3.2. Synthetic Procedures
3.2.2. General Procedure B: Removal of Boc Protecting Group
A solution of tert-butyl-carbamate derivative in CH2Cl2 (2 mL) and TFA (0.2 mL) was stirred at room temperature under N2 for 2 h, then dried in vacuo to afford the deprotected analogue. In some cases the product required no further purification, while in other cases, purification was achieved by C18 reversed-phase column chromatography eluting with 0%–50% MeOH/H2O (+0.05% TFA).
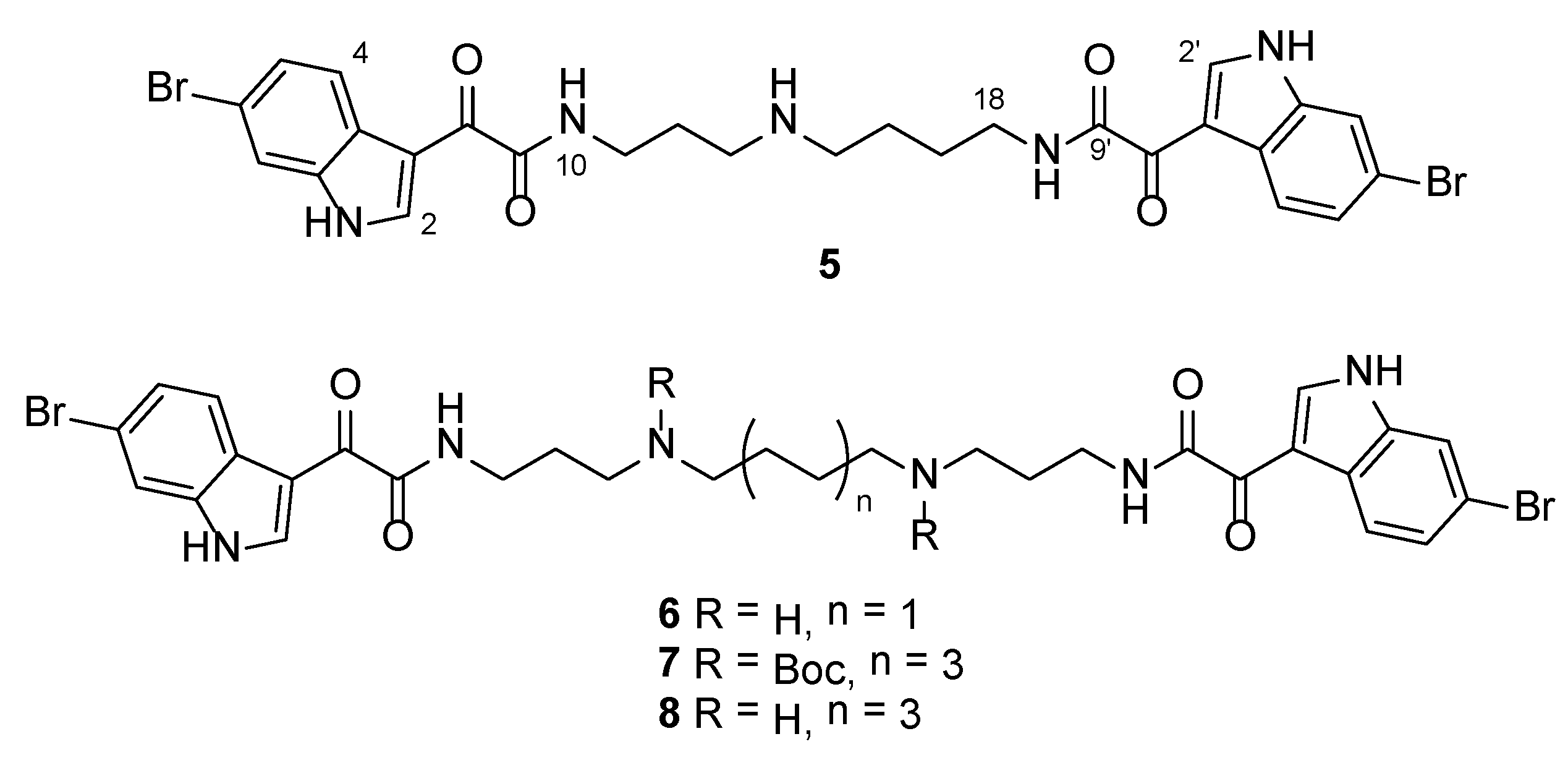
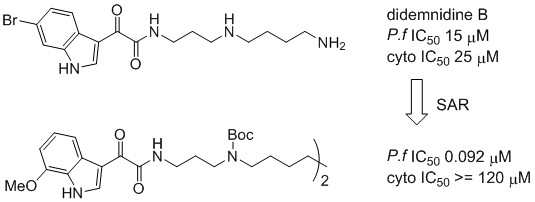
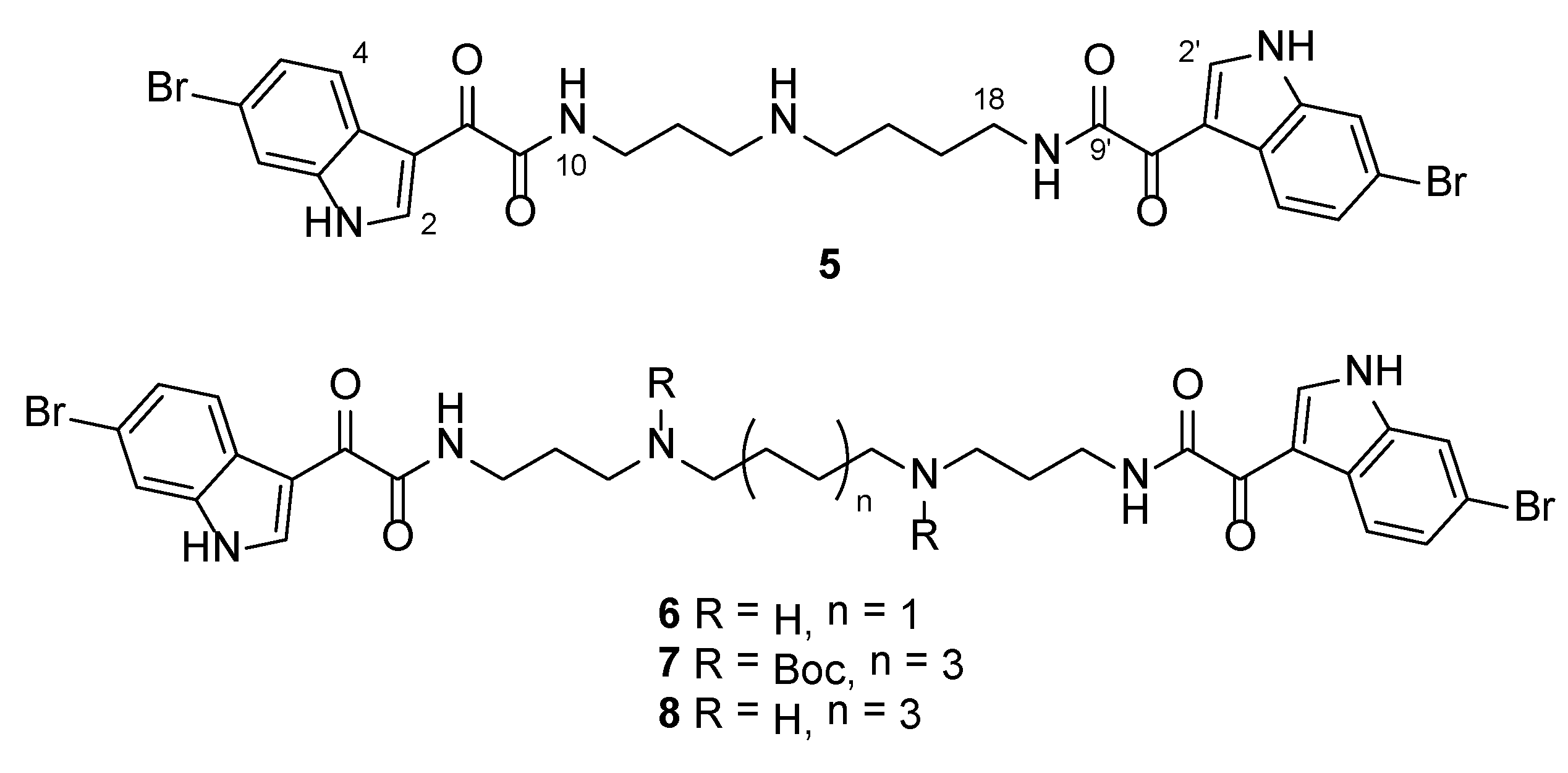
3.2.3. 4-(2-(6-Bromo-1H-indol-3-yl)-2-oxoacetamido)-N-(3-(2-(6-bromo-1H-indol-3-yl)-2-oxoacetamido)propyl)butan-1-aminium 2,2,2-trifluoroacetate (5)
Using general procedure A, 2-(6-bromo-1
H-indol-3-yl)-2-oxoacetic acid [
24] (60 mg, 0.21 mmol), spermidine (15 mg, 0.10 mmol), PyBOP (109 mg, 0.21 mmol) and Et
3N (83 μL, 0.60 mmol) afforded
5 as a yellow gum (37 mg, 58% yield).
Rf = 0.26 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3247, 1658, 1602, 1503, 1135, 841 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.38 (2H, br s, NH-1 and NH-1′), 8.91 (1H, t, J = 6.0 Hz, NH-10), 8.80 (1H, t, J = 6.1 Hz, NH-19), 8.78 (2H, d, J = 4.1 Hz, H-2 and H-2′), 8.42 (2H, br s, NH2-14), 8.15 (2H, d, J = 8.5 Hz, H-4 and H-4′), 7.75 (2H, d, J = 1.5 Hz, H-7 and H-7′), 7.40 (2H, dd, J = 8.5, 1.5 Hz, H-5 and H-5′), 3.30 (2H, td, J = 7.2, 6.0 Hz, H2-11), 3.25 (2H, td, J = 6.1, 5.8 Hz, H2-18), 3.02–2.88 (4H, m, H2-13 and H2-15), 1.85 (2H, tt, J = 7.2, 7.2 Hz, H2-12), 1.67–1.53 (4H, m, H2-16 and H2-17); 13C NMR (DMSO-d6, 100 MHz) δC 182.2 (C-8a), 181.8 (C-8′a), 163.5 (C-9), 163.4 (C-9′), 139.3 (C-2b), 139.3 (C-2′b), 137.3 (C-7a and C-7a′), 125.5 (C-5 and C-5′), 125.4 (C-3ac), 125.3 (C-3a′c), 122.9 (C-4 and C-4′), 116.0 (C-6d), 116.0 (C-6′d), 115.4 (C-7 and C-7′), 112.1 (C-3e), 112.1 (C-3′e), 46.6 (C-15), 44.8 (C-13), 37.9 (C-18), 35.8 (C-11), 25.9 (C-16f), 25.7 (C-12), 23.2 (C-17f); (+)-HRESIMS m/z 644.0506 [M + H]+ (calcd for C27H2879Br2N5O4, 644.0503).
3.2.4. N1,N4-Bis(3-(2-(6-bromo-1H-indol-3-yl)-2-oxoacetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (6)
Using general procedure A, 2-(6-bromo-1
H-indol-3-yl)-2-oxoacetic acid [
24] (50 mg, 0.18 mmol), spermine (17 mg, 0.083 mmol), PyBOP (91 mg, 0.18 mmol) and Et
3N (69 μL, 0.50 mmol) afforded
6 as a brown oil (50 mg, 86% yield).
Rf = 0.17 (CH2Cl2:MeOH:TEA 1:1:0.01); IR νmax (ATR) 3278, 1672, 1628, 1441, 1201, 1131, 799, 721, 686 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.41 (1H, br s, NH-1), 8.91 (1H, t, J = 6.3 Hz, NH-10), 8.78 (1H, d, J = 3.5 Hz, H-2), 8.58 (2H, br s, NH2-14), 8.15 (1H, d, J = 8.5 Hz, H-4), 7.76 (1H, d, J = 1.8 Hz, H-7), 7.41 (1H, dd, J = 8.5, 1.8 Hz, H-5), 3.30 (2H, td, J = 6.9, 6.3 Hz, H2-11), 2.99–2.89 (4H, m, H2-13 and H2-15), 1.85 (2H, tt, J = 6.9, 6.9 Hz, H2-12), 1.68–1.56 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 181.8 (C-8), 163.5 (C-9), 139.3 (C-2), 137.2 (C-7a), 125.5 (C-5), 125.3 (C-3a), 122.9 (C-4), 116.0 (C-6), 115.4 (C-7), 112.1 (C-3), 46.1 (C-15), 44.7 (C-13), 35.9 (C-11), 25.7 (C-12), 22.7 (C-16); (+)-HRESIMS m/z 701.1087 [M + H]+ (calcd for C30H3579Br2N6O4, 701.1081).
3.2.5. Di-tert-butyl Octane-1,8-diylbis((3-(2-(6-bromo-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (7)
Using general procedure A, 2-(6-bromo-1
H-indol-3-yl)-2-oxoacetic acid [
24] (0.12 g, 0.42 mmol), di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (91 mg, 0.20 mmol), PyBOP (0.22 g, 0.42 mmol) and Et
3N (83 μL, 0.60 mmol) afforded
7 as a peach gum (51 mg, 26% yield).
Rf = 0.60 (hexane:EtOAc 3:7); IR νmax (ATR) 3226, 2929, 1666, 1631, 1417, 1156, 793, 633 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.27 (1H, br s, NH-1), 8.78 (1H, s, H-2), 8.74 (1H, br s, NH-10), 8.15 (1H, d, J = 8.4 Hz, H-4), 7.73 (1H, d, J = 1.7 Hz, H-7), 7.39 (1H, dd, J = 8.4, 1.7 Hz, H-5), 3.18 (2H, td, J = 7.1, 6.9 Hz, H2-11), 3.13 (2H, t, J = 7.1 Hz, H2-13), 3.08 (2H, t, J = 7.2 Hz, H2-15), 1.75–1.64 (2H, m, H2-12), 1.46–1.32 (2H, m, H2-16), 1.36 (9H, s, 3H3-21), 1.26–1.11 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 100 MHz) δC 182.1 (C-8), 163.2 (C-9), 154.7 (C-19), 139.2 (C-2), 137.2 (C-7a), 125.4 (C-5), 125.3 (C-3a), 122.9 (C-4), 115.9 (C-6), 115.3 (C-7), 112.1 (C-3), 78.2 (C-20), 46.3 (C-15), 44.4, 44.0 (C-13), 36.4 (C-11), 28.7 (C-18), 28.0 (C-21), 27.7 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 979.2573 [M + Na]+ (calcd for C44H5879Br2N6NaO8, 979.2575).
3.2.6. N1,N8-Bis(3-(2-(6-bromo-1H-indol-3-yl)-2-oxoacetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (8)
Using general procedure B, reaction of 7 (9 mg, 9.4 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) afforded 8 as a yellow gum (9 mg, quant. yield) which required no further purification.
Rf = 0.19 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3321, 3180, 1717, 1597, 1184, 1133, 719, 655 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.46 (1H, br s, NH-1), 8.91 (1H, t, J = 6.3 Hz, NH-10), 8.77 (1H, s, H-2), 8.68 (2H, br s, NH2-14), 8.15 (1H, d, J = 8.4, H-4), 7.75 (1H, d, J = 1.9 Hz, H-7), 7.40 (1H, dd, J = 8.4, 1.9 Hz, H-5), 3.29 (2H, td, J = 7.3, 6.3 Hz, H2-11), 2.95–2.88 (2H, m, H2-13), 2.88–2.82 (2H, m, H2-15), 1.86 (2H, tt, J =7.3, 6.6 Hz, H2-12), 1.63–1.52 (2H, m, H2-16), 1.34–1.21 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 100 MHz) δC 181.9 (C-8), 163.5 (C-9), 139.2 (C-2), 137.2 (C-7a), 125.4 (C-5), 125.3 (C-3a), 122.9 (C-4), 116.0 (C-6), 115.4 (C-7), 112.0 (C-3), 46.7 (C-15), 44.6 (C-13), 35.9 (C-11), 28.3 (C-18), 25.8 (C-12a), 25.6 (C-17a), 25.4 (C-16a); (+)-HRESIMS m/z 757.1708 [M + H]+ (calcd for C34H4379Br2N6O4, 757.1707).
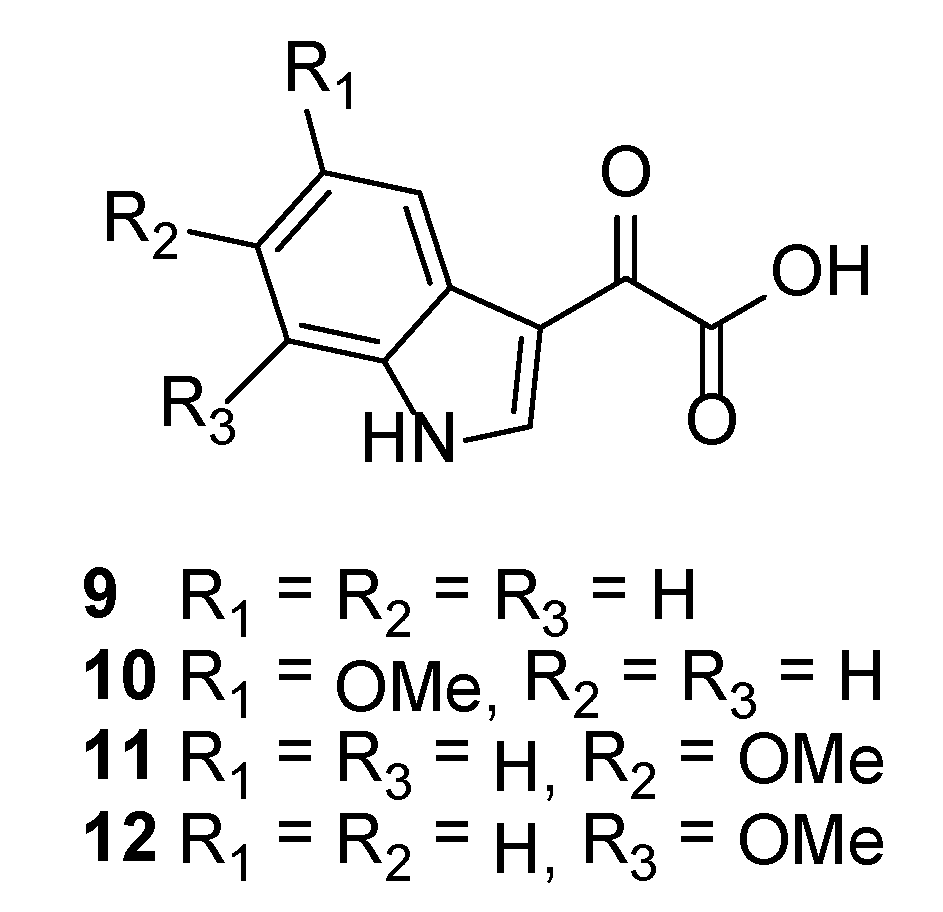
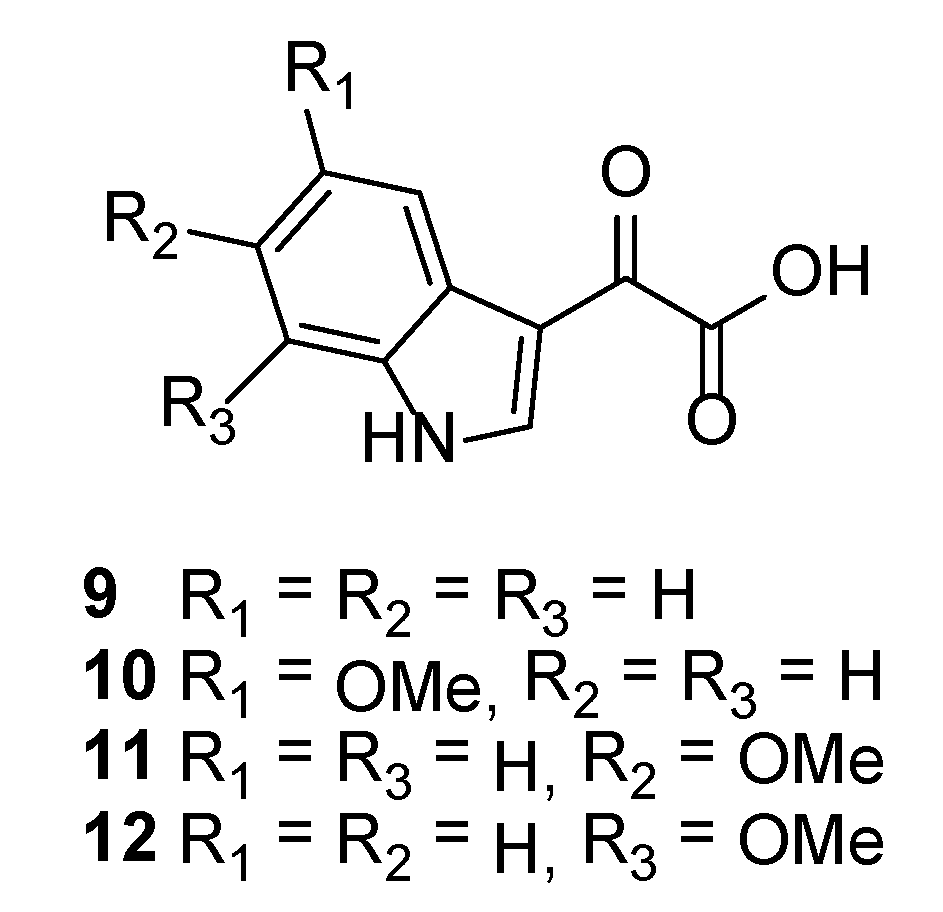
3.2.7. 2-(5-Methoxy-1H-indol-3-yl)-2-oxoacetic Acid (10)
The target compound
10 was prepared using a previously published method [
26]. To a solution of 5-methoxyindole (0.15 g, 0.985 mmol) in anhydrous diethyl ether (18 mL) was added oxalyl chloride (0.13 mL, 1.48 mmol) dropwise at 0 °C. Reaction was stirred at 0 °C for 2 h, during which time an orange precipitate was formed. Saturated aq. NaHCO
3 (6 mL) was added, and the reaction mixture heated at reflux for 2 h. After cooling to r.t., 10% HCl was added to adjust the solution to pH 1, the resulting precipitate filtered and dried under vacuum to yield
10 as an orange powder (0.20 g, 91% yield).
Mp 236 °C decomp. (lit. [
31] 248 °C); R
f = 0.09 (20% MeOH/EtOAc); IR ν
max (ATR) 3157, 2918, 1732, 1612, 1475, 1460, 1420, 1438, 1273, 1196, 1166, 913, 818, 809, 760, 709 cm
−1;
1H NMR (DMSO-
d6, 400 MHz) δ
H 12.29 (1H, br s, NH), 8.32 (1H, d,
J = 3.4 Hz, H-2), 7.67 (1H, d,
J = 2.5 Hz, H-4), 7.44 (1H, d,
J = 8.8 Hz, H-7), 6.91 (1H, dd,
J = 8.8, 2.5 Hz, H-6), 3.79 (3H, s, H
3-10), OH not observed;
13C NMR (DMSO-
d6, 100 MHz) δ
C 180.8 (C-8), 165.5 (C-9), 156.2 (C-5), 138.0 (C-2), 131.5 (C-7a), 126.6 (C-3a), 113.6 (C-7), 113.4 (C-6), 112.3 (C-3), 103.2 (C-4), 55.5 (C-10); (−)-HRESIMS
m/z 218.0470 [M − H]
− (calcd for C
11H
8NO
4, 218.0459).
3.2.8. 2-(6-Methoxy-1H-indol-3-yl)-2-oxoacetic Acid (11)
The target compound
11 was prepared using a previously published method [
26]. To a solution of 6-methoxyindole (0.13 g, 0.866 mmol) in anhydrous diethyl ether (10 mL) was added oxalyl chloride (0.11 mL, 1.30 mmol) dropwise at 0 °C. The reaction mixture was allowed to stir at 0 °C for 3 h before it was warmed to r.t. Saturated aq. NaHCO
3 (10 mL) was then added, and the reaction mixture heated at reflux for 1 h. After cooling to r.t., the pH of the reaction mixture was adjusted to 1 using 10% HCl. The resulting green precipitate was filtered, washed with cold diethyl ether (30 mL) and dried under vacuum to yield
11 as a green powder (0.18 g, 97% yield) which was used in the next step without further purification.
Mp 226 °C decomp.; Rf = 0.09 (20% MeOH/EtOAc); IR νmax (ATR) 3167, 1733, 1608, 1394, 1142, 1093, 710, 653 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.11 (1H, br s, NH), 8.30 (1H, s, H-2), 8.03 (1H, d, J = 8.6 Hz, H-4), 7.03 (1H, s, H-7), 6.90 (1H, dd, J = 8.6, 1.8 Hz, H-5), 3.80 (3H, s, H3-10), OH not observed; 13C NMR (DMSO-d6, 100 MHz) δC 180.8 (C-8), 165.3 (C-9), 156.9 (C-6), 137.7 (C-7a), 137.2 (C-2), 121.8 (C-4), 119.4 (C-3a), 112.5 (C-3), 112.3 (C-5), 95.8 (C-7), 55.3 (C-10); (+)-HRESIMS m/z 220.0615 [M + H]+ (calcd for C11H10NO4, 220.0604).
3.2.9. 2-(7-Methoxy-1H-indol-3-yl)-2-oxoacetic Acid (12)
The target compound
12 was prepared using a previously published method [
26]. To a solution of 7-methoxyindole (0.30 g, 2.04 mmol) in anhydrous diethyl ether (9 mL) was added oxalyl chloride (0.52 mL, 6.11 mmol) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 1.5 h, followed by dropwise addition of saturated aq. NaHCO
3 (10 mL), and then heated at reflux for 20.5 h. After cooling to r.t., 10% HCl was added to the reaction mixture to adjust pH to 1 and the resulting brown precipitate was filtered, washed with cold diethyl ether (20 mL), and dried under vacuum to yield
12 as a brown solid (0.45 g, quant. yield) which was used in the next step without further purification.
Mp 206 °C decomp.; Rf = 0.14 (20% MeOH/EtOAc); IR νmax (ATR) 3129, 1712, 1615, 1567, 1450, 1234, 1221, 956, 782 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 12.51 (1H, br s, NH), 8.23 (1H, d, J = 2.9 Hz, H-2), 7.74 (1H, d, J = 7.9 Hz, H-4), 7.19 (1H, t, J = 7.9 Hz, H-5), 6.87 (1H, d, J = 7.9 Hz, H-6), 3.95 (3H, s, H3-10), OH not observed; 13C NMR (DMSO-d6, 100 MHz) δC 180.8 (C-8), 165.2 (C-9), 146.5 (C-7), 136.8 (C-2), 127.2 (C-3a), 126.6 (C-7a), 123.7 (C-5), 113.6 (C-4), 112.9 (C-3), 104.6 (C-6), 55.4 (C-10); (−)-HRESIMS m/z 220.0603 [M + H]+ (calcd for C11H8NO4, 220.0604).
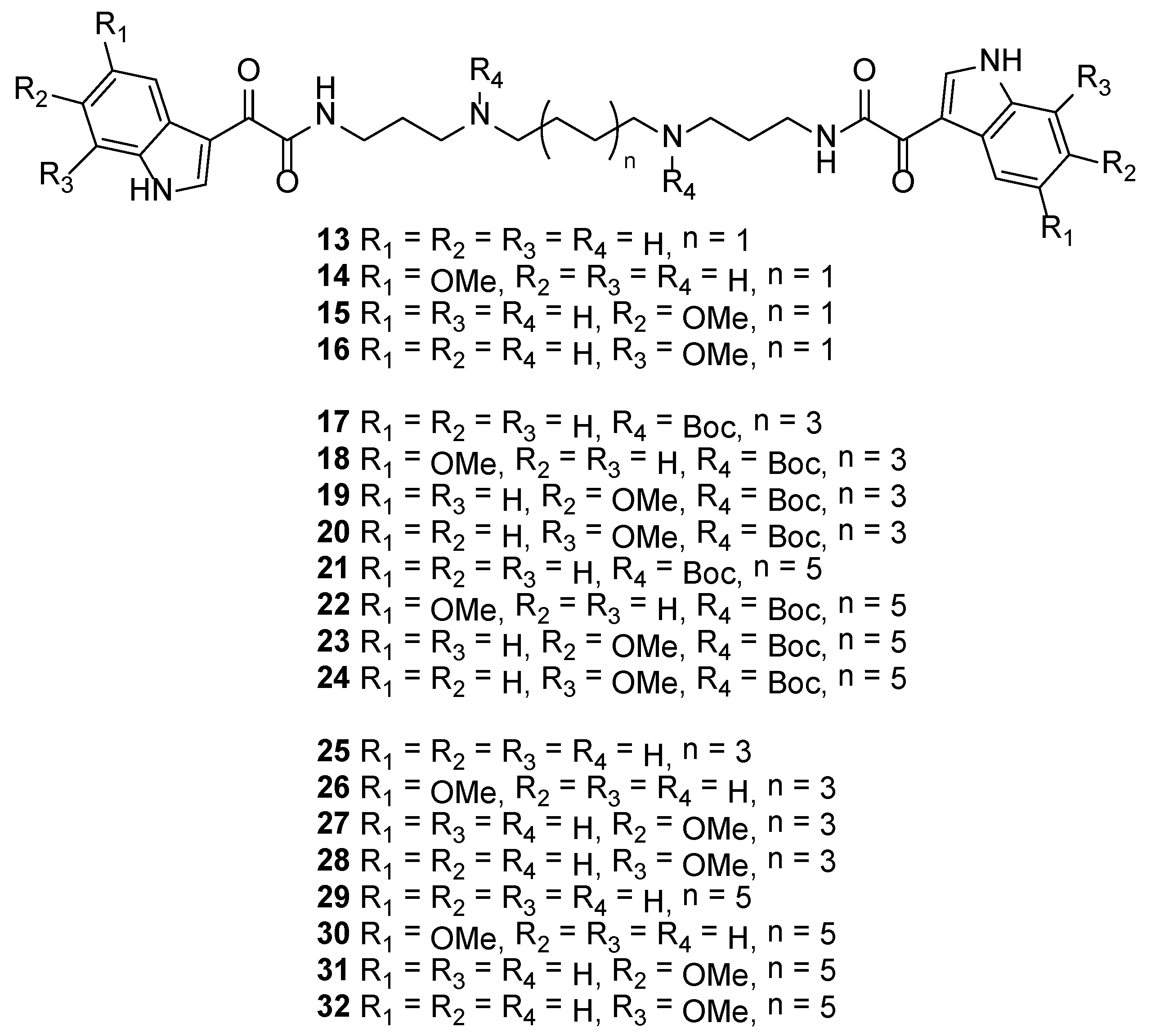
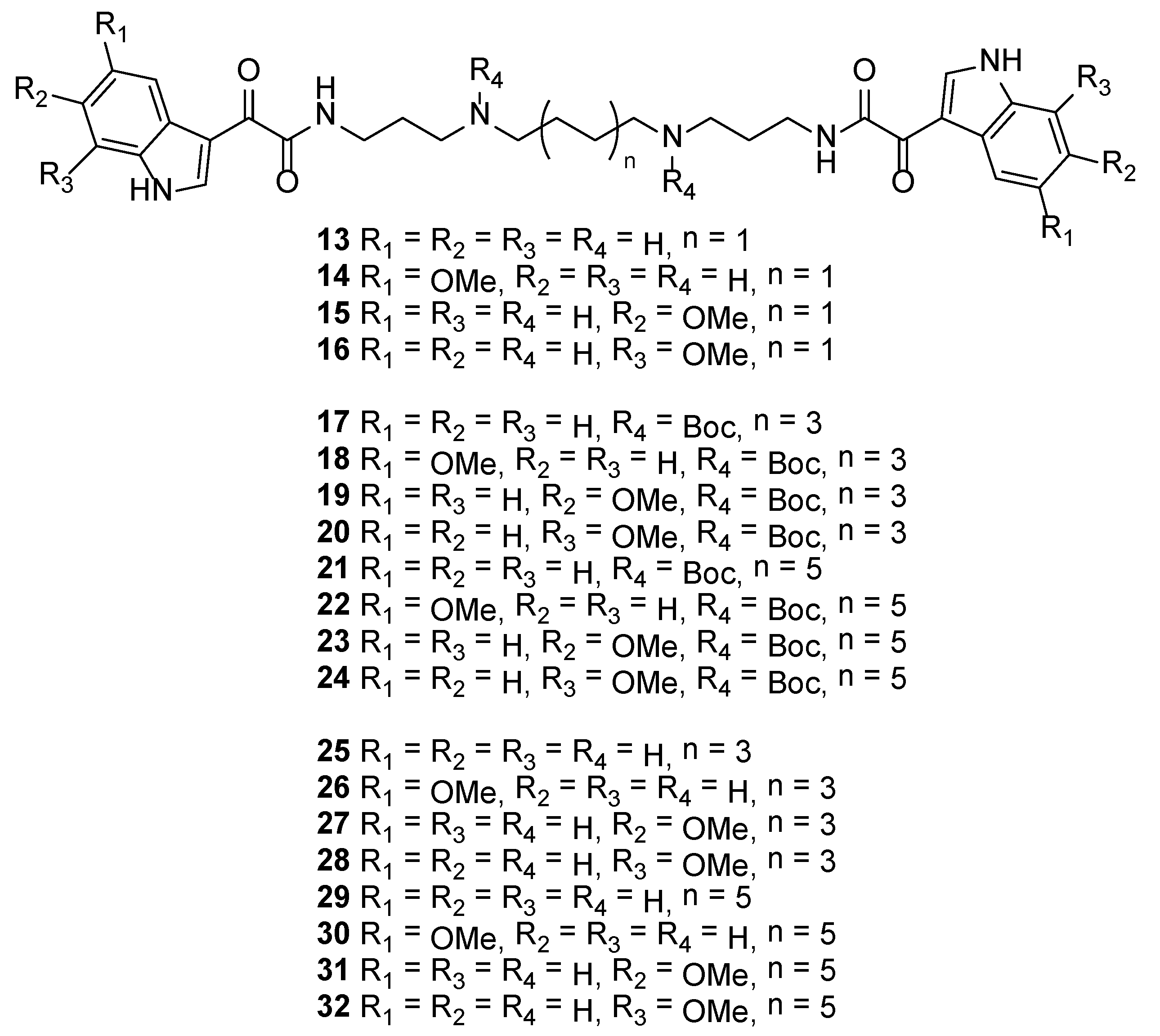
3.2.10. N1,N4-Bis(3-(2-(1H-indol-3-yl)-2-oxoacetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (13)
Using general procedure A, 2-(1H-indol-3-yl)-2-oxoacetic acid (9) (100 mg, 0.53 mmol), spermine (49 mg, 0.24 mmol), PyBOP (275 mg, 0.53 mmol) and Et3N (107 μL, 1.4 mmol) afforded 13 as a creamy gum (191 mg, quant. yield).
Rf = 0.26 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3361, 3093, 1679, 1626, 1428, 1125, 721 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.29 (1H, s, NH-1), 8.89 (1H, t, J = 6.0 Hz, NH-10), 8.76 (1H, s, H-2), 8.26–8.20 (1H, m, H-4), 7.57–7.51 (1H, m, H-7), 7.31–7.22 (2H, m, H-5 and H-6), 3.33–3.26 (2H, m, H2-11), 2.99–2.89 (4H, m, H2-13 and H2-15), 1.92–1.79 (2H, m, H2-12), 1.67–1.58 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 181.7 (C-8), 163.8 (C-9), 138.5 (C-2), 136.3 (C-7a), 126.2 (C-3a), 123.5 (C-5a), 122.6 (C-6a), 121.2 (C-4), 112.6 (C-7), 112.1 (C-3), 46.1 (C-15b), 44.8 (C-13b), 35.8 (C-11), 25.7 (C-12), 22.8 (C-16); (+)-HRESIMS m/z 545.2866 [M + H]+ (calcd for C30H37N6O4, 545.2871).
3.2.11. N1,N4-Bis(3-(2-(5-methoxy-1H-indol-3-yl)acetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (14)
Using general procedure A, 2-(5-methoxy-1H-indol-3-yl)-2-oxoacetic acid (10) (60 mg, 0.27 mmol), spermine (25 mg, 0.12 mmol), PyBOP (142 mg, 0.27 mmol), and Et3N (103 μL, 0.74 mmol) afforded 14 as a green gum (19 mg, 26% yield).
Rf = 0.06 (MeOH:TEA 5:0.01); IR νmax (ATR) 3347, 1679, 1438, 1127, 721 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.21 (1H, br s, NH-1), 8.84 (1H, t, J = 5.8 Hz, NH-10), 8.68 (1H, s, H-2), 7.74 (1H, d, J = 1.8 Hz, H-4), 7.44 (1H, d, J = 8.6 Hz, H-7), 6.90 (1H, dd, J = 8.6, 1.8 Hz, H-6), 3.79 (3H, s, H3-17), 3.33–3.25 (2H, td, J = 6.8, 5.8 Hz, H2-11), 2.91–2.80 (4H, m, H2-13 and H2-15), 1.88–1.77 (2H, m, H2-12), 1.65–1.58 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 181.7 (C-8), 163.9 (C-9), 156.1 (C-5), 138.5 (C-2), 131.1 (C-7a), 127.2 (C-3a), 113.4 (C-7), 112.9 (C-6), 112.0 (C-3), 103.5 (C-4), 55.3 (C-17), 46.7 (C-15), 45.0 (C-13), 36.1 (C-11), 26.3 (C-12), 23.8 (C-16); (+)-HRESIMS m/z 605.3089 [M + H]+ (calcd for C32H41N6O6, 605.3082).
3.2.12. N1,N4-Bis(3-(2-(6-methoxy-1H-indol-3-yl)acetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (15)
Using general procedure A, 2-(6-methoxy-1H-indol-3-yl)-2-oxoacetic acid (11) (70 mg, 0.32 mmol), spermine (29 mg, 0.15 mmol), PyBOP (116 mg, 0.32 mmol) and Et3N (121 μL, 0.87 mmol) afforded 15 as a yellow solid (44 mg, 52% yield).
Mp 223 °C decomp.; Rf = 0.03 (MeOH:TEA 5:0.01); IR νmax (ATR) 3173, 2780, 1655, 1600, 1435, 1162, 665 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH12.07 (1H, s, NH-1), 8.86 (1H, t, J = 6.3Hz, NH-10), 8.64 (1H, d, J = 3.1 Hz, H-2), 8.50 (1H, br s, NH-14), 8.07 (1H, d, J = 8.7 Hz, H-4), 7.04 (1H, d, J = 2.4 Hz, H-7), 6.89 (1H, dd, J = 8.7, 2.4 Hz, H-6), 3.79 (3H, s, H3-17), 3.29 (2H, td, J = 7.2, 6.3 Hz, H2-11), 3.02–2.86 (4H, m, H2-13 and H2-15), 1.92–1.80 (2H, m, H2-12), 1.68–1.58 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 181.6 (C-8), 163.8 (C-9), 156.8 (C-6), 137.7 (C-2), 137.3 (C-7a), 121.9 (C-4), 120.0 (C-3a), 112.2 (C-3a), 112.2 (C-5), 95.8 (C-7), 55.3 (C-17), 46.1 (C-15a), 44.7 (C-13a), 35.8 (C-11), 25.7 (C-12), 22.7 (C-16); (+)-HRESIMS m/z 605.3071 [M + H]+ (calcd for C32H41N6O6, 605.3082).
3.2.13. N1,N4-Bis(3-(2-(7-methoxy-1H-indol-3-yl)acetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (16)
Using general procedure A, 2-(7-methoxy-1H-indol-3-yl)-2-oxoacetic acid (12) (110 mg, 0.50 mmol), spermine (48 mg, 0.24 mmol), PyBOP (261 mg, 0.50 mmol) and Et3N (417 μL, 3.0 mmol) afforded 16 as a yellow gum (67 mg, 49% yield).
Rf = 0.03 (MeOH:TEA 5:0.01); IR νmax (ATR) 3191, 1671, 1623, 1432, 1179, 785 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH12.45 (1H, s, NH-1), 8.89 (1H, t, J = 6.1 Hz, NH-10), 8.62 (1H, d, J = 3.4 Hz, H-2), 8.52 (1H, br s, NH-14), 7.80 (1H, d, J = 8.1 Hz, H-4), 7.19 (1H, t, J = 8.1 Hz, H-5), 6.86 (1H, d, J = 8.1 Hz, H-6), 3.95 (3H, s, H3-17), 3.33–3.23 (2H, m, H2-11), 2.99–2.89 (4H, m, H2-13 and H2-15), 1.90–1.80 (2H, m, H2-12), 1.65–1.58 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 181.7 (C-8), 163.7 (C-9), 146.4 (C-7), 137.4 (C-2), 127.8 (C-3a), 126.1 (C-7a), 123.6 (C-5), 113.7 (C-4), 112.6 (C-3), 104.4 (C-6), 55.4 (C-17), 46.1 (C-15a), 44.7 (C-13a), 35.8 (C-11), 25.7 (C-12), 22.7 (C-16); (+)-HRESIMS m/z 605.3065 [M + H]+ (calcd for C32H41N6O6, 605.3082).
3.2.14. Di-tert-butyl Octane-1,8-diylbis((3-(2-(1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (17)
Using general procedure A, 2-(1
H-indol-3-yl)-2-oxoacetic acid (
9) (109 mg, 0.58 mmol), di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (120 mg, 0.26 mmol), PyBOP (300 mg, 0.58 mmol) and Et
3N (218 μL, 1.5 mmol) afforded
17 as a white gum (34 mg, 16% yield).
Rf = 0.66 (CH2Cl2:EtOAc 1:1); IR νmax (ATR) 3215, 2925, 1618, 1420, 1152, 746 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ 12.20 (1H, s, NH-1), 8.75 (1H, d, J = 3.0 Hz, H-2), 8.71 (1H, t, J = 6.0 Hz, NH-10), 8.26–8.19 (1H, m, H-4), 7.57–7.49 (1H, m, H-7), 7.30–7.20 (2H, m, H-5 and H-6), 3.25–3.05 (6H, m, H2-11, H2-13 and H2-15), 1.78–1.62 (2H, m, H2-12), 1.49–1.31 (2H, m, H2-16), 1.37 (9H, s, 3H3-21), 1.27–1.11 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 100 MHz) δC 182.1 (C-8), 163.5 (C-9), 155.6 (C-19), 138.4 (C-2), 136.2 (C-7a), 126.2 (C-3a), 123.4 (C-5a), 122.5 (C-6a), 121.2 (C-4), 112.5 (C-3), 112.1 (C-7), 78.2 (C-20), 46.4 (C-15), 44.4, 44.0 (C-13), 36.3 (C-11), 28.7 (C-18), 28.0 (C-21), 27.8 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 801.4510 [M + H]+ (calcd for C44H61N6O8, 801.4545).
3.2.15. Di-tert-butyl Octane-1,8-diylbis((3-(2-(5-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (18)
Using general procedure A, 2-(5-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
10) (93 mg, 0.42 mmol), di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (97 mg, 0.21 mmol), PyBOP (242 mg, 0.47 mmol) and Et
3N (176 μL, 1.3 mmol) afforded
18 as a yellow oil (83 mg, 46% yield).
Rf = 0.39 (hexane:EtOAc 2:3); IR νmax (ATR) 3371, 2929, 1619, 1420, 1153, 736 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 12.08 (1H, s, NH-1), 8.69 (1H, d, J = 2.3 Hz, H-2), 8.67 (1H, m, NH-10), 7.74 (1H, d, J = 2.6 Hz, H-4), 7.42 (1H, d, J = 8.1 Hz, H-7), 6.89 (1H, dd, J = 8.1, 2.6 Hz, H-6), 3.79 (3H, s, H3-19), 3.24–3.03 (6H, m, H2-11, H2-13 and H2-15), 1.77–1.62 (2H, m, H2-12), 1.49–1.32 (2H, m, H2-16), 1.37 (9H, s, 3H3-22), 1.27–1.11 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 75 MHz) δC 181.9 (C-8), 163.6 (C-9), 155.9 (C-5), 154.6 (C-20), 138.4 (C-2), 131.0 (C-7a), 127.2 (C-3a), 113.2 (C-6), 112.8 (C-7), 112.0 (C-3), 103.4 (C-4), 78.2 (C-21), 55.2 (C-19), 46.3 (C-15), 44.3, 44.0 (C-13), 36.3 (C-11), 28.7 (C-18), 28.0 (C-22), 27.8 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 861.4725 [M + H]+ (calcd for C46H65N6O10, 861.4757).
3.2.16. Di-tert-butyl Octane-1,8-diylbis((3-(2-(6-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (19)
Using general procedure A, 2-(6-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
11) (94 mg, 0.43 mmol), di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (98 mg, 0.21 mmol), PyBOP (245 mg, 0.47 mmol) and Et
3N (178 μL, 1.3 mmol) afforded
19 as a creamy solid (92 mg, 50% yield).
Mp 92 °C ; Rf = 0.23 (CH2Cl2:EtOAc 1:1); IR νmax (ATR) 3329, 2933, 1612, 1423, 1159, 740 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 11.99 (1H, d, J = 2.8 Hz, NH-1), 8.68 (1H, t, J = 5.7 Hz, NH-10), 8.64 (1H, d, J = 2.8 Hz, H-2), 8.07 (1H, d, J = 8.8 Hz, H-4), 7.02 (1H, d, J = 2.2 Hz, H-7), 6.88 (1H, dd, J = 8.8, 2.2 Hz, H-5), 3.79 (3H, s, H3-19), 3.23–3.05 (6H, m, H2-11, H2-13 and H2-15), 1.77–1.63 (2H, m, H2-12), 1.48–1.32 (2H, m, H2-16), 1.36 (9H, s, 3H3-22), 1.28–1.13 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 75 MHz) δC 181.9 (C-8), 163.5 (C-9), 156.7 (C-6), 154.6 (C-20), 137.6 (C-2), 137.2 (C-7a), 121.9 (C-4), 120.0 (C-3a), 112.3 (C-3), 112.0 (C-5), 95.7 (C-7), 78.2 (C-21), 55.2 (C-19), 46.3 (C-15), 44.3, 44.0 (C-13), 36.3 (C-11), 28.7 (C-18), 28.0 (C-22), 27.7 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 861.4743 [M + H]+ (calcd for C46H65N6O10, 861.4757).
3.2.17. Di-tert-butyl Octane-1,8-diylbis((3-(2-(7-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (20)
Using general procedure A, 2-(7-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
12) (86 mg, 0.39 mmol), di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (90 mg, 0.20 mmol), PyBOP (225 mg, 0.43 mmol) and Et
3N (163 μL, 1.2 mmol) afforded
20 as a green gum (94 mg, 56% yield).
Rf = 0.57 (CH2Cl2:EtOAc 1:1) 0.57; IR νmax (ATR) 3366, 2933, 1617, 1455, 1160, 778 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.39 (1H, br d, J = 3.1 Hz, NH-1), 8.71 (1H, br t, J = 5.0 Hz, NH-10), 8.61 (1H, d, J = 3.1 Hz, H-2), 7.80 (1H, d, J = 7.8 Hz, H-4), 7.17 (1H, t, J = 7.8 Hz, H-5), 6.85 (1H, d, J = 7.8 Hz, H-6), 3.94 (3H, s, H3-19), 3.22–3.05 (6H, m, H2-11, H2-13 and H2-15), 1.76–1.64 (2H, m, H2-12), 1.47–1.37 (2H, m, H2-16), 1.36 (9H, s, 3H3-22), 1.27–1.12 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 100 MHz) δC 182.1 (C-8), 163.4 (C-9), 154.7 (C-20), 146.4 (C-7), 137.3 (C-2), 127.8 (C-3a), 126.1 (C-7a), 123.4 (C-5), 113.8 (C-4), 112.7 (C-3), 104.3 (C-6), 78.2 (C-21), 55.4 (C-19), 46.3 (C-15), 44.4, 44.0 (C-13), 36.3 (C-11), 28.7 (C-18), 28.0 (C-22), 27.7 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 861.4778 [M + H]+ (calcd for C46H65N6O10, 861.4757).
3.2.18. Di-tert-butyl Dodecane-1,12-diylbis((3-(2-(1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (21)
Using general procedure A, 2-(1
H-indol-3-yl)-2-oxoacetic acid (
9) (19 mg, 0.10 mmol), di-
tert-butyl dodecane-1,12-diylbis((3-aminopropyl)carbamate) [
27,
28] (23 mg, 45 μmol), PyBOP (51 mg, 0.10 mmol) and Et
3N (82 μL, 0.60 mmol) afforded
21 as a white gum (25 mg, 65% yield).
Rf = 0.60 (CH2Cl2:EtOAc 1:1); IR νmax (ATR) 2927, 1621, 1420, 1156, 746 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.20 (1H, s, NH-1), 8.75 (1H, s, H-2), 8.72 (1H, t, J = 6.1 Hz, NH-10), 8.25–8.19 (1H, m, H-4), 7.56–7.50 (1H, m, H-7), 7.30–7.21 (2H, m, H-5 and H-6), 3.24–3.06 (6H, m, H2-11, H2-13 and H2-15), 1.77–1.64 (2H, m, H2-12), 1.49–1.33 (2H, m, H2-16), 1.37 (9H, s, 3H3-23), 1.25–1.16 (8H, m, H2-17 to H2-20); 13C NMR (DMSO-d6, 100 MHz) δC 182.1 (C-8), 163.5 (C-9), 155.0 (C-21), 138.4 (C-2), 136.2 (C-7a), 126.2 (C-3a), 123.4 (C-4), 122.5 (C-5a), 121.3 (C-6a), 112.5 (C-7), 112.2 (C-3), 78.2 (C-22), 46.3 (C-15), 44.0 (C-13), 36.3 (C-11), 28.9 (C-18b), 28.9 (C-19b), 28.7 (C-20b), 28.3 (C-16), 28.0 (C-23), 27.7 (C-12), 26.1 (C-17b); (+)-HRESIMS m/z 879.4967 [M + Na]+ (calcd for C48H68N6NaO8, 879.4991).
3.2.19. Di-tert-butyl Dodecane-1,12-diylbis((3-(2-(5-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (22)
Using general procedure A, 2-(5-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
10) (50 mg, 0.23 mmol), di-
tert-butyl dodecane-1,12-diylbis((3-aminopropyl)carbamate) [
27,
28] (53 mg, 0.10 mmol), PyBOP (117 mg, 0.23 mmol) and Et
3N (86 μL, 0.62 mmol) afforded
22 as an orange gum (53 mg, 58% yield).
Rf = 0.54 (hexane:EtOAc 3:7); IR νmax (ATR) 3237, 2927, 1621, 1420, 1139, 735 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.09 (1H, s, NH-1), 8.69 (1H, d, J = 3.0 Hz, H-2), 8.67 (1H, m, NH-10), 7.74 (1H, d, J = 2.5 Hz, H-4), 7.42 (1H, d, J = 8.8 Hz, H-7), 6.89 (1H, dd, J = 8.8, 2.5 Hz, H-6), 3.79 (3H, s, H3-21), 3.23–3.05 (6H, m, H2-11, H2-13 and H2-15), 1.75–1.64 (2H, m, H2-12), 1.47–1.32 (2H, m, H2-16), 1.37 (9H, s, 3H3-24), 1.25–1.12 (8H, m, H2-17 to H2-20); 13C NMR (DMSO-d6, 100 MHz) δC 181.9 (C-8), 163.6 (C-9), 155.9 (C-5), 154.7 (C-22), 138.4 (C-2), 131.0 (C-7a), 127.2 (C-3a), 113.2 (C-7), 112.8 (C-6), 112.0 (C-3), 103.4 (C-4), 78.2 (C-23), 55.2 (C-21), 46.3 (C-15), 44.4, 44.0 (C-13), 36.3 (C-11), 28.9 (C-18a), 28.9 (C-19a), 28.6 (C-20a), 28.0 (C-24), 27.7 (C-12 and C-16), 26.2 (C-17a); (+)-HRESIMS m/z 917.5363 [M + H]+ (calcd for C50H73N6O10, 917.5383).
3.2.20. Di-tert-butyl Dodecane-1,12-diylbis((3-(2-(6-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (23)
Using general procedure A, 2-(6-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
11) (33 mg, 0.15 mmol), di-
tert-butyl dodecane-1,12-diylbis((3-aminopropyl)carbamate) [
27,
28] (35 mg, 68 μmol), PyBOP (78 mg, 0.15 mmol) and Et
3N (57 μL, 0.41 mmol) afforded
23 as a creamy gum (35 mg, 58% yield).
Rf = 0.47 (EtOAc); IR νmax (ATR) 3641, 2929, 1625, 1421, 1150, 831 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH11.99 (1H, br d, J = 2.5 Hz, NH-1), 8.68 (1H, t, J = 5.5 Hz, NH-10), 8.63 (1H, d, J = 2.5 Hz, H-2), 8.07 (1H, d, J = 8.8 Hz, H-4), 7.02 (1H, d, J = 2.3 Hz, H-7), 6.88 (1H, dd, J = 8.8, 2.3 Hz, H-5), 3.79 (3H, s, H3-21), 3.23–3.06 (6H, m, H2-11, H2-13 and H2-15), 1.76–1.63 (2H, m, H2-12), 1.47–1.33 (2H, m, H2-16), 1.37 (9H, s, 3H3-22), 1.25–1.12 (8H, m, H2-17, H2-18, H2-19 and H2-20); 13C NMR (DMSO-d6, 100 MHz) δC181.4 (C-8), 163.0 (C-9), 156.2 (C-6), 154.2 (C-20), 137.2 (C-2), 136.7 (C-7a), 121.4 (C-4), 119.5 (C-3a), 111.8 (C-3), 111.6 (C-5), 95.2 (C-7), 77.8 (C-21), 54.8 (C-21), 45.8 (C-15), 43.9, 43.5 (C-13), 35.8 (C-11), 28.5 (C-18a), 28.4 (C-19a), 28.2 (C-20a), 27.5 (C-24), 27.2 (C-12 and C-16), 25.7 (C-17a); (+)-HRESIMS m/z 939.5161 [M + Na]+ (calcd for C50H72N6NaO10, 939.5202).
3.2.21. Di-tert-butyl Dodecane-1,12-diylbis((3-(2-(7-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)carbamate) (24)
Using general procedure A, 2-(7-methoxy-1
H-indol-3-yl)-2-oxoacetic acid (
12) (45 mg, 0.21 mmol), di-
tert-butyl dodecane-1,12-diylbis((3-aminopropyl)carbamate) [
27,
28] (48 mg, 93 μmol), PyBOP (107 mg, 0.21 mmol) and Et
3N (78 μL, 0.56 mmol) afforded
24 as a yellow oil (48 mg, 58% yield).
Rf = 0.66 (hexane:EtOAc 3:7); IR νmax (ATR) 3233, 2927, 1623, 1420, 1157, 782 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.39 (1H, s, NH-1), 8.71 (1H, br s, NH-10), 8.61 (1H, s, H-2), 7.80 (1H, d, J = 7.9 Hz, H-4), 7.17 (1H, t, J = 7.9 Hz, H-5), 6.84 (1H, d, J = 7.9 Hz, H-6), 3.94 (3H, s, H3-21), 3.22–3.06 (6H, m, H2-11, H2-13 and H2-15), 1.75–1.64 (2H, m, H2-12), 1.47–1.34 (2H, m, H2-16), 1.37 (9H, s, 3H3-24), 1.25–1.10 (8H, m, H2-17, H2-18, H2-19 and H2-20); 13C NMR (DMSO-d6, 100 MHz) δC 182.0 (C-8), 163.4 (C-9), 154.8 (C-22), 146.4 (C-7), 137.3 (C-2), 127.8 (C-3a), 126.1 (C-7a), 123.4 (C-5), 113.8 (C-4), 112.7 (C-3), 104.3 (C-6), 78.2 (C-23), 55.3 (C-21), 46.3 (C-15), 44.4, 44.0 (C-13), 36.3 (C-11), 28.9 (C-18a), 28.9 (C-19a), 28.6 (C-20a), 28.0 (C-24), 27.7 (C-12 and C-16), 26.2 (C-17a); (+)-HRESIMS m/z 917.5369 [M + H]+ (calcd for C50H73N6O10, 917.5383).
3.2.22. N1,N8-Bis(3-(2-(1H-indol-3-yl)-2-oxoacetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (25)
Using general procedure B, reaction of 17 (12 mg, 15 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) followed by purification by C18 reversed-phase column chromatography (30% MeOH/H2O (TFA)) afforded 25 as a yellow oil (12 mg, quant. yield).
Rf = 0.23 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3235, 1669, 1431, 1200, 1130, 721 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 12.29 (1H, s, NH-1), 8.88 (1H, t, J = 6.2 Hz, NH-10), 8.76 (1H, s, H-2), 8.26–8.20 (1H, m, H-4), 7.57–7.51 (1H, m, H-7), 7.30–7.23 (2H, m, H-5 and H-6), 3.30 (2H, t, J = 6.2 Hz, H2-11), 2.98–2.91 (2H, m, H2-13), 2.91–2.84 (2H, m, H2-15), 1.91–1.80 (2H, m, H2-12), 1.61–1.50 (2H, m, H2-16), 1.35–1.21 (4H, m, H2-17 and H2-18); 13C NMR (DMSO-d6, 100 MHz) δC 181.7 (C-8), 163.7 (C-9), 138.3 (C-7a), 136.2 (C-2), 126.3 (C-3a), 123.6 (C-5a), 122.7 (C-6a), 121.3 (C-4), 112.6 (C-7), 112.2 (C-3), 46.7 (C-15), 44.6 (C-13), 35.7 (C-11), 28.4 (C-18), 25.9 (C-12b), 25.7 (C-17b), 22.8 (C-16b); (+)-HRESIMS m/z 601.3488 [M + H]+ (calcd for C34H45N6O4, 601.3497).
3.2.23. N1,N8-Bis(3-(2-(5-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (26)
Using general procedure B, reaction of 18 (27 mg, 31 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) afforded 26 as a brown gum (20 mg, 96% yield) which required no further purification.
Rf = 0.20 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3407, 1674, 1478, 1181, 1025, 723 cm−1; 1H NMR (CD3OD, 400 MHz) δH8.73 (1H, s, H-2), 7.84 (1H, d, J = 2.5 Hz, H-4), 7.38 (1H, d, J = 8.8 Hz, H-7), 6.91 (1H, dd, J = 8.8, 2.5 Hz, H-6), 3.85 (3H, s, H3-19), 3.49–3.43 (2H, t, J = 6.5 Hz, H2-11), 3.08–3.02 (2H, m, H2-13), 3.01–2.95 (2H, m, H2-15), 1.99 (2H, tt, J = 7.1, 6.5 Hz, H2-12), 1.73–1.63 (2H, m, H2-16), 1.44–1.33 (4H, m, H2-17 and H2-18); 13C NMR (CD3OD, 100 MHz) δC 182.0 (C-8), 166.5 (C-9), 158.2 (C-5), 139.6 (C-2), 132.7 (C-7a), 128.9 (C-3a), 114.6 (C-6), 113.9 (C-3 and C-7), 105.1 (C-4), 56.1 (C-19), 48.8 (C-15), 46.4 (C-13), 36.9 (C-11), 29.9 (C-18a), 27.4 (C-12a), 27.4 (C-17a), 27.2 (C-16a); (+)-HRESIMS m/z 661.3690 [M + H]+ (calcd for C36H49N6O6, 661.3708).
3.2.24. N1,N8-Bis(3-(2-(6-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (27)
Using general procedure B, reaction of 19 (11 mg, 13 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) afforded 27 as a yellow oil (5 mg, 59% yield) which required no further purification.
Rf = 0.19 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3395, 1671, 1150, 1199, 1022, 722 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.67 (1H, s, H-2), 8.15 (1H, d, J = 8.8 Hz, H-4), 7.01 (1H, d, J = 2.4 Hz, H-7), 6.90 (1H, dd, J = 8.8, 2.4 Hz, H-6), 3.84 (3H, s, H3-19), 3.45 (2H, t, J = 6.6 Hz, H2-11), 3.05 (2H, t, J = 7.6, H2-13), 3.02–2.96 (2H, m, H2-15), 1.98 (2H, tt, J = 7.6, 6.6 Hz, H2-12), 1.73–1.63 (2H, m, H2-16), 1.45–1.35 (4H, m, H2-17 and H2-18); 13C NMR (CD3OD, 100 MHz) δC 182.0 (C-8), 166.5 (C-9), 159.1 (C-6), 139.0 (C-7a), 138.9 (C-2), 123.6 (C-4), 121.7 (C-3a), 114.1 (C-3), 113.5 (C-5), 96.5 (C-7), 56.0 (C-19), 49.2 (C-15), 46.5 (C-13), 36.9 (C-11), 30.0 (C-18), 27.5 (C-12a), 27.5 (C-17a), 27.3 (C-16a); (+)-HRESIMS m/z 661.3687 [M + H]+ (calcd for C36H49N6O6, 661.3708).
3.2.25. N1,N8-Bis(3-(2-(7-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (28)
Using general procedure B, reaction of 20 (20 mg, 13 μmol) in CH2Cl2 (1.8 mL) with TFA (0.2 mL) afforded 28 as a yellow oil (12 mg, quant. yield) which required no further purification.
Rf = 0.26 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3337, 2941, 1622, 1432, 1132, 721 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.70 (1H, br d, J = 1.0 Hz, H-2), 7.86 (1H, d, J = 8.2 Hz, H-4), 7.18 (1H, t, J = 8.2 Hz, H-5), 6.81 (1H, d, J = 8.2 Hz, H-6), 3.97 (3H, s, H3-19), 3.45 (2H, t, J = 6.5 Hz, H2-11), 3.04 (2H, t, J = 7.1 Hz, H2-13), 2.97 (2H, t, J = 8.0 Hz, H2-15), 1.98 (2H, tt, J = 7.1, 6.5 Hz, H2-12), 1.72–1.62 (2H, m, H2-16), 1.37–1.23 (4H, m, H2-17 and H2-18); 13C NMR (CD3OD, 100 MHz) δC182.2 (C-8), 166.4 (C-9), 148.1 (C-7), 138.6 (C-2), 129.5 (C-3a), 128.0 (C-7a), 124.8 (C-5), 115.4 (C-4), 114.4 (C-3), 105.3 (C-6), 56.0 (C-19), 49.2 (C-15), 46.4 (C-13), 36.9 (C-11), 29.9 (C-18a), 27.4 (C-12a), 27.4 (C-17a), 27.2 (C-16a); (+)-HRESIMS m/z 661.3695 [M + H]+ (calcd for C36H49N6O6, 661.3708).
3.2.26. N1,N12-Bis(3-(2-(1H-indol-3-yl)-2-oxoacetamido)propyl)dodecane-1,12-diaminium 2,2,2-trifluoroacetate (29)
Using general procedure B, reaction of 21 (14 mg, 16 μmol) in CH2Cl2 (1.8 mL) with TFA (0.2 mL) afforded 29 as a white gum (5 mg, 47% yield) which required no further purification.
Rf = 0.26 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3391, 2949, 1675, 1434, 1132, 1034, 722 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.80 (1H, d, J = 1.7 Hz, H-2), 8.34–8.28 (1H, m, H-4), 7.52–7.46 (1H, m, H-7), 7.31–7.23 (2H, m, H-5 and H-6), 3.51–3.42 (2H, m, H2-11), 3.11–3.03 (2H, m, H2-13), 3.03–2.95 (2H, m, H2-15), 2.05–1.93 (2H, m, H2-12), 1.74–1.62 (2H, m, H2-16), 1.44–1.23 (8H, m, H2-17 to H2-20); 13C NMR (CD3OD, 100 MHz) δC 182.0 (C-8), 166.4 (C-9), 139.6 (C-2), 138.0 (C-7a), 127.9 (C-3a), 124.9 (C-5a), 123.9 (C-6a), 123.0 (C-4), 114.0 (C-7), 113.2 (C-3), 48.6 (C-15), 46.4 (C-13), 36.9 (C-11), 30.6 (C-18b), 30.5 (C-19b), 30.2 (C-20b), 27.5 (C-12b), 27.5 (C-17b), 27.3 (C-16b); (+)-HRESIMS m/z 329.2098 [M + 2H]2+ (calcd for C38H54N6O4, 329.2098).
3.2.27. N1,N12-Bis(3-(2-(5-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)dodecane-1,12-diaminium 2,2,2-trifluoroacetate (30)
Using general procedure B, reaction of 22 (11 mg, 12 μmol) in CH2Cl2 (1.8 mL) with TFA (0.2 mL) afforded 30 as a yellow gum (8 mg, 90% yield) which required no further purification.
Rf = 0.29 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3033, 2930, 1670, 1618, 1434, 1178, 1130, 721 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.74 (1H, s, H-2), 7.85 (1H, d, J = 2.3 Hz, H-4), 7.38 (1H, d, J = 8.8 Hz, H-7), 6.91 (1H, dd, J = 8.8, 2.3 Hz, H-6), 3.85 (3H, s, H3-21), 3.46 (2H, t, J = 6.4 Hz, H2-11), 3.06 (2H, t, J = 7.2 Hz, H2-13), 3.00 (2H, t, J = 7.6 Hz, H2-15), 1.99 (2H, tt, J = 7.2, 6.4 Hz, H2-12), 1.74–1.63 (2H, m, H2-16), 1.43–1.25 (8H, m, H2-17 to H2-20); 13C NMR (CD3OD, 100 MHz) δC 181.9 (C-8), 166.5 (C-9), 158.2 (C-5), 139.6 (C-2), 132.7 (C-7a), 128.9 (C-3a), 114.6 (C-6), 113.9 (C-3 and C-7), 105.1 (C-4), 56.1 (C-21), 49.0 (C-15), 46.4 (C-13), 36.9 (C-11), 30.6 (C-18a), 30.5 (C-19a), 30.2 (C-20a), 27.5 (C-12b) 27.5 (C-17b), 27.3 (C-16); (+)-HRESIMS m/z 717.4304 [M + H]+ (calcd for C40H57N6O6, 717.4334).
3.2.28. N1,N12-Bis(3-(2-(6-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)dodecane-1,12-diaminium 2,2,2-trifluoroacetate (31)
Using general procedure B, reaction of 23 (14 mg, 16 μmol) in CH2Cl2 (1.8 mL) with TFA (0.2 mL) afforded 31 as a yellow gum (16 mg, quant. yield) which required no further purification.
Rf = 0.31 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3346, 1626, 1449, 1153, 518 cm−1; 1H NMR (CD3OD, 400 MHz) δH8.70 (1H, s, H-2), 8.17 (1H, d, J = 8.7 Hz, H-4), 7.05 (1H, d, J = 2.3 Hz, H-7), 6.93 (1H, dd, J = 8.7, 2.3 Hz, H-6), 3.87 (3H, s, H3-21), 3.49 (2H, t, J = 6.6 Hz, H2-11), 3.08 (2H, t, J = 7.5, H2-13), 3.02 (2H, t, J = 7.6 Hz, H2-15), 2.02 (2H, tt, J = 7.5, 6.6 Hz, H2-12), 1.76–1.66 (2H, m, H2-16), 1.47–1.23 (4H, m, H2-17 to H2-20); 13C NMR (CD3OD, 100 MHz) δC 182.0 (C-8), 166.4 (C-9), 159.0 (C-6), 139.2 (C-7a), 139.0 (C-2), 123.5 (C-6), 121.7 (C-3a), 114.0 (C-3), 113.5 (C-5), 96.6 (C-7), 56.1 (C-21), 49.0 (C-15), 46.4 (C-13), 36.9 (C-11), 30.6 (C-18a), 30.4 (C-19a), 30.2 (C-20a), 27.5 (C-12a), 27.4 (C-17a), 27.2 (C-16a); (+)-HRESIMS m/z 717.4327 [M + H]+ (calcd for C40H57N6O6, 717.4334).
3.2.29. N1,N12-Bis(3-(2-(7-methoxy-1H-indol-3-yl)-2-oxoacetamido)propyl)dodecane-1,12-diaminium 2,2,2-trifluoroacetate (32)
Using general procedure B, reaction of 24 (8 mg, 9.0 μmol) in CH2Cl2 (1.8 mL) with TFA (0.2 mL) afforded 32 as a yellow oil (5 mg, 77% yield) which required no further purification.
Rf = 0.43 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3408, 1670, 1623, 1432, 1135, 737 cm−1; 1H NMR (CD3OD, 400 MHz) δH 8.71 (1H, s, H-2), 7.87 (1H, d, J = 7.9 Hz, H-4), 7.19 (1H, t, J = 7.9 Hz, H-5), 6.82 (1H, d, J = 7.9 Hz, H-6), 3.98 (3H, s, H3-21), 3.46 (2H, t, J = 6.2 Hz, H2-11), 3.05 (2H, t, J = 7.9 Hz, H2-13), 2.99 (2H, t, J = 8.4 Hz, H2-15), 2.02–1.93 (2H, m, H2-12), 1.73–1.63 (2H, m, H2-16), 1.43–1.26 (8H, m, H2-17, H2-18, H2-19 and H2-18); 13C NMR (CD3OD, 100 MHz) δC 182.1 (C-8), 166.4 (C-9), 148.1 (C-7), 138.6 (C-2), 129.5 (C-3a), 128.0 (C-7a), 124.8 (C-5), 115.4 (C-4), 114.4 (C-3), 105.3 (C-6), 56.0 (C-21), 47.9 (C-15), 46.4 (C-13), 36.9 (C-11), 30.6 (C-18a), 30.5 (C-19a), 30.2 (C-20a), 27.5 (C-12a), 27.5 (C-17a), 27.3 (C-16a); (+)-HRESIMS m/z 717.4326 [M + H]+ (calcd for C40H57N6O6, 717.4334).
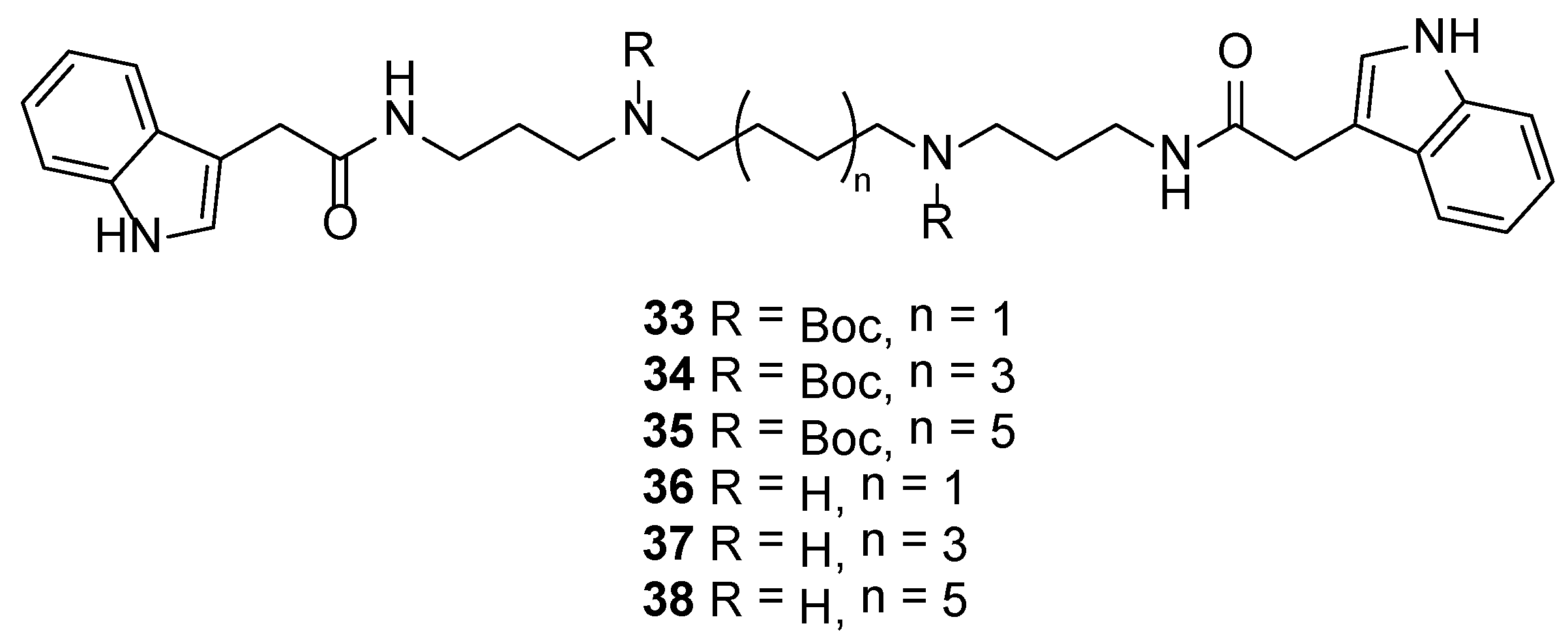
3.2.30. Di-tert-butyl Butane-1,4-diylbis((3-(2-(1H-indol-3-yl)acetamido)propyl)carbamate) (33)
Using general procedure A, 2-(1
H-indol-3-yl)acetic acid [
26] (40 mg, 0.23 mmol), di-
tert-butyl butane-1,4-diylbis((3-aminopropyl)carbamate) [
25,
27] (42 mg, 0.10 mmol), PyBOP (119 mg, 0.23 mmol) and Et
3N (87 μL, 0.63 mmol) afforded
33 as a yellow oil (29 mg, 39% yield).
Rf = 0.14 (EtOAc); IR νmax (ATR) 3320, 2942, 1660, 1421, 1162, 1025, 742 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH10.84 (1H, s, NH-1), 7.83 (1H, t, J = 5.6 Hz, NH-10), 7.54 (1H, d, J = 8.1 Hz, H-4), 7.33 (1H, d, J = 8.3 Hz, H-7), 7.17 (1H, d, J = 2.3 Hz, H-2), 7.05 (1H, ddd, J = 8.6, 8.3, 1.0 Hz, H-6), 6.95 (1H, ddd, J = 8.6, 8.1, 1.0 Hz, H-5), 3.48 (2H, s, H2-8), 3.13–2.96 (6H, m, H2-11, H2-13 and H2-15), 1.64–1.51 (2H, m, H2-12), 1.36 (9H, s, 3H3-19), 1.33–1.27 (2H, m, H2-16); 13C NMR (DMSO-d6, 100 MHz) δC 170.6 (C-9), 154.6 (C-17), 136.1 (C-7a), 127.2 (C-3a), 123.7 (C-2), 120.9 (C-6), 118.6 (C-4), 118.2 (C-5), 111.3 (C-7), 108.9 (C-3), 78.2 (C-18), 46.5, 46.1 (C-15), 44.6, 44.4 (C-13), 36.4 (C-11), 32.8 (C-8), 28.8 (C-12), 28.0 (C-19), 25.6, 25.1 (C-16); (+)-HRESIMS m/z 717.4310 [M + H]+ (calcd for C40H57N6O6, 717.4334).
3.2.31. Di-tert-butyl Octane-1,8-diylbis((3-(2-(1H-indol-3-yl)acetamido)propyl)carbamate) (34)
To a stirred solution of 2-(1
H-indol-3-yl)acetic acid [
26] (51 mg, 0.29 mmol), DIPEA (68 μL, 0.41 mmol) in DMF (1 mL) was added HATU (110 mg, 0.29 mmol). The reaction mixture was stirred under N
2 at r.t. for 80 min, followed by the addition of di-
tert-butyl octane-1,8-diylbis((3-aminopropyl)carbamate) [
25] (63 mg, 0.14 mmol). The reaction mixture was further stirred for 22 h and then partitioned between H
2O (30 mL) and CH
2Cl
2 (3 × 40 mL). The combined organic extracts were washed with brine (20 mL) and dried over MgSO
4 and concentrated
in vacuo. Purification by silica gel flash column chromatography (hexanes/EtOAc 1:1 to EtOAc/MeOH 4:1) afforded
34 as a yellow gum (79 mg, 35% yield).
Rf = 0.46 (EtOAc); IR νmax (ATR) 3283, 2930, 1658, 1419, 1156, 740 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 10.84 (1H, s, NH-1), 7.82 (1H, t, J = 5.6 Hz, NH-10), 7.53 (1H, d, J = 7.9 Hz, H-4), 7.33 (1H, m, H-7), 7.17 (1H, d, J = 2.1 Hz, H-2), 7.05 (1H, ddd, J = 8.1, 8.0, 1.0 Hz, H-6), 6.95 (1H, ddd, J = 8.1, 7.9, 1.0 Hz, H-5), 3.48 (2H, s, H2-8), 3.13–2.96 (6H, m, H2-11, H2-13 and H2-15), 1.63–1.52 (2H, m, H2-12), 1.44–1.33 (2H, m, H2-16), 1.36 (9H, s, 3H3-21), 1.26–1.19 (2H, m, H2-18), 1.17–1.11 (2H, m, H2-17); 13C NMR (DMSO-d6, 100 MHz) δC 170.6 (C-9), 154.6 (C-19), 136.1 (C-7a), 127.2 (C-3a), 123.7 (C-2), 120.8 (C-6), 118.6 (C-4), 118.2 (C-5), 111.3 (C-7), 108.9 (C-3), 78.2 (C-20), 46.4 (C-15), 44.6, 44.2 (C-13), 36.4 (C-11), 32.8 (C-8), 28.7 (C-18), 28.0 (C-21), 27.8 (C-16 and C-12), 26.1 (C-17); (+)-HRESIMS m/z 773.4937 [M + H]+ (calcd for C44H65N6O6, 773.4960).
3.2.32. Di-tert-butyl Dodecane-1,12-diylbis((3-(2-(1H-indol-3-yl)acetamido)propyl)carbamate) (35)
Using general procedure A, 2-(1
H-indol-3-yl)acetic acid [
26] (58 mg, 0.33 mmol), di-
tert-butyl dodecane-1,12-diylbis((3-aminopropyl)carbamate) [
27,
28] (78 mg, 0.15 mmol), PyBOP (174 mg, 0.33 mmol) and Et
3N (126 μL, 0.91 mmol) afforded
35 as a yellow oil (55 mg, 44% yield).
Rf = 0.60 (EtOAc); IR νmax (ATR) 3279, 2925, 1659, 1417, 1155, 740 cm−1; 1H NMR (DMSO-d6, 400 MHz) δH 10.84 (1H, s, NH-1), 7.83 (1H, t, J = 5.5 Hz, NH-10), 7.54 (1H, d, J = 8.0 Hz, H-4), 7.33 (1H, d, J = 8.1 Hz, H-7), 7.17 (1H, d, J = 1.8 Hz, H-2), 7.05 (1H, t, J = 8.1 Hz, H-6), 6.95 (1H, t, J = 8.0 Hz, H-5), 3.48 (2H, s, H2-8), 3.14–2.96 (6H, m, H2-11, H2-13 and H2-15), 1.63–1.52 (2H, m, H2-12), 1.44–1.32 (2H, m, H2-16), 1.36 (9H, s, 3H3-23), 1.27–1.20 (6H, m, H2-18 to H2-20), 1.19–1.11 (2H, m, H2-17); 13C NMR (DMSO-d6, 100 MHz) δC 170.6 (C-9), 154.6 (C-21), 136.1 (C-7a), 127.2 (C-3a), 123.7 (C-2), 120.9 (C-6), 118.6 (C-4), 118.2 (C-5), 111.3 (C-7), 108.9 (C-3), 78.2 (C-22), 46.5 (C-15), 44.5, 44.2 (C-13), 36.4 (C-11), 32.8 (C-8), 29.0 (C-18a), 28.9 (C-19a), 28.7 (C-20a), 28.0 (C-23), 27.8 (C-12 and C-16), 26.2 (C-17); (+)-HRESIMS m/z 851.5418 [M + Na]+ (calcd for C48H72N6NaO6, 851.5406).
3.2.33. N1,N4-Bis(3-(2-(1H-indol-3-yl)acetamido)propyl)butane-1,4-diaminium 2,2,2-trifluoroacetate (36)
Using general procedure B, reaction of 33 (10 mg, 14 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) and subsequent purification by C18 reversed-phase column chromatography (30% MeOH/H2O (TFA)) afforded 36 as a red oil (6 mg, 83% yield).
Rf = 0.09 (CH2Cl2:MeOH:TEA 1:1:0.01); IR νmax (ATR) 3284, 1672, 1551, 1456, 1340, 1180, 721 cm−1; 1H NMR (DMSO-d6, 300 MHz) δH 10.89 (1H, s, NH-1), 8.40 (2H, br s, NH2-14), 8.07 (1H, t, J = 6.2 Hz, NH-10), 7.54 (1H, d, J = 8.1 Hz, H-4), 7.35 (1H, ddd, J = 8.0, 0.9, 0.7 Hz, H-7), 7.19 (1H, d, J = 2.2 Hz, H-2) 7.07 (1H, ddd, J = 8.0, 8.0, 1.2 Hz, H-6), 6.97 (1H, ddd, J = 8.1, 8.0, 0.9 Hz, H-5), 3.52 (2H, s, H2-8), 3.13 (2H, td, J = 6.9, 6.2 Hz, H2-11), 2.89–2.70 (4H, m, H2-13 and H2-15), 1.78–1.65 (2H, m, H2-12), 1.60–1.46 (2H, m, H2-16); 13C NMR (DMSO-d6, 75 MHz) δC 171.5 (C-9), 136.1 (C-7a), 127.1 (C-3a), 123.9 (C-2), 121.0 (C-6), 118.5 (C-5), 118.3 (C-4), 111.4 (C-7), 108.6 (C-3), 46.1 (C-15a), 44.5 (C-13a), 35.7 (C-11), 32.7 (C-8), 26.2 (C-12), 22.7 (C-16); (+)-HRESIMS m/z 517.3277 [M + H]+ (calcd for C30H41N6O2, 517.3286).
3.2.34. N1,N8-Bis(3-(2-(1H-indol-3-yl)acetamido)propyl)octane-1,8-diaminium 2,2,2-trifluoroacetate (37)
Using general procedure B, reaction of 34 (9 mg, 12 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) followed by purification by LH20 column chromatography (MeOH) afforded 37 as a brown oil (6 mg, 90% yield).
Rf = 0.46 (EtOAc); IR νmax (ATR) 3277, 2940, 1672, 1132, 1023, 721 cm−1; 1H NMR (CD3OD, 400 MHz) δH7.57 (1H, d, J = 8.0 Hz, H-4), 7.37 (1H, d, J = 8.2 Hz, H-7), 7.21 (1H, s, H-2), 7.12 (1H, ddd, J = 8.2, 8.2, 1.0 Hz, H-6), 7.03 (1H, ddd, J = 8.2, 8.0, 1.0 Hz, H-5), 3.69 (2H, s, H2-8), 3.31–3.27 (2H, m, H2-11), 2.78 (2H, t, J = 6.8 Hz, H2-13), 2.75–2.70 (2H, m, H2-15), 1.79 (2H, tt, J = 6.8, 6.8 Hz, H2-12), 1.60–1.50 (2H, m, H2-16), 1.43–1.27 (4H, m, H2-17 and H2-18); 13C NMR (CD3OD, 100 MHz) δC 176.5 (C-9), 138.2 (C-7a), 128.4 (C-3a), 125.2 (C-2), 122.7 (C-6), 120.1 (C-5), 119.3 (C-4), 112.6 (C-7), 109.4 (C-3), 48.8 (C-15), 46.0 (C-13), 36.7 (C-11), 34.0 (C-8), 29.9 (C-18), 27.6 (C-12), 27.3 (C-17a), 27.1 (C-16a); (+)-HRESIMS m/z 573.3899 [M + H]+ (calcd for C34H49N6O2, 573.3912).
3.2.35. N1,N12-Bis(3-(2-(1H-indol-3-yl)acetamido)propyl)dodecane-1,12-diaminium 2,2,2-trifluoroacetate (38)
Using general procedure B, reaction of 35 (10 mg, 12 μmol) in CH2Cl2 (1.7 mL) with TFA (0.3 mL) followed by purification using LH20 column chromatography to afford 38 as a pink oil (8 mg, 92% yield).
Rf = 0.20 (CH2Cl2:MeOH:TEA 4:1:0.01); IR νmax (ATR) 3319, 2929, 1672, 1433, 1133, 721 cm−1; 1H NMR (CD3OD, 400 MHz) δH 7.59–7.56 (1H, m, H-4), 7.39–7.35 (1H, m, H-7), 7.21 (1H, s, H-2), 7.12 (1H, ddd, J = 8.3, 8.0, 1.2 Hz, H-6), 7.03 (1H, ddd, J = 8.3, 8.0, 1.0 Hz, H-5), 3.69 (2H, s, H2-8), 3.31–3.26 (2H, m, H2-11), 2.77 (2H, t, J = 7.1 Hz, H2-13), 2.74–2.68 (2H, m, H2-15), 1.78 (2H, tt, J = 7.1, 6.8 Hz, H2-12), 1.59–1.49 (2H, m, H2-16), 1.36–1.29 (8H, m, H2-17 to H2-20); 13C NMR (CD3OD, 100 MHz) δC 176.4 (C-9), 138.2 (C-7a), 128.4 (C-3a), 125.2 (C-2), 122.7 (C-6), 120.1 (C-5), 119.3 (C-4), 112.6 (C-7), 109.4 (C-3), 48.8 (C-15), 46.0 (C-13), 36.7 (C-11), 34.0 (C-8), 30.6 (C-18a), 30.5 (C-19a), 30.2 (C-20a), 27.6 (C-12), 27.5 (C-17a), 27.2 (C-16); (+)-HRESIMS m/z 629.4553 [M + H]+ (calcd for C38H57N6O2, 629.4538).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}