Marinopyrrole Derivatives with Sulfide Spacers as Selective Disruptors of Mcl-1 Binding to Pro-Apoptotic Protein Bim

Abstract
:1. Introduction
2. Results and Discussion
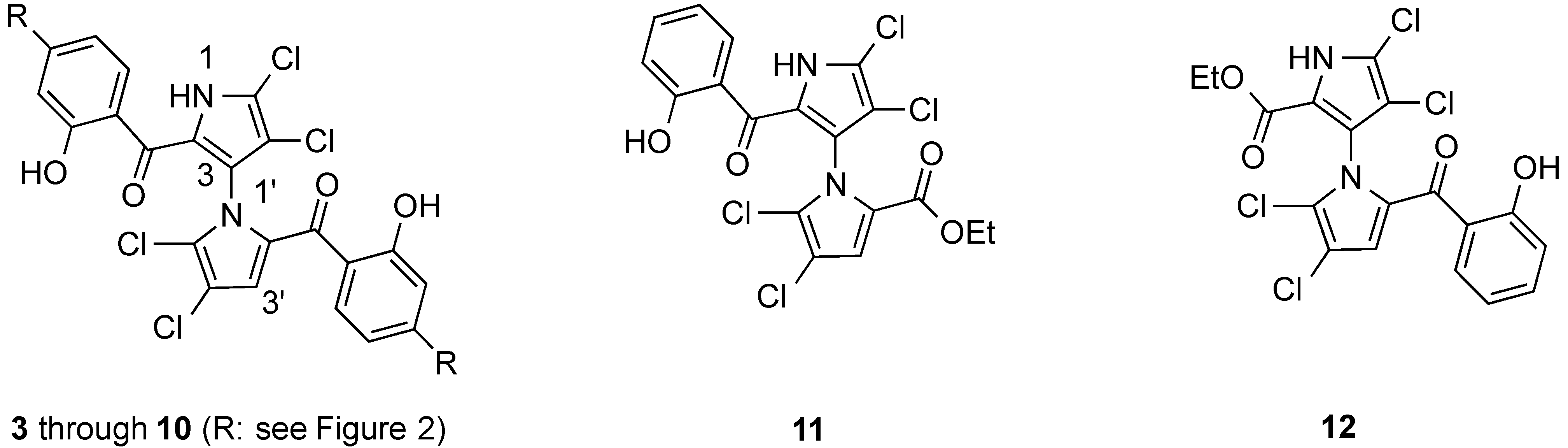
2.1. Design of Marinopyrrole Derivatives

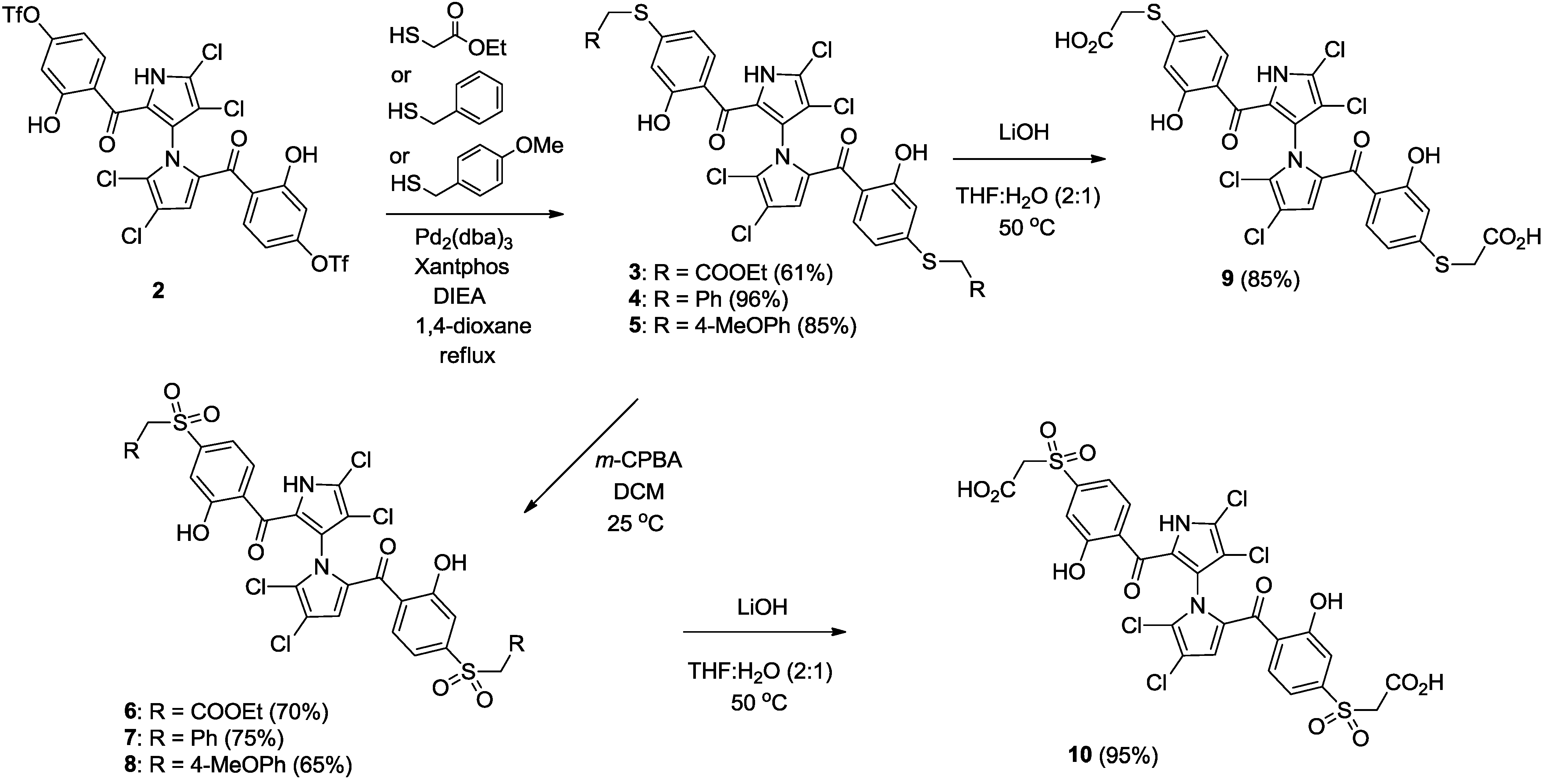
2.2. Synthesis of Marinopyrrole Derivatives

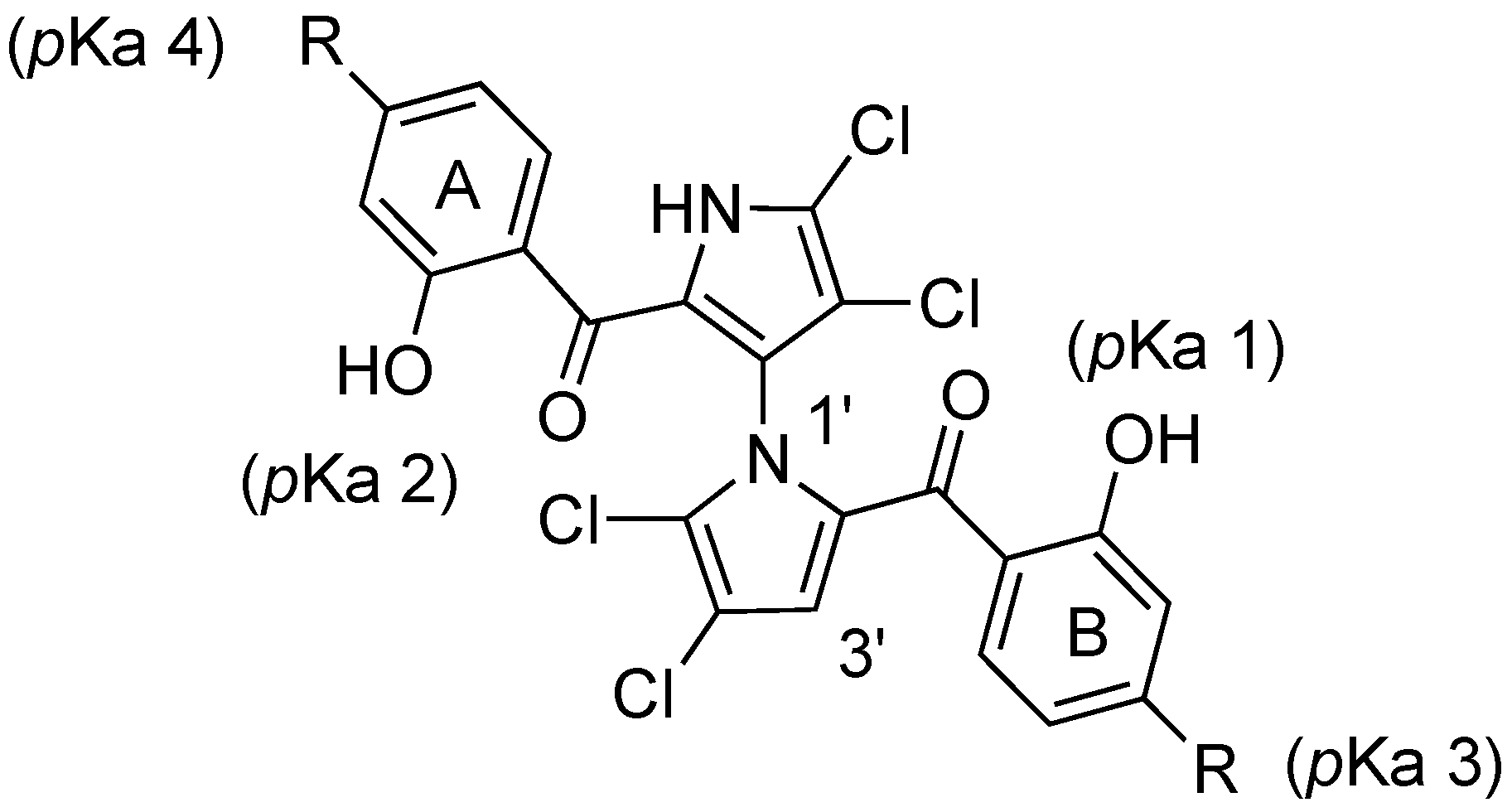
2.3. Physicochemical Properties and SAR of the Marinopyrroles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substituent (R) | Mcl-1/Bim a | Bcl-xL/Bim a | pKa 1 b | pKa 2 b | pKa 3 b,c | pKa 4 b,c | Clog p b |
|---|---|---|---|---|---|---|---|---|
| 1 | H | 8.9 ± 1.0 d | 16.4 ± 3.3 d | 7.8 | 8.4 | - | - | 5.6 |
| 3 | SCH2CO2Et | 1.8 ± 0.3 | 1.2 ± 0.2 | 7.8 | 8.4 | - | - | 6.1 |
| 4 | SCH2Ph | 0.7 ± 0.2 | 0.6 ± 0.2 | 7.8 | 8.4 | - | - | 10.2 |
| 5 | SCH2 (p-MeOPh) | 0.7 ± 0.1 | 0.6 ± 0.1 | 7.8 | 8.4 | - | - | 9.7 |
| 6 | SO2CH2CO2Et | 37.3 ± 3.1 | >100 | 6.7 | 7.3 | - | - | 3.7 |
| 7 | SO2CH2Ph | 7.3 ± 1.4 | 69.3 ± 15.8 | 6.7 | 7.3 | - | - | 6.9 |
| 8 | SO2CH2 (p-MeOPh) | 17.4 ± 3.1 | >100 | 6.7 | 7.3 | - | - | 6.4 |
| 9 | SCH2CO2H | 6.1 ± 1.3 | >100 | 7.8 | 8.4 | 2.9 | 3.5 | 5.3 |
| 10 | SO2CH2CO2H | 63.0 ± 5.4 | >100 | 6.7 | 7.3 | 2.2 | 2.9 | 2.9 |
| 1 | e See Figure 1 | 25.1 ± 4.7 | 96.6 (n = 2) | - | 8.1 | - | - | 4.5 |
| 12 | e See Figure 1 | 11.5 ± 1.9 | 17.6 ± 4.5 | 8.1 | - | - | - | 4.5 |
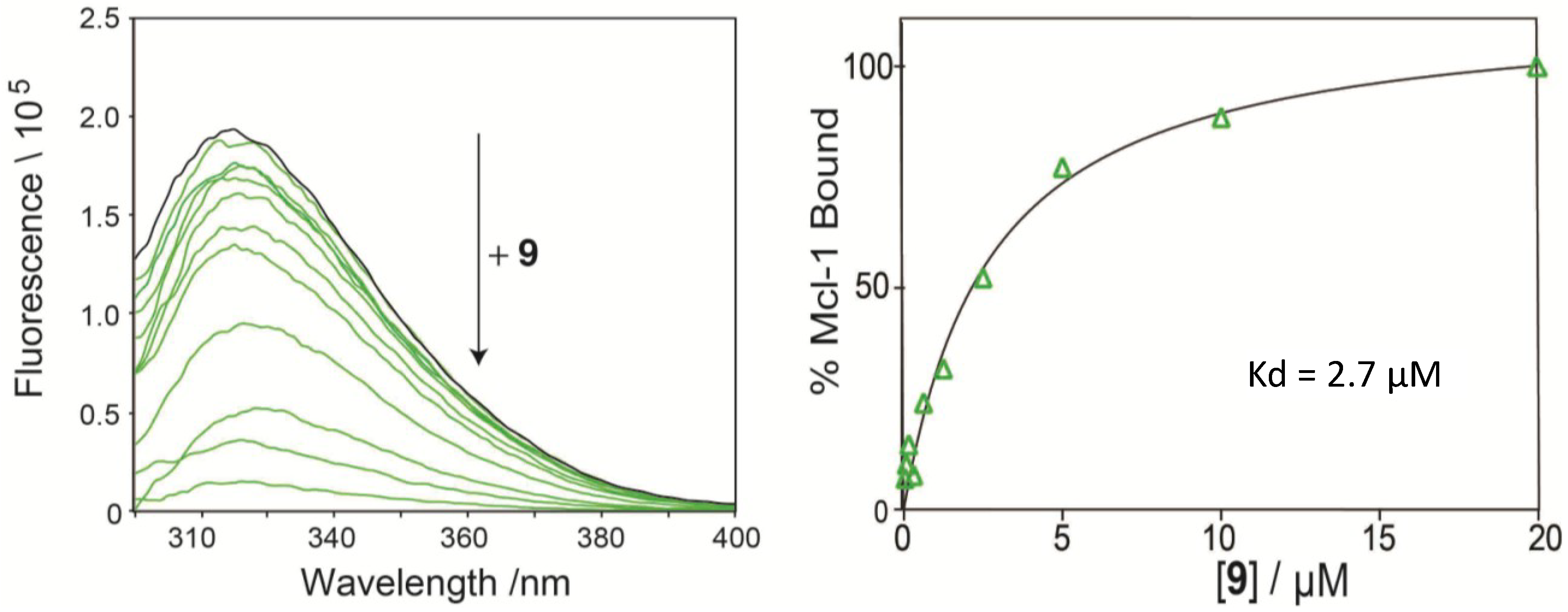
2.4. Direct Binding Measurement by Fluorescence Quenching

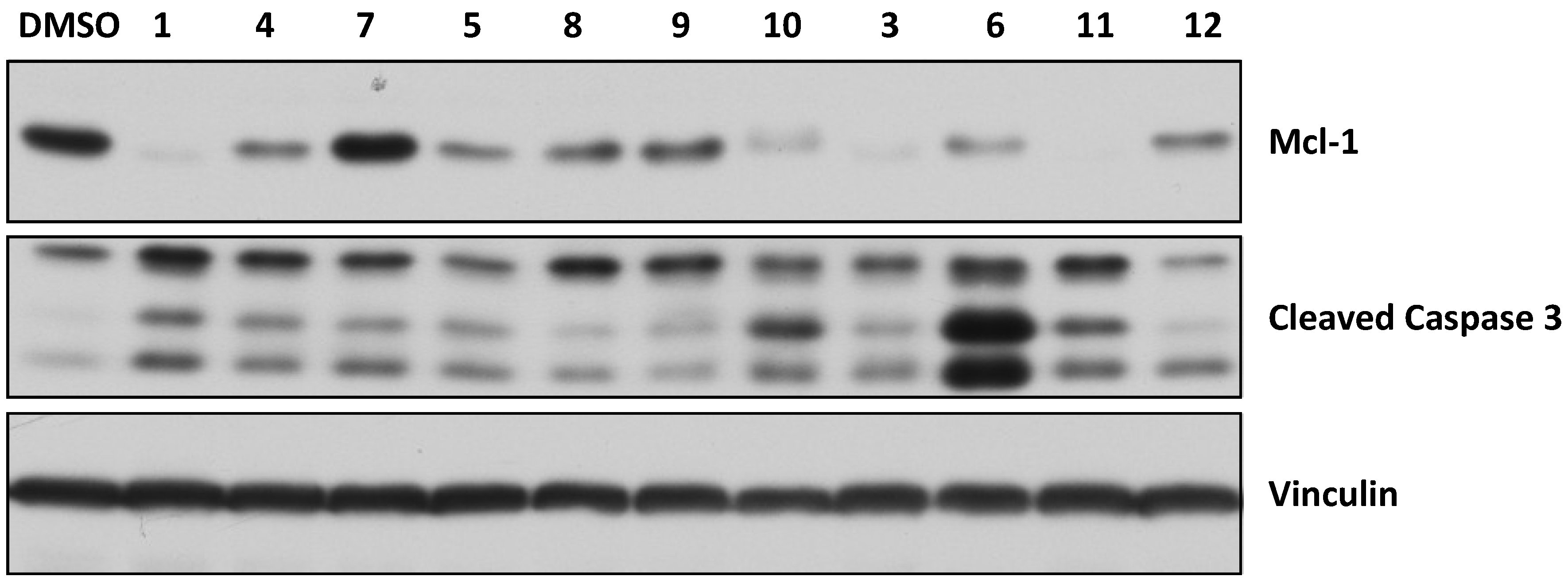
2.5. Activity in Intact Human Breast Cancer Cells

3. Experimental Section
3.1. Synthesis of Marinopyrrole Derivatives
3.2. Fluorescence Quenching, Enzyme-Linked Immunosorbent Assay (ELISA) and Western Blotting Following Treatment of Intact Human Breast Cancer Cells
3.2.1. Fluorescence Quenching
3.2.2. Enzyme-Linked Immunosorbent Assay
3.2.3. Western Blotting Following Treatment of Intact Human Breast Cancer Cells
3.2.4. MTT Assays, Nuclear and Cellular Morphology
4. Conclusions
Supplementary Files
Supplementary File 1Abbreviations
| ADME | absorption, distribution, metabolism and excretion |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma extra large |
| BH3 | Bcl-2 homology domain 3 |
| calcd. | calculated |
| m-CPBA | m-chloroperbenzoic acid |
| DCM | dichloromethane |
| dd | double doublet |
| br | broad |
| DPPP | bis(diphenylphosphino)propane |
| DIEA | diisopropylethylamine |
| DMF | dimethylformamide |
| DMSO | dimethyl sulfoxide |
| EtOAc | ethyl acetate |
| ESI | electrospray ionization |
| HPLC | high performance liquid chromatography |
| HRMS | high resolution mass spectrometry |
| HRP | horseradish peroxidase |
| IR | infrared |
| KBr | potassium bromide |
| KF | potassium fluoride |
| LC | liquid chromatography |
| LiOH | lithium hydroxide |
| Mcl-1 | Myeloid cell leukemia 1 |
| MeCN | acetonitrile |
| MeOH | methyl alcohol |
| MDA-MB-468 | breast cancer cell line |
| mp | melting point |
| MRSA | methicillin-resistant Staphylococcus aureus |
| NCS | N-chlorosuccinimide |
| NaI | sodium iodide |
| NMR | nuclear magnetic resonance |
| PBS | phosphate-buffered saline |
| RO5 | Rule of Five |
| SAR | structure activity relationship; s, singlet |
| THF | tetrahydrofuran |
| TMB | 3,3′,5,5′-Tetramethylbenzidine; |
| tox | toxicity |
| Pd2(dba)3 | tris(dibenzylideneacetone)dipalladium |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Structures, reactivities, and antibiotic properties of the marinopyrroles A–F. J. Org. Chem. 2010, 75, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Pan, L.; Chen, Y.; Song, H.; Qin, Y.; Li, R. Total synthesis of (±)-marinopyrrole a and its library as potential antibiotic and anticancer agents. J. Comb. Chem. 2010, 12, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Kanakis, A.A.; Sarli, V. Total synthesis of (±)-marinopyrrole A via copper-mediated N-arylation. Org. Lett. 2010, 12, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Simmons, N.L.; Chen, J.S.; Haste, N.M.; Nizet, V. Total synthesis and biological evaluation of marinopyrrole A and analogues. Tetrahedron Lett. 2011, 52, 2041–2043. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Haste, N.M.; Thienphrapa, W.; Nizet, V.; Hensler, M.; Li, R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (I). Mar. Drugs 2012, 10, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Cheng, C.; Song, H. Optimization of synthetic method of marinopyrrole A derivatives. Chem. J. Chin. Univ. 2012, 33, 1476–1480. [Google Scholar]
- Hughes, C.C.; Yang, Y.L.; Liu, W.T.; Dorrestein, P.C.; la Clair, J.J.; Fenical, W. Marinopyrrole A target elucidation by acyl dye transfer. J. Am. Chem. Soc. 2009, 131, 12094–12096. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Ryan, K.S.; Gulder, T.A.; Hughes, C.C.; Moore, B.S. Flavoenzyme-catalyzed atropo-selective N,C-bipyrrole homocoupling in marinopyrrole biosynthesis. J. Am. Chem. Soc. 2012, 134, 12434–12437. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Li, R.; Sung, S.S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X.; et al. Discovery of marinopyrrole A (Maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar]
- Cheng, P.; Clive, D.L.; Fernandopulle, S.; Chen, Z. Racemic marinopyrrole B by total synthesis. Chem. Commun. 2013, 49, 558–560. [Google Scholar] [CrossRef]
- Clive, D.L.J.; Cheng, P. The marinopyrroles. Tetrahedron 2013, 69, 5067–5078. [Google Scholar] [CrossRef]
- Cheng, P.; Shao, W.; Clive, D.L. A general route to 1,3′-bipyrroles. J. Org. Chem. 2013, 78, 11860–11873. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Liu, Y.; Song, H.; Pan, L.; Li, J.; Qin, Y.; Li, R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (II). Mar. Drugs 2013, 11, 2927–2948. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Haste, N.H.; Thienphrapa, W.; Li, J.; Nizet, V.; Hensler, M.; Li, R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (III). Mar. Drugs 2014, 12, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Liu, Y.; Balasis, M.E.; Simmons, N.L.; Li, J.; Song, H.; Pan, L.; Qin, Y.; Nicolaou, K.C.; Sebti, S.M.; et al. Cyclic marinopyrrole derivatives as disruptors of Mcl-1 and Bcl-xL binding to Bim. Mar. Drugs 2014, 12, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Goustin, A.S.; Aboukameel, A.; Chen, B.; Banerjee, S.; Wang, G.; Nikolovska-Coleska, Z.; Wang, S.; Al-Katib, A. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin. Cancer Res. 2007, 13, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Paoluzzi, L.; Gonen, M.; Gardner, J.R.; Mastrella, J.; Yang, D.; Holmlund, J.; Sorensen, M.; Leopold, L.; Manova, K.; Marcucci, G.; et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 2008, 111, 5350–5358. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Shore, G.C. Unique biology of Mcl-1: Therapeutic opportunities in cancer. Curr. Mol. Med. 2008, 8, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Ding, K.; Nikolovska-Coleska, Z.; Yang, C.Y.; Qiu, S.; Shangary, S.; Wang, R.; Guo, J.; Gao, W.; Meagher, J. Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic Bcl-2 proteins. J. Med. Chem. 2007, 50, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.H.; Sivaraman, T.; Wan, K.F.; Xu, J.; Krishnamoorthy, J.; Song, C.M.; Tian, L.; Chin, J.S.; Lim, D.S.; Mok, H.Y.; et al. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J. Med. Chem. 2010, 53, 2314–2318. [Google Scholar] [CrossRef] [PubMed]
- Rega, M.F.; Wu, B.; Wei, J.; Zhang, Z.; Cellitti, J.F.; Pellecchia, M. SAR by interligand nuclear overhauser effects (ILOEs) based discovery of acylsulfonamide compounds active against Bcl-x(L) and Mcl-1. J. Med. Chem. 2011, 54, 6000–6013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, C.; Li, X.; Song, T.; Wu, Z.; Liang, X.; Zhao, Y.; Shen, X.; Chen, H. Fragment-based design, synthesis, and biological evaluation of N-substituted-5-(4-isopropylthiophenol)-2-hydroxynicotinamide derivatives as novel Mcl-1 inhibitors. Eur. J. Med. Chem. 2013, 60, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Friberg, A.; Vigil, D.; Zhao, B.; Daniels, R.N.; Burke, J.P.; Garcia-Barrantes, P.M.; Camper, D.; Chauder, B.A.; Lee, T.; Olejniczak, E.T.; et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Li, Y.; Lv, L.; Zhou, M.; Han, L.; Zhang, Z.; Ba, Q.; Li, J.; Wang, H.; Liu, H.; et al. De novo design, synthesis and evaluation of benzylpiperazine derivatives as highly selective binders of Mcl-1. ChemMedChem 2013, 8, 1986–2014. [Google Scholar] [CrossRef] [PubMed]
- Abulwerdi, F.; Laio, C.; Liu, M.; Azmi, A.S.; Aboukameel, A.; Mady, A.S.A.; Gulappa, T.; Cierpicki, T.; Owens, S.; Zhang, T.; et al. A novel small-molecule inhibitor of Mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Jurs, P.C. Estimation of pKa for organic oxyacids using calculated atomic charge. J. Comput. Chem. 1993, 14, 1460–1467. [Google Scholar] [CrossRef]
- Csizmadia, F.; Tsantili-Kakoulidou, A.; Panderi, I.; Darvas, F. Prediction of distribution coefficient from structure. 1. Estimation method. J. Pharm. Sci. 1997, 86, 865–871. [Google Scholar] [CrossRef]
- Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldgus, S.; Shields, G.C. Absolute pK(a) determinations for substituted phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Fire, E.; Gulla, S.V.; Grant, R.A.; Keating, A.E. Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Sci. 2010, 19, 507–519. [Google Scholar] [PubMed]
- Balasis, M.E.; Forinash, K.D.; Chen, Y.A.; Fulp, W.J.; Coppola, D.; Hamilton, A.D.; Cheng, J.Q.; Sebti, S.M. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin. Cancer Res. 2011, 17, 2852–2862. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Sun, J.; Doi, K.; Sung, S.S.; Takahashi, Y.; Yin, H.; Rodriguez, J.M.; Becerril, J.; Berndt, N.; Hamilton, A.D. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L),Bcl-2,and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J. Biol. Chem. 2011, 18, 9382–9392. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, C.; Liu, Y.; Balasis, M.E.; Garner, T.P.; Li, J.; Simmons, N.L.; Berndt, N.; Song, H.; Pan, L.; Qin, Y.; et al. Marinopyrrole Derivatives with Sulfide Spacers as Selective Disruptors of Mcl-1 Binding to Pro-Apoptotic Protein Bim. Mar. Drugs 2014, 12, 4311-4325. https://doi.org/10.3390/md12084311
Cheng C, Liu Y, Balasis ME, Garner TP, Li J, Simmons NL, Berndt N, Song H, Pan L, Qin Y, et al. Marinopyrrole Derivatives with Sulfide Spacers as Selective Disruptors of Mcl-1 Binding to Pro-Apoptotic Protein Bim. Marine Drugs. 2014; 12(8):4311-4325. https://doi.org/10.3390/md12084311
Chicago/Turabian StyleCheng, Chunwei, Yan Liu, Maria E. Balasis, Thomas P. Garner, Jerry Li, Nicholas L. Simmons, Norbert Berndt, Hao Song, Lili Pan, Yong Qin, and et al. 2014. "Marinopyrrole Derivatives with Sulfide Spacers as Selective Disruptors of Mcl-1 Binding to Pro-Apoptotic Protein Bim" Marine Drugs 12, no. 8: 4311-4325. https://doi.org/10.3390/md12084311