
Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK

 and
and
Abstract
:1. Introduction


2. Results and Discussion
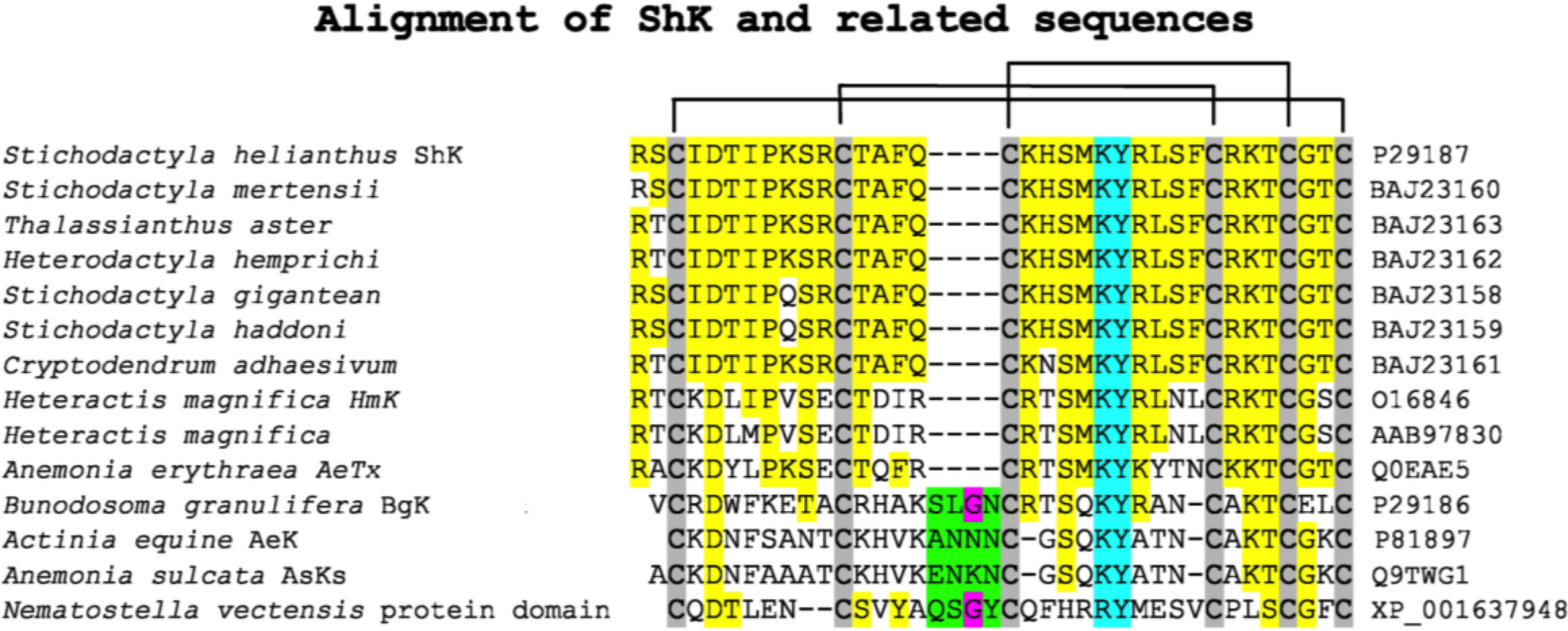
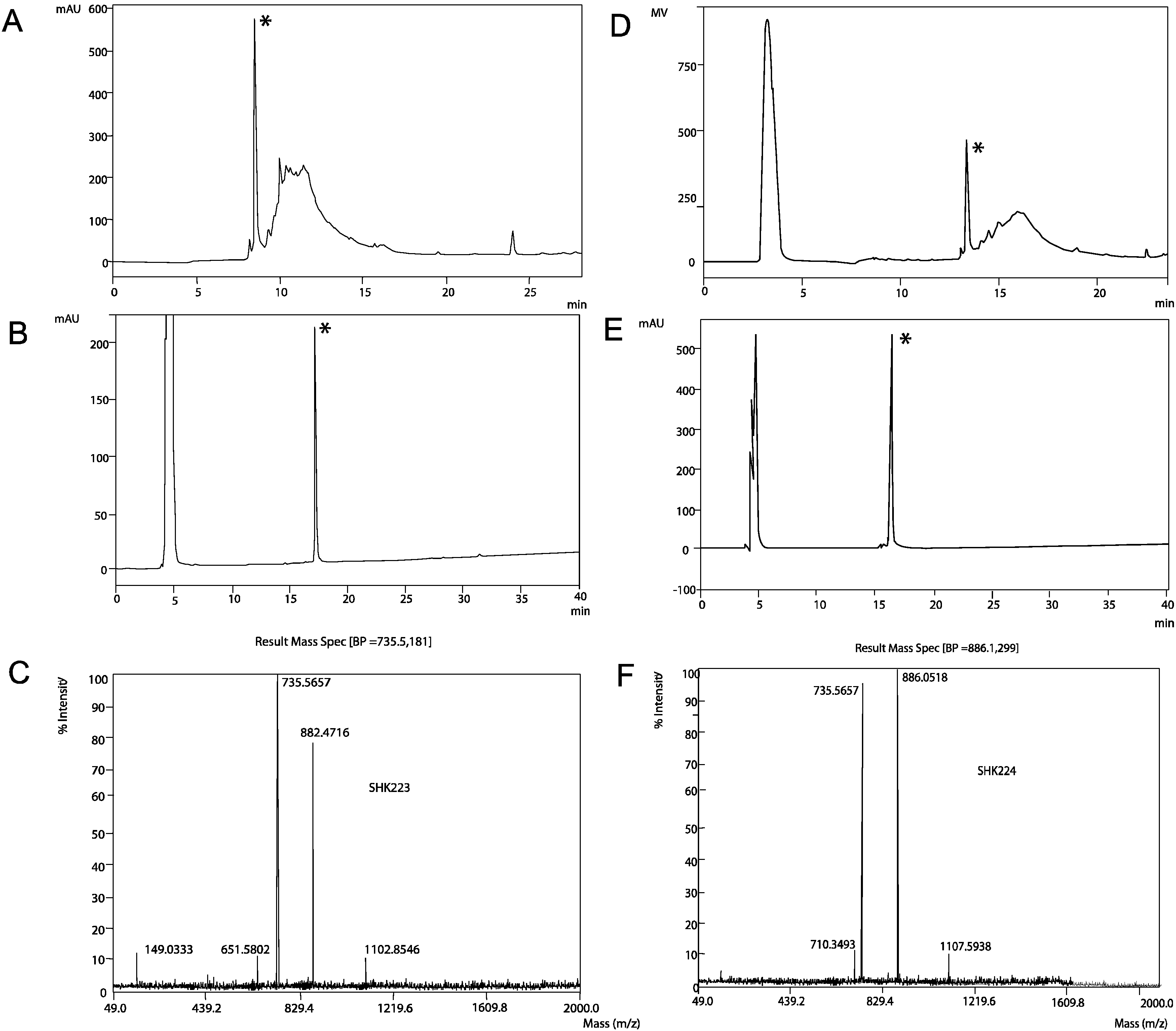
2.1. Synthesis, Expression and Folding


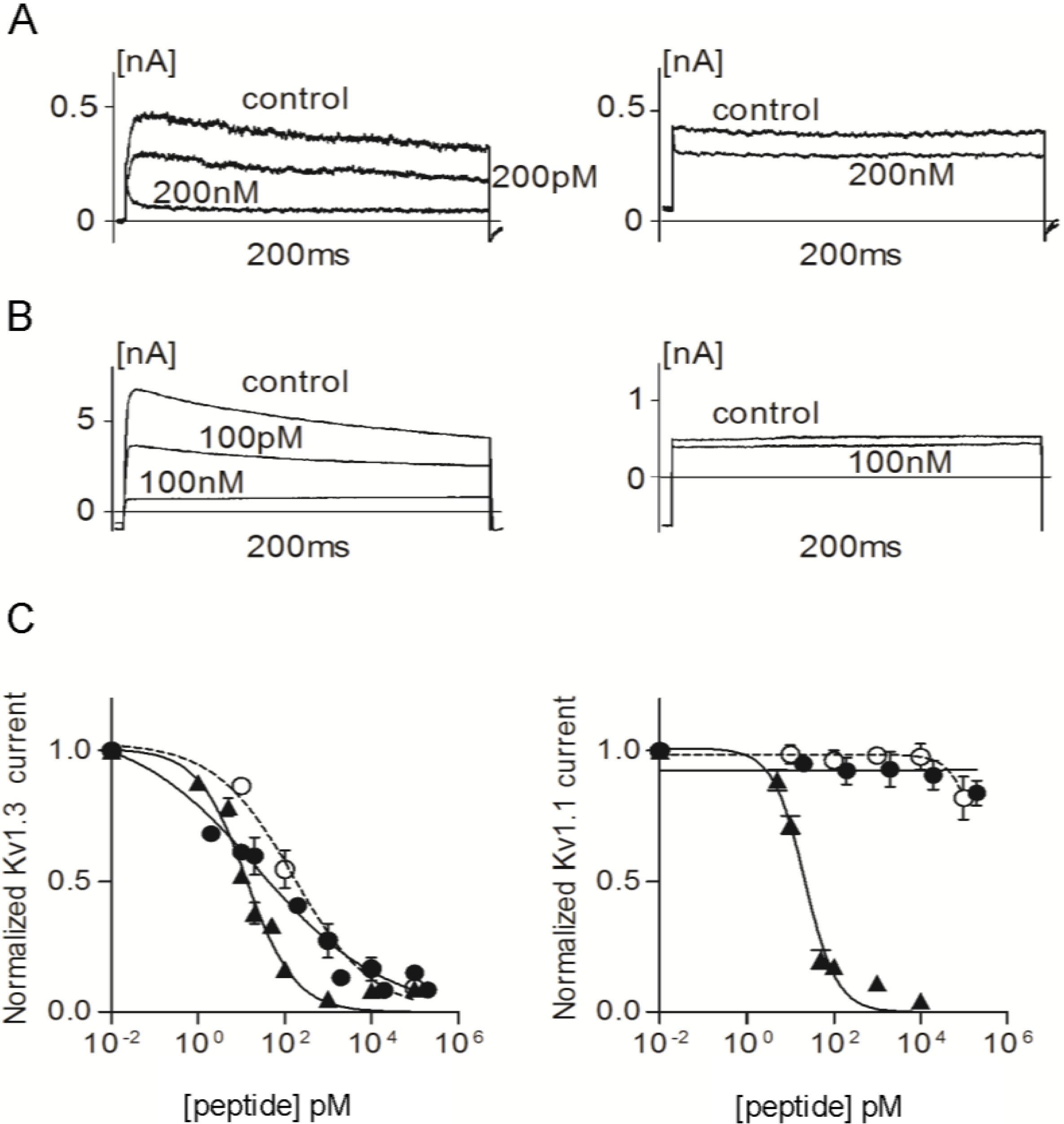
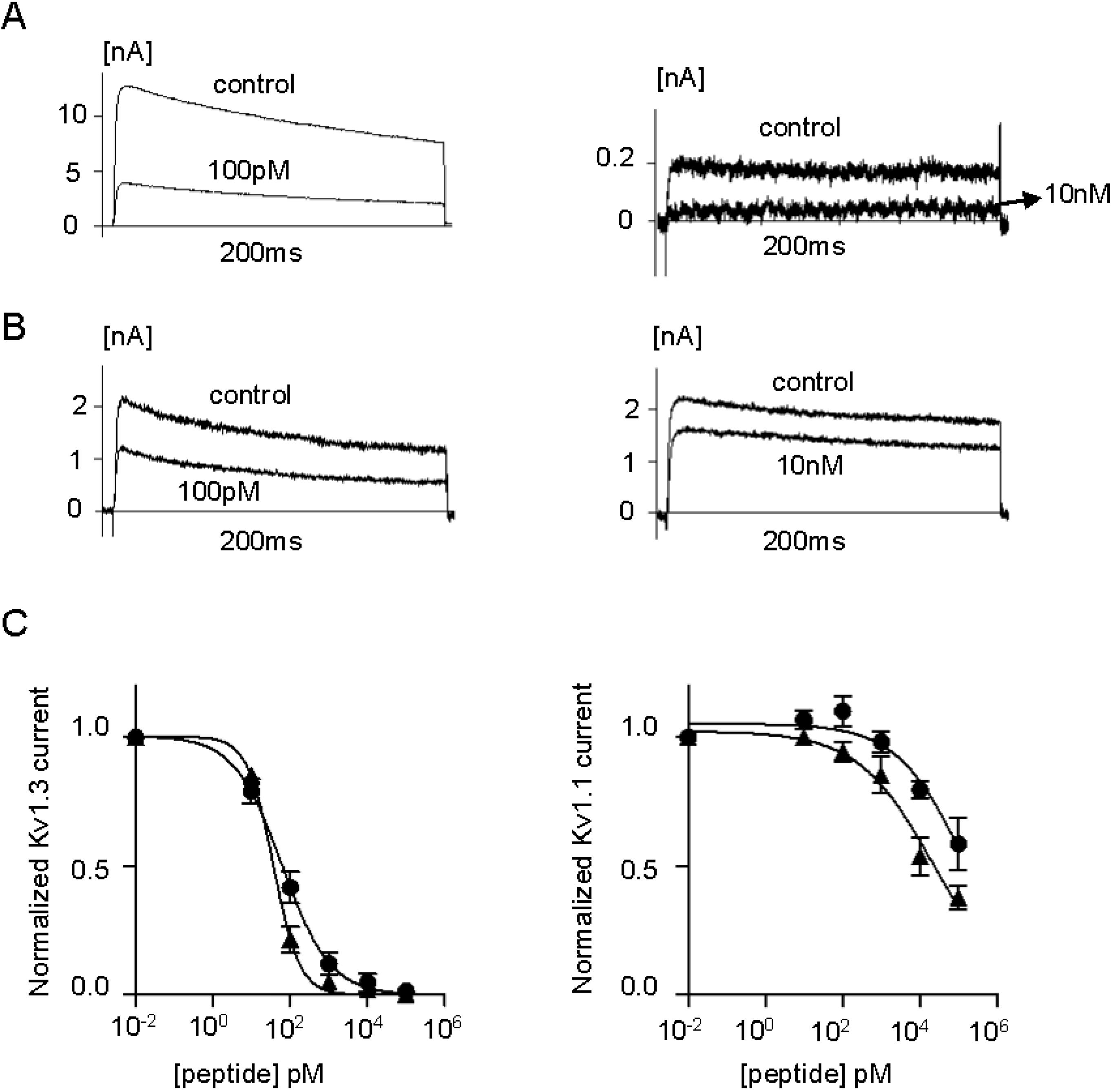
2.2. Efficacy and Selectivity of ShK Analogs on K+ Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | Kv1.1 | Kv1.3 | Kv1.4 | Kv1.6 |
|---|---|---|---|---|
| ShK | IC50 = 21.5 ± 2.2 pM | IC50 = 13.3 ± 1.4 pM | IC50 = 312 ± 14 pM | IC50 = 165 ± 8 nM |
| ShK-223 | <10% block at 100 nM | IC50 = 25 ± 14 pM | <5% block at 100 nM | <10% block at 100 nM |
| ShK-224 | <10% block at 100 nM | IC50 = 164 ± 59 pM | <20% block at 100 nM | ~25% block at 100 nM |
| ShK-234 | IC50 = 21.5 ± 8 nM | IC50 = 37 ± 4.6 pM | <15% block at 100 nM | ~25% block at 100 nM |
| ShK-235 | IC50 = 140 ± 62 nM | IC50 = 62 ± 13.2 pM | <15% block at 100 nM | ~25% block at 100 nM |

3. Experimental Section
3.1. Peptide Synthesis
3.2. Recombinant Peptide Production
3.3. Patch-Clamp Electrophysiology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frazão, B.; Vasconcelos, V.; Antunes, A. Sea anemone (cnidaria, anthozoa, actiniaria) toxins: An overview. Mar. Drugs 2012, 10, 1812–1851. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S. Structures of sea anemone toxins. Toxicon 2009, 54, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Kem, W.R.; Karlsson, E. Sea anemone potassium channel toxins. In Guidebook to Protein Toxins and Their Use in Cell Biology; Montecucco, C.A.R., Ed.; Oxford University Press: Oxford, UK, 1997; pp. 159–161. [Google Scholar]
- Kem, W.R.; Dunn, B.M. Separation and characterization of four different amino acid sequence variants of a sea anemone (Stichodactyla helianthus) protein cytolysin. Toxicon 1988, 26, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Kem, W.R.; Parten, B.; Pennington, M.W.; Price, D.A.; Dunn, B.M. Isolation, characterization, and amino acid sequence of a polypeptide neurotoxin occurring in the sea anemone Stichodactyla helianthus. Biochemistry 1989, 28, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, O.; Sotolongo, V.; Amor, A.M.; Stöcklin, R.; Anderson, A.J.; Harvey, A.L.; Engström, Å.; Wernstedt, C.; Karlsson, E. Characterization of a potassium channel toxin from the caribbean sea anemone Stichodactyla helianthus. Toxicon 1995, 33, 603–613. [Google Scholar] [CrossRef]
- Pennington, M.W.; Byrnes, M.E.; Zaydenberg, I.; Khaytin, I.; de Chastonay, J.; Krafte, D.S.; Hill, R.; Mahnir, V.M.; Volberg, W.A.; Gorczyca, W.; et al. Chemical synthesis and characterization of ShK toxin: A potent potassium channel inhibitor from a sea anemone. Int. J. Pept. Protein Res. 1995, 46, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Hubalek, F.; Byrnes, M.E.; Nielsen, K.R.; Woods, A.; Pennington, M.W. Assignment of the three disulfide bonds in ShK toxin: A potent potassium channel inhibitor from the sea anemone Stichodactyla helianthus. Lett. Pept. Sci. 1995, 1, 291–297. [Google Scholar] [CrossRef]
- Pennington, M.; Mahnir, V.; Khaytin, I.; Zaydenberg, I.; Byrnes, M.; Kem, W. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry 1996, 35, 16407–16411. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.; Mahnir, V.; Krafte, D.; Zaydenberg, I.; Byrnes, M.; Khaytin, I.; Crowley, K.; Kem, W. Identification of three separate binding sites on ShK toxin, a potent inhibitor of voltage-dependent potassium channels in human T-lymphocytes and rat brain. Biochem. Biophys. Res. Commun. 1996, 219, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Rauer, H.; Pennington, M.; Cahalan, M.; Chandy, K.G. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 1999, 274, 21885–12892. [Google Scholar] [CrossRef] [PubMed]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Tudor, J.E.; Pennington, M.W.; Norton, R.S. Ionisation behaviour and solution properties of the potassium-channel blocker ShK toxin. Eur. J. Biochem. 1998, 251, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.C.; Rowan, E.G.; et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef] [PubMed]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [CrossRef] [PubMed]
- Middleton, R.E.; Sanchez, M.; Linde, A.R.; Bugianesi, R.M.; Dai, G.; Felix, J.P.; Koprak, S.L.; Staruch, M.J.; Bruguera, M.; Cox, R.; et al. Substitution of a single residue in Stichodactyla helianthus peptide, ShK-Dap22, reveals a novel pharmacological profile. Biochemistry 2003, 42, 13698–13707. [Google Scholar] [CrossRef]
- Beeton, C.; Pennington, M.W.; Norton, R.S. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm. Allergy Drug Targets 2011, 10, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef] [PubMed]
- DeCoursey, T.E.; Chandy, K.G.; Gupta, S.; Cahalan, M.D. Voltage-gated K+ channels in human T lymphocytes: A role in mitogenesis? Nature 1984, 307, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Matteson, D.R.; Deutsch, C. K channels in T lymphocytes: A patch clamp study using monoclonal antibody adhesion. Nature 1984, 307, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Chandy, K.G. Potassium channels, memory T cells and multiple sclerosis. Neuroscientist 2005, 11, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, P.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Koshy, S.; Huq, R.; Tanner, M.R.; Atik, M.A.; Porter, P.C.; Khan, F.S.; Pennington, M.W.; Hanania, N.A.; Corry, D.B.; Beeton, C. Blocking Kv1.3 channels inhibits Th2 lymphocyte function and treats a rat model of asthma. J. Biol. Chem. 2014, 289, 12623–12632. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.; Pardo, C.A.; Hu, L.; Darrah, E.; Cudrici, C.; Niculescu, T.; Niculescu, F.; Mullen, K.M.; Allie, R.; Gao, L.; et al. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc. Natl. Acad. Sci. USA 2005, 102, 11094–11099. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Calabresi, P.; Allie, R.; Yun, S.; Pennington, M.W.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J. Clin. Investig. 2003, 111, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Breland, A.E.; Currier, R.D. Scorpion venom and multiple sclerosis. Lancet 1983, 2, 1021. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Barbaria, J.; Giraud, P.; Devaux, J.; Benoliel, A.; Gola, M.; Sabatier, J.M.; Bernard, D.; Crest, M.; Beraud, E. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J. Immunol. 2001, 166, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Pennington, M.W.; Wulff, H.; Singh, S.; Nugent, D.; Crossley, G.; Khaytin, I.; Calabresi, P.A.; Chen, C.-Y.; Gutman, G.A.; et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol. Pharmacol. 2005, 67, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Wulff, H.; Barbaria, J.; Clot-Faybesse, O.; Pennington, M.W.; Bernard, D.; Cahalan, M.D.; Chandy, K.G.; Beraud, E. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2001, 98, 13942–13947. [Google Scholar] [CrossRef] [PubMed]
- Matheu, M.P.; Beeton, C.; Garcia, A.; Chi, V.; Rangaraju, S.; Safrina, O.; Monaghan, K.; Uemura, M.I.; Li, D.; Pal, S.; et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008, 29, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Tarcha, E.J.; Chi, V.; Munoz-Elias, E.; Bailey, D.; Londono, L.M.; Upadhyay, S.K.; Norton, K.; Olson, A.; Tjong, I.; Nguyen, H.; et al. Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune diseases. J. Pharmacol. Exp. Ther. 2012, 342, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Beeton, C.; Galea, C.A.; Smith, B.J.; Chi, V.; Monaghan, K.P.; Garcia, A.; Rangaraju, S.; Giuffrida, A.; Plank, D.; et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol. Pharmacol. 2009, 75, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Pennington, M.W.; Beeton, C. Transforming a toxin into a therapeutic: The sea anemone potassium channel blocker ShK toxin for treatment of autoimmune diseases. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; King, G.F., Ed.; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Pennington, M.W.; Rashid, M.H.; Tajhya, R.B.; Beeton, C.; Kuyucak, S.; Norton, R.S. A C-terminally amidated analogue of ShK is a potent and selective blocker of the voltage-gated potassium channel Kv1.3. FEBS Lett. 2012, 586, 3996–4001. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A potent and selective peptide blocker of the Kv1.3 channel: Prediction from free-energy simulations and experimental confirmation. PLoS One 2013, 8, e78712. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.K.; Miranda, L.P.; Gegg, C.V.; Hu, S.F.S.; Belouski, E.J.; Murray, J.K.; Nguyen, H.; Walker, K.W.; Arora, T.; Jacobsen, F.W.; et al. Selective and potent peptide inhibitors of Kv1.3. Patent WO2010108154, 3 March 2011. [Google Scholar]
- Chang, S.C.; Galea, C.A.; Leung, E.W.; Tajhya, R.B.; Beeton, C.; Pennington, M.W.; Norton, R.S. Expression and isotopic labelling of the potassium channel blocker ShK toxin as a thioredoxin fusion protein in bacteria. Toxicon 2012, 60, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Huq, R.; Tanner, M.R.; Chhabra, S.; Estrada, R.; Dhawan, V.; Chauhan, S.; Pennington, M.W.; Beeton, C.; Kuyucak, S.; et al. A potent and Kv1.3-selective analog of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennington, M.W.; Chang, S.C.; Chauhan, S.; Huq, R.; Tajhya, R.B.; Chhabra, S.; Norton, R.S.; Beeton, C. Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK. Mar. Drugs 2015, 13, 529-542. https://doi.org/10.3390/md13010529
Pennington MW, Chang SC, Chauhan S, Huq R, Tajhya RB, Chhabra S, Norton RS, Beeton C. Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK. Marine Drugs. 2015; 13(1):529-542. https://doi.org/10.3390/md13010529
Chicago/Turabian StylePennington, Michael W., Shih Chieh Chang, Satendra Chauhan, Redwan Huq, Rajeev B. Tajhya, Sandeep Chhabra, Raymond S. Norton, and Christine Beeton. 2015. "Development of Highly Selective Kv1.3-Blocking Peptides Based on the Sea Anemone Peptide ShK" Marine Drugs 13, no. 1: 529-542. https://doi.org/10.3390/md13010529