
Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs

 ,
,
Abstract
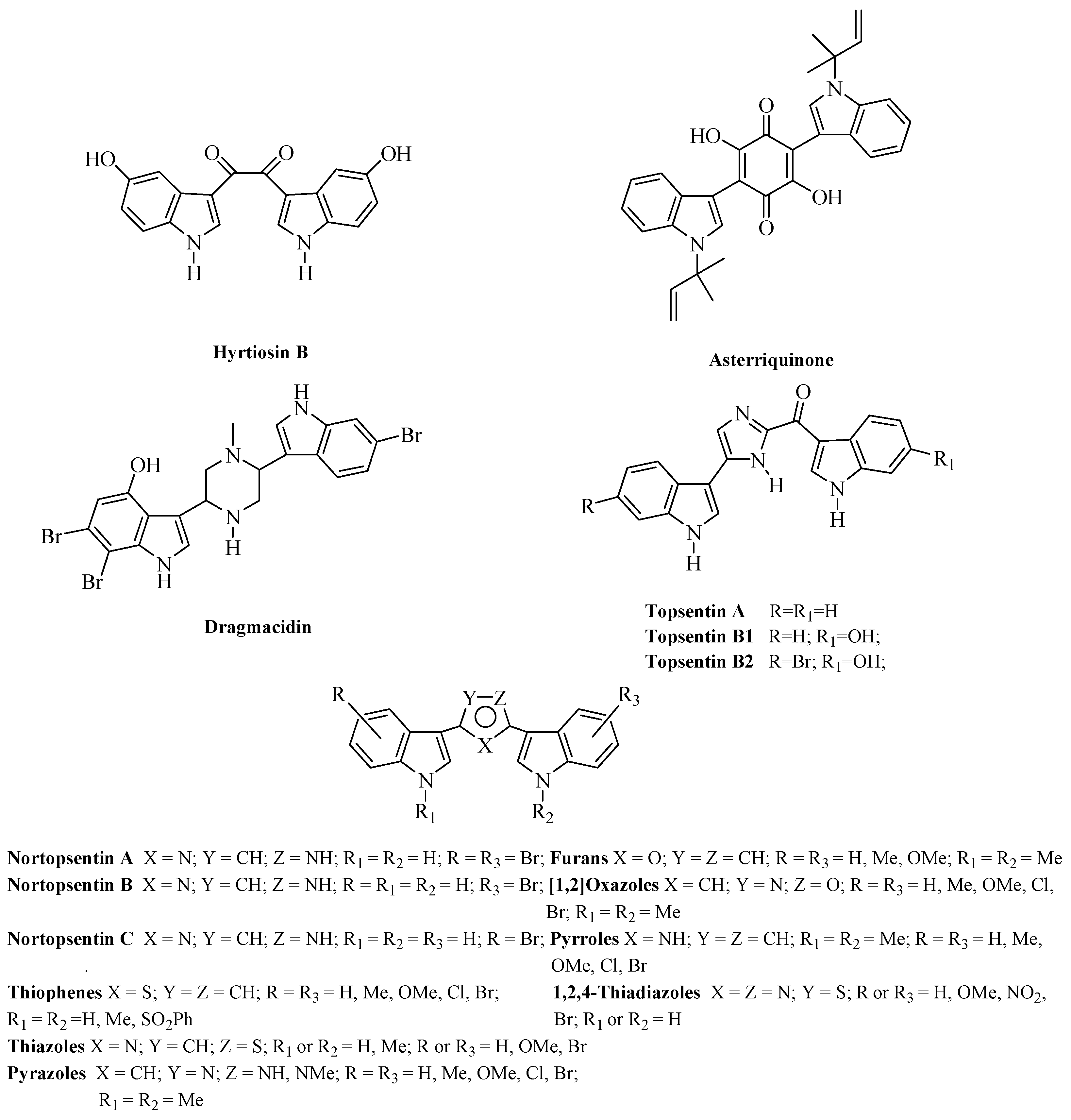
:1. Introduction
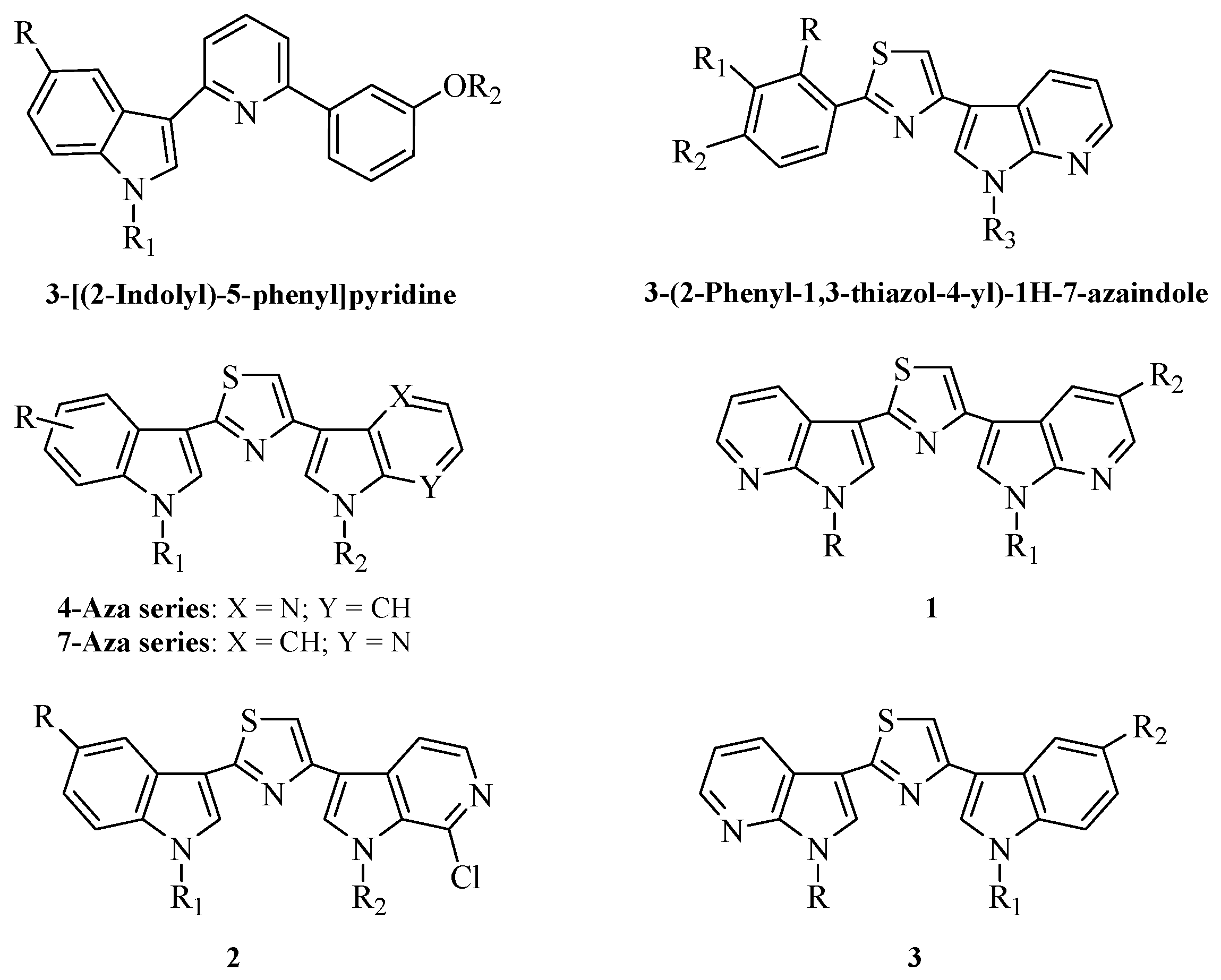
2. Results and Discussion
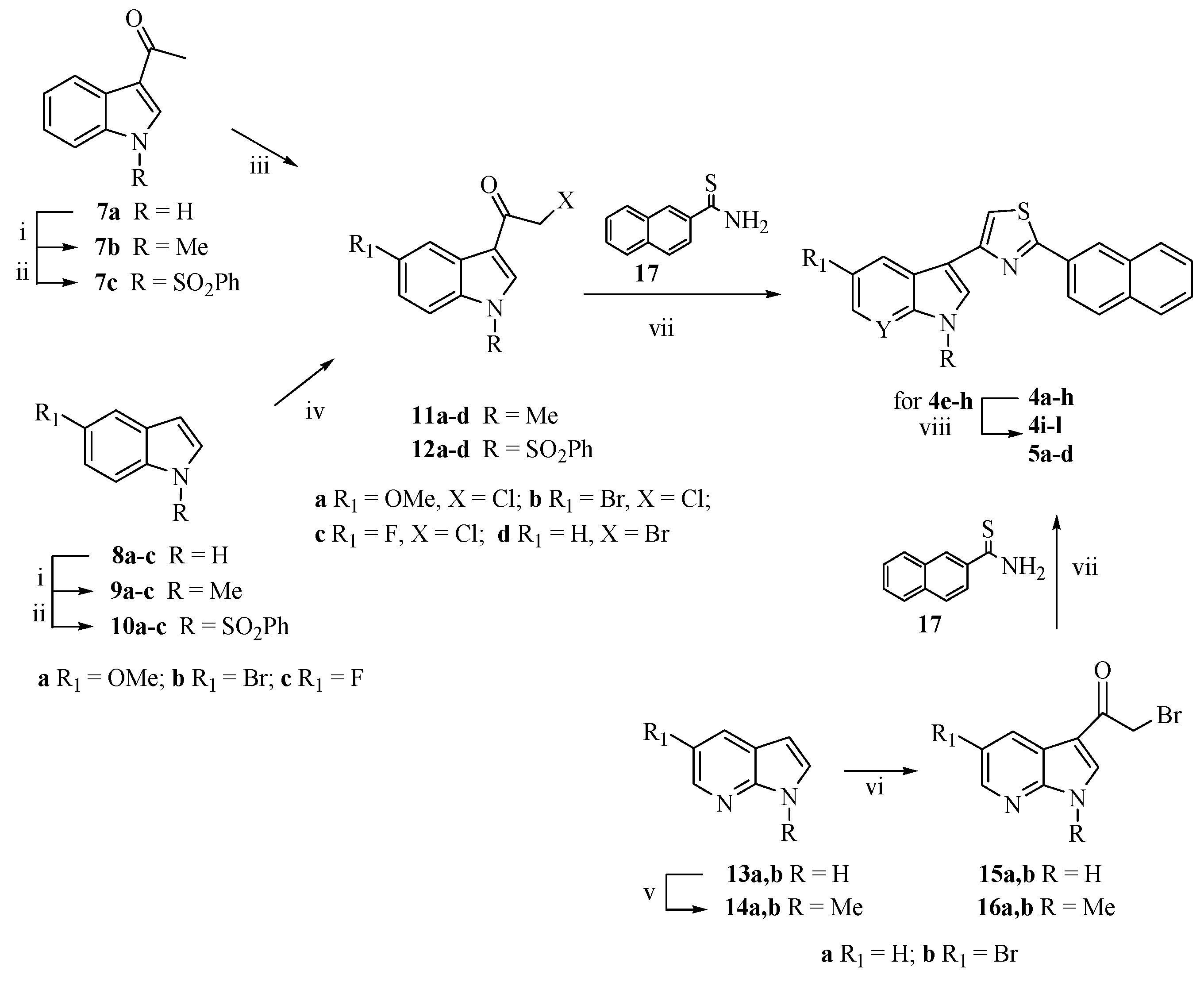
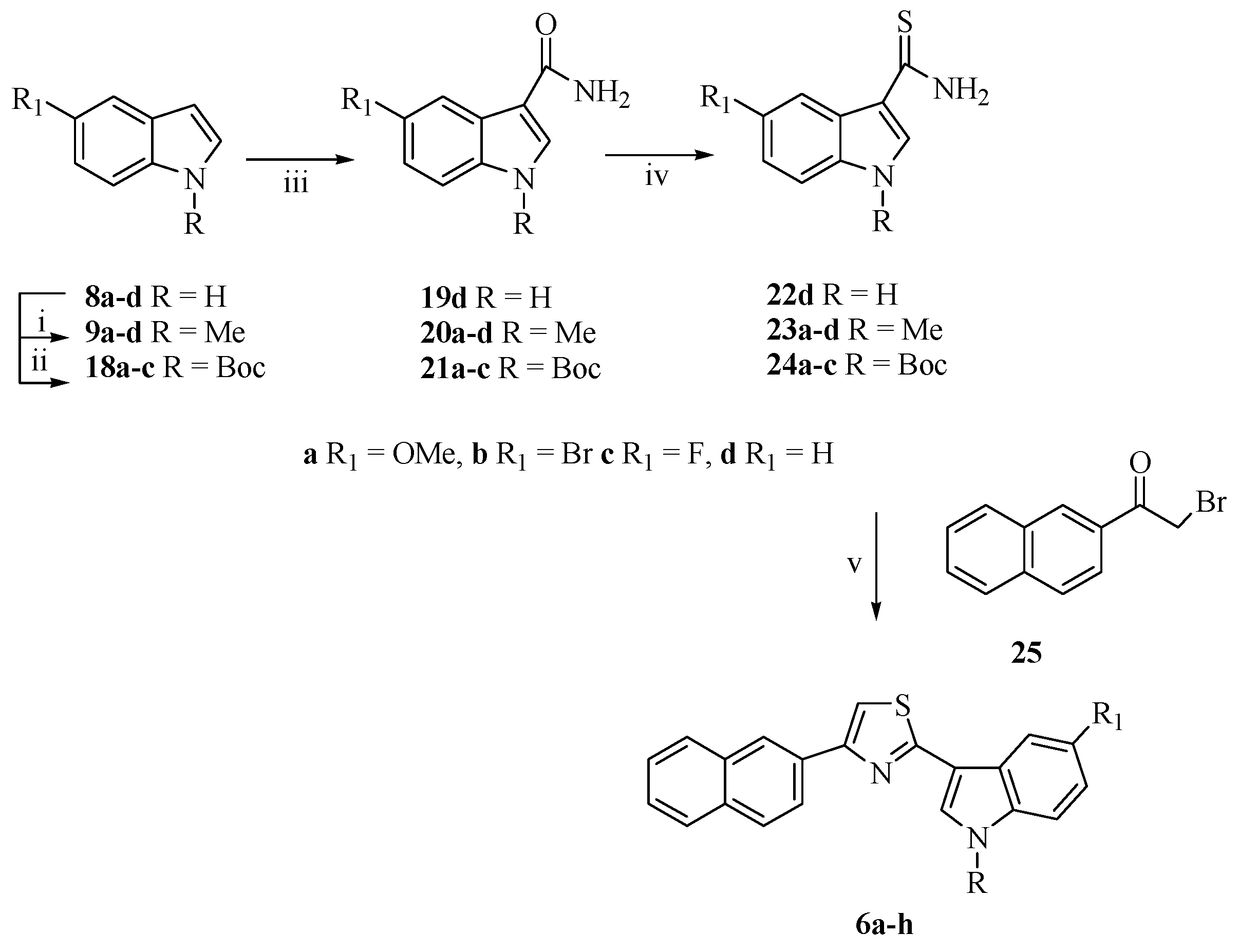
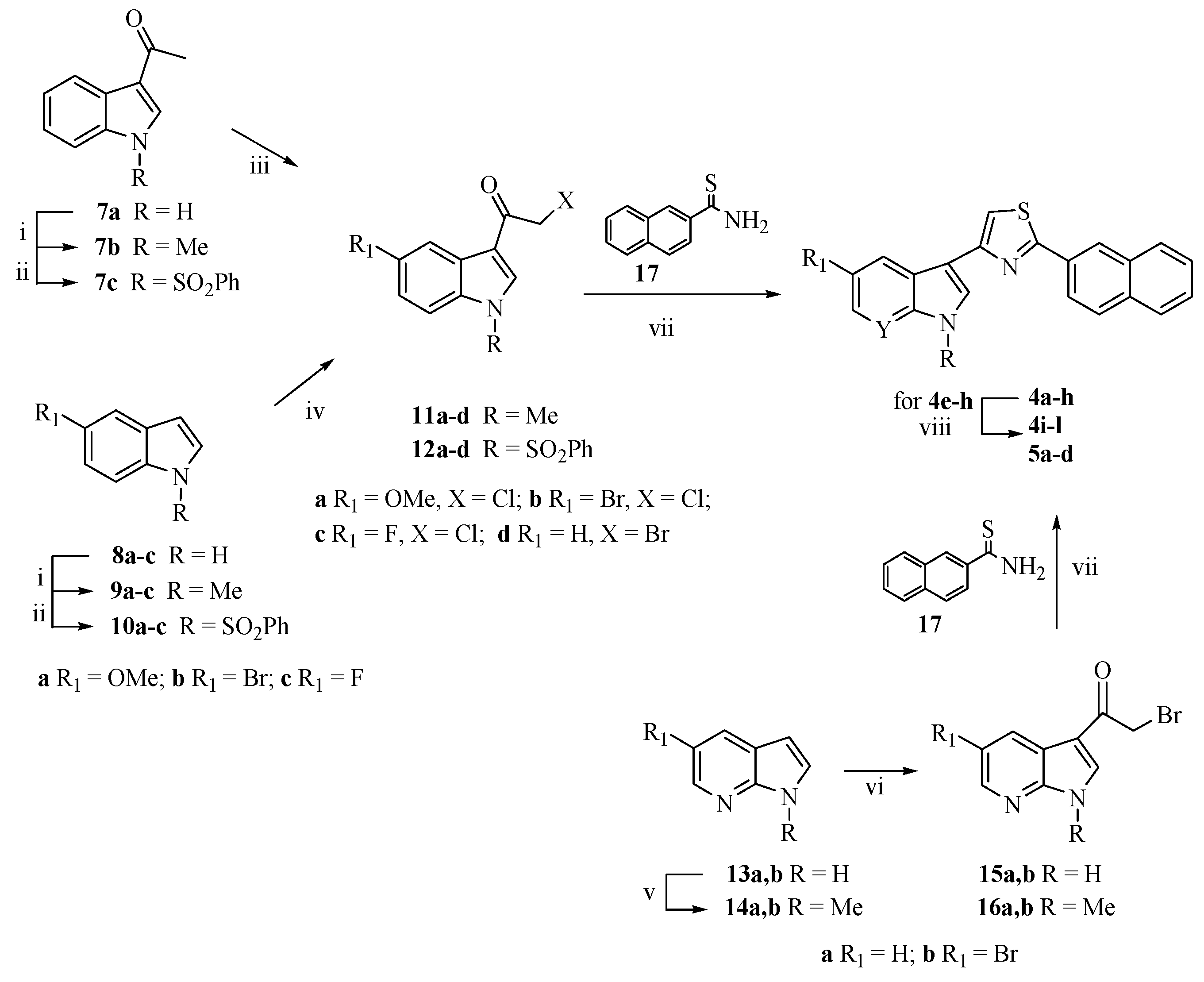
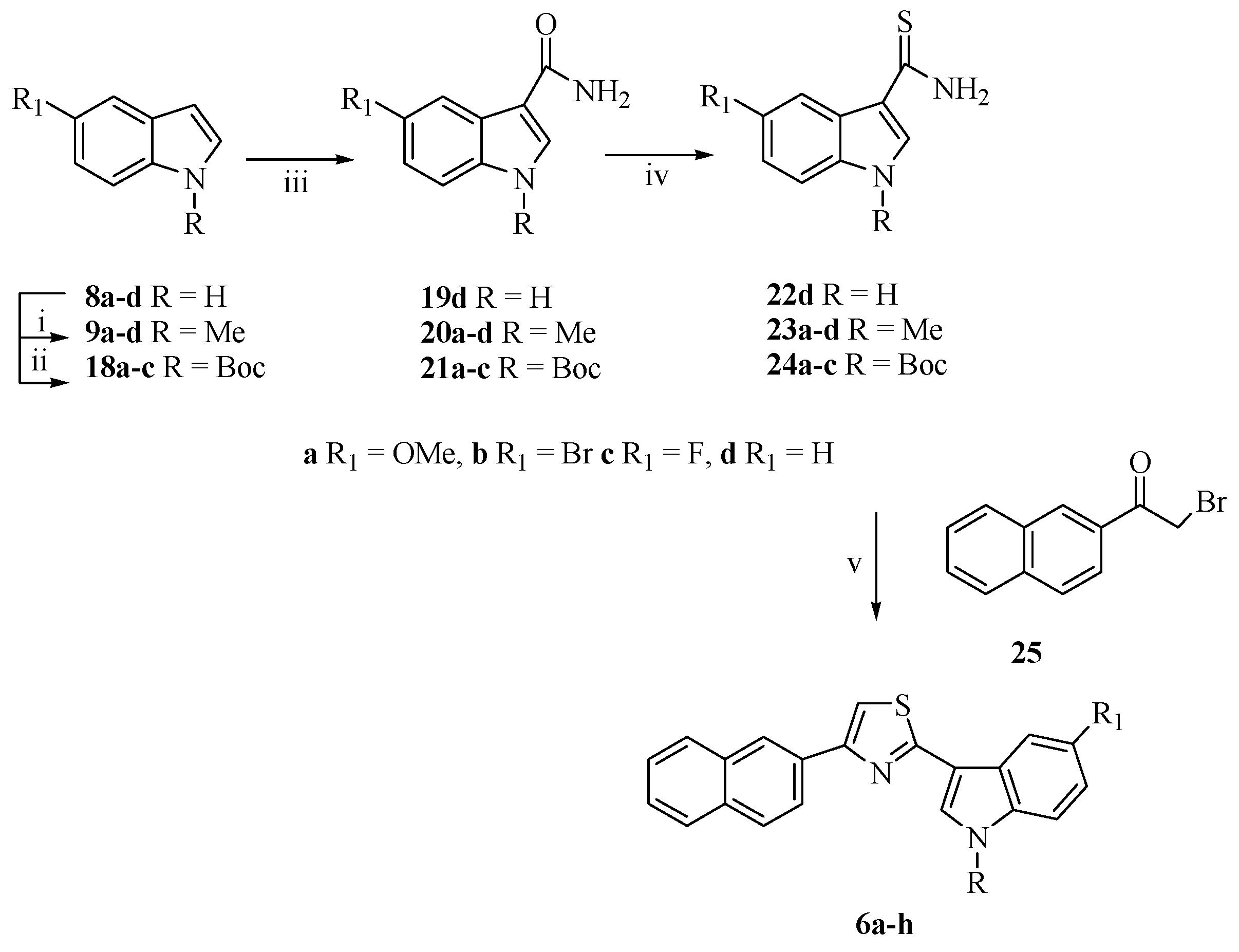
2.1. Chemistry
2.2. Biology
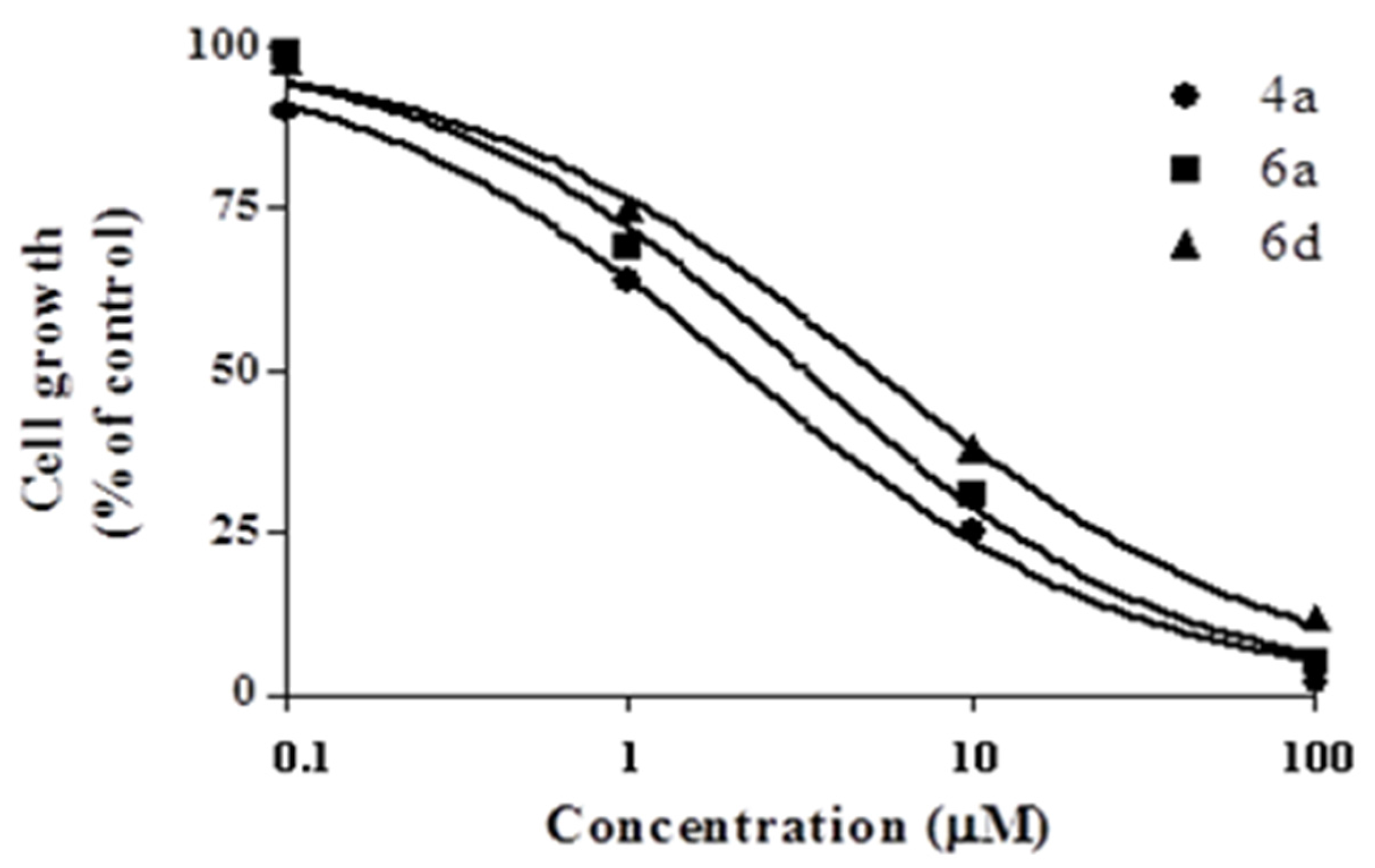
2.2.1. Cytotoxic Activity
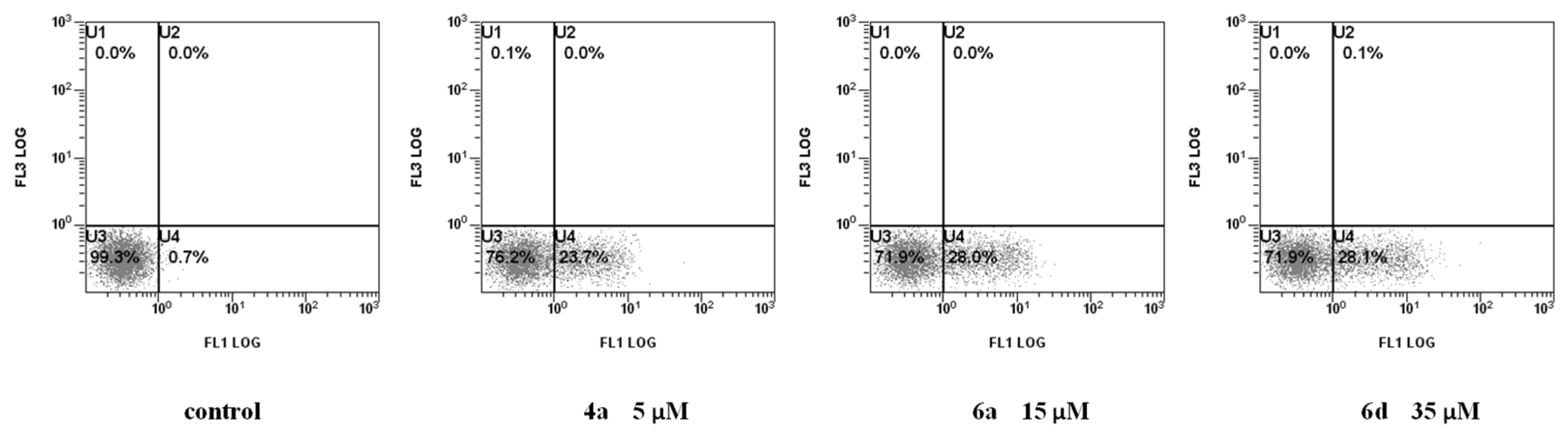
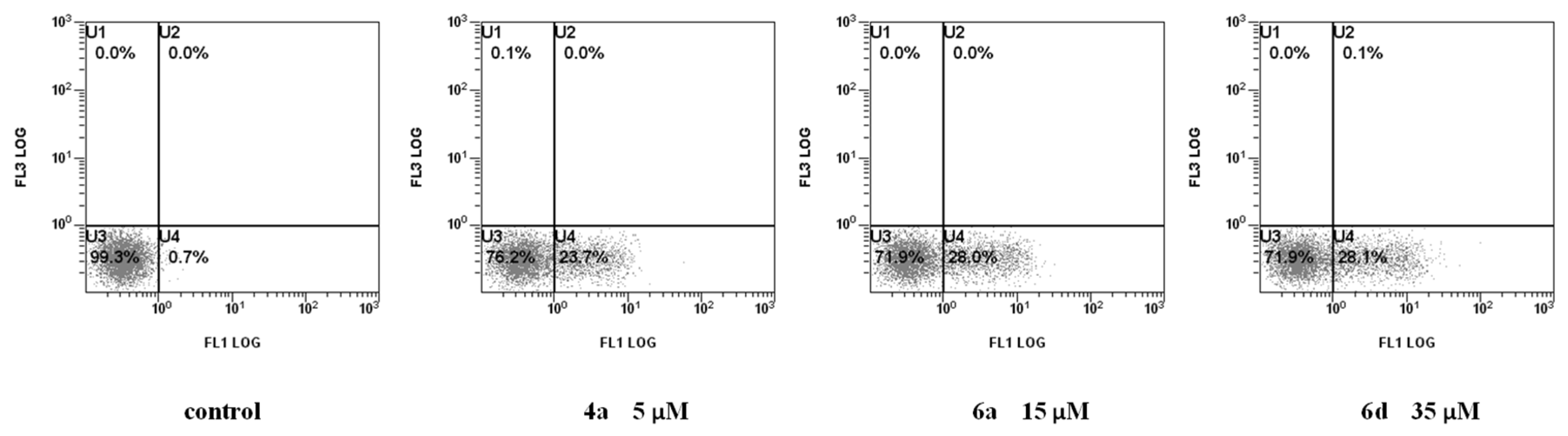
2.2.2. Cell Death Mechanism
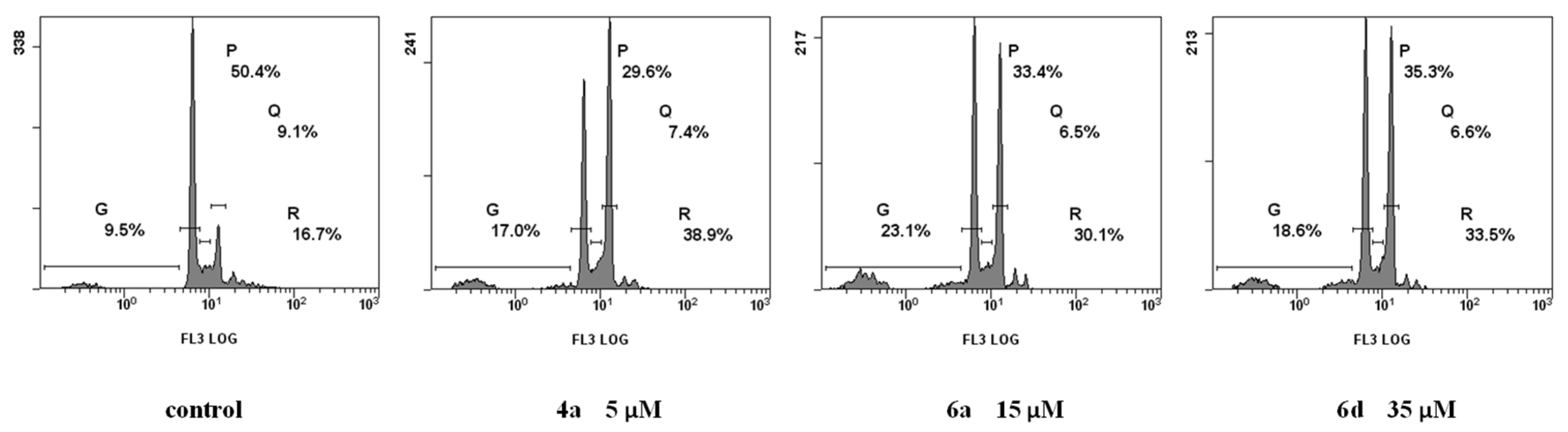
2.2.3. Cell Cycle Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of 1-[1-(Phenylsulfonyl)-1H-indol-3-yl]ethanone (7c)
3.1.3. Synthesis of Substituted 2-Chloro-1-(1-methyl-1H-indol-3-yl)ethanones (11a,b) and 2-Chloro-1-[1-(phenylsulfonyl)-1H-indol-3-yl]ethanones (12a–c)
2-Chloro-1-(5-methoxy-1-methyl-1H-indol-3-yl)ethanone (11a)
1-(5-Bromo-1-methyl-1H-indol-3-yl)-2-chloroethanone (11b)
2-Chloro-1-[5-methoxy-1-(phenylsulfonyl)-1H-indol-3-yl]ethanone (12a)
1-[5-Bromo-1-(phenylsulfonyl)-1H-indol-3-yl]-2-chloroethanone (12b)
2-Chloro-1-[5-fluoro-1-(phenylsulfonyl)-1H-indol-3-yl]ethanone (12c)
3.1.4. Synthesis of 3-(1-Benzenesulfonyl-1H-indol-3-yl)-2-bromoethanone (12d)
3.1.5. Synthesis of 5-Substituted-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1-(protected)-1H-indoles (4a–h)
5-Methoxy-1-methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4a)
5-Bromo-1-methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4b)
5-Fluoro-1-methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4c)
1-Methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4d)
5-Methoxy-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1-(phenylsulfonyl)-1H-indole (4e)
5-Bromo-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1-(phenylsulfonyl)-1H-indole (4f)
5-Fluoro-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1-(phenylsulfonyl)-1H-indole (4g)
3-[2-(Naphthalen-2-yl)-1,3-thiazol-4-yl]-1-(phenylsulfonyl)-1H-indole (4h)
3.1.6. Synthesis of 5-Substituted-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indoles (4i–l)
5-Methoxy-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (4i)
5-Bromo-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4j)
5-Fluoro-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (4k)
3-[2-(Naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-indole (4l)
3.1.7. Synthesis of 3-[2-(Naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridines (5a–d)
1-Methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (5a)
5-Bromo-1-methyl-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (5b)
3-[2-(Naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (5c)
5-Bromo-3-[2-(naphthalen-2-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (5d)
3.1.8. Synthesis of 3-[4-(Naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indoles (6a–h)
5-Methoxy-1-methyl-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6a)
5-Bromo-1-methyl-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6b)
5-Fluoro-1-methyl-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6c)
1-Methyl-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6d)
5-Methoxy-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6e)
5-Bromo-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6f)
5-Fluoro-3-[4-(naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6g)
3-[4-(Naphthalen-2-yl)-1,3-thiazol-2-yl]-1H-indole (6h)
3.2. Biology Studies
3.2.1. Biology
3.2.2. Viability Assay In Vitro
3.2.3. Measurement of Phosphatidylserine (PS) Exposure
3.2.4. Cell Cycle Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zheng, L.-H.; Wang, Y.-J.; Sheng, J.; Wang, F.; Zheng, Y.; Lin, X.-K.; Sun, M. Antitumor peptides from marine organism. Mar. Drugs 2011, 9, 1840–1859. [Google Scholar] [CrossRef] [PubMed]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New bisindole alkaloids of the topsentin and hamacanthin classes from the Mediterranean marina sponge Rhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.; Sim, C.J.; Im, K.S.; Jung, J.H. Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J. Nat. Prod. 2005, 68, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Novel antitumor agents: Marine sponge alkaloids, their synthetic analogs and derivatives. Mini-Rev. Med. Chem. 2005, 5, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.-R.; Chen, K.; Zhu, H.-L. A Functional Scaffold in Marine Alkaloid: An Anticancer Moiety for Humans. Curr. Med. Chem. 2013, 20, 3903–3922. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural product scaffolds as leads to drugs. Future Med. Chem. 2009, 1, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Negi, A.; Singh, V. Indole based alkaloid in cancer: An overview. PharmaTutor Mag. 2014, 2, 76–82. [Google Scholar]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.H.; Sakemi, S.; Gunasekera, S.; Kashman, Y.; Lui, M.; Burres, N.; McCarthy, P. Bis-Indole Imidazole Compounds Which Are Useful Antitumor and Antimicrobial Agents. U.S. Patent 4970226, 13 November 1990. [Google Scholar]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Sasaki, T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Shimizu, S.; Yamamoto, Y.; Inagaki, L.; Koshimura, S. Antitumor effect and structure-activity relationship of asterriquinone analogs. Gann 1982, 73, 642–648. [Google Scholar] [PubMed]
- Kohmoto, S.; Kashman, Y.; McConnel, O.J.; Rinehart, K.L., Jr.; Wrigh, A.; Koehn, F. Dragmacidin, a new cytotoxic bis(indole)alkaloid from a deep water marine sponge, Dragmacidon sp. J. Org. Chem. 1988, 53, 3116–3118. [Google Scholar] [CrossRef]
- Bartik, K.; Braekman, J.C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentin, new toxic bis-indole alkaloids from the marine sponge Topsentia genitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Kawasaki, I.; Yamashita, M.; Ohta, S. Total synthesis of nortopsentins A–D marine alkaloids. Chem. Pharm. Bull. 1996, 44, 1831–1839. [Google Scholar] [CrossRef]
- Moody, C.J.; Roffey, J.R.A. Synthesis of N-protected Nortopsentins B and D. Arkivoc 2000, 1, 393–401. [Google Scholar]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. A concise synthesis of topsentin A and nortopsentin B and D. Org. Lett. 2000, 2, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Fresneda, P.M.; Molina, P.; Sanz, M.A. Microwave-assisted regioselective synthesis of 2,4-disubstituted imidazoles: Nortopsentin D synthesized by minimal effort. Synlett 2001, 2, 218–221. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Martorana, A.; Dattolo, G.; Gia, O.; Dalla Via, L.; Cirrincione, G. Synthesis and antitumor properties of 2,5-bis(3′-indolyl)thiophenes: Analogues of marine alkaloid nortopsentin. Bioorg. Med. Chem. Lett. 2007, 17, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Martorana, A.; Gia, O.; Dalla Via, L.; Cirrincione, G. 3,5-Bis(3′-indolyl)pyrazoles, analogues of marine alkaloid nortopsentin: Synthesis and antitumor properties. Bioorg. Med. Chem. Lett. 2007, 17, 6134–6137. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.H. Synthesis and antiproliferative activity of 2,5-bis(3′-indolyl)pyrroles, analogues of the marine alkaloid nortopsentin. Mar. Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumar, N.M.; Chang, K.H.; Gupta, R.; Shah, K. Synthesis and in vitro anticancer activity of 3,5-bis(indolyl)-1,2,4-thiadiazoles. Bioorg. Med. Chem. Lett. 2011, 21, 5897–5900. [Google Scholar] [CrossRef] [PubMed]
- Jacquemard, U.; Dias, N.; Lansiaux, A.; Bailly, C.; Logè, C.; Robert, J.M.; Lozach, O.; Meijer, L.; Merour, J.Y.; Routier, S. Synthesis of 3,5-bis(2-indolyl)pyridine and 3-[(2-indolyl)-5-phenyl]pyridine derivatives as CDK inhibitors and cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 4932–4953. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Parrino, B.; Lopergolo, A.; Pennati, M.; Zaffaroni, N.; Cirrincione, G. Synthesis and antitumor activity of 3-(2-phenyl-1,3-thiazol-4-yl)-1H-indoles and 3-(2-phenyl-1,3-thiazol-4-yl)-1H-7-azaindoles. ChemMedChem 2011, 6, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spanò, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and antiproliferative activity of substituted 3[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-b]pyridines, marine alkaloid nortopsentin analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; de Cesare, M.; et al. Novel 1H-pyrrolo[2,3-b]pyridine derivatives nortopsentin analogues: Synthesis and antitumor activity in peritoneal mesothelioma experimental models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; di Vita, G.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Livrea, M.A.; Diana, P.; et al. Synthesis and antiproliferative activity of thiazolyl-bis-pyrrolo[2,3-b]pyridines and indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, nortopsentin analogues. Mar. Drugs 2015, 13, 460–492. [Google Scholar] [CrossRef] [PubMed]
- Parrino, B.; Carbone, A.; Di Vita, G.; Ciancimino, C.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Diana, P.; et al. 3-[4-(1H-Indol-3-yl)-1,3-thiazol-2-yl]-1H-pyrrolo[2,3-b]pyridines, nortopsentin analogues with antiproliferative activity. Mar. Drugs 2015, 13, 1901–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahboobi, S.; Uecker, A.; Sellmer, A.; Cenac, C.; Hoecher, H.; Pongratz, H.; Eichhorn, E.; Hufsky, H.; Truempler, A.; Sicker, M.; et al. Novel Bis(1H-indol-2-yl)-methanones as potent inhibitors of FLT3 and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2006, 49, 3101–3115. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.N.; Meyer, E.B.; Stahl, S.S. Regiocontrolled aerobic oxidative coupling of indoles and benzene using Pd catalysts with 4,5-diazafluorene ligands. Chem. Commun. 2011, 47, 10257–10259. [Google Scholar] [CrossRef] [PubMed]
- Ottoni, O.; Cruz, R.; Alves, R. Efficient and simple methods for the introduction of the sulfonyl, acyl and alkyl protecting groups on the nitrogen of indole and its derivatives. Tetrahedron 1998, 54, 13915–13928. [Google Scholar] [CrossRef]
- Johnson, A.L.; Bergmann, J. Synthetic approaches towards an indole alkaloid isolated from the marine sponge Halichondria melanodocia. Tetrahedron 2006, 62, 10815–10820. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substrate | R | R1 | Y | Yields (%) |
|---|---|---|---|---|---|
 | |||||
| 4a | 11a | Me | OMe | CH | 95 |
| 4b | 11b | Me | Br | CH | 72 |
| 4c | 11c | Me | F | CH | 48 |
| 4d | 11d | Me | H | CH | 75 |
| 4e | 12a | SO2Ph | OMe | CH | 90 |
| 4f | 12b | SO2Ph | Br | CH | 70 |
| 4g | 12c | SO2Ph | F | CH | 60 |
| 4h | 12d | SO2Ph | H | CH | 94 |
| 4i | 4e | H | OMe | CH | 50 |
| 4j | 4f | H | Br | CH | 68 |
| 4k | 4g | H | F | CH | 75 |
| 4l | 4h | H | H | CH | 80 |
| 5a | 16a | Me | H | N | 75 |
| 5b | 16b | Me | Br | N | 55 |
| 5c | 15a | H | H | N | 80 |
| 5d | 15b | H | Br | N | 85 |
| Compound | Substrate | R | R1 | Yields (%) |
|---|---|---|---|---|
 | ||||
| 6a | 23a | Me | OMe | 98 |
| 6b | 23b | Me | Br | 98 |
| 6c | 23c | Me | F | 75 |
| 6d | 23d | Me | H | 99 |
| 6e | 24a | H | OMe | 48 |
| 6f | 24b | H | Br | 75 |
| 6g | 24c | H | F | 60 |
| 6h | 22d | H | H | 60 |
| Growth Percent 1 | |||
|---|---|---|---|
| Compound | HCT116 | MCF-7 | MDA-MB-435 |
| 4a | 85.6 ± 4.3 | 24.9 ± 1.9 | 87.7 ± 4.1 |
| 4c | 87.6 ± 5.2 | 74.5 ± 4.3 | 87.9 ± 5.3 |
| 4i | 86.5 ± 4.8 | 84.8 ± 5.4 | 94.8 ± 5.5 |
| 5b | 91.7 ± 5.4 | 62.9 ± 4.2 | 96.4 ± 4.9 |
| 5d | 103.5 ± 2.3 | 47.1 ± 3.0 | 101.4 ± 3.2 |
| 6a | 87.9 ± 3.8 | 30.7 ± 3.1 | 70.1 ± 4.2 |
| 6c | 83.1 ± 4.1 | 50.3 ± 4.8 | 39.1 ± 2.7 |
| 6d | 95.3 ± 4.5 | 37.5 ± 2.2 | 82.2 ± 5.1 |
| 6g | 91.4 ± 6.4 | 41.6 ± 2.9 | 98.4 ± 4.6 |
| Compound | GI50 (µM) 1 |
|---|---|
| 4a | 2.13±0.12 |
| 6a | 3.26±0.19 |
| 6d | 5.14±0.34 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spanò, V.; Attanzio, A.; Cascioferro, S.; Carbone, A.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs. Mar. Drugs 2016, 14, 226. https://doi.org/10.3390/md14120226
Spanò V, Attanzio A, Cascioferro S, Carbone A, Montalbano A, Barraja P, Tesoriere L, Cirrincione G, Diana P, Parrino B. Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs. Marine Drugs. 2016; 14(12):226. https://doi.org/10.3390/md14120226
Chicago/Turabian StyleSpanò, Virginia, Alessandro Attanzio, Stella Cascioferro, Anna Carbone, Alessandra Montalbano, Paola Barraja, Luisa Tesoriere, Girolamo Cirrincione, Patrizia Diana, and Barbara Parrino. 2016. "Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs" Marine Drugs 14, no. 12: 226. https://doi.org/10.3390/md14120226