Astaxanthin Alleviates Early Brain Injury Following Subarachnoid Hemorrhage in Rats: Possible Involvement of Akt/Bad Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. General Observation
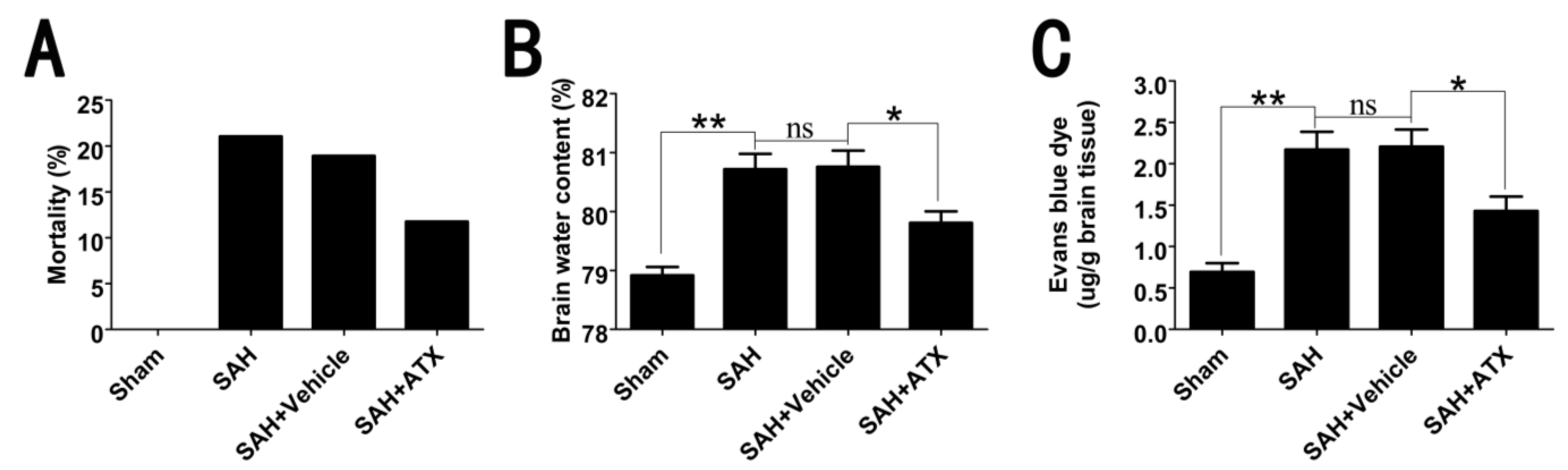
2.2. Mortality, Brain Water Content and BBB Permeability

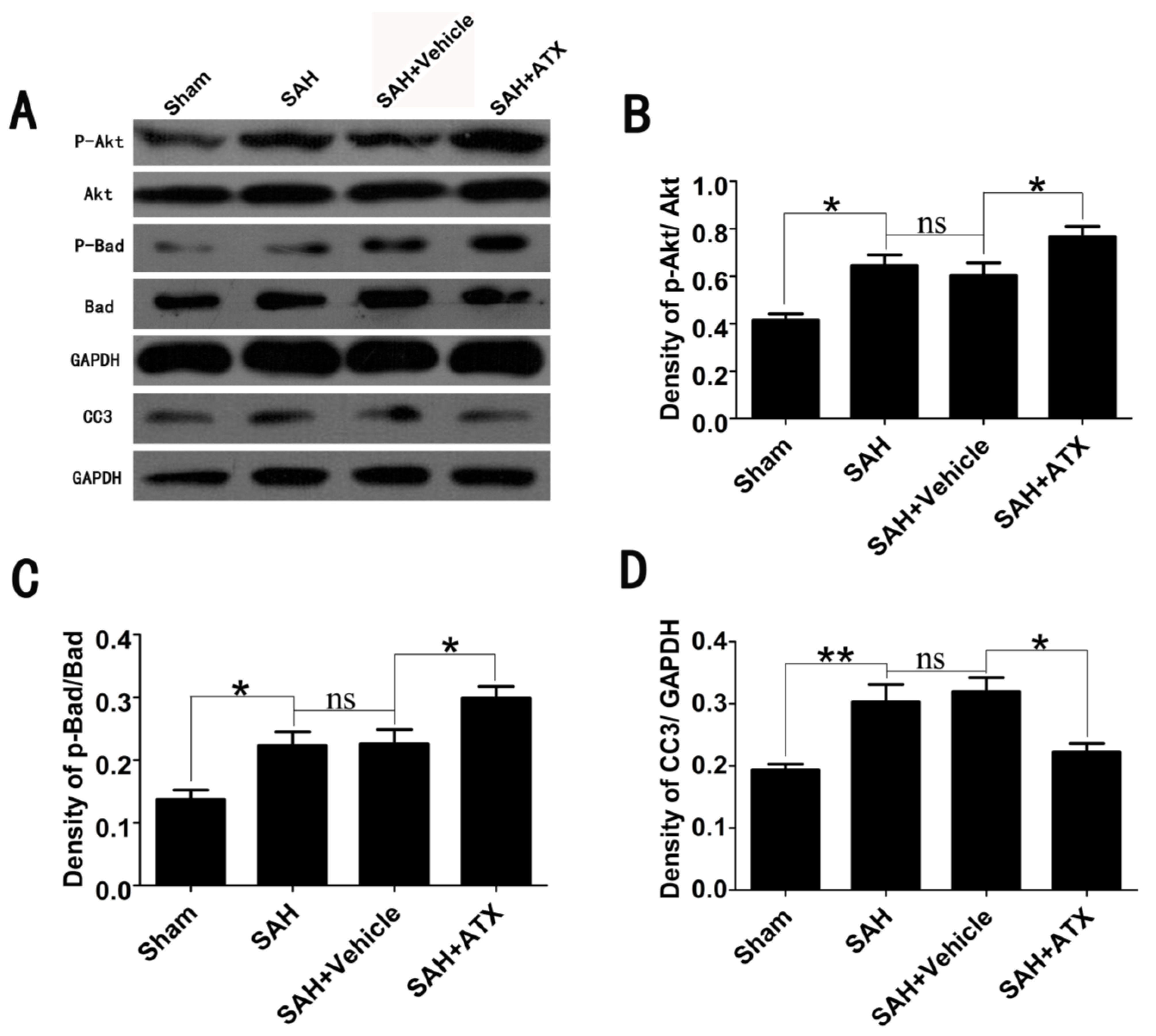
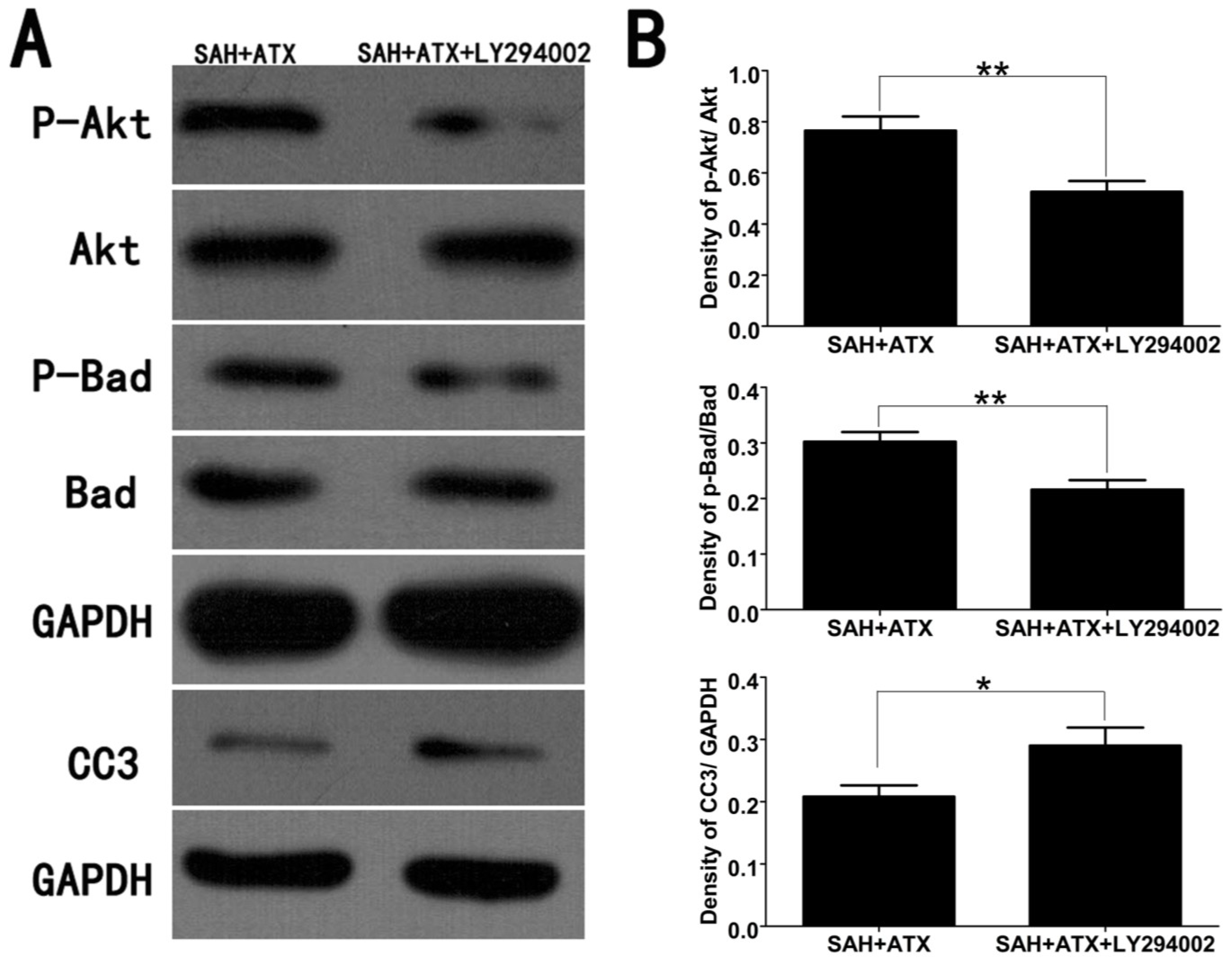
2.3. Effects of ATX on p-Akt, p-Bad and Caspase-3 Expression

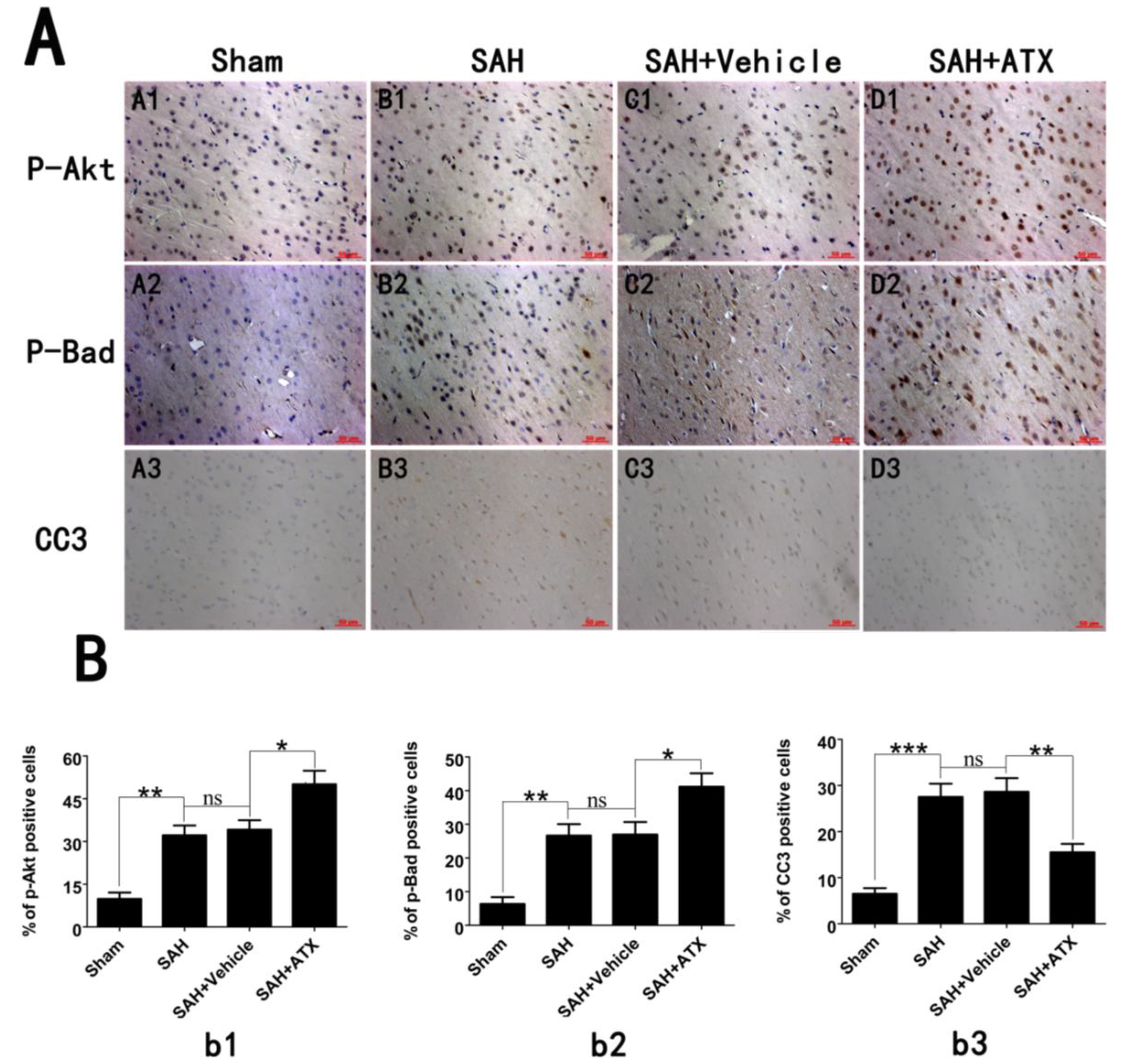
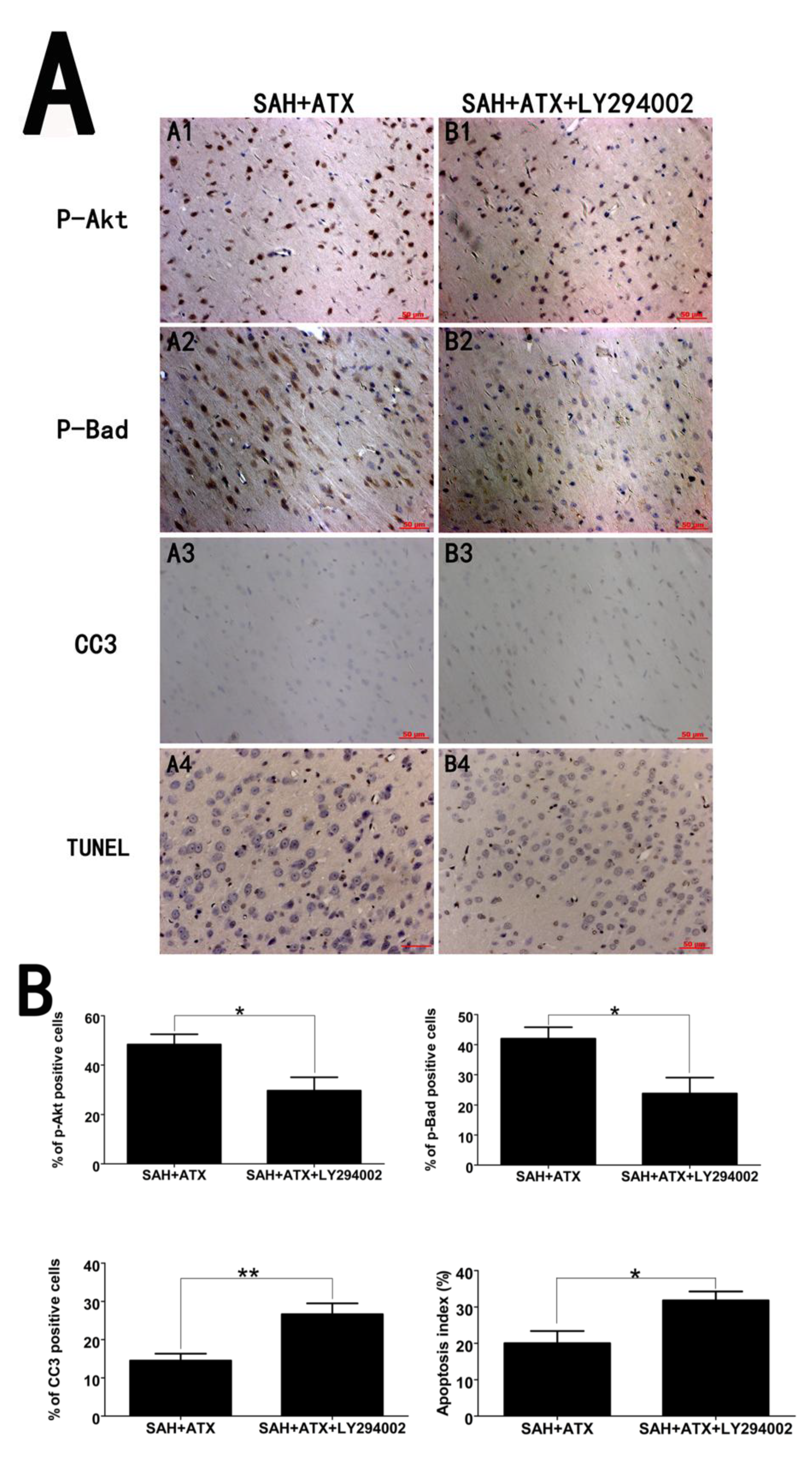
2.4. Effects of ATX on p-Akt, p-Bad and Caspase-3 Distribution

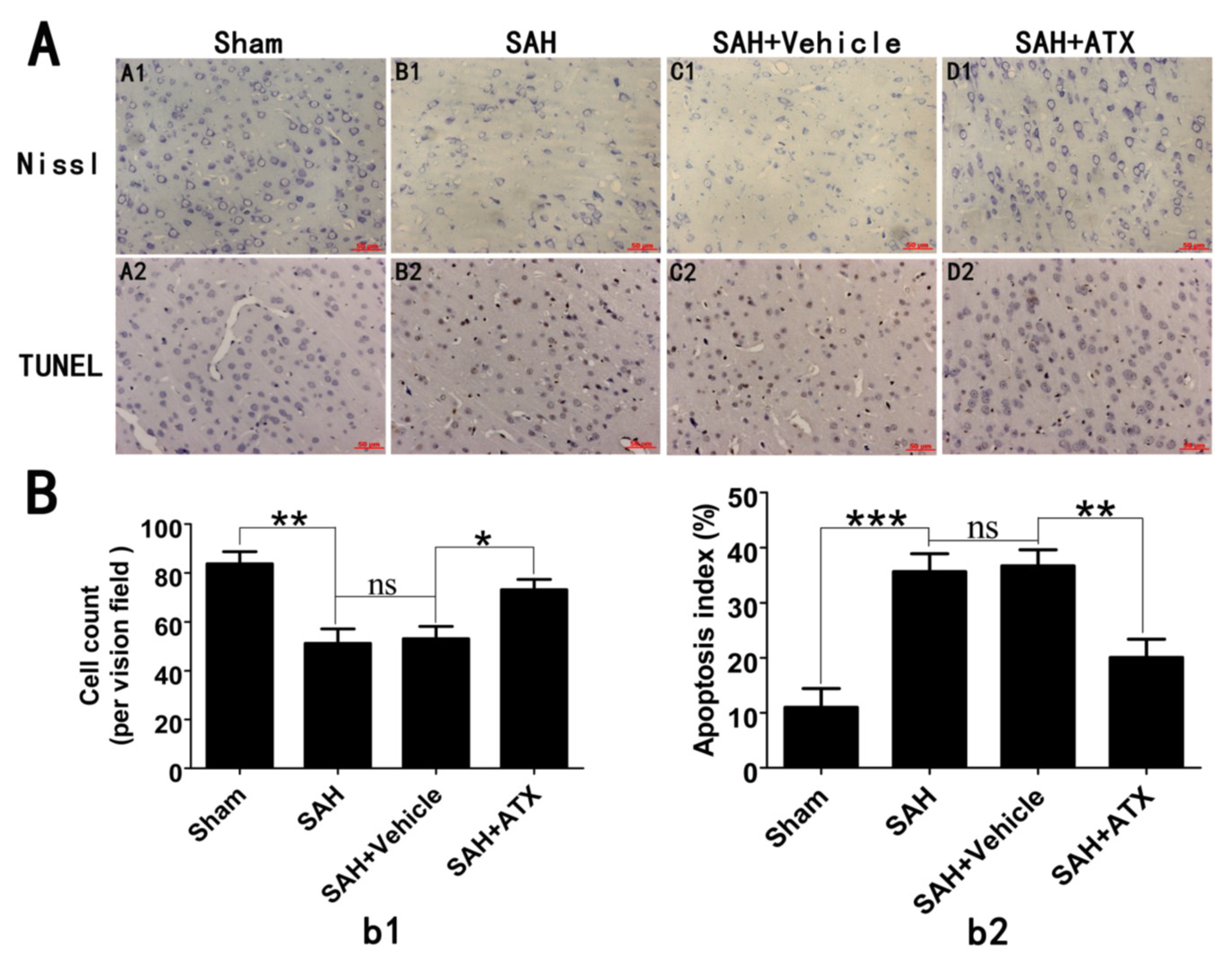
2.5. Effects of ATX on Neural Survival and Cell Apoptosis at 24 h after SAH
2.6. LY294002 Abolished the Protective Effects of ATX at 24 h after SAH


2.7. Influence of ATX on Neuronal Survival and Neurological Function within 72 h after SAH


3. Discussion

4. Experimental Section
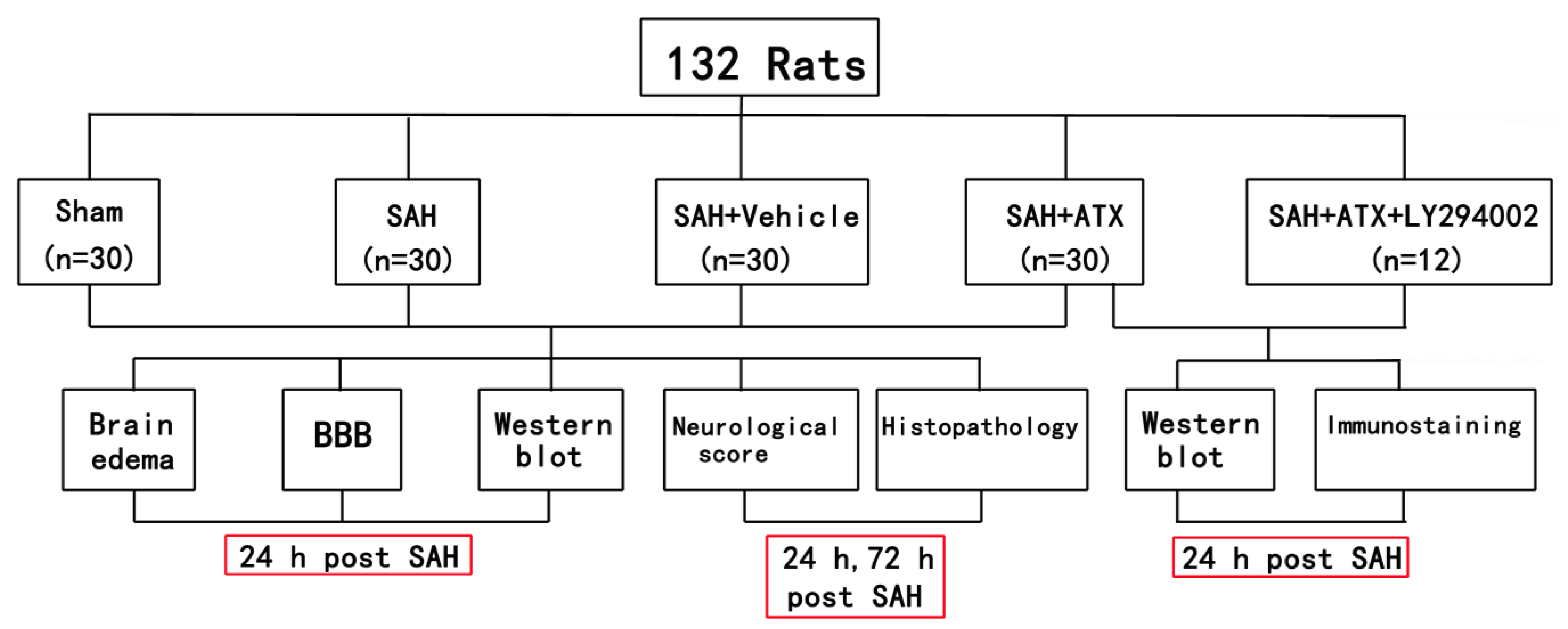
4.1. Animals
4.2. Experimental Groups

4.3. Pre-Chiasmatic Cistern SAH Model

4.4. Brain Water Content
4.5. Blood-Brain Barrier (BBB) Permeability
4.6. Western Blot Analysis
4.7. Immunohistochemistry
4.8. Nissl Staining
4.9. TUNEL Staining
4.10. Cell Counting
4.11. Neurological Scores
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.; Calvert, J.W.; Zhang, J.H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2006, 26, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Haase, S.; Bunkowski, S.; Bruck, W. Neuronal apoptosis in the dentate gyrus in humans with subarachnoid hemorrhage and cerebral hypoxia. Brain Pathol. 2002, 12, 329–336. [Google Scholar] [PubMed]
- Park, S.; Yamaguchi, M.; Zhou, C.; Calvert, J.W.; Tang, J.; Zhang, J.H. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke 2004, 35, 2412–2417. [Google Scholar] [CrossRef] [PubMed]
- Duris, K.; Manaenko, A.; Suzuki, H.; Rolland, W.B.; Krafft, P.R.; Zhang, J.H. alpha7 nicotinic acetylcholine receptor agonist PNU-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke 2011, 42, 3530–3536. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [PubMed]
- Springer, J.E.; Azbill, R.D.; Nottingham, S.A.; Kennedy, S.E. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J. Neurosci. 2000, 20, 7246–7251. [Google Scholar] [PubMed]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001, 21, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Akt phosphorylation and neuronal survival after traumatic brain injury in mice. Neurobiol. Dis. 2002, 9, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Sugawara, T.; Maier, C.M.; Hsieh, L.B.; Chan, P.H. Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol. Dis. 2005, 20, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Yu, F.; Chan, P.H. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke 2006, 37, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Fassett, R.G.; Coombes, J.S. Astaxanthin, oxidative stress, inflammation and cardiovascular disease. Future Cardiol. 2009, 5, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Kuo, C.C.; Chou, J.; Delvolve, A.; Jackson, S.N.; Post, J.; Woods, A.S.; Hoffer, B.J.; Wang, Y.; Harvey, B.K. Astaxanthin reduces ischemic brain injury in adult rats. FASEB J. 2009, 23, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, Y.J.; Kwon, K.H. Neuroprotective Effects of Astaxanthin in Oxygen-Glucose Deprivation in SH-SY5Y Cells and Global Cerebral Ischemia in Rat. J. Clin. Biochem. Nutr. 2010, 47, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Nakamura, M.; Zhao, Q.; Iguchi, T.; Goto, H.; Sankawa, U.; Watanabe, H. Antihypertensive and neuroprotective effects of astaxanthin in experimental animals. Biol. Pharm. Bull. 2005, 28, 47–52. [Google Scholar] [PubMed]
- Kim, J.H.; Choi, W.; Lee, J.H.; Jeon, S.J.; Choi, Y.H.; Kim, B.W.; Chang, H.I.; Nam, S.W. Astaxanthin inhibits H2O2-mediated apoptotic cell death in mouse neural progenitor cells via modulation of P38 and MEK signaling pathways. J. Microbiol. Biotechnol. 2009, 19, 1355–1363. [Google Scholar] [PubMed]
- Nagendraprabhu, P.; Sudhandiran, G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Investig. New Drugs 2011, 29, 207–224. [Google Scholar] [CrossRef]
- Arunkumar, E.; Bhuvaneswari, S.; Anuradha, C.V. An intervention study in obese mice with astaxanthin, a marine carotenoid—Effects on insulin signaling and pro-inflammatory cytokines. Food Funct. 2012, 3, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, K.; Kowshik, J.; Kishore, T.K.; Baba, A.B.; Nagini, S. Astaxanthin inhibits NF-kappaB and Wnt/beta-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochim. Biophys. Acta 2013, 1830, 4433–4444. [Google Scholar]
- Dong, L.Y.; Jin, J.; Lu, G.; Kang, X.L. Astaxanthin attenuates the apoptosis of retinal ganglion cells in db/db mice by inhibition of oxidative stress. Mar. Drugs 2013, 11, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Shao, A.; Wang, J.; Chen, S.; Wu, H.; McBride, D.W.; Wu, Q.; Sun, X.; Zhang, J. Neuroprotective effect of hydrogen-rich saline against neurologic damage and apoptosis in early brain injury following subarachnoid hemorrhage: Possible role of the Akt/GSK3beta signaling pathway. PLoS One 2014, 9, e96212. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.N.; Jena, G.B. Astaxanthin intervention ameliorates cyclophosphamide-induced oxidative stress, DNA damage and early hepatocarcinogenesis in rat: Role of Nrf2, p53, p38 and phase-II enzymes. Mutat. Res. 2010, 696, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Lu, C.W.; Wang, S.J. Astaxanthin inhibits glutamate release in rat cerebral cortex nerve terminals via suppression of voltage-dependent Ca(2+) entry and mitogen-activated protein kinase signaling pathway. J. Agric. Food Chem. 2010, 58, 8271–8278. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Bai, S.K.; Lee, K.S.; Namkoong, S.; Na, H.J.; Ha, K.S.; Han, J.A.; Yim, S.V.; Chang, K.; Kwon, Y.G.; et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol. Cells 2003, 16, 97–105. [Google Scholar]
- Zhang, X.S.; Zhang, X.; Zhou, M.L.; Zhou, X.M.; Li, N.; Li, W.; Cong, Z.X.; Sun, Q.; Zhuang, Z.; Wang, C.X.; et al. Amelioration of oxidative stress and protection against early brain injury by astaxanthin after experimental subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 42–54. [Google Scholar]
- Lang, F.; Stournaras, C. Serum and glucocorticoid inducible kinase, metabolic syndrome, inflammation, and tumor growth. Hormones (Athens) 2013, 12, 160–171. [Google Scholar] [CrossRef]
- Krafft, P.R.; Altay, O.; Rolland, W.B.; Duris, K.; Lekic, T.; Tang, J.; Zhang, J.H. Alpha7 nicotinic acetylcholine receptor agonism confers neuroprotection through GSK-3beta inhibition in a mouse model of intracerebral hemorrhage. Stroke 2012, 43, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shi, X.Y.; Yin, J.; Zuo, G.; Zhang, J.; Chen, G. Role of autophagy in early brain injury after experimental subarachnoid hemorrhage. J. Mol. Neurosci. 2012, 46, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Zhou, M.L.; You, W.C.; Zhu, L.; Ma, C.Y.; Sun, X.J.; Shi, J.X. Hydrogen-rich saline alleviates early brain injury via reducing oxidative stress and brain edema following experimental subarachnoid hemorrhage in rabbits. BMC Neurosci. 2012, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995, 26, 627–634; discussion 635. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.-S.; Zhang, X.; Wu, Q.; Li, W.; Zhang, Q.-R.; Wang, C.-X.; Zhou, X.-M.; Li, H.; Shi, J.-X.; Zhou, M.-L. Astaxanthin Alleviates Early Brain Injury Following Subarachnoid Hemorrhage in Rats: Possible Involvement of Akt/Bad Signaling. Mar. Drugs 2014, 12, 4291-4310. https://doi.org/10.3390/md12084291
Zhang X-S, Zhang X, Wu Q, Li W, Zhang Q-R, Wang C-X, Zhou X-M, Li H, Shi J-X, Zhou M-L. Astaxanthin Alleviates Early Brain Injury Following Subarachnoid Hemorrhage in Rats: Possible Involvement of Akt/Bad Signaling. Marine Drugs. 2014; 12(8):4291-4310. https://doi.org/10.3390/md12084291
Chicago/Turabian StyleZhang, Xiang-Sheng, Xin Zhang, Qi Wu, Wei Li, Qing-Rong Zhang, Chun-Xi Wang, Xiao-Ming Zhou, Hua Li, Ji-Xin Shi, and Meng-Liang Zhou. 2014. "Astaxanthin Alleviates Early Brain Injury Following Subarachnoid Hemorrhage in Rats: Possible Involvement of Akt/Bad Signaling" Marine Drugs 12, no. 8: 4291-4310. https://doi.org/10.3390/md12084291