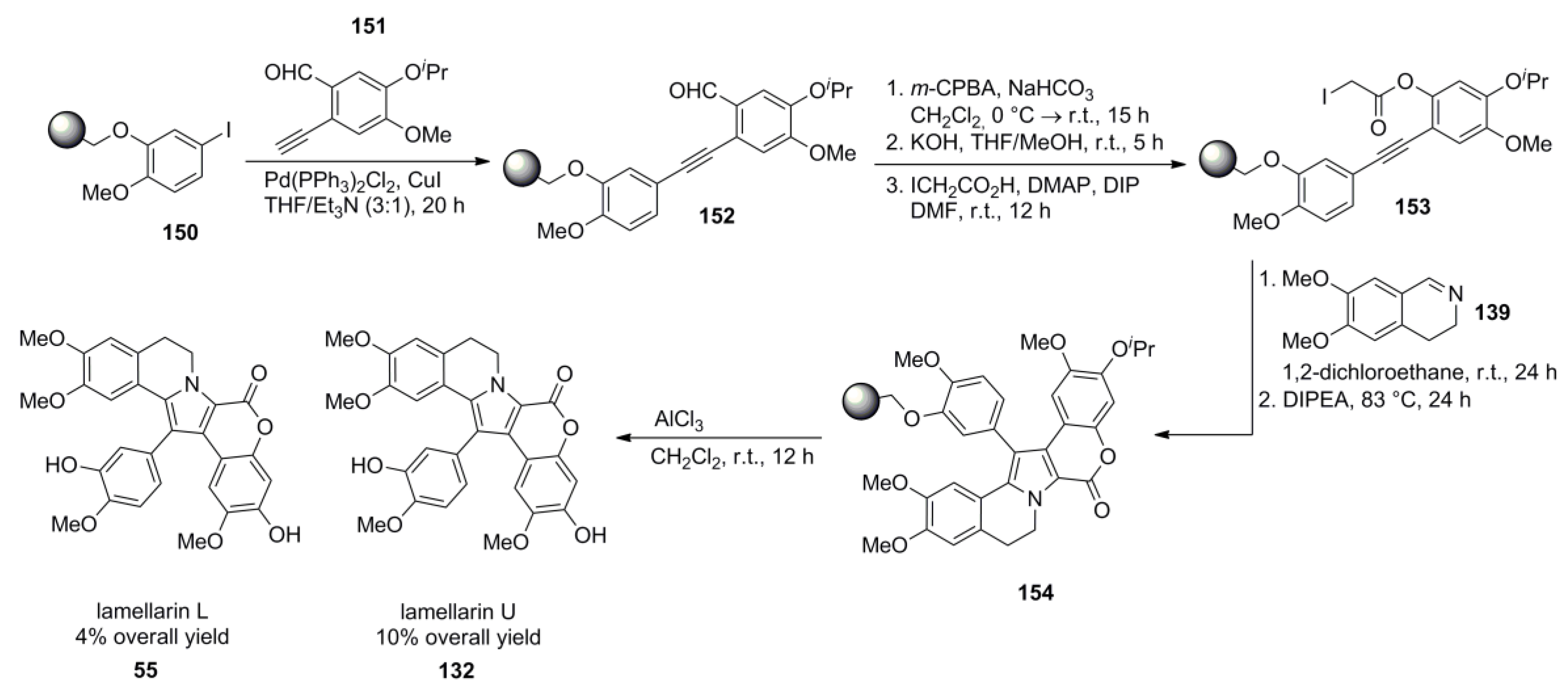
4.4. Ruchirawat 2004 (lamellarin K&L) [85]-2006 (28 different lamellarins) [86]-Opatz 2008 (lamellarin G trimethyl ether & lamellarin U) [87]


5. Synthesis of type-I Lamellarins—Miscellaneous Approaches
5.1. Banwell 1997 [88]//Faulkner 2002 [89]//Alvarez 2003 [90]



5.2. Steglich 1997 (lamellarin G trimethyl ether) [92]-2000 (lamellarin L) [77]-2006 (lamellarin G & K) [93]-2009 Guptón (Steglich Synthon) [94]




5.3. Yadav 2009 [97]

5.4. Jia 2011 [70]

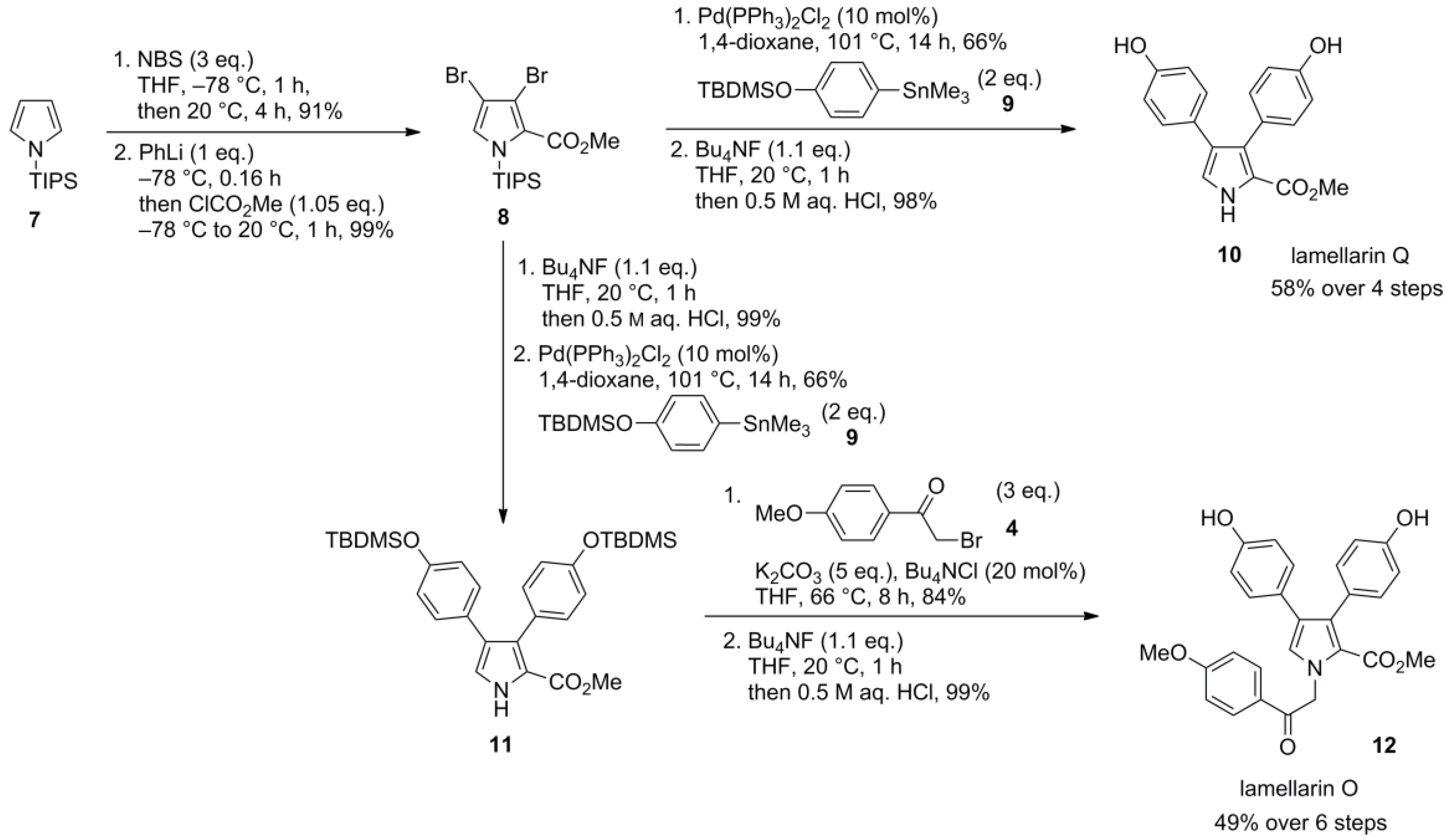
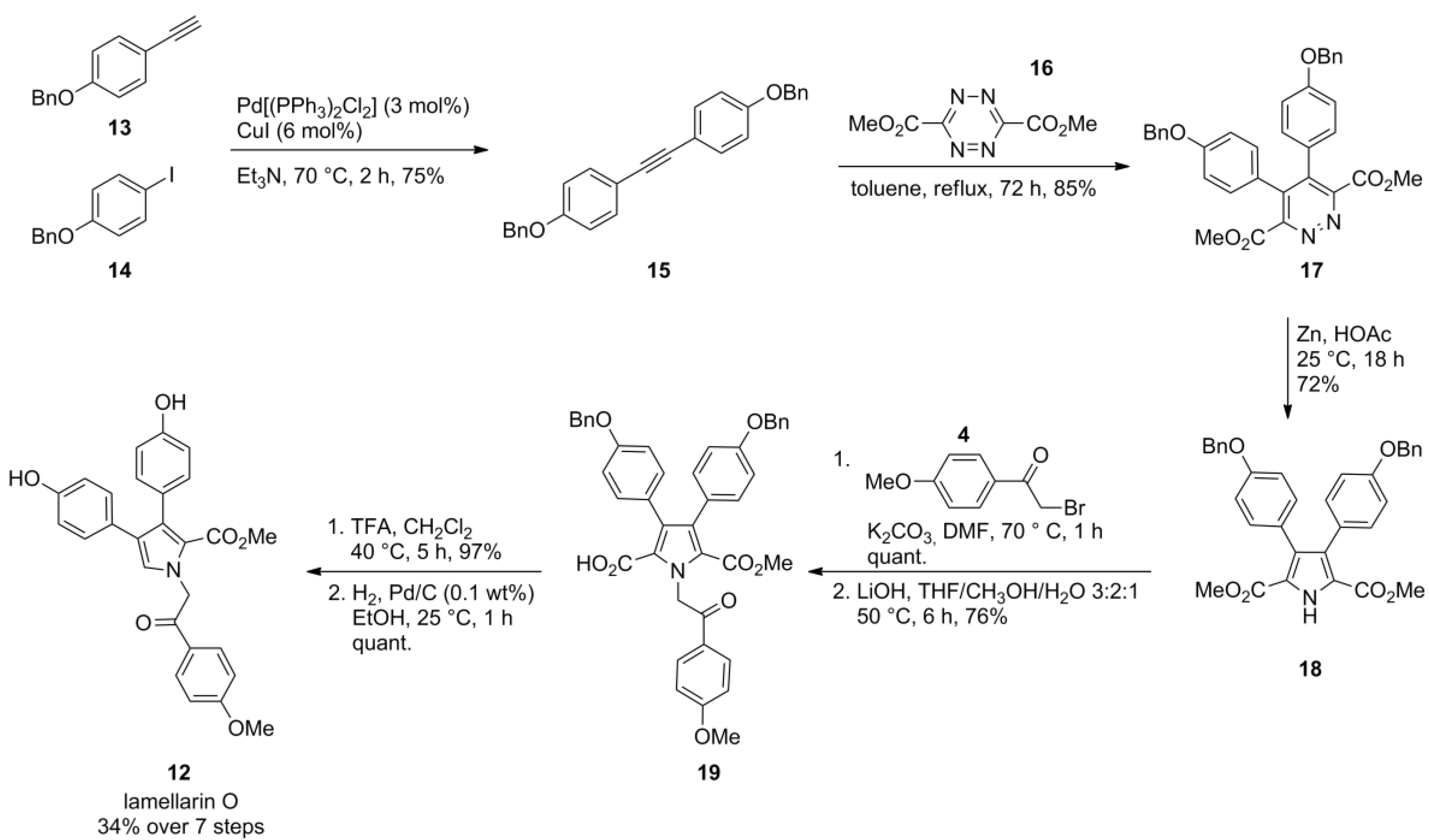
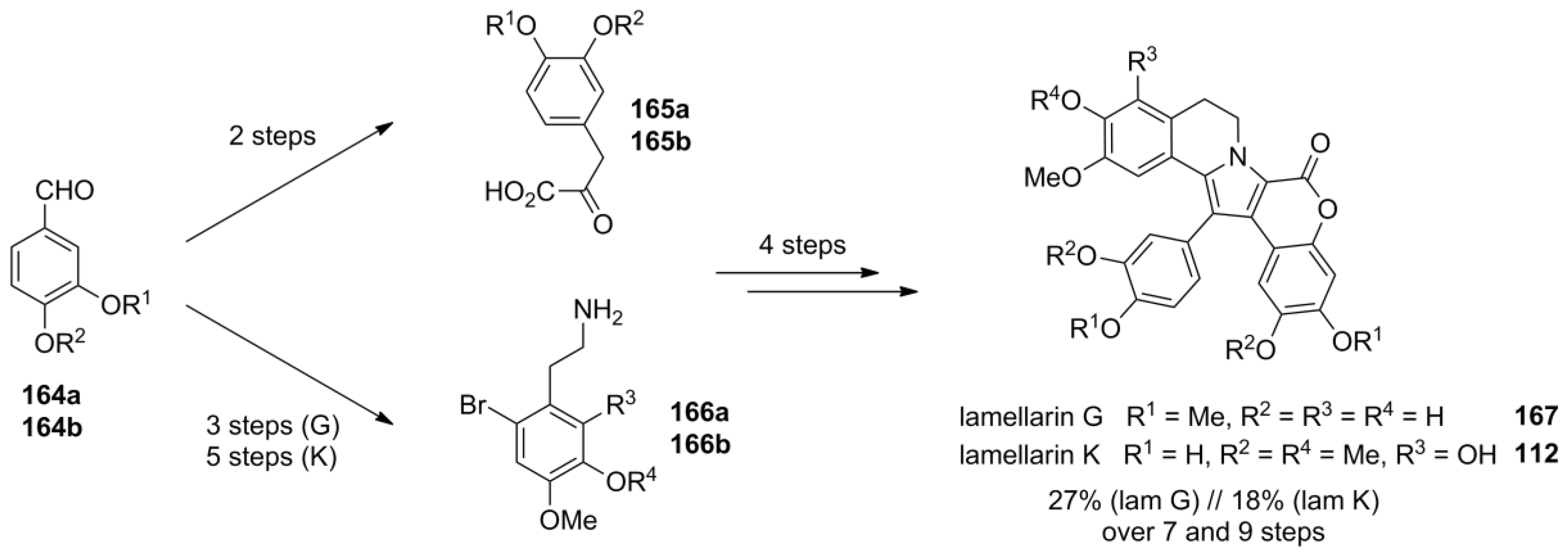
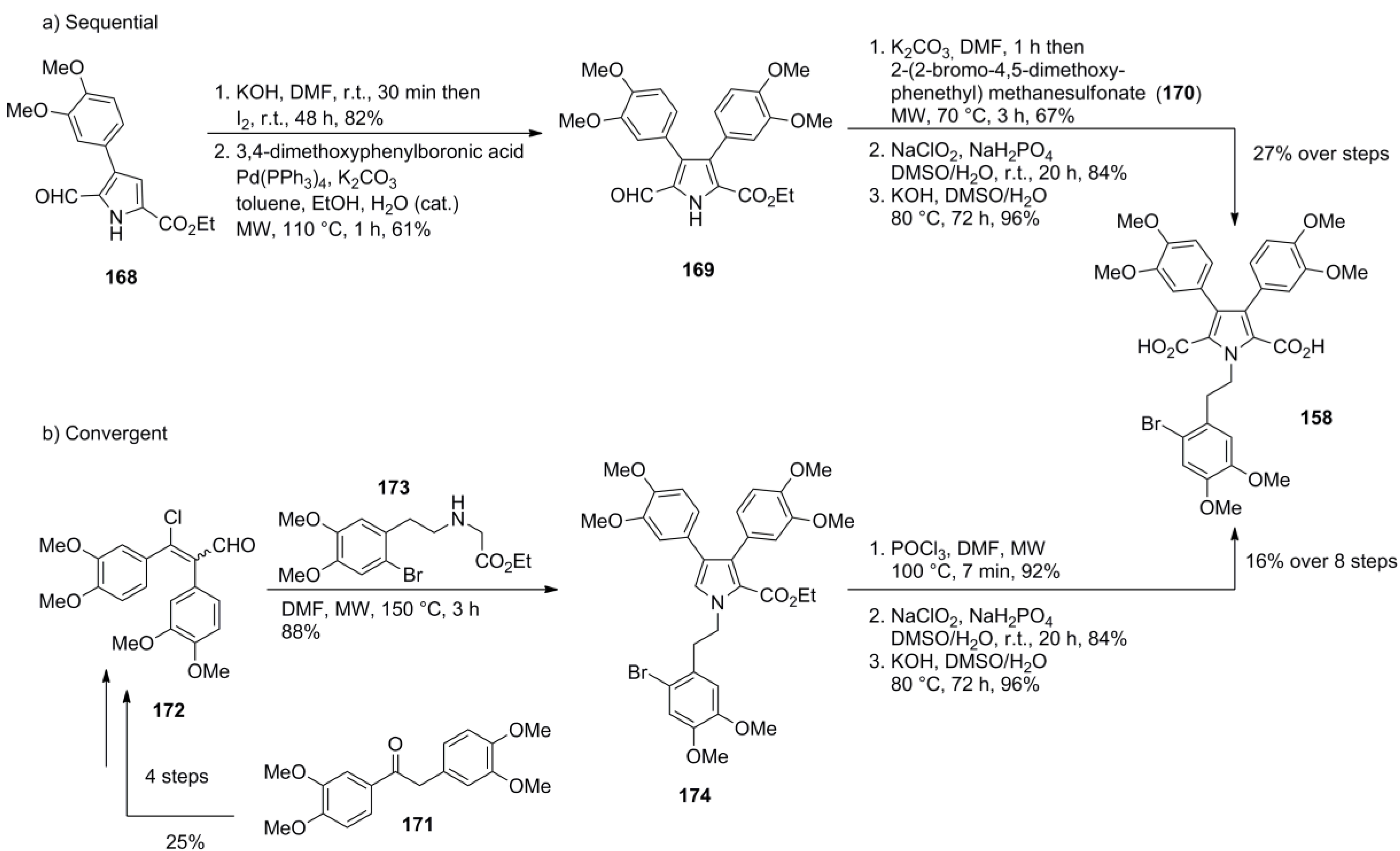
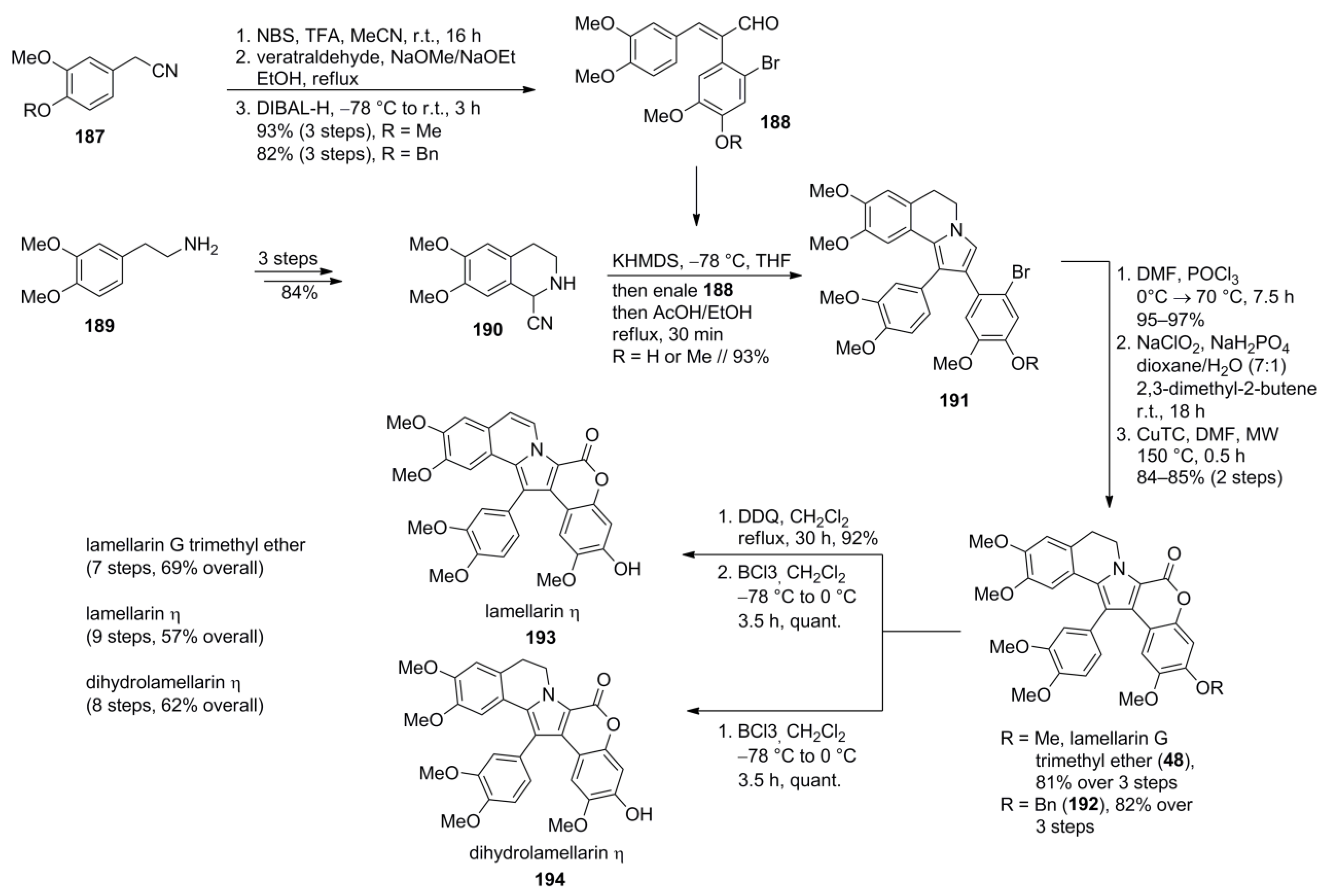
5.5. Opatz 2013 [16]

5.6. Yamaguchi 2014 [98]
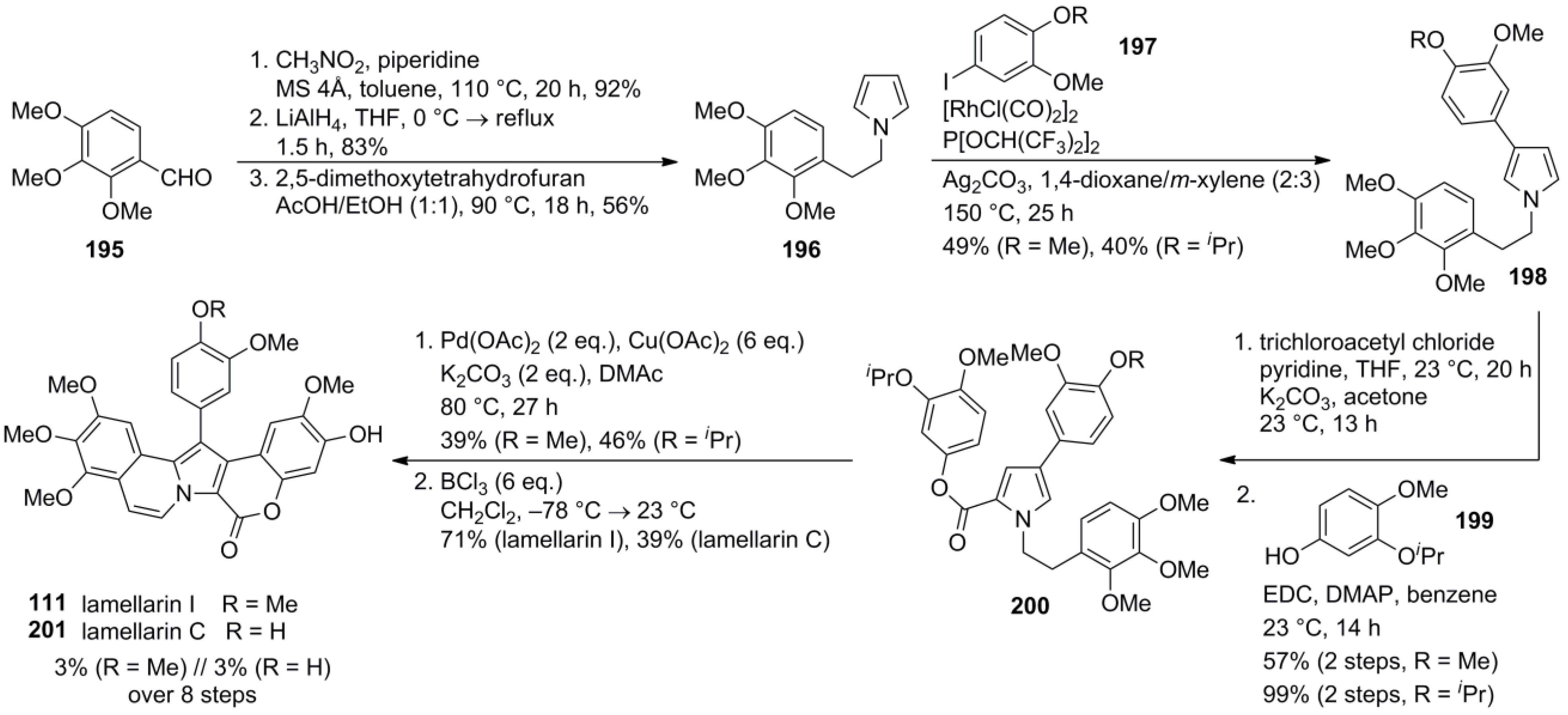
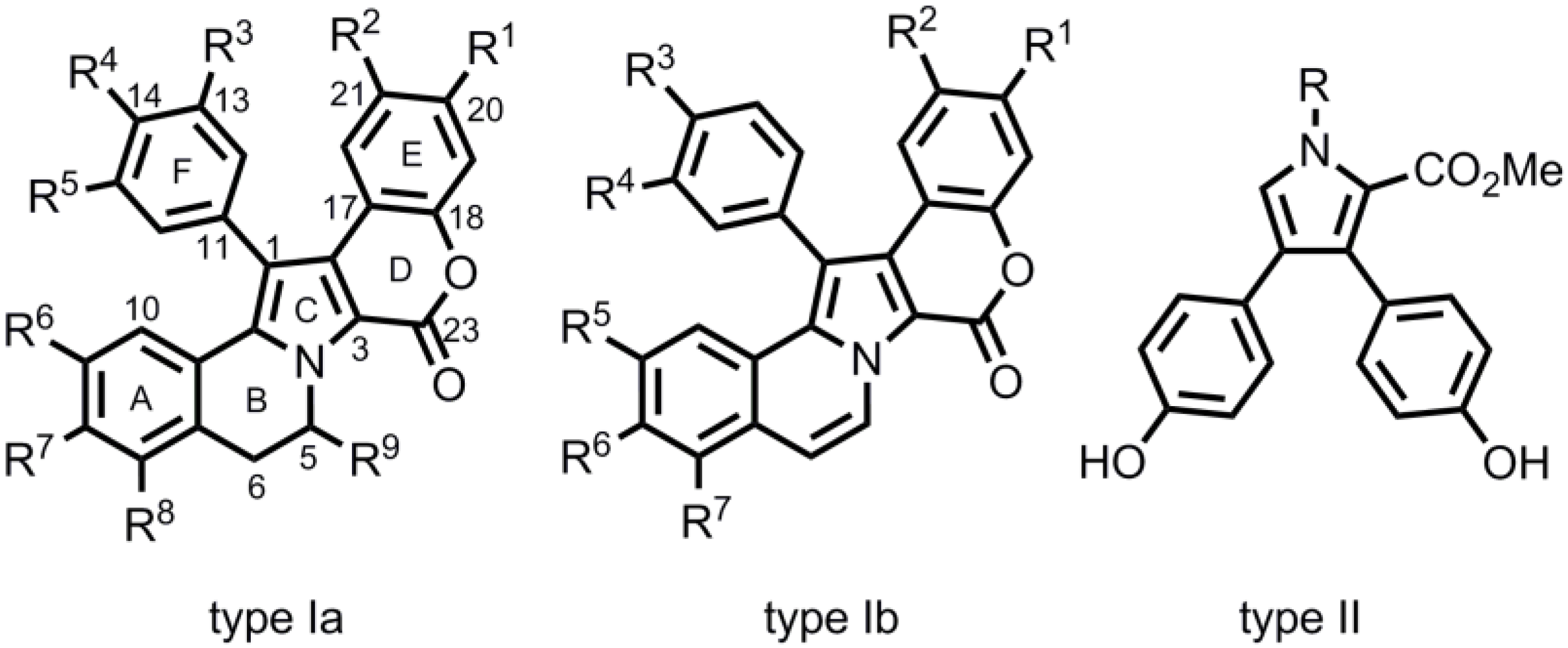
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lead Author | Year | Lamellarin | Overall Yield | Linear Steps | Starting from |
|---|---|---|---|---|---|
| Steglich | 1997 | G trimethyl ether | 33% | 4 | 3-(3,4-Dimethoxyphenyl)pyruvic acid |
| Banwell | 1997 | K | 58% | 7 | 4-Isopropoxy-3-methoxybenzaldehyde |
| Iwao | 1997 | H | 4% | 10 | 3-Benzyloxy-4-methoxybenzaldehyde |
| D | 5% | 10 | 3-Benzyloxy-4-methoxybenzaldehyde | ||
| Steglich | 2000 | L | 18% | 9 | 3-Isopropoxy-4-methoxybenzaldeyde |
| Guitián | 2001 | I | 4% | 9 | 3-Isopropoxy-4-methoxybenzaldeyde |
| K | 6% | 9 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| Ruchirawat | 2001 | G trimethyl ether | 27% | 5 | 2‘-Hydroxy-4′,5′-dimethoxyacetophenone |
| Faulkner | 2002 | H | 17% | 9 | 3-Isopropoxy-4-methoxybenzaldehyde |
| α | 17% | 9 | 3-Isopropoxy-4-methoxybenzaldehyde | ||
| α 13,20-disulfate | 14% | 10 | 3-Isopropoxy-4-methoxybenzaldehyde | ||
| Iwao | 2003 | G trimethyl ether | 12% | 9 | Homoveratrylamine |
| Handy | 2004 | G trimethyl ether | 10% | 8 | Ethyl 4-bromopyrrole-2-carboxylate |
| Álvarez | 2005 | D | 3% | 16 | 3-Isopropoxy-4-methoxybenzaldehyde |
| Iwao | 2006 | N | 16% | 12 | 3-Isopropoxy-4-methoxybenzaldeyde |
| D | 18% | 12 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| L | 19% | 11 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| 2006 | α 20-sulfate | 24% | 14 | Homoveratrylamine | |
| Steglich | 2006 | K | 18% | 9 | 4-Bromo-2,3-dimethoxybenzaldehyde |
| G | 27% | 7 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| Ruchirawat | 2006 | I, C, T, F, K, E, dihydro‑η, G trimethyl ether, J, U, L, G, χ, Y ξ, B, W, ε, M, X, η, J‑DB, α, N, G‑DB, D, Y‑DB | 5%–34% (saturated) 4%–30% (unsaturated) | 8–9 (saturated) 11–12 (unsaturated) | 3-Benzyloxy-4-methoxybenzaldehyde 4-Benzyloxy-3-methoxybenzaldehyde Veratraldehyde 2,3,4-Trimethoxybenzaldehyde 2,4-Dibenzyloxy-5-methoxybenzaldehyde 2,5-Dibenzyloxy-4-methoxybenzaldehyde 2-Benzyloxy-4,5-dimethoxybenzaldehyde |
| Opatz | 2008 | U | 14% | 9 | 3-Benzyloxy-4-methoxybenzaldehyde |
| G trimethyl ether | 18% | 8 | Veratraldehyde | ||
| Yadav | 2009 | G trimethyl ether | 44% | 4 | Veratrole |
| Iwao | 2010 | α 20-sulfate | 6% | 15 | 3-Isopropoxy-4-methoxybenzaldeyde |
| α 13-sulfate | 4% | 15 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| α 13,20-disulfate | 9% | 14 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| Banwell | 2011 | G trimethyl ether | 3% | 10 | N-Boc-pyrrole |
| S | 7% | 11 | 3,4-Diisopropoxybenzaldehyde | ||
| Jia | 2011 | D | 13% | 9 | 4-Isopropoxy-3-methoxybenzaldehyde |
| H | 13% | 9 | 4-Isopropoxy-3-methoxybenzaldehyde | ||
| Banwell | 2012 | T | 41% | 6 | 3-Isopropoxy-4-methoxybenzaldeyde |
| W | 42% | 7 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| U | 45% | 6 | 3-Isopropoxy-4-methoxybenzaldeyde | ||
| Opatz | 2013 | η | 57% | 9 | 4-Benzyloxy-3-methoxyphenylacetonitrile |
| dihydro-η | 62% | 8 | 4-Benzyloxy-3-methoxyphenylacetonitrile | ||
| G trimethyl ether | 69% | 7 | 3,4-Dimethoxyphenylacetonitrile | ||
| Iwao | 2014 | L | 30% | 13 | N-Boc-2,5-dibromopyrrole |
| N | 42% | 11 | N-Boc-2,5-dibromopyrrole | ||
| Yamaguchi | 2014 | C | 3% | 8 | 2,3,4-Trimethoxybenzaldehyde |
| I | 3% | 8 | 2,3,4-Trimethoxybenzaldehyde |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andersen, R.J.; Faulkner, D.J.; He, C.H.; Van Duyne, G.D.; Clardy, J. Metabolites of the marine prosobranch mollusk Lamellaria sp. J. Am. Chem. Soc. 1985, 107, 5492–5495. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; van Duyne, G.D.; Clardy, J. New alkaloids of the lamellarin class from the marine ascidian Didemnum chartaceum. J. Org. Chem. 1988, 53, 4570–4574. [Google Scholar] [CrossRef]
- Carroll, A.R.; Bowden, B.F.; Coll, J.C. Studies of Australian ascidians. I. Six new lamellarin-class alkaloids from a colonial ascidian, Didemnum sp. Aust. J. Chem. 1993, 46, 489–501. [Google Scholar] [CrossRef]
- Urban, S.; Butler, M.S.; Capon, R.J. Lamellarins O and P: New aromatic metabolites from the Australian marine sponge Dendrilla cactos. Aust. J. Chem. 1994, 47, 1919–1924. [Google Scholar] [CrossRef]
- Urban, S.; Hobbs, L.; Hooper, J.N.A.; Capon, R.J. Lamellarins Q and R: New aromatic metabolites from an Australian marine sponge, Dendrilla cactos. Aust. J. Chem. 1995, 48, 1491–1494. [Google Scholar] [CrossRef]
- Urban, S.; Capon, R.J. Lamellarin-s: A new aromatic metabolite from an Australian tunicate, Didemnum sp. Aust. J. Chem. 1996, 49, 711–713. [Google Scholar]
- Reddy, M.V.R.; Faulkner, D.J.; Venkateswarlu, Y.; Rao, M.R. New lamellarin alkaloids from an unidentified ascidian from the Arabian Sea. Tetrahedron 1997, 53, 3457–3466. [Google Scholar] [CrossRef]
- Davis, R.A.; Carroll, A.R.; Pierens, G.K.; Quinn, R.J. New lamellarin alkaloids from the Australian ascidian, Didemnum chartaceum. J. Nat. Prod. 1999, 62, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Kang, H. A novel cytotoxic alkaloid of lamellarin class from a marine ascidian Didemnum sp. Bull. Korean Chem. Soc. 2002, 23, 163–166. [Google Scholar] [CrossRef]
- Krishnaiah, P.; Reddy, V.L.N.; Venkataramana, G.; Ravinder, K.; Srinivasulu, M.; Raju, T.V.; Ravikumar, K.; Chandrasekar, D.; Ramakrishna, S.; Venkateswarlu, Y. New lamellarin alkaloids from the Indian ascidian Didemnum obscurum and their antioxidant properties. J. Nat. Prod. 2004, 67, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Srinivasulu, M.; Satyanarayana, N.; Kondapi, A.K.; Venkateswarlu, Y. New potent cytotoxic lamellarin alkaloids from Indian ascidian Didemnum obscurum. Tetrahedron 2005, 61, 9242–9247. [Google Scholar] [CrossRef]
- Koeck, M.; Reif, B.; Fenical, W.; Griesinger, C. Differentiation of HMBC two- and three-bond correlations: A method to simplify the structure determination of natural products. Tetrahedron Lett. 1996, 37, 363–366. [Google Scholar] [CrossRef]
- Reif, B.; Kock, M.; Kerssebaum, R.; Kang, H.; Fenical, W.; Griesinger, C. Adequate, a new set of experiments to determine the constitution of small molecules at natural abundance. J. Magn. Reson. Ser. A 1996, 118, 282–285. [Google Scholar] [CrossRef]
- Reif, B.; Koeck, M.; Kerssebaum, R.; Schleucher, J.; Griesinger, C. Determination of 1J, 2J, and 3J carbon-carbon coupling constants at natural abundance. J. Magn. Reson. Ser. B 1996, 112, 295–301. [Google Scholar] [CrossRef]
- Fukuda, T.; Itoyama, R.; Minagawa, T.; Iwao, M. Rotational energy barrier around the C1-C11 single bond in lamellarins: A study by variable-temperature NMR. Heterocycles 2014, 88, 1121–1133. [Google Scholar] [CrossRef]
- Imbri, D.; Tauber, J.; Opatz, T. A high-yielding modular access to the lamellarins: Synthesis of lamellarin G trimethyl ether, lamellarin η and dihydrolamellarin η. Chem. Eur. J. 2013, 19, 15080–15083. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.R.; Gravalos, M.D.G.; Puentes, J.L.F. Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br. J. Cancer 1996, 74, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Burnham, B.S.; Gupton, J.T.; Krumpe, K.; Webb, T.; Shuford, J.; Bowers, B.; Warren, A.E.; Barnes, C.; Hall, I.H. Cytotoxicity of substituted alkyl-3,4-bis(4-methoxyphenyl)pyrrole-2-carboxylates in L1210 lymphoid leukemia cells. Arch. Pharm. 1998, 331, 337–341. [Google Scholar] [CrossRef]
- Reddy, M.V.R.; Rao, M.R.; Rhodes, D.; Hansen, M.S.T.; Rubins, K.; Bushman, F.D.; Venkateswarlu, Y.; Faulkner, D.J. Lamellarin α 20-sulfate, an inhibitor of HIV-1 integrase active against HIV-1 virus in cell culture. J. Med. Chem. 1999, 42, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, Y.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents: Neolamellarins from the marine sponge Dendrilla nigra inhibit hypoxia-inducible factor-1 activation and secreted vascular endothelial growth factor production in breast tumor cells. J. Nat. Prod. 2007, 70, 1741–1745. [Google Scholar] [CrossRef] [PubMed]
- Baunbaek, D.; Trinkler, N.; Ferandin, Y.; Lozach, O.; Ploypradith, P.; Rucirawat, S.; Ishibashi, F.; Iwao, M.; Meijer, L. Anticancer alkaloid lamellarins inhibit protein kinases. Mar. Drugs 2008, 6, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Gallego, M.A.; Ballot, C.; Kluza, J.; Hajji, N.; Martoriati, A.; Castera, L.; Cuevas, C.; Formstecher, P.; Joseph, B.; Kroemer, G.; et al. Overcoming chemoresistance of non-small cell lung carcinoma through restoration of an AIF-dependent apoptotic pathway. Oncogene 2008, 27, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-C.; Xiao, X.; Salim, A.A.; Capon, R.J.; Zhang, Y.-K.; Talele, T.T.; Chen, Z.-S. Lamellarin O, a pyrrole alkaloid from an Australian marine sponge, Ianthella sp., reverses BCRP‑mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Vanhuyse, M.; Kluza, J.; Tardy, C.; Otero, G.; Cuevas, C.; Bailly, C.; Lansiaux, A. Lamellarin D: A novel pro-apoptotic agent from marine origin insensitive to P-glycoprotein-mediated drug efflux. Cancer Lett. 2005, 221, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Gallego, M.-A.; Loyens, A.; Beauvillain, J.-C.; Fernandez Sousa-Faro, J.-M.; Cuevas, C.; Marchetti, P.; Bailly, C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin D. Cancer Res. 2006, 66, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Ballot, C.; Kluza, J.; Martoriati, A.; Nyman, U.; Formstecher, P.; Joseph, B.; Bailly, C.; Marchetti, P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid lamellarin D. Mol. Cancer Ther. 2009, 8, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Ballot, C.; Kluza, J.; Lancel, S.; Martoriati, A.; Hassoun, S.M.; Mortier, L.; Vienne, J.-C.; Briand, G.; Formstecher, P.; Bailly, C.; et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010, 15, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Khiati, S.; Seol, Y.; Agama, K.; Dalla Rosa, I.; Agrawal, S.; Fesen, K.; Zhang, H.; Neuman, K.C.; Pommie, Y. Poisoning of mitochondrial topoisomerase I by lamellarin D. Mol. Pharmacol. 2014, 86, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Facompre, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin D: A novel potent inhibitor of topoisomerase I. Cancer Res. 2003, 63, 7392–7399. [Google Scholar] [PubMed]
- Tardy, C.; Facompre, M.; Laine, W.; Baldeyrou, B.; Garcia-Gravalos, D.; Francesch, A.; Mateo, C.; Pastor, A.; Jimenez, J.A.; Manzanares, I.; et al. Topoisomerase I-mediated DNA cleavage as a guide to the development of antitumor agents derived from the marine alkaloid lamellarin D: Triester derivatives incorporating amino acid residues. Bioorg. Med. Chem. 2004, 12, 1697–1712. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.; Laine, W.; Tardy, C.; Lansiaux, A.; Iwao, M.; Ishibashi, F.; Bailly, C.; Gago, F. Molecular determinants of topoisomerase I poisoning by lamellarins: Comparison with camptothecin and structure-activity relationships. J. Med. Chem. 2005, 48, 3796–3807. [Google Scholar] [CrossRef] [PubMed]
- Chittchang, M.; Batsomboon, P.; Ruchirawat, S.; Ploypradith, P. Cytotoxicities and structure-activity relationships of natural and unnatural lamellarins toward cancer cell lines. ChemMedChem 2009, 4, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pla, D.; Sischka, A.; Albericio, F.; Alvarez, M.; Fernandez-Busquets, X.; Anselmetti, D. Optical-tweezers study of topoisomerase inhibition. Small 2009, 5, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Lim, S.; Barthomeuf, C.; Morvan, D. Biochemical disorders induced by cytotoxic marine natural products in breast cancer cells as revealed by proton NMR spectroscopy-based metabolomics. Biochem. Pharmacol. 2010, 80, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadka, D.B.; Cho, W.-J. Topoisomerase inhibitors as anticancer agents: A patent update. Expert Opin. Ther. Pat. 2013, 23, 1033–1056. [Google Scholar] [CrossRef] [PubMed]
- Ballot, C.; Martoriati, A.; Jendoubi, M.; Buche, S.; Formstecher, P.; Mortier, L.; Kluza, J.; Marchetti, P. Another facet to the anticancer response to lamellarin D: Induction of cellular senescence through inhibition of topoisomerase I and intracellular ROS production. Mar. Drugs 2014, 12, 779–798. [Google Scholar] [CrossRef] [PubMed]
- Pungkham, H.; Swatdipakdi, N.; Theerasilp, M.; Karnkla, S.; Chittchang, M.; Ploypradith, P.; Nasongkla, N. PEG-b-PCL and PEG-b-PLA polymeric micelles as nanocarrieres for lamellarin N delivery. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 3245–3248. [Google Scholar] [PubMed]
- Chittchang, M.; Paul Gleeson, M.; Ploypradith, P.; Ruchirawat, S. Assessing the drug-likeness of lamellarins, a marine-derived natural product class with diverse oncological activities. Eur. J. Med. Chem. 2010, 45, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Thipnate, P.; Chittchang, M.; Thasana, N.; Saparpakorn, P.; Ploypradith, P.; Hannongbua, S. Exploring the molecular basis for selective cytotoxicity of lamellarins against human hormone-dependent T47D and hormone-independent MDA-MB-231 breast cancer cells. Monatsh. Chem. 2011, 142, 97–109. [Google Scholar] [CrossRef]
- Plisson, F.; Conte, M.; Khalil, Z.; Huang, X.-C.; Piggott, A.M.; Capon, R.J. Kinase inhibitor scaffolds against neurodegenerative diseases from a southern Australian ascidian, Didemnum sp. ChemMedChem 2012, 7, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Vullo, D.; Maresca, A.; Supuran, C.T.; Poulsen, S.-A. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 363–378. [Google Scholar] [CrossRef]
- Cironi, P.; Albericio, F.; Alvarez, M. Lamellarins: Isolation, activity and synthesis. Prog. Heterocycl. Chem. 2004, 16, 1–26. [Google Scholar]
- Handy, S.T.; Zhang, Y. Approaches to the synthesis of the lamellarins and related natural products. A review. Org. Prep. Proced. Int. 2005, 37, 411–445. [Google Scholar] [CrossRef]
- Yang, G.; Wang, A.-L.; Chen, H.-L.; You, Y.-C. Recent research progresses on lamellarins and analogous alkaloids of bioactive marine mollusc. Chin. J. Org. Chem. 2005, 25, 641–653. (In Chinese) [Google Scholar]
- He, Y.-R.; Li, D.-P.; Yang, G.; Wang, A.-L.; You, Y.-C.; Hu, D.-H.; Yu, X.-S. Recent research progress on biological activity of lamellarins and analogous alkaloids. Chin. J. Nat. Med. 2007, 5, 150–156. (In Chinese) [Google Scholar]
- Fan, H.; Peng, J.; Hamann, M.T.; Hu, J.-F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem. Rev. 2008, 108, 264–287. [Google Scholar] [CrossRef] [PubMed]
- Pla, D.; Albericio, F.; Alvarez, M. Recent advances in lamellarin alkaloids: Isolation, synthesis and activity. Anti-Cancer Agents Med. Chem. 2008, 8, 746–760. [Google Scholar] [CrossRef]
- You, Y.-C.; Hu, D.-H.; Li, D.-P.; Wang, A.-L.; Yu, X.-S. Recent research progress in lamellarin D and its derivatives. Chin. J. Org. Chem. 2008, 28, 797–803. (In Chinese) [Google Scholar]
- Shen, L.; Hu, Y. Recent advances in the synthesis of lamellarins as anticancer alkaloids. Chin. J. Org. Chem. 2009, 29, 867–875. (In Chinese) [Google Scholar]
- Fan, A.-L.; Lin, W.-H.; Jia, Y.-X. Recent progress in the research on lamellarins and related pyrrole-derived alkaloids from marine organisms. J. Chin. Pharm. Sci. 2011, 20, 425–441. (In Chinese) [Google Scholar]
- Fukuda, T.; Ishibashi, F.; Iwao, M. Synthesis and biological activity of lamellarin alkaloids. Heterocycles 2011, 83, 491–529. [Google Scholar] [CrossRef]
- Liao, S.; Li, D.; Liu, R.; Wang, A. Research progress in lamellarin H. Chem. Res. Appl. 2011, 23, 961–966. (In Chinese) [Google Scholar]
- Pla, D.; Albericio, F.; Alvarez, M. Progress on lamellarins. Med. Chem. Commun. 2011, 2, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Zhao, Z.; Zheng, X.; Cao, H. Recent research progress in anticancer alkaloid lamellarin N and lamellarin L. Chin. J. Org. Chem. 2013, 33, 483–491. (In Chinese) [Google Scholar] [CrossRef]
- Urban, S.; Hickford, S.J.H.; Blunt, J.W.; Munro, M.H.G. Bioactive marine alkaloids. Curr. Org. Chem. 2000, 4, 765–807. [Google Scholar] [CrossRef]
- Fernandez, D.; Ahaidar, A.; Danelon, G.; Cironi, P.; Marfil, M.; Perez, O.; Cuevas, C.; Albericio, F.; Joule, J.A.; Alvarez, M. Synthesis of polyheterocyclic nitrogen-containing marine natural products. Monatsh. Chem. 2004, 135, 615–627. [Google Scholar] [CrossRef]
- Dias, N.; Vezin, H.; Lansiaux, H.; Lansiaux, A.; Bailly, C. Topoisomerase inhibitors of marine origin and their potential use as anticancer agents. Top. Curr. Chem. 2005, 253, 89–108. [Google Scholar]
- Wang, W.; Namikoshi, M. Bioactive nitrogenous metabolites from ascidians. Heterocycles 2007, 74, 53–88. [Google Scholar] [CrossRef]
- Barthomeuf, C.; Bourguet-Kondracki, M.-L.; Kornprobst, J.-M. Marine metabolites overcoming or circumventing multidrug resistance mediated by ATP-dependent transporters: A new hope for patient with tumors resistant to conventional chemotherapy. Anti-Cancer Agents Med. Chem. 2008, 8, 886–903. [Google Scholar] [CrossRef]
- Gong, W.; Li, L.; Yi, Y.; Zhang, D. Research progress in the studies on the synthesis of marine alkaloids. Chin. Pharm. J. 2008, 26, 327–333. (In Chinese) [Google Scholar]
- Elyashberg, M.; Williams, A.J.; Blinov, K. Structural revisions of natural products by computer-assisted structure elucidation (CASE) systems. Nat. Prod. Rep. 2010, 27, 1296–1328. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Fattorusso, E.; Imperatore, C. Alkaloids from marine ascidians. Molecules 2011, 16, 8694–8732. [Google Scholar] [CrossRef] [Green Version]
- Menna, M. Important classes of bioactive alkaloids from marine ascidians: Structures, isolation and bioactivity. Curr. Top. Med. Chem. 2014, 14, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Fuerstner, A.; Weintritt, H.; Hupperts, A. A new, titanium-mediated approach to pyrroles: First synthesis of lukianol A and lamellarin O dimethyl ether. J. Org. Chem. 1995, 60, 6637–6641. [Google Scholar] [CrossRef]
- Banwell, M.G.; Flynn, B.L.; Hamel, E.; Hockless, D.C.R. Convergent syntheses of the pyrrolic marine natural products lamellarin O, lamellarin Q, lukianol A and some more highly oxygenated congeners. Chem. Commun. 1997, 2, 207–208. [Google Scholar] [CrossRef]
- Boger, D.L.; Boyce, C.W.; Labroli, M.A.; Sehon, C.A.; Jin, Q. Total syntheses of ningalin A, lamellarin O, lukianol A, and permethyl storniamide A utilizing heterocyclic azadiene diels-alder reactions. J. Am. Chem. Soc. 1999, 121, 54–62. [Google Scholar] [CrossRef]
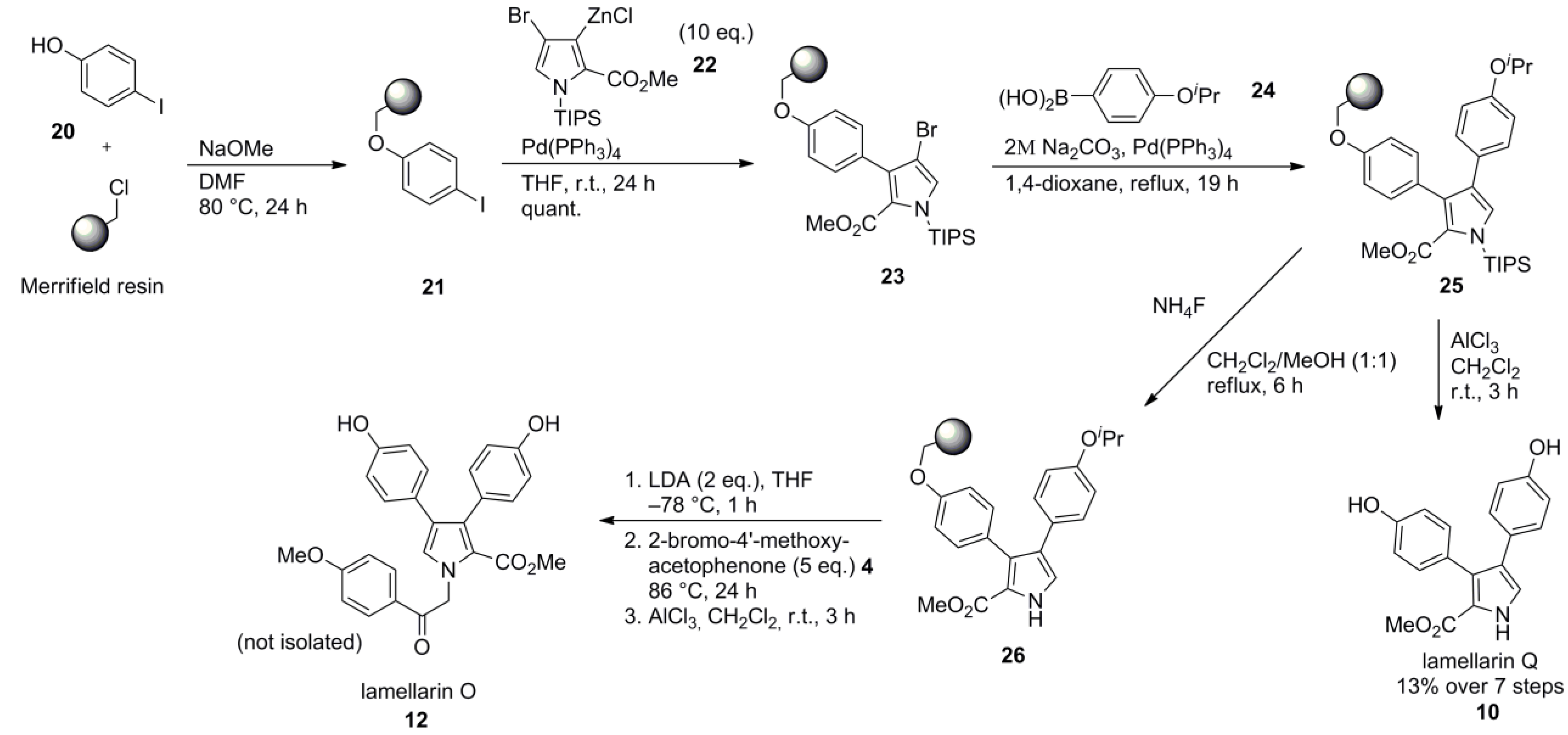
- Marfil, M.; Albericio, F.; Alvarez, M. Solid-phase synthesis of lamellarins Q and O. Tetrahedron 2004, 60, 8659–8668. [Google Scholar] [CrossRef]
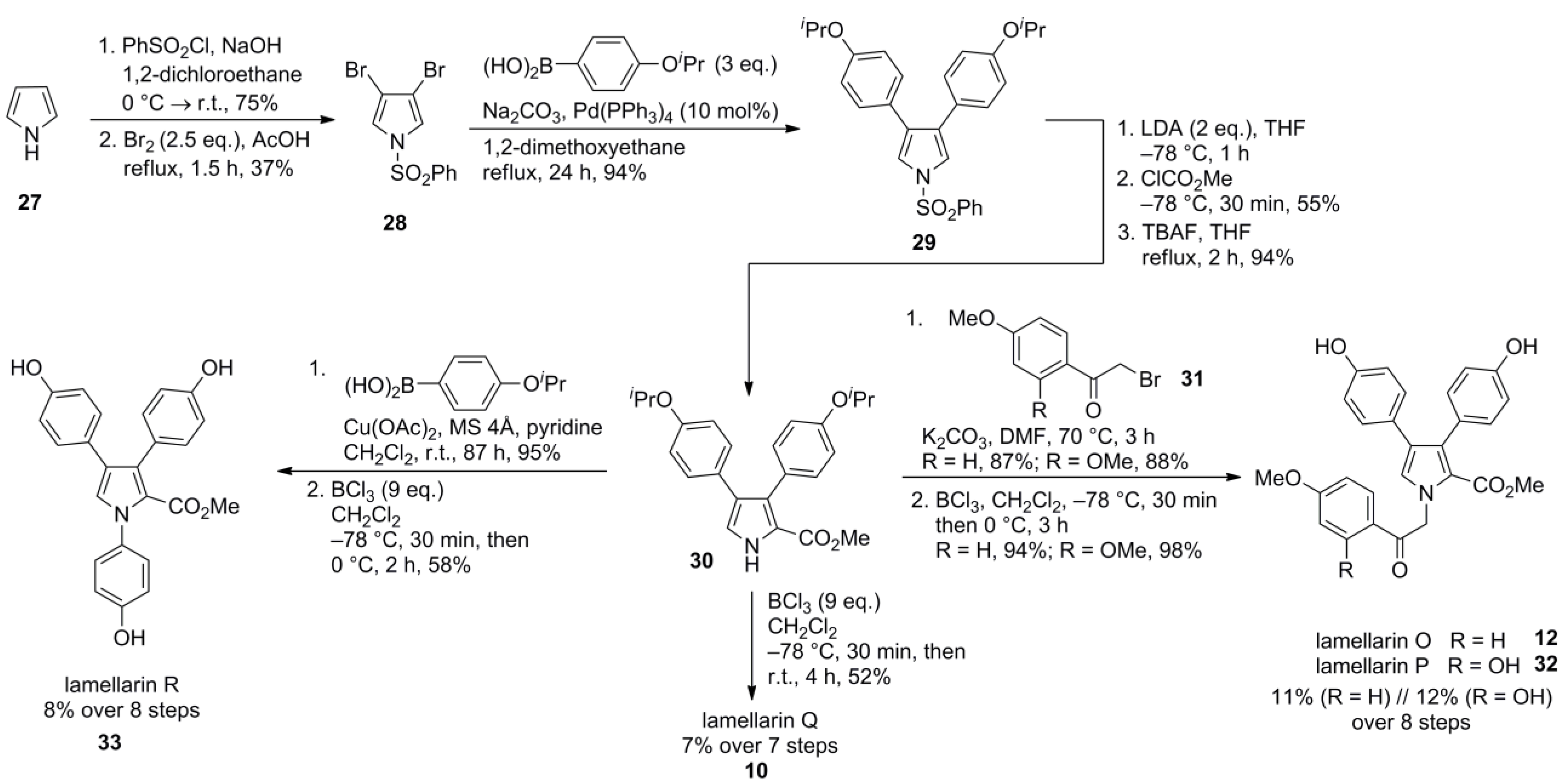
- Fukuda, T.; Sudo, E.-I.; Shimokawa, K.; Iwao, M. Palladium-catalyzed cross-coupling of N-benzenesulfonyl-3,4-dibromopyrrole and its application to the total syntheses of lamellarins O, P, Q and R. Tetrahedron 2008, 64, 328–338. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total synthesis of lamellarins D, H, and R and ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Rodriguez, A.; Mendez, J.M.; Jimenez, C.C.; Leon, F.; Vazquez, A. A Paal-Knorr approach to 3,4-diaryl-substituted pyrroles: Facile synthesis of lamellarins O and Q. Synthesis 2012, 44, 3321–3326. [Google Scholar] [CrossRef]
- Iwao, M.; Takeuchi, T.; Fujikawa, N.; Fukuda, T.; Ishibashi, F. Short and flexible route to 3,4-diarylpyrrole marine alkaloids: Syntheses of permethyl storniamide A, ningalin B, and lamellarin G trimethyl ether. Tetrahedron Lett. 2003, 44, 4443–4446. [Google Scholar] [CrossRef]
- Fujikawa, N.; Ohta, T.; Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. Total synthesis of lamellarins D, L, and N. Tetrahedron 2006, 62, 594–604. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fukuda, T.; Ishibashi, F.; Iwao, M. The first total synthesis of lamellarin α 20‑sulfate, a selective inhibitor of HIV-1 integrase. Tetrahedron Lett. 2006, 47, 3755–3757. [Google Scholar] [CrossRef]
- Fukuda, T.; Ohta, T.; Saeki, S.; Iwao, M. Divergent synthesis of lamellarin α 13-sulfate, 20-sulfate, and 13,20-disulfate. Heterocycles 2010, 80, 841–846. [Google Scholar] [CrossRef]
- Takada, T.; Arisawa, M.; Gyoten, M.; Hamada, R.; Tohma, H.; Kita, Y. Oxidative biaryl coupling reaction of phenol ether derivatives using a hypervalent iodine(III) reagent. J. Org. Chem. 1998, 63, 7698–7706. [Google Scholar] [CrossRef]
- Peschko, C.; Winklhofer, C.; Steglich, W. Alkaloids from marine organisms, part 5: Biomimetic total synthesis of lamellarin L by coupling of two different arylpyruvic acid units. Chem. Eur. J. 2000, 6, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Handy, S.T.; Zhang, Y.; Bregman, H. A modular synthesis of the lamellarins: Total synthesis of lamellarin G trimethyl ether. J. Org. Chem. 2004, 69, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
- Pla, D.; Marchal, A.; Olsen, C.A.; Albericio, F.; Alvarez, M. Modular total synthesis of lamellarin D. J. Org. Chem. 2005, 70, 8231–8234. [Google Scholar] [CrossRef] [PubMed]
- Hasse, K.; Willis, A.C.; Banwell, M.G. Modular total syntheses of lamellarin G trimethyl ether and lamellarin S. Eur. J. Org. Chem. 2011, 88–99. [Google Scholar]
- Komatsubara, M.; Umeki, T.; Fukuda, T.; Iwao, M. Modular synthesis of lamellarins via regioselective assembly of 3,4,5-differentially arylated pyrrole-2-carboxylates. J. Org. Chem. 2014, 79, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, F.; Miyazaki, Y.; Iwao, M. Total syntheses of lamellarin D and H. The first synthesis of lamellarin-class marine alkaloids. Tetrahedron 1997, 53, 5951–5962. [Google Scholar] [CrossRef]
- Diaz, M.; Guitian, E.; Castedo, L. Syntheses of lamellarins I and K by [3+2] cycloaddition of a nitrone to an alkyne. Synlett 2001, 1164–1166. [Google Scholar]
- Ruchirawat, S.; Mutarapat, T. An efficient synthesis of lamellarin alkaloids: Synthesis of lamellarin G trimethyl ether. Tetrahedron Lett. 2001, 42, 1205–1208. [Google Scholar] [CrossRef]
- Ploypradith, P.; Mahidol, C.; Sahakitpichan, P.; Wongbundit, S.; Ruchirawat, S. A highly efficient synthesis of lamellarins K and L by the Michael addition/ring-closure reaction of benzyldihydroisoquinoline derivatives with ethoxycarbonyl-β-nitrostyrenes. Angew. Chem. Int. Ed. 2004, 43, 866–868. [Google Scholar] [CrossRef]
- Ploypradith, P.; Petchmanee, T.; Sahakitpichan, P.; Litvinas, N.D.; Ruchirawat, S. Total synthesis of natural and unnatural lamellarins with saturated and unsaturated D-rings. J. Org. Chem. 2006, 71, 9440–9448. [Google Scholar] [CrossRef] [PubMed]
- Liermann, J.C.; Opatz, T. Synthesis of lamellarin U and lamellarin G trimethyl ether by alkylation of a deprotonated α-aminonitrile. J. Org. Chem. 2008, 73, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Banwell, M.G.; Flynn, B.L.; Hockless, D.C.R. Convergent total synthesis of lamellarin K. Chem. Commun. 1997, 23, 2259–2260. [Google Scholar] [CrossRef]
- Ridley, C.P.; Reddy, M.V.R.; Rocha, G.; Bushman, F.D.; Faulkner, D.J. Total synthesis and evaluation of lamellarin α 20-sulfate analogues. Bioorg. Med. Chem. 2002, 10, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Cironi, P.; Manzanares, I.; Albericio, F.; Alvarez, M. Solid-phase total synthesis of the pentacyclic system lamellarins U and L. Org. Lett. 2003, 5, 2959–2962. [Google Scholar] [CrossRef] [PubMed]
- Flynn, B.L.; Banwell, M.G. Convergent total syntheses of the pentacyclic lamellarins K, T, U and W via the addition of azomethine ylides to tethered tolans. Heterocycles 2012, 84, 1141–1170. [Google Scholar] [CrossRef]
- Heim, A.; Terpin, A.; Steglich, W. Alkaloids from marine organisms. 2. Biomimetic synthesis of lamellarin G trimethyl ether. Angew. Chem. Int. Ed. 1997, 36, 155–156. [Google Scholar] [CrossRef]
- Peschko, C.; Winklhofer, C.; Terpin, A.; Steglich, W. Biomimetic syntheses of lamellarin and lukianol-type alkaloids. Synthesis 2006, 18, 3048–3057. [Google Scholar]
- Gupton, J.T.; Giglio, B.C.; Eaton, J.E.; Rieck, E.A.; Smith, K.L.; Keough, M.J.; Barelli, P.J.; Firich, L.T.; Hempel, J.E.; Smith, T.M.; et al. The application of vinylogous iminium salt derivatives to efficient formal syntheses of the marine alkaloids lamellarin G trimethyl ether and ningalin B. Tetrahedron 2009, 65, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Gupton, J.T.; Krumpe, K.E.; Burnham, B.S.; Webb, T.M.; Shuford, J.S.; Sikorski, J.A. The application of vinylogous iminium salt derivatives to a regiocontrolled and efficient relay synthesis of lukianol A and related marine natural products. Tetrahedron 1999, 55, 14515–14522. [Google Scholar] [CrossRef]
- Gupton, J.T.; Telang, N.; Patteson, J.; Lescalleet, K.; Yeudall, S.; Sobieski, J.; Harrison, A.; Curry, W. The application of formyl group activation of bromopyrrole esters to formal syntheses of lycogarubin C, permethyl storniamide A and lamellarin G trimethyl ether. Tetrahedron 2014, 70, 9759–9767. [Google Scholar] [CrossRef]
- Yadav, J.S.; Gayathri, K.U.; Reddy, B.V.S.; Prasad, A.R. Modular total synthesis of lamellarin G trimethyl ether. Synlett 2009, 1, 43–46. [Google Scholar] [CrossRef]
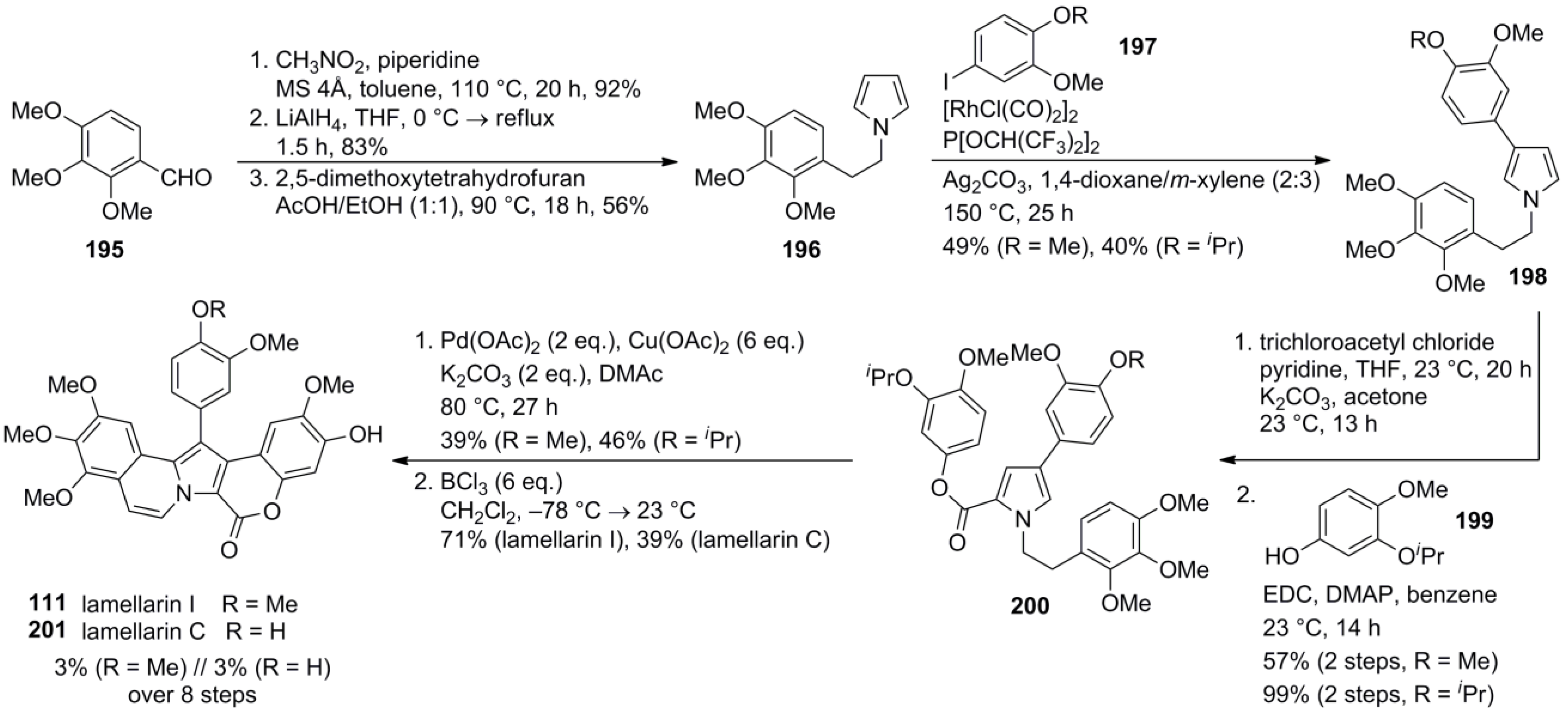
- Ueda, K.; Amaike, K.; Maceiczyk, R.M.; Itami, K.; Yamaguchi, J. β-selective C-H arylation of pyrroles leading to concise syntheses of lamellarins C and I. J. Am. Chem. Soc. 2014, 136, 13226–13232. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imbri, D.; Tauber, J.; Opatz, T. Synthetic Approaches to the Lamellarins—A Comprehensive Review. Mar. Drugs 2014, 12, 6142-6177. https://doi.org/10.3390/md12126142
Imbri D, Tauber J, Opatz T. Synthetic Approaches to the Lamellarins—A Comprehensive Review. Marine Drugs. 2014; 12(12):6142-6177. https://doi.org/10.3390/md12126142
Chicago/Turabian StyleImbri, Dennis, Johannes Tauber, and Till Opatz. 2014. "Synthetic Approaches to the Lamellarins—A Comprehensive Review" Marine Drugs 12, no. 12: 6142-6177. https://doi.org/10.3390/md12126142