
Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products


Abstract
:
1. Introduction
2. A Short Walk through NGS Technologies
3. Challenges of Actinobacterial Genomics
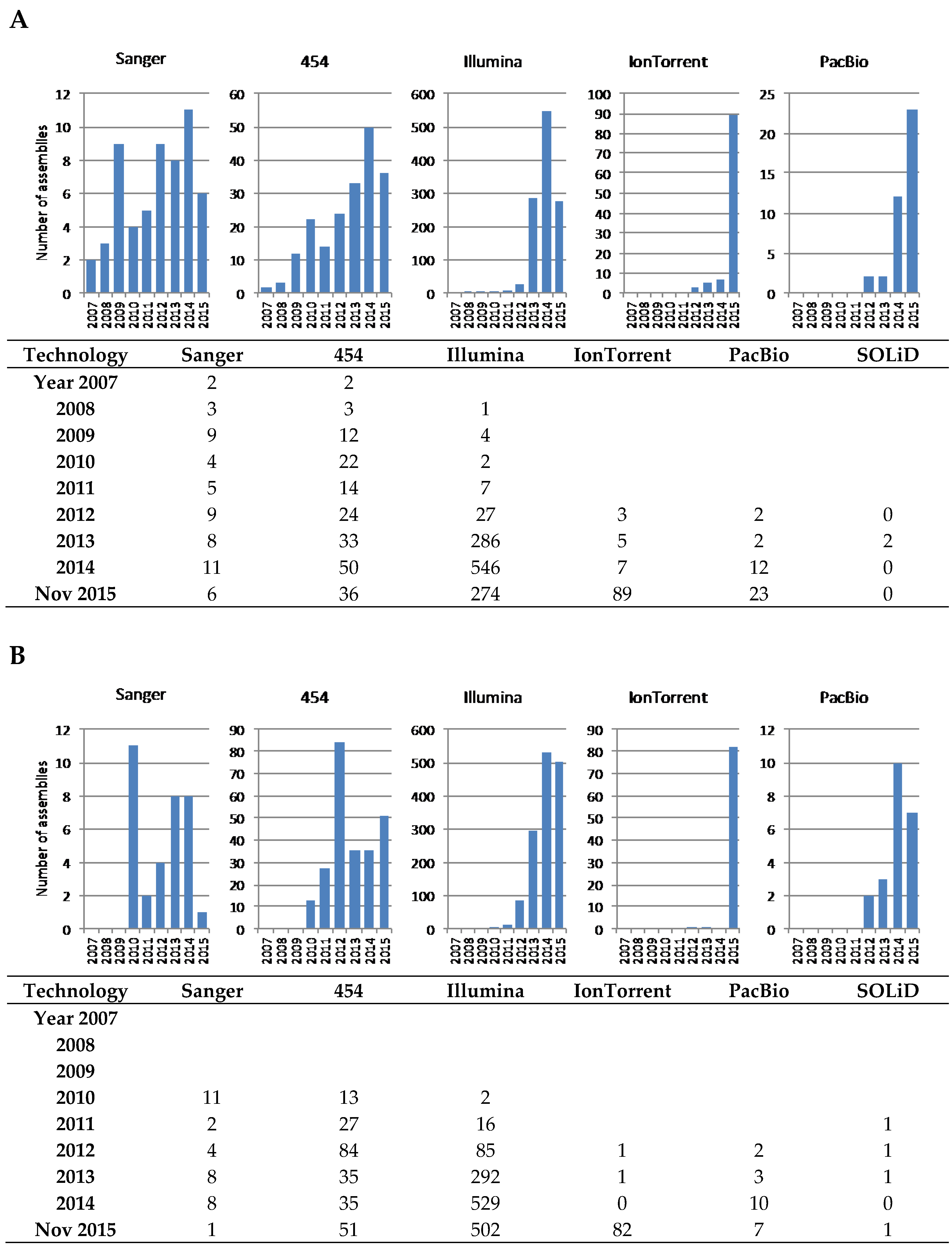
4. The Read-Length Problem
5. Historical Perspective of Actinobacterial Genome Sequencing
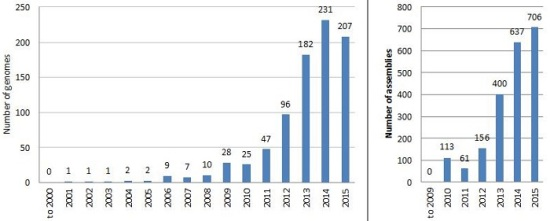
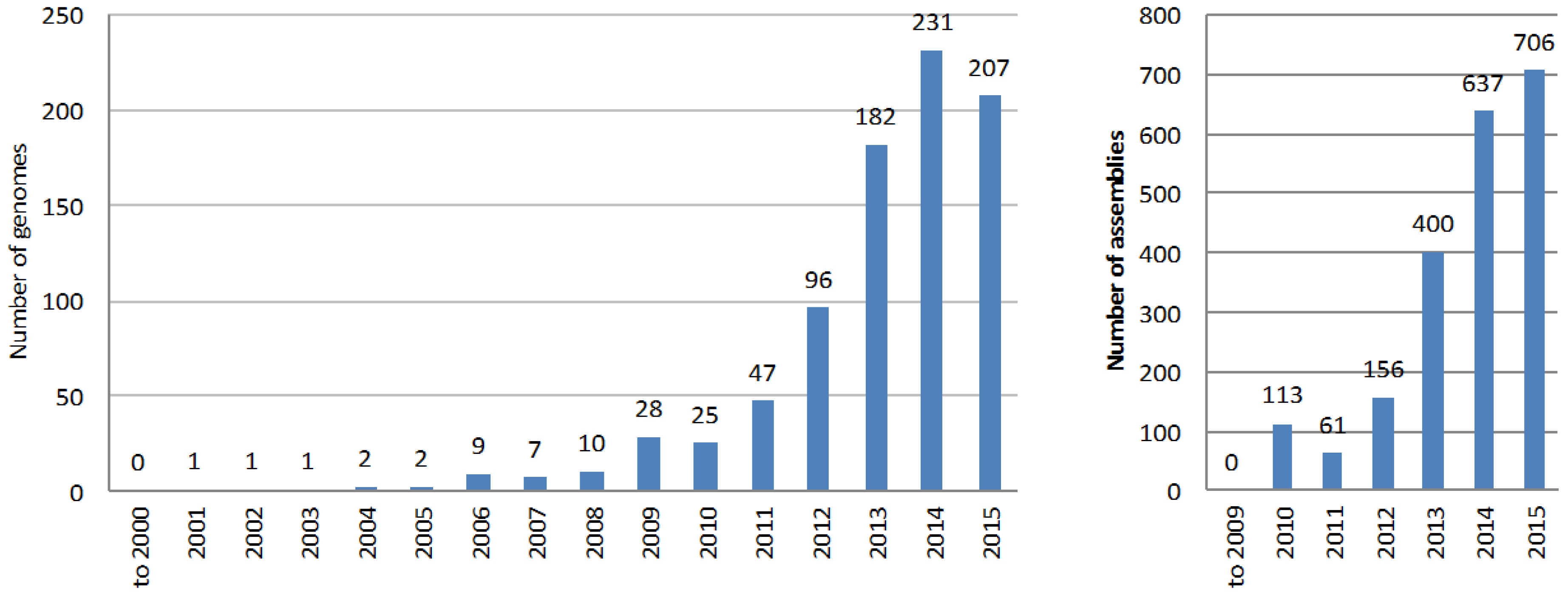
6. The Explosion of Genome Mining
7. Pacific Biosciences SMRT Platform
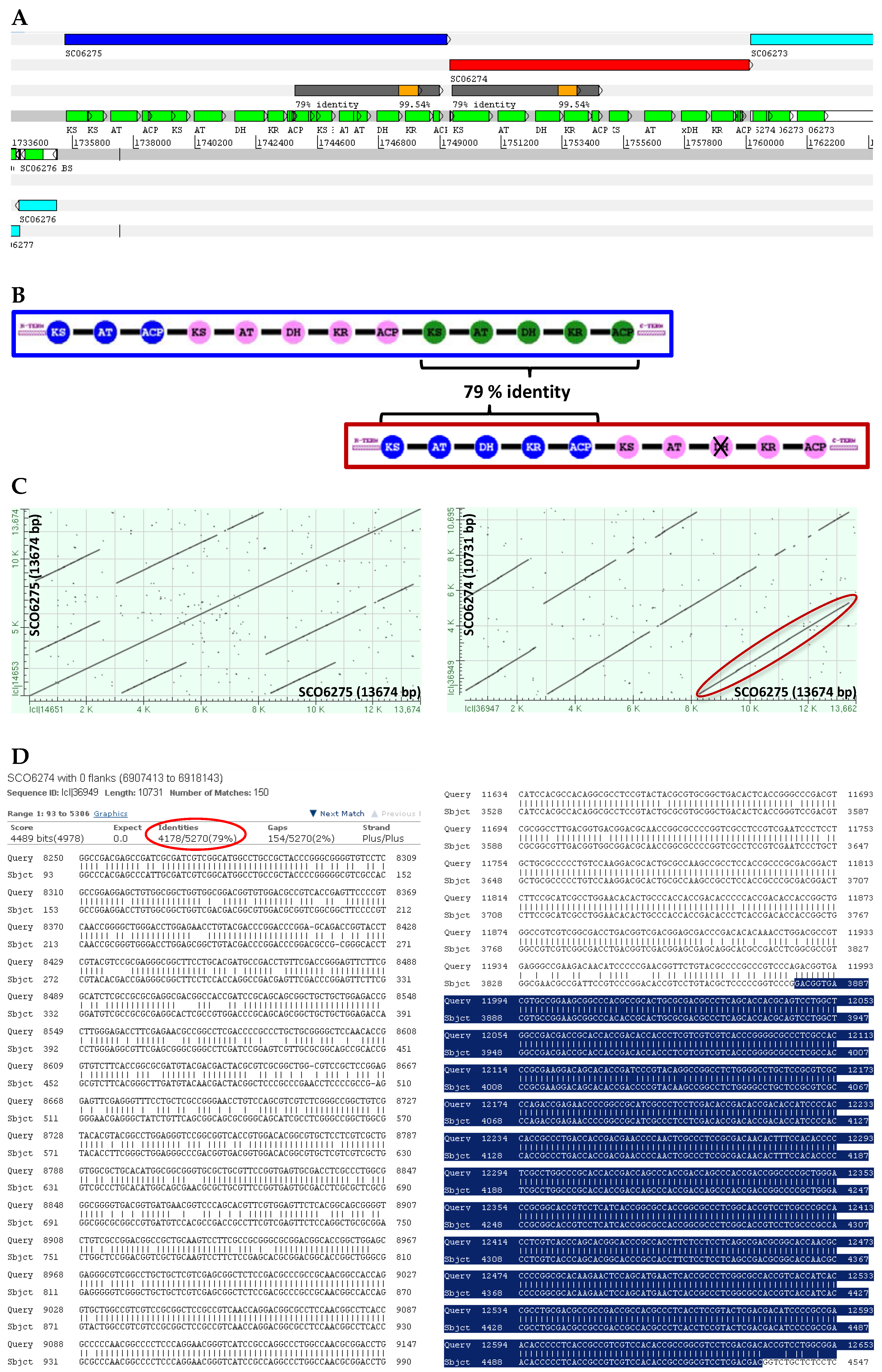
8. PKS Modularity can be Resolved with PacBio
9. Identification of Circular or Linear Replicons
10. Limitations of PacBio
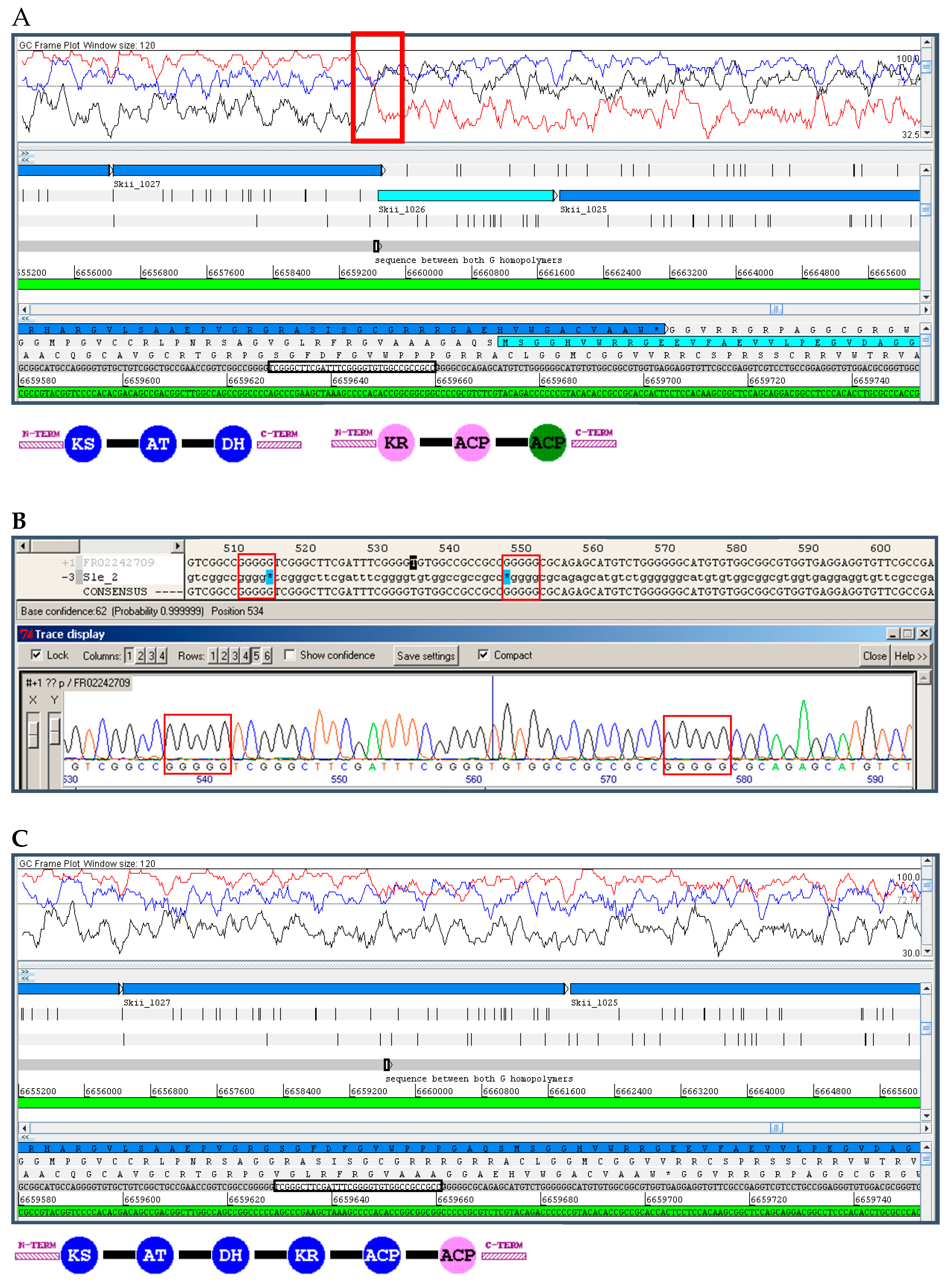
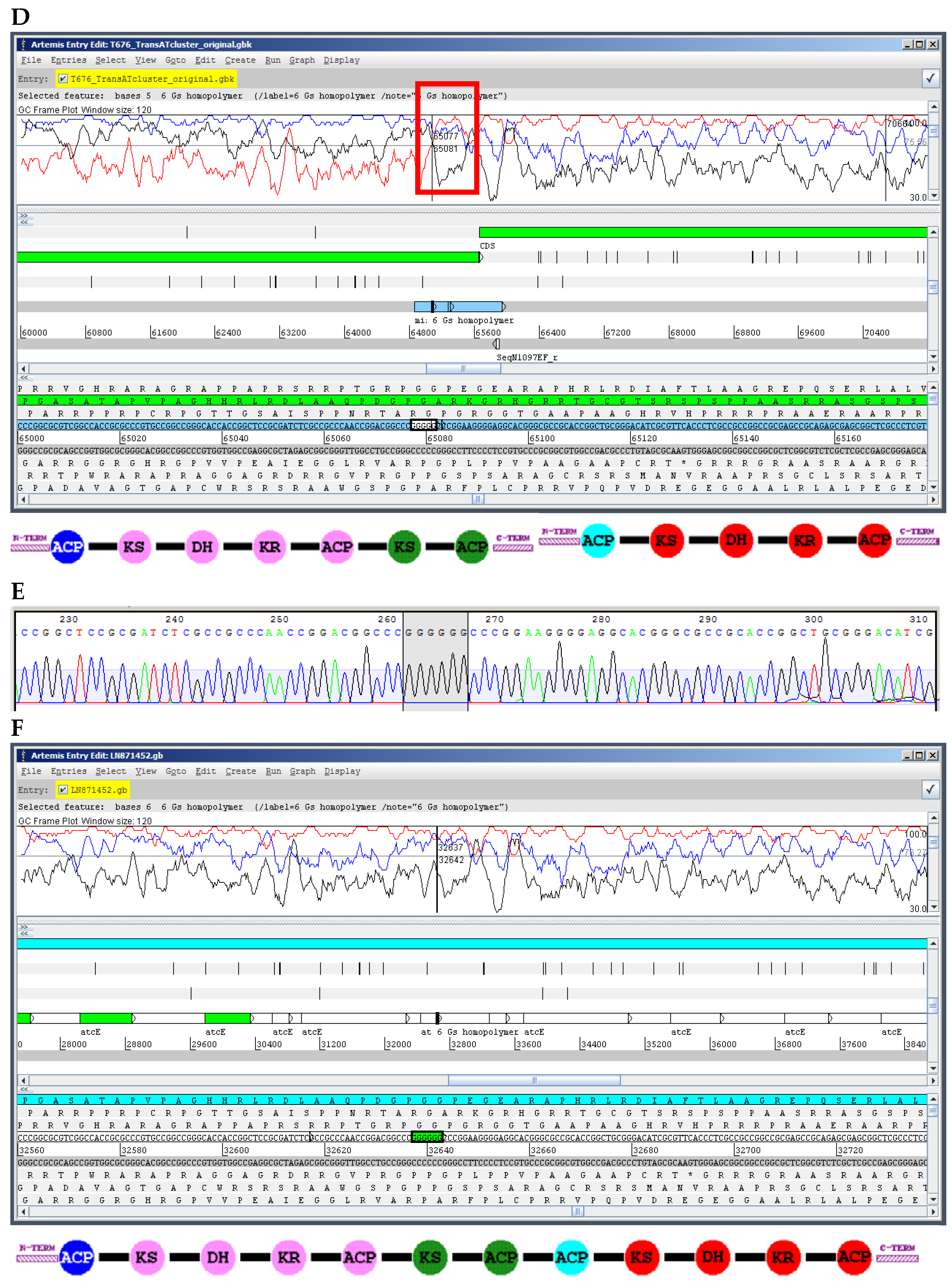
10.1. Insertions and Deletions: Shifts in the Reading Frame
10.2. Sequence Missing from the Final Assembly
11. Application to Actinobacteria from Marine Environments
12. Concluding Remarks
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| NRPS | Non-Ribosomal Peptide Synthetase |
| PKS | PolyKetide Synthase |
| PacBio | Pacific Biosciences SMRT technology |
| SGS | Second Generation Sequencing |
| TGS | Third Generation Sequencing |
| TIR | Terminal Inverted Repeat |
References
- Gomez-Escribano, J.P.; Bibb, M.J. Heterologous expression of natural product biosynthetic gene clusters in Streptomyces coelicolor: From genome mining to manipulation of biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 2014, 41, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-Q.; Wang, J.-F.; Hao, Y.-Y.; Wang, Y. Recent advances in the discovery and development of marine microbial natural products. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H. Renaissance in antibacterial discovery from actinomycetes. Curr. Opin. Pharmacol. 2008, 8, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.-M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.L. Exploitation of the Streptomyces coelicolor A3(2) genome sequence for discovery of new natural products and biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 2014, 41, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Zerikly, M.; Challis, G.L. Strategies for the discovery of new natural products by genome mining. ChemBioChem 2009, 10, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escribano, J.P.; Bibb, M.J. Streptomyces coelicolor as an expression host for heterologous gene clusters. Methods Enzymol. 2012, 517, 279–300. [Google Scholar] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Next-generation-sequencing. Available online: http://www.nature.com/subjects/next-generation-sequencing (accessed on 1 February 2016).
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0—Comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Minion-access-programme. Available online: https://nanoporetech.com/community/the-minion-access-programme (accessed on 1 February 2016).
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.; Karoonuthaisiri, N.; Tsai, H.-H.; Huang, C.-H.; Ho, M.-L.; Gai, S.; Patel, K.G.; Huang, J.; Cohen, S.N.; Hopwood, D.A.; et al. Genome plasticity in Streptomyces: Identification of 1 Mb TIRs in the S. coelicolor A3(2) chromosome. Mol. Microbiol. 2004, 51, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Doroghazi, J.R.; Metcalf, W.W. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics 2013, 14, 611. [Google Scholar] [CrossRef] [PubMed]
- Commins, J.; Toft, C.; Fares, M.A. Computational biology methods and their application to the comparative genomics of endocellular symbiotic bacteria of insects. Biol. Proced. Online 2009, 11, 52–78. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Thermofisher. Available online: http://www.thermofisher.com (accessed on 1 February 2016).
- Pacific Biosciences. Available online: http://www.pacb.com/smrt-science/smrt-sequencing/read-lengths (accessed on 1 February 2016).
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Shinose, M.; Kikuchi, H.; Shiba, T.; Sakaki, Y.; Hattori, M.; Omura, S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003, 21, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Qu, S.; Lu, C.; Zheng, H.; Zhou, X.; Bai, L.; Deng, Z. Genomic and transcriptomic insights into the thermo-regulated biosynthesis of validamycin in Streptomyces hygroscopicus 5008. BMC Genomics 2012, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Zaburannyi, N.; Rabyk, M.; Ostash, B.; Fedorenko, V.; Luzhetskyy, A. Insights into naturally minimised Streptomyces albus J1074 genome. BMC Genomics 2014, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Foulston, L.C.; Bibb, M.J. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. USA 2010, 107, 13461–13466. [Google Scholar] [CrossRef] [PubMed]
- Foulston, L. Cloning and analysis of the microbisporicin lantibiotic gene cluster from Microbispora corallina. Ph.D. Thesis, University of East Anglia, Norwich, UK, 2010. [Google Scholar]
- Claesen, J.; Bibb, M. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc. Natl. Acad. Sci. USA 2010, 107, 16297–16302. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, E.J.; Hesketh, A.R.; Bibb, M.J. Cloning and analysis of the planosporicin lantibiotic biosynthetic gene cluster of Planomonospora alba. J. Bacteriol. 2013, 195, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, E. The planosporicin gene cluster from Planomonospora alba. Ph.D. Thesis, University of East Anglia, Norwich, UK, 2011. [Google Scholar]
- Wyszynski, F.J.; Hesketh, A.R.; Bibb, M.J.; Davis, B.G. Dissecting tunicamycin biosynthesis by genome mining: Cloning and heterologous expression of a minimal gene cluster. Chem. Sci. 2010, 1, 581–589. [Google Scholar] [CrossRef]
- Gomez-Escribano, J.P.; Song, L.; Bibb, M.J.; Challis, G.L. Posttranslational [small beta]-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics. Chem. Sci. 2012, 3, 3522–3525. [Google Scholar] [CrossRef]
- Gomez-Escribano, J.P.; Castro, J.F.; Razmilic, V.; Chandra, G.; Andrews, B.; Asenjo, J.A.; Bibb, M.J. The Streptomyces leeuwenhoekii genome: De novo sequencing and assembly in single contigs of the chromosome, circular plasmid pSLE1 and linear plasmid pSLE2. BMC Genomics 2015, 16, 485. [Google Scholar] [CrossRef] [PubMed]
- Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G.L.; Aigle, B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. USA 2011, 108, 6258–6263. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Y.; Lu, C.; Zhang, J.; Zhu, J.; Wang, H.; Shen, Y. Activating a cryptic ansamycin biosynthetic gene cluster to produce three new naphthalenic octaketide ansamycins with n-pentyl and n-butyl side chains. Org. Lett. 2015, 17, 3706–3709. [Google Scholar] [CrossRef] [PubMed]
- Claesen, J.; Bibb, M.J. Biosynthesis and regulation of grisemycin, a new member of the linaridin family of ribosomally synthesized peptides produced by Streptomyces griseus IFO 13350. J. Bacteriol. 2011, 193, 2510–2516. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, H.; Lu, C.; Ding, Y.; Li, Y.; Shen, Y. Identification and characterization of the cuevaene A biosynthetic gene cluster in Streptomyces sp. LZ35. ChemBioChem 2013, 14, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Schorn, M.; Zettler, J.; Noel, J.P.; Dorrestein, P.C.; Moore, B.S.; Kaysser, L. Genetic basis for the biosynthesis of the pharmaceutically important class of epoxyketone proteasome inhibitors. ACS Chem. Biol. 2014, 9, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Bragg, L.M.; Stone, G.; Butler, M.K.; Hugenholtz, P.; Tyson, G.W. Shining a light on dark sequencing: Characterising errors in Ion Torrent PGM data. PLoS Comput. Biol. 2013, 9, e1003031. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Morales, P.; Vijgenboom, E.; Iruegas-Bocardo, F.; Girard, G.; Yáсez-Guerra, L.A.; Ramos-Aboites, H.E.; Pernodet, J.-L.; Anné, J.; van Wezel, G.P.; Barona-Gómez, F. The genome sequence of Streptomyces lividans 66 reveals a novel tRNA-dependent peptide biosynthetic system within a metal-related genomic island. Genome Biol. Evol. 2013, 5, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Girard, G.; Willemse, J.; Zhu, H.; Claessen, D.; Bukarasam, K.; Goodfellow, M.; van Wezel, G.P. Analysis of novel Kitasatosporae reveals significant evolutionary changes in conserved developmental genes between Kitasatospora and Streptomyces. Antonie Van Leeuwenhoek 2014, 106, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Hoefler, B.C.; Konganti, K.; Straight, P.D. De Novo Assembly of the Streptomyces sp. Strain Mg1 Genome Using PacBio Single-Molecule Sequencing. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.; Studholme, D.J. Recently published Streptomyces genome sequences. Microb. Biotechnol. 2014, 7, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.F.; Razmilic, V.; Gomez-Escribano, J.P.; Andrews, B.; Asenjo, J.A.; Bibb, M.J. Identification and heterologous expression of the chaxamycin biosynthesis gene cluster from Streptomyces leeuwenhoekii. Appl. Environ. Microbiol. 2015, 81, 5820–5831. [Google Scholar] [CrossRef] [PubMed]
- Busarakam, K.; Bull, A.T.; Girard, G.; Labeda, D.P.; van Wezel, G.P.; Goodfellow, M. Streptomyces leeuwenhoekii sp. nov., the producer of chaxalactins and chaxamycins, forms a distinct branch in Streptomyces gene trees. Antonie Van Leeuwenhoek 2014, 105, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Alt, S.; Wilkinson, B. Biosynthesis of the novel macrolide antibiotic anthracimycin. ACS Chem. Biol. 2015, 10, 2468–2479. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.; Bibb, M.J. Codon usage in the G+C-rich Streptomyces genome. Gene 1992, 113, 55–65. [Google Scholar] [CrossRef]
- Bibb, M.J.; Findlay, P.R.; Johnson, M.W. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 1984, 30, 157–166. [Google Scholar] [CrossRef]
- Otto, T.D.; Sanders, M.; Berriman, M.; Newbold, C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 2010, 26, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Prasad, M.V.R.; Yadav, G.; Kumar, N.; Shehara, J.; Ansari, M.Z.; Mohanty, D. SBSPKS: Structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010, 38, W487–W496. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F.; Labes, A.; Wiese, J. Bio-mining the microbial treasures of the ocean: New natural products. Biotechnol. Adv. 2011, 29, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Manivasagan, P.; Venkatesan, J.; Sivakumar, K.; Kim, S.-K. Pharmaceutically active secondary metabolites of marine actinobacteria. Microbiol. Res. 2014, 169, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.-B.; Ye, W.-W.; Han, Y.; Deng, Z.-X.; Hong, K. Natural products from mangrove actinomycetes. Mar. Drugs 2014, 12, 2590–2613. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Flemer, B.; Jackson, S.A.; Lejon, D.P.H.; Morrissey, J.P.; O’Gara, F.; Dobson, A.D.W. Marine metagenomics: New tools for the study and exploitation of marine microbial metabolism. Mar. Drugs 2010, 8, 608–628. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Marchesi, J.R.; Dobson, A.D.W. Metagenomic approaches to exploit the biotechnological potential of the microbial consortia of marine sponges. Appl. Microbiol. Biotechnol. 2007, 75, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kleigrewe, K.; Almaliti, J.; Tian, I.Y.; Kinnel, R.B.; Korobeynikov, A.; Monroe, E.A.; Duggan, B.M.; Di Marzo, V.; Sherman, D.H.; Dorrestein, P.C.; et al. Combining mass spectrometric metabolic profiling with genomic analysis: A powerful approach for discovering natural products from cyanobacteria. J. Nat. Prod. 2015, 78, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. Approaches to capturing and designing biologically active small molecules produced by uncultured microbes. Annu. Rev. Microbiol. 2011, 65, 431–453. [Google Scholar] [CrossRef] [PubMed]
- Reen, F.J.; Romano, S.; Dobson, A.D.W.; O’Gara, F. The sound of silence: Activating silent biosynthetic gene clusters in marine microorganisms. Mar. Drugs 2015, 13, 4754–4783. [Google Scholar] [CrossRef] [PubMed]
- Trindade, M.; van Zyl, L.J.; Navarro-Fernández, J.; Abd Elrazak, A. Targeted metagenomics as a tool to tap into marine natural product diversity for the discovery and production of drug candidates. Front. Microbiol. 2015, 6, 890. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Moore, B.S.; Fenical, W. The marine actinomycete genus Salinispora: A model organism for secondary metabolite discovery. Nat. Prod. Rep. 2015, 32, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. Engl. 2003, 42, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Mincer, T.J.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef] [PubMed]
- Bonet, B.; Teufel, R.; Crüsemann, M.; Ziemert, N.; Moore, B.S. Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 2015, 78, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Yorke, J.A. Beware of mis-assembled genomes. Bioinformatics 2005, 21, 4320–4321. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.R.; O’Connell, J.; Cox, A.J.; Schulz-Trieglaff, O. NxRepair: Error correction in de novo sequence assembly using Nextera mate pairs. PeerJ 2015, 3, e996. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Yurkovich, M.E.; Wen, S.; Lebe, K.E.; Samborskyy, M.; Liu, Y.; Yang, A.; Liu, Y.; Ju, Y.; Deng, Z.; et al. A genomics-led approach to deciphering the mechanism of thiotetronate antibiotic biosynthesis. Chem. Sci. 2016, 7, 376–385. [Google Scholar] [CrossRef]
- Latreille, P.; Norton, S.; Goldman, B.S.; Henkhaus, J.; Miller, N.; Barbazuk, B.; Bode, H.B.; Darby, C.; Du, Z.; Forst, S.; et al. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics 2007, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Muggli, M.D.; Puglisi, S.J.; Ronen, R.; Boucher, C. Misassembly detection using paired-end sequence reads and optical mapping data. Bioinformatics 2015, 31, i80–i88. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Ishikawa, J.; Hara, H.; Suzuki, H.; Ikenoya, M.; Ikeda, H.; Yamashita, A.; Hattori, M.; Horinouchi, S. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 2008, 190, 4050–4060. [Google Scholar] [CrossRef] [PubMed]
- Seven days. The news in brief. Business. End sequence. Available online: http://www.nature.com/news/seven-days-18-24-october-2013-1.13994 (accessed on 8 April 2016).
- Frasch, H.-J.; Medema, M.H.; Takano, E.; Breitling, R. Design-based re-engineering of biosynthetic gene clusters: Plug-and-play in practice. Curr. Opin. Biotechnol. 2013, 24, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.J.; Donia, M.S.; Siqueira-Neto, J.L.; Ray, D.; Raskatov, J.A.; Green, R.E.; McKerrow, J.H.; Fischbach, M.A.; Linington, R.G. Genome-directed lead discovery: Biosynthesis, structure elucidation, and biological evaluation of two families of polyene macrolactams against Trypanosoma brucei. ACS Chem. Biol. 2015, 10, 2373–2381. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | Technology | Year | Number of contigs1 | N50 contig (nt) | Longest contig (nt) | Total sum of contigs | Total data | Lanes/ runs | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Microbispora corallina NRRL 30420 | Solexa/ Illumina | Mid 2007 | 14395 | 163 | 4436 | 2.93 Mb | 881 Mb | 7 | [37] |
| Microbispora corallina NRRL 30420 | 454 | Mid 2008 | 7580 (3027) | 1219 | 8913 | 4.64 Mb | 28 Mb | 1/4 | [37] |
| Streptomyces sp. OH-4156 | Solexa/ Illumina | Mid 2007 | 15,471 | 378 | 7830 | 8.5 Mb | [38] | ||
| Planomonospora alba NRRL 18924 | 454 | Mid 2009 | 3066 (1618) | 756 | 14,767 | 2.32 Mb | 13 Mb | 1/8 | [40] |
| Planomonospora alba NRRL 18924 | 454 | Mid 2011 | 1017 (944) | 17,314 | 141,100 | 7.3 Mb | 72 Mb | 1/4 | [40] |
| Streptomyces chartreusis NRRL 3882 | 454 | 2008 | 3112 | 4582 | 53,916 | 7.95 Mb | 286 Mb | 1/2 | [41] |
| Streptomyces bottropensis DSM 40262 | 454 | End 2010 | 463 (427) | 40,440 | 183,403 | 8.85 Mb | 115 Mb | 1/4 | [42] |
| Streptomyces leeuwenhoekii DSM 42122 | Illumina MiSeq | Mid 2013 | 387 (279) (175 scaf.)2 | 59,284 | 157,225 | 8.1 Mb | 712 Mb (1.25 Gb) | Full (500 cycles) | [43] |
| Streptomyces leeuwenhoekii DSM 42122 | PacBio | End 2013 | 3 | 7,895,833 | 7,895,833 | 8 Mb | 966 Mb | 2 (3) cells | [43] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Escribano, J.P.; Alt, S.; Bibb, M.J. Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products. Mar. Drugs 2016, 14, 78. https://doi.org/10.3390/md14040078
Gomez-Escribano JP, Alt S, Bibb MJ. Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products. Marine Drugs. 2016; 14(4):78. https://doi.org/10.3390/md14040078
Chicago/Turabian StyleGomez-Escribano, Juan Pablo, Silke Alt, and Mervyn J. Bibb. 2016. "Next Generation Sequencing of Actinobacteria for the Discovery of Novel Natural Products" Marine Drugs 14, no. 4: 78. https://doi.org/10.3390/md14040078