
Marine Invertebrate Metabolites with Anticancer Activities: Solutions to the “Supply Problem”


Abstract
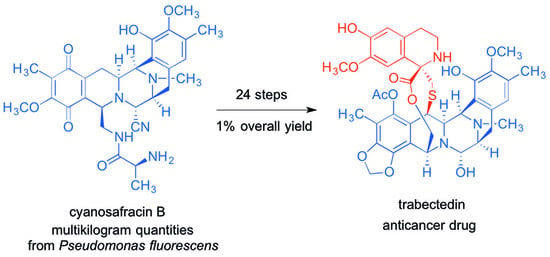
:
1. Introduction
2. Solutions to the “Supply Problem”
2.1. Biosynthetic Origin of Marine Invertebrate Metabolites
2.2. Marine Invertebrate Aquaculture
2.3. Invertebrate and Symbiont Cell Culture
2.4. Culture-Independent Strategies
2.5. Total Chemical Synthesis
2.6. Semisynthesis
2.7. Synthetic Access to Marine Invertebrate Metabolites with Promising Anticancer Activities
2.8. Hybrid Strategies
3. Future Prospects
Acknowledgments
Conflicts of Interest
Abbreviations
| Alloc | Allyloxycarbonyl |
| Bn | Benzyl |
| Boc | tert-Butyloxycarbonyl |
| BuLi | n-Butyllithium |
| Cbz | Carboxybenzyl |
| CIP | 2-Chloro-1,3-dimethylimidazolidinium hexafluorophosphate |
| DIBAL | Diisobutylaluminium hydride |
| DMSO | Dimethyl sulfoxide |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| HOAt | 1-Hydroxy-7-azabenzotriazole |
| MOM | Methoxymethyl |
| NCI | US National Cancer Institute |
| NHK | Nozaki-Hiyama-Kishi reaction |
| NRPS | Non-ribosomal peptide synthetase |
| PKC | Protein kinase C |
| PLD | Pegylated liposomal doxoribucin |
| PPTS | Pyridinium p-toluenesulfonate |
| TBAF | Tetra-n-butylammonium fluoride |
| TBDPS | tert-Butyldiphenylsilyl |
| TBS | tert-Butyldimethylsilyl |
| Tf2O | Trifluoromethanesulfonic anhydride |
| THF | Tetrahydrofuran |
| trans-AT-PKS | trans Acyltransferase-polyketide synthase |
| Troc | Trichloroethyl chloroformate |
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Ready for a comeback of natural products in oncology. Biochem. Pharmacol. 2009, 77, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A.D.; Chin, Y.-W.; Swanson, S.M. Discovery of natural product anticancer agents from biodiverse organisms. Curr. Opin. Drug. Discov. Dev. 2009, 12, 189–196. [Google Scholar]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2014, 13, 123–137. [Google Scholar] [CrossRef]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer. Ther. 2005, 4, 333–342. [Google Scholar] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Sawadogo, W.R.; Schumacher, M.; Teiten, M.H.; Cerella, C.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2011. Molecules 2013, 18, 3641–3673. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.A.; Fedorov, S.N. Marine low molecular weight natural products as potential cancer preventive compounds. Mar. Drugs 2014, 12, 636–671. [Google Scholar] [CrossRef] [PubMed]
- Sawadogo, W.R.; Boly, R.; Cerella, C.; Teiten, M.H.; Dicato, M.; Diederich, M. A Survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2012. Molecules 2015, 20, 7097–7142. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Gustafson, K.R. Marine pharmacology in 2001–2002: Antitumour and cytotoxic compounds. Eur. J. Cancer 2004, 40, 2676–2704. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Gustafson, K.R. Marine pharmacology in 2003–2004: Anti-tumour and cytotoxic compounds. Eur. J. Cancer 2006, 42, 2241–2270. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Gustafson, K.R. Marine pharmacology in 2005–2006: Antitumour and cytotoxic compounds. Eur. J. Cancer 2008, 44, 2357–2387. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.H.; Stone, M.J.; Hauck, P.R.; Rahman, S.K. Why are secondary metabolites (natural products) biosynthesized? J. Nat. Prod. 1989, 52, 1189–1208. [Google Scholar] [CrossRef] [PubMed]
- Firn, R.D.; Jones, C.G. Natural products: A simple model to explain chemical diversity. Nat. Prod. Rep. 2003, 20, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.J.; Puglisi, M.P. Chemical mediation of interactions among marine organisms. Nat. Prod. Rep. 2004, 21, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.J.; Puglisi, M.P.; Ritson-Williams, R. Marine chemical ecology. Nat. Prod. Rep. 2006, 23, 153–180. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.J.; Ritson-Williams, R.; Sharp, K. Marine chemical ecology in benthic environments. Nat. Prod. Rep. 2011, 28, 345–387. [Google Scholar] [CrossRef] [PubMed]
- Cooper, E.L.; Yao, D. Diving for drugs: Tunicate anticancer compounds. Drug Discov. Today 2012, 17, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from marine invertebrates as important leads for anticancer drugs discovery and development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejin, B.; Mojovic, M.; Savic, A.G. Novel and highly potent antitumour natural products from cnidarians of marine origin. Nat. Prod. Res. 2014, 28, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, W.; Feeney, R.J. The isolation of a new thymine pentoside from sponges 1. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar] [CrossRef]
- Bergmann, W.; Feeney, R.J. Contributions to the study of marine products. XXXII. The nucleosides of sponges I. J. Org. Chem. 1951, 16, 981–987. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Gloer, J.B.; Hughes, R.G., Jr.; Renis, H.E.; McGovren, J.P.; Swynenberg, E.B.; Stringfellow, D.A.; Kuentzel, S.L.; Li, L.H. Didemnins: Antiviral and antitumor depsipeptides from a Caribbean tunicate. Science 1981, 212, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Nuijen, B.; Bouma, M.; Manada, C.; Jimeno, J.M.; Schellens, J.H.M.; Bult, A.; Beijnen, J.H. Pharmaceutical development of anticancer agents derived from marine sources. Anticancer Drugs 2000, 11, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Puga, J.; Serôdio, J.; Gomes, N.C.; Calado, R. Trends in the discovery of new marine natural products from invertebrates over the last two decades—Where and what are we bioprospecting? PLoS ONE 2012, 7, e30580. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.-K. Marine antitumor drugs: Status, shortfalls and strategies. Mar. Drugs 2010, 8, 2702–2720. [Google Scholar] [CrossRef] [PubMed]
- Marine Pharmaceuticals. The Clinical Pipeline. Available online: http://marinepharmacology.midwestern.edu/clinPipeline.htm (accessed on 10 March 2016).
- National Cancer Institute. Cytarabine. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/cytarabine (accessed on 10 March 2016).
- European Medicines Agency. Cytarabine EU/3/11/942. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/orphans/2012/02/human_orphan_001014.jsp&mid=WC0b01ac058001d12b&source=homeMedSearch (accessed on 10 March 2016).
- National Cancer Institute. Cytarabine Liposome. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/cytarabineliposome (accessed on 10 March 2016).
- European Medicines Agency. DepoCyte. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000317/human_med_000740.jsp&mid=WC0b01ac058001d124 (accessed on 10 March 2016).
- ClinicalTrials.gov. Cytarabine (Open Studies). Available online: https://www.clinicaltrials.gov/ct2/results?term=cytarabine&recr=Open&no_unk=Y (accessed on 10 March 2016).
- EU Clinical Trials Register. Clinical trials for Cytarabine. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=Cytarabine+&status=ongoing (accessed on 10 March 2016).
- European Medicines Agency. Trabectedin. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000773/human_med_001165.jsp&mid=WC0b01ac058001d124 (accessed on 10 March 2016).
- National Cancer Institute. Trabectedin. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/trabectedin (accessed on 10 March 2016).
- Uemura, D.; Takahashi, K.; Yamamoto, T.; Katayama, C.; Tanaka, J.; Okumura, Y.; Hirata, Y. Norhalichondrin A: An antitumor polyether macrolide from a marine sponge. J. Am. Chem. Soc. 1985, 107, 4796–4798. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- European Medicines Agency. Halaven. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002084/human_med_001427.jsp&mid=WC0b01ac058001d124 (accessed on 10 March 2016).
- National Cancer Institute. Eribulin Mesylate. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/eribulinmesylate (accessed on 10 March 2016).
- EU Clinical Trials Register. Clinical trials for Eribulin Mesylate. Available online: https://www.clinicaltrialsregister.eu/ctr-search/search?query=Eribulin+Mesylate+&status=ongoing (accessed on 10 March 2016).
- ClinicalTrials.gov. Eribulin Mesylate (ongoing). Available online: https://www.clinicaltrials.gov/ct2/results?term=Eribulin+Mesylate&recr=Open&no_unk=Y (accessed on 10 March 2016).
- National Cancer Institute. Brentuximab Vedotin. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/brentuximabvedotin (accessed on 10 March 2016).
- European Medicines Agency. Adcetris. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002455/human_med_001588.jsp&mid=WC0b01ac058001d124 (accessed on 10 March 2016).
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Vaishampayan, U.; Glode, M.; Du, W.; Kraft, A.; Hudes, G.; Wright, J.; Hussain, M. Phase II study of dolastatin 10 in patients with hormone-refractory metastatic prostate adenocarcinoma. Clin. Cancer Res. 2000, 6, 4205–4208. [Google Scholar] [PubMed]
- Hoffman, M.; Blessing, J.; Lentz, S. A phase II trial of dolastatin-10 in recurrent platinum-sensitive ovarian carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2003, 89, 95–98. [Google Scholar] [CrossRef]
- Thoms, C.; Schupp, P. Biotechnological potential of marine sponges and their associated bacteria as producers of new pharmaceuticals (Part II). JIBL 2005, 2, 257–264. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Bowden, B.F.; Toth, S.I. Antitumor and cytotoxic compounds from marine organisms. In Marine Biotechnolog. Pharmaceutical and Bioactive Natural Products; Attaway, D.H., Zaborsky, O.R., Eds.; Plenum: New York, NY, USA, 1993; Volume 1, pp. 197–308. [Google Scholar]
- Allen, M.J.; Jaspars, M. Realizing the potential of marine biotechnology: Challenges & opportunities. Ind. Biotechnol. 2009, 5, 77–83. [Google Scholar]
- Desbois, A.P. How might we increase success in marine-based drug discovery? Expert Opin. Drug Discov. 2014, 9, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Clardy, J. Bacterial symbionts and natural products. Chem. Commun. 2011, 47, 7559–7566. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, T.; Niederkrüger, H.; Gernert, C.; Siegl, A.; Taudien, S.; Platzer, M.; Crews, P.; Hentschel, U.; Piel, J. Linking chemical and microbial diversity in marine sponges: Possible role for poribacteria as producers of methyl-branched fatty acids. ChemBioChem 2010, 11, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.S.; Taylor, M.W.; Benham, F.; Lücker, S.; Rattei, T.; Whalan, S.; Horn, M.; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 2010, 12, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, U.; Hopke, J.; Horn, M.; Friedrich, A.B.; Wagner, M.; Hacker, J.; Moore, B.S. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbiol. 2002, 68, 4431–4440. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Wehrl, M.; Bayer, K.; Siegl, A.; Hentschel, U. Marine sponges as models for commensal microbe-host interactions. Symbiosis 2007, 44, 43–50. [Google Scholar]
- Thacker, W.; Starnes, S. Host specificity of the symbiotic cyanobacterium Oscillatoria spongeliae in marine sponges, Dysidea spp. Mar. Biol. 2003, 142, 643–648. [Google Scholar]
- Usher, K.M.; Fromont, J.; Sutton, D.C.; Toze, S. The biogeography and phylogeny of unicellular cyanobacterial symbionts in sponges from Australia and the Mediterranean. Microb. Ecol. 2004, 48, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gandelman, J.F.; Giambiagi-deMarval, M.; Oelemann, W.M.R.; Laport, M.S. Biotechnological potential of sponge-associated bacteria. Curr. Pharm. Biotechnol. 2014, 15, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Hathaway, B.J.; Sudek, S.; Haygood, M.G.; Rosovitz, M.J.; Ravel, J.; Schmidt, E.W. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat. Chem. Biol. 2006, 2, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Fricke, W.F.; Ravel, J.; Schmidt, E.W. Variation in tropical reef symbiont metagenomes defined by secondary metabolism. PLoS ONE 2011, 6, e17897. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Donia, M.S.; McIntosh, J.A.; Fricke, W.F.; Ravel, J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod. 2012, 75, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S. Biosynthesis of marine natural products: Microorganisms and macroalgae. Nat. Prod. Rep. 1999, 16, 653–674. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S. Biosynthesis of marine natural products: Macroorganisms (Part B). Nat. Prod. Rep. 2006, 23, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Donia, M.S. Life in cellulose houses: Symbiotic bacterial biosynthesis of ascidian drugs and drug leads. Curr. Opin. Biotechnol. 2010, 21, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.L.; Hill, R.T.; Place, A.R.; Hamann, M.T. The expanding role of marine microbes in pharmaceutical development. Curr. Opin. Biotechnol. 2010, 21, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.M.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can some marine-derived fungal metabolites become actual anticancer agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
- König, G.M.; Kehraus, S.; Seibert, S.F.; Abdel-Lateff, A.; Müller, D. Natural products from marine organisms and their associated microbes. ChemBioChem 2006, 7, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Uria, A.; Piel, J. Cultivation-independent approaches to investigate the chemistry of marine symbiotic bacteria. Phytochem. Rev. 2009, 8, 401–414. [Google Scholar] [CrossRef]
- Giddings, L.-A.; Newman, D.J. Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 2013, 40, 1181–1210. [Google Scholar] [CrossRef] [PubMed]
- Tsukimoto, M.; Nagaoka, M.; Shishido, Y.; Fujimoto, J.; Nishisaka, F.; Matsumoto, S.; Harunari, E.; Imada, C.; Matsuzaki, T. Bacterial production of the tunicate-derived antitumor cyclic depsipeptide didemnin B. J. Nat. Prod. 2011, 74, 2329–2331. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.M.; Mironenko, T.; Sun, Y.; Hong, H.; Deng, Z.; Leadlay, P.F.; Weissman, K.J.; Haydock, S.F. Insights into polyether biosynthesis from analysis of the nigericin biosynthetic gene cluster in Streptomyces sp. DSM4137. Chem. Biol. 2007, 14, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Demydchuk, Y.; Sun, Y.; Hong, H.; Staunton, J.; Spencer, J.B.; Leadlay, P.F. Analysis of the tetronomycin gene cluster: Insights into the biosynthesis of a polyether tetronate antibiotic. ChemBioChem 2008, 9, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Yoshida, W.Y.; Moore, R.E.; Nagle, D.G.; Park, P.U.; Biggs, J.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H.; Valeriote, F.A. Isolation, structure determination, and biological activity of dolastatin 12 and lyngbyastatin 1 from Lyngbya majuscula/Schizothrix calcicola cyanobacterial assemblages. J. Nat. Prod. 1998, 61, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Barr, P.M.; Lazarus, H.M.; Cooper, B.W.; Schluchter, M.D.; Panneerselvam, A.; Jacobberger, J.W.; Hsu, J.W.; Janakiraman, N.; Simic, A.; Dowlati, A.; et al. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009, 84, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.-J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.K.; Allen, S.W.; Lim, G.E.; Anderson, C.M.; Haygood, M.G. Evidence for the biosynthesis of bryostatins by the bacterial symbiont, Candidatus Endobugula sertula, of the bryozoan Bugula neritina. Appl. Environ. Microbiol. 2001, 67, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Sudek, S.; Lopanik, N.B.; Waggoner, L.E.; Hildebrand, M.; Anderson, C.; Liu, H.; Patel, A.; Sherman, D.H.; Haygood, M.G. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoans Bugula neritina. J. Nat. Prod. 2007, 70, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lopanik, N.B.; Lindquist, N.; Targett, N. Potent cytotoxins produced by a microbial symbiont protect host larvae from predation. Oecologia 2004, 139, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Scheuer, P.J. Kahalalide F: A bioactive depsipeptide from the sacoglossan mollusk Elysia rufescens and the green alga Bryopsis sp. J. Am. Chem. Soc. 1993, 115, 5825–5826. [Google Scholar] [CrossRef]
- Hamann, M.T.; Otto, C.S.; Scheuer, P.J.; Dunbar, D.C. Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp. J. Org. Chem. 1996, 61, 6594–6600. [Google Scholar] [CrossRef] [PubMed]
- Enticknap, J.; Hamann, M.T.; Hill, R.T.; Rao, K.V. Kahalalide-Producing Bacteria. WO2005042720 A2, 12 May 2005. [Google Scholar]
- Long, B.H.; Carboni, J.M.; Wasserman, A.J.; Cornell, L.A.; Casazza, A.M.; Jensen, P.R.; Lindel, T.; Fenical, W.; Fairchild, C.R. Eleutherobin, a novel cytotoxic agent that induces tubulin polymerization, is similar to paclitaxel (Taxol). Cancer Res. 1998, 58, 1111–1115. [Google Scholar] [PubMed]
- Cinel, B.; Roberge, M.; Behrisch, H.; Van Ofwegen, L.; Castro, C.B.; Andersen, R.J. Antimitotic diterpenes from Erythropodium caribaeorum test pharmacophore models for microtubule stabilization. Org. Lett. 2000, 2, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. Metabolites from symbiotic bacteria. Nat. Prod. Rep. 2004, 21, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W. Trading molecules and tracking targets in symbiotic interactions. Nat. Chem. Biol. 2008, 4, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. Metabolites from symbiotic bacteria. Nat. Prod. Rep. 2009, 26, 338–362. [Google Scholar] [CrossRef] [PubMed]
- Radjasa, O.K.; Vaske, Y.M.; Navarro, G.; Vervoort, H.C.; Tenney, K.; Linington, R.G.; Crews, P. Highlights of marine invertebrate-derived biosynthetic products: Their biomedical potential and possible production by microbial associants. Bioorg. Med. Chem. 2011, 19, 6658–6674. [Google Scholar] [CrossRef] [PubMed]
- Garson, M.J.; Simpson, J.S. Marine isocyanides and related natural products–structure, biosynthesis and ecology. Nat. Prod. Rep. 2004, 21, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Chasing the treasures of the sea—bacterial marine natural products. Curr. Opin. Microbiol. 2009, 12, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. Bacterial symbionts: Prospects for the sustainable production of invertebrate-derived pharmaceuticals. Curr. Med. Chem. 2006, 13, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Coates, R.C.; Clark, B.R.; Engene, N.; Gonzalez, D.; Esquenazi, E.; Dorrestein, P.C.; Gerwick, W.H. Biosynthetic origin of natural products isolated from marine microorganism-invertebrate assemblages. Proc. Natl. Acad. Sci. USA 2008, 105, 4587–4594. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Waggoner, L.E.; Lim, G.E.; Sharp, K.H.; Ridley, C.P.; Haygood, M.G. Approaches to identifify, clone, and express symbiont bioactive metabolite genes. Nat. Prod. Rep. 2004, 21, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Pomponi, S.A. The bioprocess—Technological potential of the sea. J. Biotechnol. 1999, 70, 5–13. [Google Scholar] [CrossRef]
- Munro, M.H.G.; Blunt, J.W.; Dumdei, E.J.; Hickford, S.J.H.; Lill, R.E.; Li, S.X.; Battershill, C.N.; Duckworth, A.R. The discovery and development of marine compounds with pharmaceutical potential. J. Biotechnol. 1999, 70, 15–25. [Google Scholar] [CrossRef]
- Schaufelberger, D.E.; Koleck, M.P.; Beutler, J.A.; Vatakis, A.M.; Alvarado, A.B.; Andrews, P.; Marzo, L.V.; Muschik, G.M.; Roach, J.; Ross, J.T.; et al. The large-scale isolation of bryostatin 1 from Bugula neritina following good manufacturing practices. J. Nat. Prod. 1991, 54, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, A.; Battershill, C.N. Sponge aquaculture for the production of biologically active metabolites: The influence of farming protocols and environment. Aquaculture 2003, 221, 311–329. [Google Scholar] [CrossRef]
- Van Treeck, P.; Eisinger, M.; Müller, J.; Paster, M.; Schuhmacher, H. Mariculture trials with Mediterranean sponge species: The exploitation of an old natural resource with sustainable and novel methods. Aquaculture 2003, 218, 439–455. [Google Scholar] [CrossRef]
- Page, M.J.; Northcote, P.T.; Webb, V.L.; Mackey, S.; Handley, S.J. Aquaculture trials for the production of biologically active metabolites in the New Zealand sponge Mycale hentscheli (Demospongiae: Poecilosclerida). Aquaculture 2005, 250, 256–269. [Google Scholar] [CrossRef]
- Handley, S.J.; Page, M.J.; Northcote, P.T. Anti-cancer sponge: The race is on for aquaculture supply. Water Atmos. 2006, 14, 14–15. [Google Scholar]
- Taglialatela-Scafati, O.; Deo-Jangra, U.; Campbell, M.; Roberge, M.; Andersen, R.J. Diterpenoids from cultured Erythropodium caribaeorum. Org. Lett. 2002, 4, 4085–4088. [Google Scholar] [CrossRef] [PubMed]
- Osinga, R.; Tramper, J.; Wijffels, R.H. Cultivation of marine sponges for metabolite production: Applications for biotechnology? Trends Biotechnol. 1998, 16, 130–134. [Google Scholar] [CrossRef]
- Belarbi, E.H.; Gómez, A.C.; Chisti, Y.; Camacho, F.C.; Grima, E.M. Producing drugs from marine sponges. Biotechnol. Adv. 2003, 21, 585–598. [Google Scholar] [CrossRef]
- Duckworth, A. Farming sponges to supply bioactive metabolites and bath sponges: A review. Mar. Biotechnol. 2009, 11, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Thornton, R.S.; Kerr, R.G. Induction of pseudopterosin biosynthesis in the gorgonian Pseudopterogorgia elisabethea. J. Chem. Ecol. 2002, 28, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, E.H.; Dominguez, M.R.; Garcia, M.C.C.; Gómez, A.C.; Camacho, F.G.; Grima, E.M. Cultivation of explants of the marine sponge Crambe crambe in closed systems. Biomol. Eng. 2003, 20, 333–337. [Google Scholar] [CrossRef]
- De Caralt, S.; Agell, G.; Uriz, M.J. Long-term culture of sponge explants: Conditions enhancing survival and growth, and assessment of bioactivity. Biomol. Eng. 2003, 20, 339–347. [Google Scholar] [CrossRef]
- Duckworth, A.R.; Samples, G.A.; Wright, A.E.; Pomponi, S.A. In vitro culture of the tropical sponge Axinella corrugata (Demospongia): Effect of food cell concentration on growth, clearance rate and biosynthesis of stevensine. Mar. Biotechnol. 2003, 5, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Mendola, D. Aquaculture of three phyla of marine invertebrates to yield bioactive metabolites: Process developments and economics. Biomol. Eng. 2003, 20, 441–458. [Google Scholar] [CrossRef]
- Osinga, R.; de Beukelaer, P.B.; Meijer, E.M.; Tramper, J.; Wijffels, R.H. Growth of the sponge Pseudosuberites (aff.) andrewsi in a closed system. J. Biotechnol. 1999, 70, 155–161. [Google Scholar] [CrossRef]
- Koopmans, M.; Martens, D.; Wijffels, R.H. Towards commercial production of sponge medicines. Mar. Drugs 2009, 7, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Zahn, R.K.; Gasic, M.J.; Dogovic, N.; Maidhof, A.; Becker, C.; Diehl-Seifert, B.; Eich, E. Avarol, a cytostatically active compound from the marine sponge Dysidea avara. Comp. Biochem. Physiol. C 1985, 80, 47–52. [Google Scholar] [CrossRef]
- Sipkema, D.; Osinga, R.; Schatton, W.; Mendola, D.; Tramper, J.; Wijffels, R.H. Large-scale production of pharmaceuticals by marine sponges: Sea, cell, or synthesis. Biotechnol. Bioeng. 2005, 90, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Grebenjuk, V.A.; Le Pennec, G.; Schröder, H.-C.; Brümmer, F.; Hentschel, U.; Müller, I.M.; Breter, H.-J. Sustainable production of bioactive compounds by sponges—Cell culture and gene cluster approach: A review. Mar. Biotechnol. 2004, 6, 105–117. [Google Scholar] [CrossRef] [PubMed]
- De Caralt, S.; Uriz, M.J.; Wijffels, R.H. Cell culture from sponges: Pluripotency and immortality. Trends Biotechnol. 2008, 25, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Sipkema, D.; van Wielink, R.; van Lammeren, A.A.M.; Tramper, J.; Osinga, R.; Wijffels, R.H. Primmorphs from seven marine sponges: Formation and structure. J. Biotechnol. 2003, 100, 127–139. [Google Scholar] [CrossRef]
- Sipkema, D.; Heilig, H.G.H.; Akkermans, A.D.L.; Osinga, R.; Tramper, J.; Wijffels, R.H. Sponge cell culture? A molecular identification method for sponge cells. Mar. Biotechnol. 2003, 5, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Krasko, A.; Le Pennec, G.; Steffen, R.; Wiens, M.; Ammar, M.S.A.; Müller, I.M.; Schröder, H.-C. Molecular mechanism of spicule formation in the demosponge Suberites domuncula—Silicatein-myotrophin-collagen. Silicon Biominer. 2003, 33, 195–222. [Google Scholar]
- Müller, W.E.G.; Böhm, M.; Batel, R.; De Rosa, S.; Tommonaro, G.; Müller, I.M.; Schröder, H.C. Application of cell culture for the production of bioactive compounds from sponges: Synthesis of avarol by primmorphs from Dysidea avara. J. Nat. Proc. 2000, 63, 1077–1082. [Google Scholar] [CrossRef]
- Penesyan, A.; Kjelleberg, S.; Egan, S. Development of novel drugs from marine surface associated microorganisms. Mar. Drugs 2010, 8, 438–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; An, R.; Wang, J.; Sun, N.; Zhang, S.; Hu, J.; Kuai, J. Exploring novel bioactive compounds from marine microbes. Curr. Opin. Microbiol. 2005, 8, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Proksch, P.; Putz, A.; Ortlepp, S.; Kjer, J.; Bayer, M. Bioactive natural products from marine sponges and fungal endophytes. Phytochem. Rev. 2010, 9, 475–489. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Fenner, A.M. Drug discovery from marine microbes. Microb. Ecol. 2013, 65, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.Q.; Wang, J.F.; Hao, Y.Y.; Wang, Y. Recent advances in the discovery and development of marine microbial natural products. Mar. Drugs 2013, 11, 700–717. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.M.; Weller, R.; Bateson, M.M. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 1990, 345, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Osinga, R.; Armstrong, E.; Burgess, J.G.; Hoffmann, F.; Reitner, J.; Schumann-Kindel, G. Sponge-microbe associations and their importance for sponge bioprocess engineering. Hydrobiologia 2001, 461, 55–62. [Google Scholar] [CrossRef]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Barberel, S.I.; Walker, J.R.L. The effect of aeration upon the secondary metabolism of microorganisms. Biotechnol. Genet. Eng. Rev. 2000, 17, 281–323. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, C.; Theobald, U.; Gurtler, H.; Fiedler, H. Improved secondary metabolite production in the genus Streptosporangium by optimization of the fermentation conditions. J. Biotechnol. 2001, 23, 135–142. [Google Scholar] [CrossRef]
- Yan, L.; Boyd, K.G.; Burgess, J.G. Surface attachment induced production of antimicrobial compounds by marine epiphytic bacteria using modified roller bottle cultivation. Mar. Biotechnol. 2002, 4, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Marman, A.; Aly, A.H.; Lin, W.; Wang, B.; Proksch, P. Co-cultivation—A powerful emerging tool for enhancing the chemical diversity of microorganisms. Mar. Drugs 2014, 12, 1043–1065. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Peraud, O.; Hamann, M.; Kasanah, N. Manzamine-Producing Actinomycetes. U.S. Patent 20,050,244,938 A1, 3 November 2005. [Google Scholar]
- Dunlap, W.C.; Battershill, C.N.; Liptrot, C.H.; Cobb, R.E.; Bourne, D.G.; Jaspars, M.; Long, P.F.; Newman, D.J. Biomedicinals from the photosymbionts of marine invertebrates: A molecular approach. Methods 2007, 42, 358–376. [Google Scholar] [CrossRef] [PubMed]
- Salomon, C.E.; Magarvey, N.A.; Sherman, D.H. Merging the potential of microbial genetics with biological and chemical diversity: An even brighter future for marine natural product drug discovery. Nat. Prod. Rep. 2004, 21, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.; Gabor, E.M.; Liebeton, K.; Meurer, G.; Niehaus, F.; Schulze, R.; Eck, J.; Lorenz, P. Metagenomics: An inexhaustible access to nature’s diversity. Biotechnol. J. 2006, 1, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Piel, J. Metagenomic approaches for exploiting uncultivated bacteria as a resource for novel biosynthetic enzymology. Chem. Biol. 2013, 20, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Fortman, J.L.; Sherman, D.H. Utilizing the power of microbial genetics to bridge the gap between the promise and the application of marine natural products. ChemBioChem 2005, 6, 960–978. [Google Scholar] [CrossRef] [PubMed]
- Schofield, M.M.; Sherman, D.H. Meta-omic characterization of prokaryotic gene clusters for natural product biosynthesis. Curr. Opin. Biotechnol. 2013, 24, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Sorting out metagenomes. Nat. Biotechnol. 2005, 23, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, B.A.; Khosla, C. Biosynthesis of polyketides in heterologous hosts. Microbiol. Mol. Biol. Rev. 2001, 65, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, B.A.; Admiraal, S.J.; Gramajo, H.; Cane, D.E.; Khosla, C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 2001, 291, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Khosla, C.; Keasling, J.D. Metabolic engineering for drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Gross, F.; Zhang, Y.; Fu, J.; Stewart, A.F.; Muller, R. Heterologous expression of a myxobacterial natural products assembly line in pseudomonads via red/ET recombineering. Chem. Biol. 2005, 12, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Sarovich, D.S.; Pemberton, J.M. pPSX: A novel vector for the cloning and heterologous expression of antitumor antibiotic gene clusters. Plasmid 2007, 57, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Butzin, N.C.; Owen, H.A.; Collins, M.L. A new system for heterologous expression of membrane proteins: Rhodospirillum rubrum. Protein Expr. Purif. 2010, 70, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [Google Scholar] [CrossRef] [PubMed]
- Piel, J.; Butzke, D.; Fusetani, N.; Hui, D.; Platzer, M.; Wen, G.; Matsunaga, S. Exploring the chemistry of uncultivated bacterial symbionts: Antitumor polyketides of the Pederin family. J. Nat. Prod. 2005, 68, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kersten, R.D.; Nam, S.-J.; Lu, L.; Al-Suwailem, A.M.; Zheng, H.; Fenical, W.; Dorrestein, P.C.; Moore, B.S.; Qian, P.-Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012, 134, 8625–8632. [Google Scholar] [CrossRef] [PubMed]
- Long, P.F.; Dunlap, W.C.; Battershill, C.N.; Jaspars, M. Shotgun cloning and heterologous expression of the patellamide gene cluster as a strategy to achieving sustained metabolite production. ChemBioChem 2005, 6, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. USA 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Marchesi, J.R.; Dobson, A.D.W. Metagenomic approaches to exploit the biotechnological potential of the microbial consortia of marine sponges. Appl. Microbiol. Biotechnol. 2007, 75, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, J.E.; van der Donk, W.A. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011, 15, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Schulte, G.K. Discodermolide: A new bioactive polyhydroxylated lactone from the marine sponge Discodermia dissoluta. J. Org. Chem. 1990, 55, 4912–4915. [Google Scholar] [CrossRef]
- Florence, G.J.; Gardner, N.M.; Paterson, I. Development of practical syntheses of the marine anticancer agents discodermolide and dictyostatin. Nat. Prod. Rep. 2008, 25, 342–375. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Freeze, B.S. (+)-Discodermolide: Total Synthesis, Construction of Novel Analogues, and Biological Evaluation. Tetrahedron 2007, 64, 261–298. [Google Scholar] [CrossRef] [PubMed]
- Mickel, S.J.; Sedelmeier, G.H.; Niederer, D.; Schuerch, F.; Grimler, D.; Koch, G.; Daeffler, R.; Osmani, A.; Hirni, A.; Schaer, K.; et al. Large-Scale Synthesis of the Anti-Cancer Marine Natural Product (+)-Discodermolide. Part 2: Synthesis of Fragments C1–6 and C9–14. Org. Proc. Res. Dev. 2004, 8, 101–106. [Google Scholar] [CrossRef]
- Mickel, S.J.; Sedelmeier, G.H.; Niederer, D.; Schuerch, F.; Koch, G.; Kuesters, E.; Daeffler, R.; Osmani, A.; Seeger-Weibel, M.; Schmid, E.; Hirni, A.; et al. Large-Scale Synthesis of the Anti-Cancer Marine Natural Product (+)-Discodermolide. Part 3: Synthesis of Fragment C15–21. Org. Proc. Res. Dev. 2004, 8, 107–112. [Google Scholar] [CrossRef]
- Mickel, S.J.; Sedelmeier, G.H.; Niederer, D.; Schuerch, F.; Seger, M.; Schreiner, K.; Daeffler, R.; Osmani, A.; Bixel, D.; Loiseleur, O.; et al. Large-Scale Synthesis of the Anti-Cancer Marine Natural Product (+)-Discodermolide. Part 4: Preparation of Fragment C7–24. Org. Proc. Res. Dev. 2004, 8, 113–121. [Google Scholar] [CrossRef]
- Mickel, S.J.; Niederer, D.; Daeffler, R.; Osmani, A.; Kuesters, E.; Schmid, E.; Schaer, K.; Gamboni, R. Large-Scale Synthesis of the Anti-Cancer Marine Natural Product (+)-Discodermolide. Part 5: Linkage of Fragments C1–6 and C7–24 and Finale. Org. Proc. Res. Dev. 2004, 8, 122–130. [Google Scholar]
- Jackson, K.L.; Henderson, J.A.; Phillips, A.J. The Halichondrins and E7389. Chem. Rev. 2009, 109, 3044–3079. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinate. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinate [Erratum to document cited in CA113(9):75189d]. J. Org. Chem. 1991, 56, 1676. [Google Scholar] [CrossRef]
- Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, A.H. Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11456–11460. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Sakai, R.; Rinehart, K.L.; Wang, A.H. Molecular and crystal structures of ecteinascidins: Potent antitumor compounds from the Caribbean tunicate Ecteinascidia turbinate. J. Biomol. Struct. Dyn. 1993, 10, 793–818. [Google Scholar] [PubMed]
- Izbicka, E.; Lawrence, R.; Raymond, E.; Eckhardt, G.; Faircloth, G.; Jimeno, J.; Clark, G.; Von Hoff, D.D. In vitro antitumor activity of the novel marine agent, Ecteinascidin-743 (ET-743, NSC-648766) against human tumors explanted from patients. Ann. Oncol. 1998, 9, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, H.R.; Fiebig, H.H.; Giavazzi, R.; Langdon, S.P.; Jimeno, J.M.; Faircloth, G.T. High antitumor activity of ET743 against human tumor xenografts from melanoma, non-small-cell lung and ovarian cancer. Ann. Oncol. 1999, 10, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Taamma, A.; Misset, J.L.; Riofrio, M.; Guzman, C.; Brain, E.; Lopez-Lazaro, L.; Rosing, H.; Jimeno, J.M.; Cvitkovic, E. Phase I and Pharmacokinetic Study of Ecteinascidin-743, a New Marine Compound, Administered as a 24-hour Continuous Infusion in Patients With Solid Tumors. J. Clin. Oncol. 2001, 19, 1256–1265. [Google Scholar] [PubMed]
- Villalona-Calero, M.A.; Eckhardt, S.G.; Weiss, G.; Hidalgo, M.; Beijnen, J.H.; Van Kesteren, C.; Rosing, H.; Campbell, E.; Lopez-Lazaro, L.; Guzman, C.; et al. A Phase I and Pharmacokinetic Study of Ecteinascidin-743 on Daily × 5 Schedule in Patients with Solid Malignancies. Clin. Cancer. Res. 2002, 8, 75–85. [Google Scholar] [PubMed]
- D’Incalci, M.; Jimeno, J. Preclinical and clinical results with the natural marine product ET-743. Expert. Opin. Investig. Drugs 2003, 12, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Janssen Products, L.P. Yondelis (trabectedin). Available online: http://www.yondelis.com (accessed on 11 May 2016).
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin Plus Pegylated Liposomal Doxorubicin in Recurrent Ovarian Cancer. J. Clin. Oncol. 2010, 28, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Krasner, C.N.; Poveda, A.; Herzog, T.J.; Vermorken, J.B.; Kaye, S.B.; Nieto, A.; Claret, P.L.; Park, Y.C.; Parekh, T.; Monk, B.J. Patient-reported outcomes in relapsed ovarian cancer: Results from a randomized Phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD Alone. Gynecol. Oncol. 2012, 127, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Lebedinsky, C.; Gomez, J.; Park, Y.C.; Nieto, A.; Soto-Matos, A.; Parekh, T.; Alfaro, V.; Roy, E.; Lardelli, P.; Kahatt, C. Trabectedin has a low cardiac risk profile: A comprehensive cardiac safety analysis. Cancer Chemother. Pharmacol. 2011, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhen, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2015, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Cancer Network Staff. FDA Approves Trabectedin (Yondelis) for Advanced Soft-tissue Sarcoma. Cancer Network 2015. Available online: http://www.cancernetwork.com/sarcoma/fda-approves-trabectedin-yondelis-advanced-soft-tissue-sarcoma (accessed on 11 May 2016).
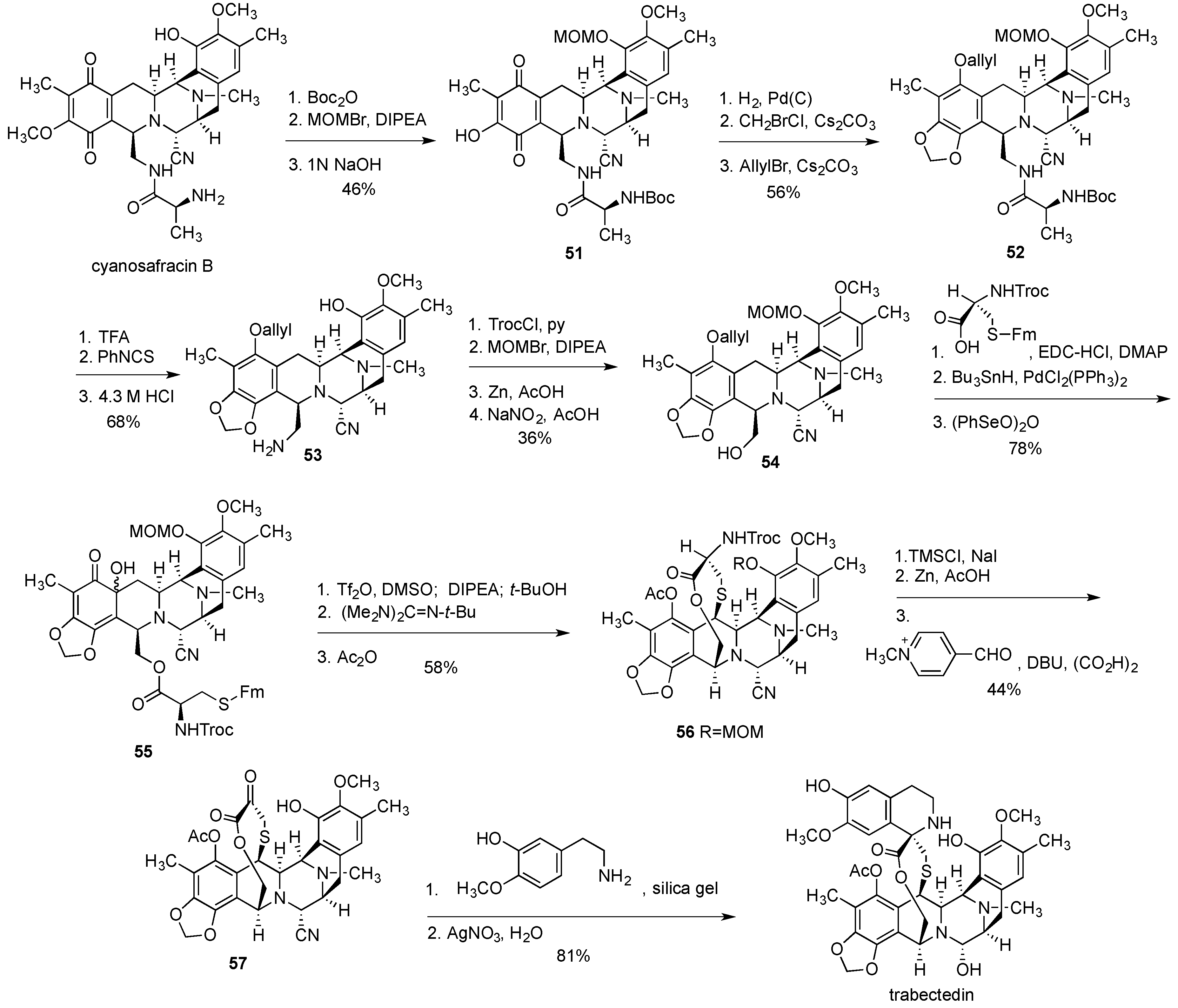
- Cuevas, C.; Francesch, A. Development of Yondelis (R) (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Carballo, J.L.; Hernandez-Zanuy, A.; Naranjo, S.; Kukurtzu, B.; Cagide, A.G. Recovery of Ecteinascidia turbinate Herman 1880 (Ascidiacea: Perophoridae) populations after different levels of harvesting on a sustainable basis. Bull. Mar. Sci. 1999, 65, 755–760. [Google Scholar]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective Total Synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- Martinez, E.J.; Corey, E.J. A new, more efficient, and effective process for the synthesis of a key pentacyclic intermediate for production of ecteinascidin and phthalascidin antitumor agents. Org. Lett. 2000, 2, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.G.; Miranda, N.F. Biosynthetic Studies of Ecteinascidins in the Marine Tunicate Ecteinascidia turbinate. J. Nat. Prod. 1995, 58, 1618–1621. [Google Scholar] [CrossRef]
- Jeedigunta, S.; Krenisky, J.M.; Kerr, R.G. Diketopiperazines as Advanced Intermediates in the Biosynthesis of Ecteinascidins. Tetrahedron 2000, 56, 3303–3307. [Google Scholar] [CrossRef]
- Corey, E.J.; Gin, D.Y. A Convergent Enantioselective Synthesis of the Tetrahydroisoquinoline Unit in the Spiro Ring of Ecteinascidin 743. Tetrahedron Lett. 1996, 37, 7163–7166. [Google Scholar] [CrossRef]
- Le, V.H.; Inai, M.; Williams, R.M.; Kan, T. Ecteinascidins. A review of the chemistry, biology and clinical utility of potent tetrahydroisoquinoline antitumor antibiotics. Nat. Prod. Rep. 2015, 32, 328–347. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.C.; Perez, M.; Martin, M.J.; Chicharro, C.F.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; de la Calle, F.; Garcia, J.; et al. Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Matsuki, H.; Ogawa, T.; Munakata, T. Safracins, new antitumor antibiotics. 2. Physicochemical properties and chemical structures. J. Antibiot. 1983, 36, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Idemoto, H.; Hirayama, F.; Yamamoto, K.; Iwao, K.; Asao, T.; Munakata, T. Safracins, new antitumor antibiotics. I. Producing organism, fermentation and isolation. J. Antibiot. 1983, 36, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Croatt, M.P.; Witulski, B. New reactions and step economy: The total synthesis of (±)-salsolene oxide based on the type II transition metal-catalyzed intramolecular [4+4] cycloaddition. Tetrahedron 2006, 62, 7505–7511. [Google Scholar] [CrossRef]
- Wender, P.A.; Miller, B.L. Synthesis at the molecular frontier. Nature 2009, 460, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Balczewski, P.; Mallon, M.K.J.; Street, J.D.; Joule, J.A. A synthesis of aaptamine from 6,7-dimethoxy-1-methylisoquinoline. Tetrahedron Lett. 1990, 31, 569–572. [Google Scholar] [CrossRef]
- Harada, N.; Sugioka, T.; Soutome, T.; Hiyoshi, N.; Uda, H.; Kuriki, T. Synthesis and Absolute Stereochemistry of (+)-Adociaquinones A and B. Tetrahedron Asymmetry 1995, 6, 375–376. [Google Scholar] [CrossRef]
- Han, S.; Siegel, D.S.; Morrison, K.C.; Hergenrother, P.J.; Movassaghi, M. Synthesis and Anticancer Activity of All Known (–)-Agelastatin Alkaloids. J. Org. Chem. 2013, 78, 11970–11984. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Sugimoto, M.; Morita, M.; Kobayashi, M. Total Synthesis of Agosterol A: An MDR-Modulator from a Marine Sponge. Chem. Eur. J. 2001, 7, 2663–2670. [Google Scholar] [CrossRef]
- Jou, G.; González, I.; Albericio, F.; Lloyd-Williams, P.; Giralt, E. Total Synthesis of Dehydrodidemnin B. Use of Uronium and Phosphonium Salt Coupling Reagents in Peptide Synthesis in Solution. J. Org. Chem. 1997, 62, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Kigoshi, H.; Ojika, M.; Ishigaki, T.; Suenaga, K.; Mutou, T.; Sakakura, A.; Ogawa, T.; Yamada, K. Total Synthesis of Aplyronine A, a Potent Antitumor Substance of Marine Origin. J. Am. Chem. Soc. 1994, 116, 1443–1444. [Google Scholar] [CrossRef]
- Wang, J.; Pagenkopf, B.L. First Total Synthesis and Structural Reassignment of (–)-Aplysiallene. Org. Lett. 2007, 9, 3703–3706. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Purnima, K.V.; Reddy, B.V.S.; Nagaiah, K.; Ghamdi, A.K. Total synthesis of cryptophycin-24 (arenastatin A) via Prins cyclization. Tetrahedron Lett. 2011, 52, 6709–6712. [Google Scholar] [CrossRef]
- Sakurai, J.; Oguchi, T.; Watanabe, K.; Abe, H.; Kanno, S.; Ishikawa, M.; Katoh, T. Highly Efficient Total Synthesis of the Marine Natural Products (+)-Avarone, (+)-Avarol, (–)-Neoavarone, (–)-Neoavarol and (+)-Aureol. Chem. Eur. J. 2008, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, N.; Tsujita, H.; Hiramatsu, A.; Mori, C.; Koizumi, N.; Kobayashi, M. Efficient total synthesis of bastadin 6, an anti-angiogenic brominated tyrosine-derived metabolite from marine sponge. Tetrahedron 2005, 61, 7211–7218. [Google Scholar] [CrossRef]
- Couladouros, E.A.; Pitsinos, E.N.; Moutsos, V.I.; Sarakinos, G. A General Method for the Synthesis of Bastaranes and Isobastaranes: First Total Synthesis of Bastadins 5, 10, 12, 16, 20, and 21. Chem. Eur. J. 2005, 11, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Bros, M.A.; Gras, G.; Ajana, W.; Joule, J.A. Syntheses of Batzelline A, Batzeline B, Isobatzelline A, and Isobatzelline B. Eur. J. Org. Chem. 1999, 1995, 1173–1183. [Google Scholar] [CrossRef]
- Manaviazar, S.; Hale, K.J. Total Synthesis of Bryostatin1: A Short Route. Angew. Chem. Int. Ed. 2011, 50, 8786–8789. [Google Scholar] [CrossRef] [PubMed]
- Keck, G.E.; Poudel, Y.B.; Cummins, T.J.; Rudra, A.; Covel, J.A. Total Synthesis of Bryostatin 1. J. Am. Chem. Soc. 2011, 133, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.S.; Nagarajan, R. Total synthesis of the marine alkaloids Caulibugulones A and D. Tetrahedron 2015, 71, 801–804. [Google Scholar] [CrossRef]
- Fortner, K.C.; Kato, D.; Tanaka, Y.; Shair, M.D. Enantioselective Synthesis of (+)-Cephalostatin 1. J. Am. Chem. Soc. 2010, 132, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Forestieri, R.; Donohue, E.; Balgi, A.; Roberge, M.; Andersen, R.J. Synthesis of Clionamine B, an Autophagy Stimulating Aminosteroid Isolated from the Sponge Cliona celata. Org. Lett. 2013, 15, 3918–3921. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Manolikakes, G.; Yeh, C.-H.; Guerrero, C.A.; Shenvi, R.A.; Shigehisa, H.; Baran, P.S. Scalable Synthesis of Cortistatin A and Related Structures. J. Am. Chem. Soc. 2011, 133, 8014–8027. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.; Julian, L.D.; Newcom, J.S.; Roush, W.R. Total Syntheses of (+)-Tedanolide and (+)-13-Deoxytedanolide. J. Am. Chem. Soc. 2008, 130, 16407–16416. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.R.; Carpenter, J.; Blakey, S.B.; Kayano, A.; Mangion, I.K.; Sinz, C.J.; MacMillan, D.W.C. Total Synthesis of Diazonamide A. Chem. Sci. 2011, 2, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Sumii, Y.; Kotoku, N.; Fukuda, A.; Kawachi, T.; Sumii, Y.; Arai, M.; Kobayashi, M. Enantioselective synthesis of dictyoceratin-A (smenospondiol) and -C, hypoxia-selective growth inhibitors from marine sponge. Bioorg. Med. Chem. 2015, 23, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.; Bucher, C.; Leighton, J.L. A Highly Step-Economical Synthesis of Dictyostatin. Angew. Chem. Int. Ed. 2013, 52, 6757–6761. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-R.; Ewing, W.R.; Harris, B.D.; Joullié, M.M. Total Synthesis and Structural Investigations of Didemnins A, B, and C. J. Am. Chem. Soc. 1990, 112, 7659–7672. [Google Scholar] [CrossRef]
- Yu, Z.; Ely, R.J.; Morken, J.P. Synthesis of (+)-Discodermolide by Catalytic Stereoselective Borylation Reactions. Angew. Chem. Int. Ed. 2014, 53, 9632–9636. [Google Scholar] [CrossRef] [PubMed]
- Mordant, C.; Reymond, S.; Tone, H.; Lavergne, D.; Touati, R.; Hassine, B.B.; Ratovelomanana-Vidal, V.; Genet, J.-P. Total Synthesis of dolastatin 10 through ruthenium-catalyzed asymmetric hydrogenations. Tetrahedron 2007, 63, 6115–6123. [Google Scholar] [CrossRef]
- Akaji, K.; Hayashi, Y.; Kiso, Y.; Kuriyama, N. Convergent Synthesis of Dolastatin 15 by Solid Phase Coupling of an N-Methylamino Acid. J. Org. Chem. 1999, 64, 405–411. [Google Scholar] [CrossRef]
- Chen, X.-T.; Bhattacharya, S.K.; Zhou, B.; Gutteridge, C.E.; Pettus, T.R.R.; Danishefsky, S.J. The Total Synthesis of Eleutherobin. J. Am. Chem. Soc. 1999, 121, 6563–6579. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R.A. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. Med. Chem. Commun. 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Kotoku, N.; Fujioka, S.; Nakata, C.; Yamada, M.; Sumii, Y.; Kawachi, T.; Arai, M.; Kobayashi, M. Concise synthesis and structure-activity relationship of furospinosulin-1, a hypoxia-selective growth inhibitor from marine sponge. Tetrahedron 2011, 67, 6673–6678. [Google Scholar] [CrossRef]
- Boukouvalas, J.; Albert, V. Synthesis of the Hypoxic Signaling Inhibitor Furospongolide. Syn. Lett. 2011, 17, 2541–2544. [Google Scholar]
- White, J.D.; Amedio, J.C., Jr. Total Synthesis of Geodiamolide A, a Novel Cyclodepsipeptide of Marine Origin. J. Org. Chem. 1989, 54, 736–738. [Google Scholar] [CrossRef]
- Hirai, Y.; Yokota, K.; Yamzaki, T.; Momose, T. A Total Synthesis of (+)-Geodiamolides A and B, the Novel Cyclodesipeptides. Heterocycles 1990, 30, 1101–1119. [Google Scholar]
- Fung, S.-Y.; Sofiyev, V.; Schneiderman, J.; Hirschfeld, A.F.; Victor, R.E.; Woods, K.; Piotrowski, J.S.; Deshpande, R.; Li, S.C.; de Voogd, N.J.; et al. Unbiased Screening of Marine Sponge Extracts for Anti-inflammatory Agents Combined with Chemical Genomics Identifies Girolline as an Inhibitor of Protein Synthesis. ACS Chem. Biol. 2014, 9, 247–257. [Google Scholar]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total Synthesis of Halichondrin B and Norbalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Somaiah, R.; Ravindar, K.; Cencic, R.; Pelletier, J.; Deslongchamps, P. Synthesis of the Antiproliferative Agent Hippuristanol and Its Analogues from Hydrocortisone via Hg(II)-Catalyzed Spiroketalization: Structure-Activity Relationship. J. Med. Chem. 2014, 57, 2511–2523. [Google Scholar] [CrossRef] [PubMed]
- Shearman, J.W.; Myers, R.M.; Beale, T.M.; Brenton, J.D.; Ley, S.V. Total syntheses of the bromotyrosine-derived natural products ianthelline, 5-bromoverongamine and JBIR-44. Tetrahedron Lett. 2010, 51, 4812–4814. [Google Scholar] [CrossRef]
- Fürstner, A.; Nevado, C.; Waser, M.; Tremblay, M.; Chevrier, C.; Teplý, F.; Aïssa, C.; Moulin, E.; Müller, O. Total Synthesis of Iejimalide A-D and Assessment of the Remarkable Actin-Depolymerizing Capacity of These Polyene Macrolides. J. Am. Chem. Soc. 2007, 129, 9150–9161. [Google Scholar] [CrossRef] [PubMed]
- Ling, T.; Poupon, E.; Rueden, E.J.; Theodorakis, E.A. Synthesis of (–)-Ilimaquinone via a Radical Decarboxylation and Quinone Addition Reaction. Org. Lett. 2002, 4, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, S.; Watanabe, T.; Imaizumi, T.; Ota, Y.; Yoshida, K.; Ebisu, H.; Chinen, T.; Nagumo, Y.; Shibuya, M.; Kanoh, N.; et al. Total Synthesis and Biological Evaluation of Irciniastatin A (a.k.a. Psymberin) and Irciniastatin B. J. Org. Chem. 2015, 80, 12333–12350. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, A.P.; Venturelli, A.; Butler, L.A.; Parker, R.J. First Total Synthesis of the Irciniasulfonic Acids. Syn. Lett. 2005, 652–654. [Google Scholar]
- López-Macia, A.; Jiménez, J.C.; Royo, M.; Giralt, E.; Albericio, F. Synthesis and Structure Determination of Kahalalide F. J. Am. Chem. Soc. 2001, 123, 11398–11401. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total Synthesis of Lamellarins D, H, and R and Ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Stivala, C.E.; Hull, K.L.; Huang, A.; Fandrick, D.R. A Concise Synthesis of (–)-Lasonolide A. J. Am. Chem. Soc. 2014, 136, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; De Souza, D.; Turet, L.; Fenster, M.D.B.; Parra-Rapado, L.; Wirtz, C.; Mynott, R.; Lehmann, C.W. Total Syntheses of the Actin-Binding Macrolides Latrunculin A, B, C, M, S and 16-epi-Latrunculin B. Chem. Eur. J. 2007, 13, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Amans, D.; Seganish, W.M.; Chung, C.K. Total Synthesis of Laulimalide: Assembly of the Fragments and Completion of the Synthesis of the Natural Product and Potent Analogue. Chem. Eur. J. 2012, 18, 2961–2971. [Google Scholar] [CrossRef] [PubMed]
- Boukouvalas, J.; Robichaud, J.; Maltais, F. A Unified Strategy for the Regiospecific Assembly of Homoallyl-Substituted Butenolides and y-Hydroxybutenolides: First Synthesis of Luffariellolide. Synlett 2006, 15, 2480–2482. [Google Scholar] [CrossRef]
- Oshiyama, T.; Satoh, T.; Okano, K.; Tokuyama, H. Total Synthesis of makaluvamine A/D, damirone B, batzelline C, makaluvone, and isobatzelline C featuring one-pot benzyne-mediated cyclization-functionalization. Tetrahedron 2012, 68, 9376–9383. [Google Scholar] [CrossRef]
- Jakubec, P.; Hawkins, A.; Felzmann, W.; Dixon, D.J. Total Synthesis of Manzamine A and Related Alkaloids. J. Am. Chem. Soc. 2012, 134, 17482–17485. [Google Scholar] [CrossRef] [PubMed]
- Fresneda, P.M.; Molina, P.; Bleda, J.A. Synthesis of the indole alkaloids meridianins from tunicate Aplidium meridianum. Tetrahedron 2001, 57, 2355–2363. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Sultana, S.S. Stereoselective synthesis of the C1-C20 segment of the microsclerodermins A and B. Tetrahedron Lett. 2006, 47, 7255–7258. [Google Scholar] [CrossRef]
- Shi, Y.; Pierce, J.G. Synthesis of the 5,6-Dihydroxymorpholin-3-one Fragment of Monanchocidin A. Org. Lett. 2015, 17, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.H.; Waizumi, N.; Zhong, M.; Rawal, V.H. Total synthesis of mycalamide A. J. Am. Chem. Soc. 2005, 127, 7290–7291. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hale, K.J. Total Synthesis of the Potent HIF-1 Inhibitory Antitumor Natural Product, (8R)-Mycothiazole, via Baldwin-Lee CsF/CuI sp3-sp2-Stille Cross-Coupling. Confirmation of the Crews Reassignment. Org. Lett. 2015, 17, 4200–4203. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; del Poz, C.; Reyes, F.; Rodríguez, A.; Francesch, A.; Echavarren, A.M.; Cuevas, C. Total Synthesis of Natural Myriaporones. Angew. Chem. Int. Ed. 2004, 43, 1728–1730. [Google Scholar]
- Gao, S.; Wang, Q.; Chen, C. Synthesis and Structure Revision of Nakiterpiosin. J. Am. Chem. Soc. 2009, 131, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Abraham, A.D.; Zhou, Q.; Ali, H.; O’Brien, J.V.; Hamill, B.D.; Arcaroli, J.J.; Messersmith, W.A.; LaBarbera, D.V. An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase Iiα and Potent Anticancer Agent. Mar. Drugs. 2014, 12, 4833–4850. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Boudreau, M.A.; Zheng, J.; Whittal, R.M.; Austin, P.; Roskelley, C.D.; Roberge, M.; Andersen, R.J.; Vederas, J.C. Chemical Synthesis and Biological Activity of the Neopetrosiamides and Their Analogues: Revision of Disulfide Bond Connectivity. J. Am. Chem. Soc. 2010, 132, 1486–1487. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Boyce, C.W.; Labroli, M.A.; Sehon, C.A.; Jin, Q. Total Syntheses of Ningalin A, Lamellarin O, Lukianol A, and Permethyl Storniamide A Utilizing Heterocyclic Azadiene Diels-Alder Reactions. J. Am. Chem. Soc. 1999, 121, 54–62. [Google Scholar] [CrossRef]
- Hong, C.Y.; Kishi, Y. Total Synthesis of Onnamide A. J. Am. Chem. Soc. 1991, 113, 9694–9696. [Google Scholar] [CrossRef]
- Dhand, V.; Chang, S.; Britton, R. Total Synthesis of the Cytotoxic Anhydrophytosphingosine Pachastrissamine (Jaspine B). J. Org. Chem. 2013, 78, 8208–8213. [Google Scholar] [CrossRef] [PubMed]
- Seiple, I.B.; Su, S.; Young, I.S.; Nakamura, A.; Yamaguchi, J.; Jørgensen, L.; Rodriguez, R.A.; O’Malley, D.P.; Gaich, T.; Köck, M.; et al. Enantioselective Total Syntheses of (–)-Palau’amine, (–)-Axinellamines, and (–)-Massadines. J. Am. Chem. Soc. 2011, 133, 14710–14726. [Google Scholar] [CrossRef] [PubMed]
- Pattenden, G.; Critcher, D.J.; Remuiñán, M. Total synthesis of (-)-pateamine A, a novel immunosuppressive agent from Mycale sp1. Can. J. Chem. 2004, 82, 353–365. [Google Scholar] [CrossRef]
- Trost, B.M.; Michaelis, D.J.; Malhotra, S. Total Synthesis of (–)-18-epi-Peloruside A: An Alkyne Linchpin Strategy. Org. Lett. 2013, 15, 5274–5277. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.J.; Coello, L.; Fernández, R.; Reyes, F.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez-Sancho, F.; Bueno, S.; et al. Isolation and First Total Synthesis of PM050489 and PM060184, Two New Marine Anticancer Compounds. J. Am. Chem. Soc. 2013, 135, 10164–10171. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Shin, Y.; Jung, M.; Ha, M.W.; Park, Y.; Lee, Y.-J.; Shin, J.; Oh, K.B.; Lee, S.K.; Park, H.-G. Efficient synthesis and biological activity of Psammaplin A and its analogues as antitumor agents. Eur. J. Med. Chem. 2015, 96, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, S.N.; Clardy, J. Total Synthesis of Psammaplysenes A and B, Naturally Occurring Inhibitors of FOXO1a Nuclear Export. Org. Lett. 2005, 7, 4091–4094. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wu, Y.-C. Asymmetric Total Syntheses of (–)-Renieramycin M and G and (–)-Jorumycin Using Aziridine as a Lynchpin. Org. Lett. 2009, 11, 5558–5561. [Google Scholar]
- Herb, C.; Bayer, A.; Maier, M.E. Total Synthesis of Salicylihalamides A and B. Chem. Eur. J. 2004, 10, 5649–5660. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Xu, J.Y.; Kim, S.; Pfefferkorn, J.; Ohshima, T.; Vourloumis, D.; Hosokawa, S. Total Synthesis of Sarcodictyins A and B. J. Am. Chem. Soc. 1998, 120, 8661–8673. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science 2014, 346, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Shinkre, B.A.; Velu, S.E. Total Synthesis of Secobatzelline B. Synth. Commun. 2007, 37, 2399–2409. [Google Scholar] [CrossRef]
- Ghosal, P.; Shaw, A.K. An efficient total synthesis of the anticancer agent (+)-spisulosine (ES-285) from Garner’s aldehyde. Tetrahedron Lett. 2010, 51, 4140–4142. [Google Scholar] [CrossRef]
- Smith, A.B.; Sfouggatakis, C.; Risatti, C.A.; Sperry, J.B.; Zhu, W.; Doughty, V.A.; Tomioka, T.; Gotchev, D.B.; Bennett, C.S.; Sakamoto, S.; et al. Spongipyran synthetic studies. Evolution of a scalable total synthesis of (+)-spongistatin 1. Tetrahedron 2009, 65, 6489–6509. [Google Scholar] [CrossRef] [PubMed]
- Shearman, J.W.; Myers, R.M.; Brenton, J.D.; Ley, S.V. Total Syntheses of subereamollines A and B. Org. Biomol. Chem. 2011, 9, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, F.; Toma, T.; Inui, T.; Yokoshima, S.; Fukuyama, T. Total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 2013, 135, 13684–13687. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.J.; Morris, J.C. Total synthesis of variolin B. Tetrahedron Lett. 2001, 42, 8697–8699. [Google Scholar] [CrossRef]
- Zurwerra, D.; Glaus, F.; Betschart, L.; Schuster, J.; Gertsch, J.; Ganci, W.; Altmann, K.-H. Total Synthesis of (–)-Zampanolide and Structure-Activity Relationship Studies on (–)-Dactylolide Derivatives. Chem. Eur. J. 2012, 18, 16868–16883. [Google Scholar] [CrossRef] [PubMed]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar] [CrossRef]
- Burres, N.S.; Clement, J.J. Antitumor activity and mechanism of action of the novel marine natural products mycalamide-A and -B and onnamide. Cancer Res. 1989, 49, 2935–2940. [Google Scholar] [PubMed]
- Gürel, G.; Blaha, G.; Steitz, T.A.; Moore, P.B. Structures of triacetyloleandomycin and mycalamide A bind to the large ribosomal subunit of Haloarcula marismortui. Antimicrob. Agents Chemother. 2009, 53, 5010–5014. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.S.; Garson, M.J.; Blunt, J.W.; Munro, M.H.G.; Hooper, J.N.A. Mycalamides C and D, cytotoxic compounds from the marine sponge Stylinos n. species. J. Nat. Prod. 2000, 63, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Venturi, V.; Davies, C.; Singh, A.J.; Matthews, J.H.; Bellows, D.S.; Northcote, P.T.; Keyzers, R.A.; Teesdale-Spittle, P.H. The protein synthesis inhibitors mycalamides A and E have limited susceptibility toward the drug efflux network. J. Biochem. Mol. Toxicol. 2012, 26, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Kalinovsky, A.I.; Shubina, L.K.; Bokemeyer, C.; Stonik, V.A.; Honecker, F. Mycalamide A shows cytotoxic properties and prevents EGF-induced neoplastic transformation through inhibition of nuclear factors. Mar. Drugs 2012, 10, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooper, J.N. Isolation and structure of spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Sakai, H.; Kawazoe, K.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtin A, a potent antitumor macrolide from the Okinawan marine sponge Hyrtios altum. Tetrahedron Lett. 1993, 34, 2795–2798. [Google Scholar] [CrossRef]
- Bai, R.; Taylor, G.F.; Cichacz, Z.A.; Herald, C.L.; Kepler, J.A.; Pettit, G.R.; Hamel, E. The spongistatins, potently cytotoxic inhibitors of tubulin polymerization, bind in a distinct region of the vinca domain. Biochemistry 1995, 34, 9714–9721. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ischenko, I.; Joore, J.; Garczarczyk, D.; Fürst, R.; Bruns, C.J.; Vollmar, A.M.; Zahler, S. Investigation of the marine compound spongistatin 1 links the inhibition of PKCα translocation to nonmitotic effects of tubulin antagonism in angiogenesis. FASEB J. 2009, 23, 1127–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneiders, U.M.; Schyschka, L.; Rudy, A.; Vollmar, A.M. BH3-only proteins Mcl-1 and Bim as well as endonuclease G are targeted in spongistatin 1-induced apoptosis in breast cancer cells. Mol. Cancer Ther. 2009, 8, 2914–2925. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Y.; Meng, Y.H.; Zeng, L.M.; Fu, X.; Schmitz, F.J. Stellettin A, a new triterpenoid pigment from the marine sponge Stelletta tenuis. J. Nat. Prod. 1994, 57, 1450–1451. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchera pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Ling, Y.H.; Cheung, F.W.; Che, C.T. Stellettin A induces endoplasmic reticulum stress in murine B16 melanoma cells. J. Nat. Prod. 2012, 75, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Hauschild, J.; Amann, K.; Tabakmakher, K.M.; Venz, S.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; Fedorov, S.N.; et al. Marine alkaloid monanchocidin A overcomes drug resistance by induction of autophagy and lysosomal membrane permeabilization. Oncotarget 2015, 6, 17328–17341. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.C.; Bokesch, H.R.; McCormick, J.L.; Rashid, M.A.; Spielvogel, D.; Gustafson, K.R.; Alavanja, M.M.; Boyd, M.R. Isolation and characterization of new anti-HIV and cytotoxic leads from plants, marine, and microbial organisms. J. Nat. Prod. 1997, 60, 431–438. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.L.; McKee, T.C.; Cardellina, J.H., II; Leid, M.; Boyd, M.R. Cytotoxic triterpenes from a marine sponge, Stelletta sp. J. Nat. Prod. 1996, 59, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Deng, Z.; Li, J.; Fu, H.; van Soest, R.W.M.; Proksch, P.; Lin, W. Isomalabaricane-type compounds from the marine sponge Rhabdastrella aff. distincta. J. Nat. Prod. 2004, 67, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchocidins B–E: Polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Mudit, M.; Khanfar, M.; Muralidharan, A.; Thomas, S.; Shah, G.V.; van Soest, R.W.; El Sayed, K.A. Discovery, design, and synthesis of anti-metastatic lead phenylmethylene hydantoins inspired by marine natural products. Bioorg. Med. Chem. 2009, 17, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.V.; Muralidharan, A.; Thomas, S.; Gokulgandhi, M.; Mudit, M.; Khanfar, M.; El Sayed, K. Identification of a small molecule class to enhance cell-cell adhesion and attenuate prostate tumor growth and metastasis. Mol. Cancer Ther. 2009, 8, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Sallam, A.A.; Mohyeldin, M.M.; Foudah, A.I.; Akl, M.R.; Nazzal, S.; Meyer, S.A.; Liu, Y.Y.; El Sayed, K.A. Marine natural products-inspired phenylmethylene hydantoins with potent in vitro and in vivo antitumor activities via suppression of Brk and FAK signaling. Org. Biomol. Chem. 2014, 12, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Shastina, V.V.; Shin, S.W.; Xu, Q.; Park, J.-I.; Rasskazov, V.A.; Avilov, S.A.; Fedorov, S.N.; Stonik, V.A.; Kwak, J.-Y. Differential effects of triterpene glycosides, frondoside A and cucumarioside A2–2 isolated from sea cucumbers on caspase activation and apoptosis of human leukemia cells. FEBS Lett. 2009, 583, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Mohammed, A.; Zhang, Y.; Choi, C.-I.; Woodward, C.; Collin, P.; Steele, V.E.; Rao, C.V. Chemopreventive effects of Frondanol A5, a Cucumaria frondosa extract, against rat colon carcinogenesis and inhibition of human colon cancer cell growth. Cancer Prev. Res. 2010, 3, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Kundu, N.; Collin, P.D.; Goloubeva, O.; Fulton, A.M. Frondoside A inhibits breast cancer metastasis and antagonizes prostaglandin E receptors EP4 and EP2. Breast Cancer Res. Treat. 2012, 132, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, Y.H.; Kim, Y.; Lee, S.-J. Frondoside A has an anti-invasive effect by inhibiting TPA-induced MMP-9 activation via NF-κB and AP-1 signaling in human breast cancer cells. Int. J. Oncol. 2012, 41, 933–940. [Google Scholar] [PubMed]
- Attoub, S.; Arafat, K.; Gélaude, A.; Al Sultan, M.A.; Bracke, M.; Collin, P.; Takahashi, T.; Adrian, T.E.; de Wever, O. Frondoside A suppressive effects on lung cancer survival, tumor growth, angiogenesis, invasion, and metastasis. PLoS ONE 2013, 8, e53087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avilov, S.A.; Kalinin, V.I.; Prozdova, O.A.; Kalinovskii, A.I.; Stonik, V.A.; Gudimova, E.N. Triterpene glycosides from the holothurian Cucumaria frondosa. Chem. Nat. Comp. 1993, 29, 216–218. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I.; Kalinovsky, A.I.; Dmitrenok, P.S.; Fedorov, S.N.; Stepanov, V.G.; Dong, Z.; Stonik, V.A. Constituents of the sea cucumber Cucumaria okhotensis. Structures of okhotosides B1-B3 and cytotoxic activities of some glycosides from this species. J. Nat. Prod. 2008, 71, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Floss, H.G. Combinatorial biosynthesis—Potential and problems. J. Biotechnol. 2006, 124, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Menzella, H.G.; Reeves, C.D. Combinatorial biosynthesis for drug development. Curr. Opin. Microbiol. 2007, 10, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J. Mutasynthesis, chemobiosynthesis, and back to semi-synthesis: Combining synthetic chemistry and biosynthetic engineering for diversifying natural products. Nat. Prod. Rep. 2008, 25, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kirschning, A.; Hahn, F. Merging chemical synthesis and biosynthesis: A new chapter in the total synthesis of natural products and natural product libraries. Angew. Chem. Int. Ed. Engl. 2012, 51, 4012–4022. [Google Scholar] [CrossRef] [PubMed]
- Regentin, R.; Kennedy, J.; Wu, N.; Carney, J.R.; Licari, P.; Galazzo, J.; Desai, R. Precursor-directed biosynthesis of novel triketide lactones. Biotechnol. Prog. 2004, 20, 122–127. [Google Scholar]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Li, R. Marinopyrroles: Unique drug discoveries based on marine natural products. Med. Res. Rev. 2016, 36, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, S.; Parameswaran, P.S. Recent advances in marine drug research. Biotechnol. Adv. 2013, 31, 1826–1845. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
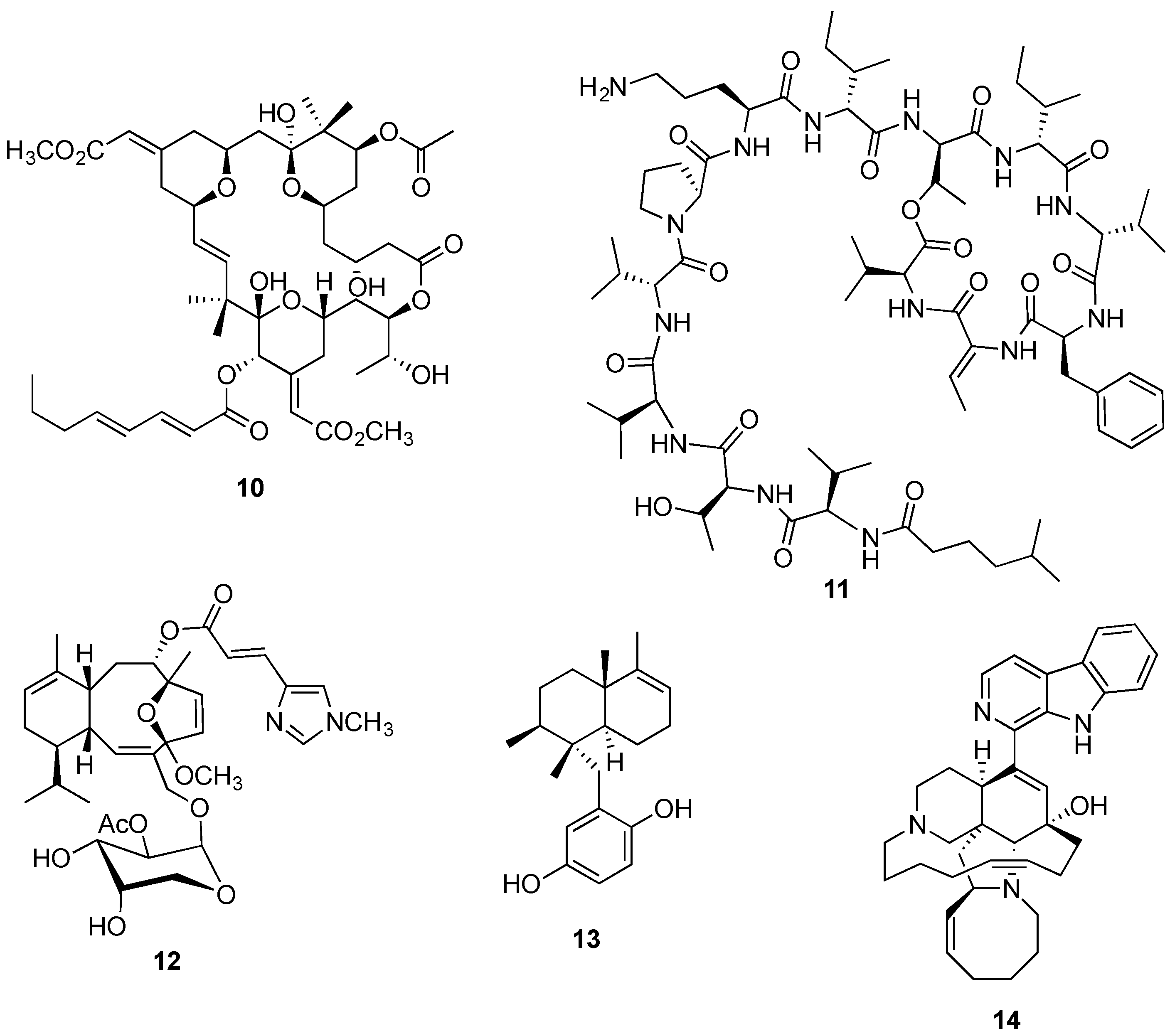{kind=link}
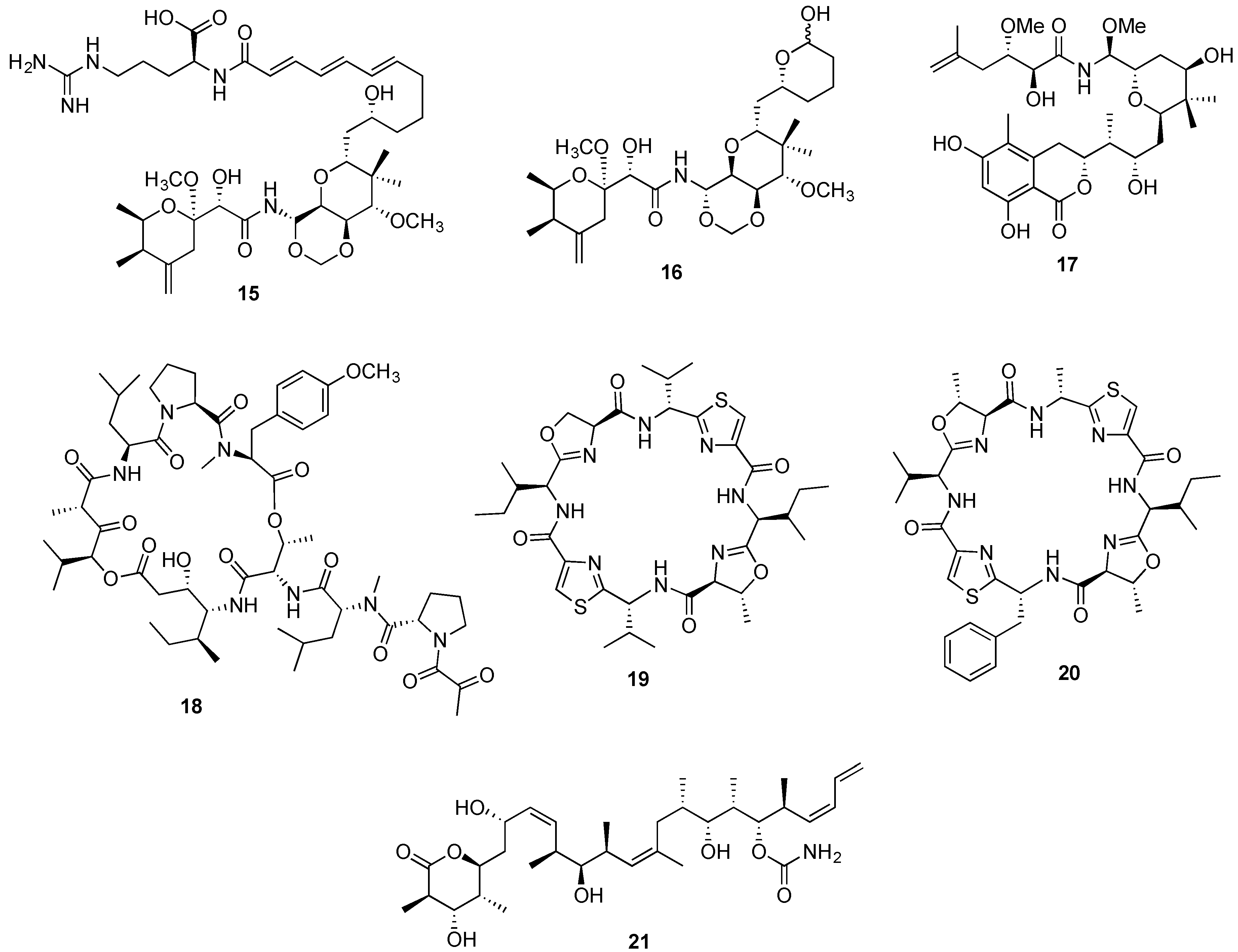{kind=link}
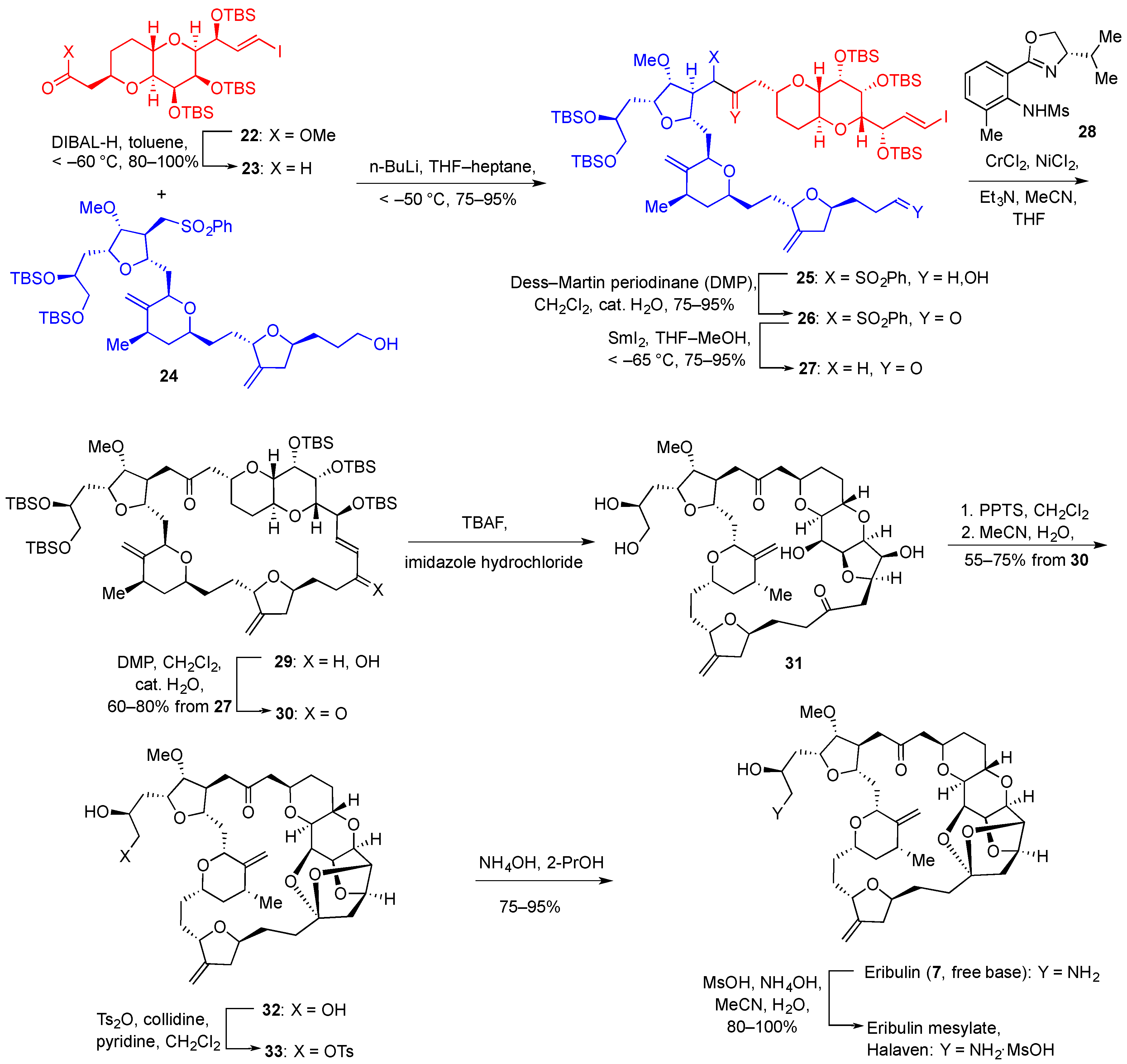{kind=link}
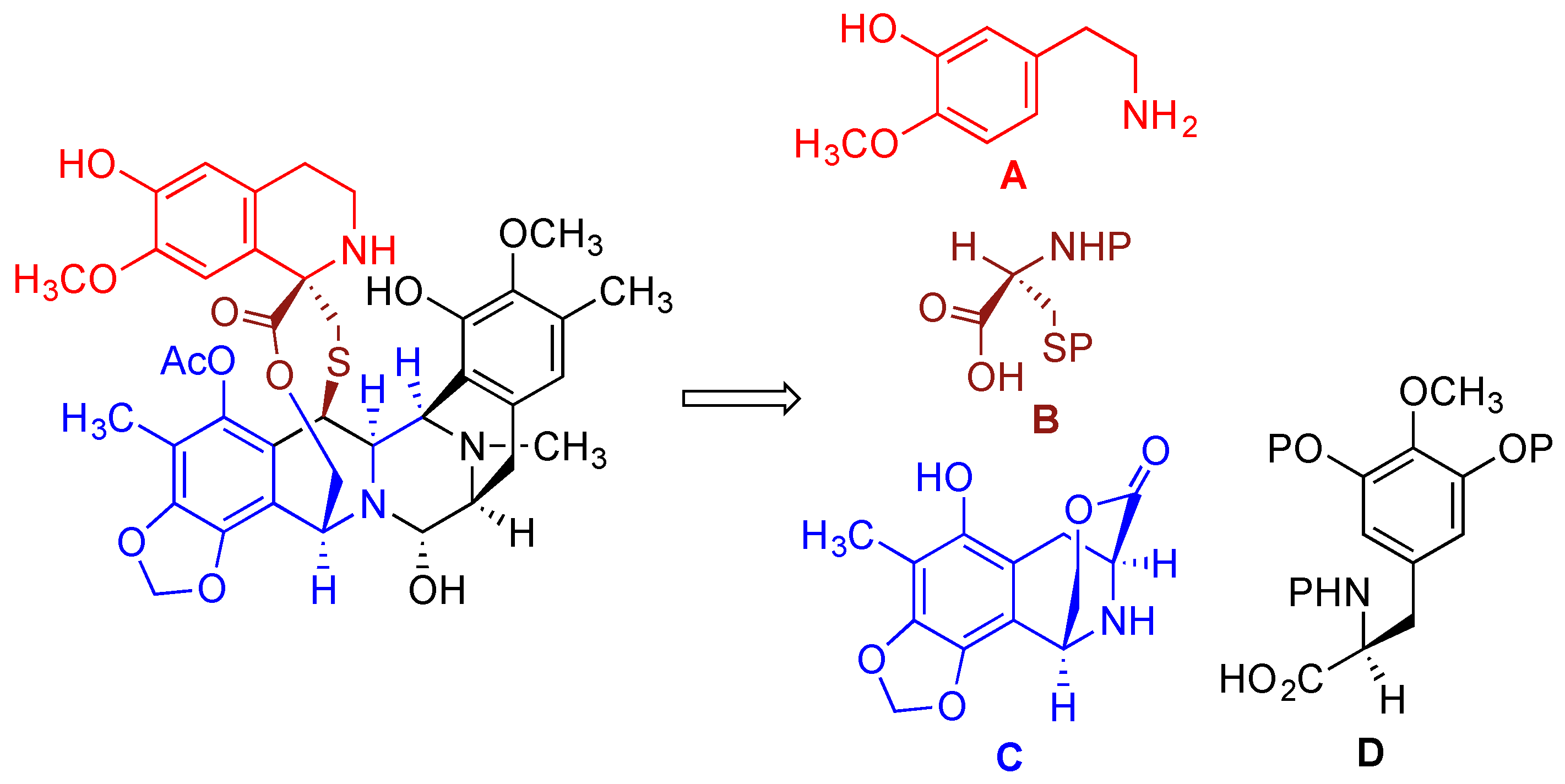{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Product | Total or Semisynthesis | |||
|---|---|---|---|---|
| T/S a | # of Steps | Starting Material | Reference | |
| Aaptamine | T | 5 | 6,7-dimethoxy-1-methylisoquinoline | [202] |
| 4-Acetoxythorectidaeolide A | N/A | N/A | N/A | N/A |
| Adociaquinones A and B | T | 3 | 2,5-dimethoxy-bicyclo[4.2.0]octa-1,3,5-triene | [203] |
| Agelastatin A | T | 9 | d-Aspartic acid | [204] |
| Agosterol A | T/S | 23 | Ergosterol | [205] |
| Aplidine (plitidepsin or dehydrodidemnin B) | T | 3 | d-Proline | [206] |
| Aplyronine A | T | 15 | (R)-3-(benzyloxy)-2 methylpropanal | [207] |
| Aplysiallene | T | 16 | (S,S)-Diepoxybutane | [208] |
| Arenastatin A (Cryptophycin-24) | T | 14 | (d)-N-Boc-tyrosine methyl ether | [209] |
| Austrasulfone | N/A | N/A | N/A | N/A |
| Avarol | T | 11 | Wieland-Miescher ketone | [210] |
| Avarone | T | 10 | Wieland-Miescher ketone | [210] |
| Bastadin 6 | T | 7 | N-Boc-3,5-dibromotyramine | [211] |
| Bastadin 9 | N/A | N/A | N/A | N/A |
| Bastadin 16 | T | 16 | p-benzyloxybenzaldehyde | [212] |
| Batzellines A-D | T | 12 | 4-aminoveratrol | [213] |
| Botryllamide A | N/A | N/A | N/A | N/A |
| Bryostatin-1 | T | 31 | 1,3-propanediol | [214] |
| Bryostatin-1 | T | 46 | (R)-Isobutyl lactate | [215] |
| Candidaspongiolides A and B | N/A | N/A | N/A | N/A |
| Caulibugulone A | T | 5 | 2,5-dihydroxybenzaldehyde | [216] |
| Cephalostatin 1 | T | 33 | trans-androsterone | [217] |
| Chondropsin A | N/A | N/A | N/A | N/A |
| Clionamine B | S | 11 | Tigogenin | [218] |
| Comaparvin | N/A | N/A | NA | N/A |
| Cortistatin A | T | 19 | Prednisone | [219] |
| Crambescidin-816 | N/A | N/A | N/A | N/A |
| Cytarabine | N/A b | N/A | N/A | N/A |
| 13-Deoxytedanolide | T | 27 | Methyl 3-oxopentanoate | [220] |
| Diacarnoxide B | N/A | N/A | N/A | N/A |
| Diazonamide A | T | 20 | 7-benzyloxyindol | [221] |
| Dictyoceratin C | T | 11 | 4-Hydroxy-3-methylbenzoic acid | [222] |
| Dictyostatin-1 | T | 19 | 2-Vinyl-1,3-dioxolane | [223] |
| Didemnin B | T | 9 | Ethyl lactate | [224] |
| Discodermolide | T | 36 | trans-Pentadiene | [225] |
| Dolastatin-10 | T | 11 | (S)-Boc-proline | [226] |
| Dolastatin-15 | T | 10 | l-Hydroxyisovalelic acid | [227] |
| Eleutherobin | T | 27 | (R)-(−)-α-Phellandrene | [228] |
| Ethylsmenoquinone | N/A | N/A | N/A | N/A |
| Fascaplysin | T | 2 | Tryptamine | [229] |
| Fijianolides A and B (Laulimalide) | N/A | N/A | N/A | N/A |
| Frondoside A | N/A | N/A | N/A | N/A |
| Furospinosulin-1 | T | 4 | 3-(3-furyl)propan-1-ol | [230] |
| Furospongolide | T | 8 | Geranyl acetate | [231] |
| Geodiamolide A | T | 15 | (−)-4-methylbutyrolactone | [232] |
| Geodiamolide B | T | 13 | d-tyrosine benzyl ester | [233] |
| Geodiamolide H | N/A | N/A | N/A | N/A |
| Girodazole | T | 8 | [234] | |
| Halichondramide | N/A | N/A | N/A | N/A |
| Halichondrin B | T | 38 | 2-deoxy-l-arabinose diethyl thioacetal 4,5-acetonide | [235] |
| Hemiasterlin A | N/A | N/A | N/A | N/A |
| Heteronemin | N/A | N/A | N/A | N/A |
| Hippuristanol | S | 15 | Hydrocortisone | [236] |
| 14-Hydroxymethylxestoquinone | N/A | N/A | N/A | N/A |
| 7-Hydroxyneolamellarin A | N/A | N/A | N/A | N/A |
| Z-4-Hydroxyphenylmethylene | CA d | N/A | N/A | N/A |
| Hyrtioreticulins A and B | N/A | N/A | N/A | N/A |
| Ianthelline | T | 5 | 3,5-dibromo-4-hydroxybenzaldehyde | [237] |
| Iejimalides B and C | T | 15 | Methyl (E)-3-bromo-2-methylacrylate | [238] |
| Ilimaquinone | T | 17 | Wieland-Miescher enone | [239] |
| Irciniastatin A (psymberin) | T | 31 | (−)-pantolactone | [240] |
| Irciniasulfonic acid | T | 11 | Hex-1-yne | [241] |
| Isobatzellines A-D | T | 13 | 4-aminoveratrol | [213] |
| Kahalalide F | T | 10 | 2-chlorotrityl chloride-resin | [242] |
| Kendarimide A | N/A | N/A | N/A | N/A |
| Lamellarin D | T | 11 | Vanillin and isovanillin | [243] |
| Lasonolide A | T | 34 | 2,2,5-Trimethyl-1,3-dioxane-5-carbaldehyde | [244] |
| Latrunculin A | T | 13 | l-cysteine ethyl ester hydrochloride | [245] |
| Laulimalide | T | 7 | 3,4-dihydro-2[2-methyl-4-pentyn-1-yl]-4-(phenylmethoxy) | [246] |
| Leucettamol A | N/A | N/A | N/A | N/A |
| Luffariellolide | T | 8 | Geranyl Bromide | [247] |
| Makaluvamines A, C | T | 15 | 2-bromo-5-methoxy-benzenamine | [248] |
| Manadosterols A and B | N/A | N/A | N/A | N/A |
| Manzamine A | T | 18 | 2-(bromomethyl)-2-ethenyl-1,3-dioxolane | [249] |
| Meridianin E | T | 11 | 5-bromo-2-hydroxybenzaldehyde | [250] |
| Microsclerodermin A | N/A | N/A | S-citronellol | [251] c |
| Mirabilin G | N/A | N/A | N/A | N/A |
| Monanchocidin A | N/A | N/A | 3-azidopropanoic acid | [252] c |
| Mycalamide A | T | 33 | Diethyl-D-tartrate | [253] |
| Mycothiazole | T | 16 | 4,4-dimethyl-5-(phenylmethoxy)-2-penten-1-ol | [254] |
| Myriaporone 3 | T | 27 | Methyl-(S)-(+)-3-hydroxy-2-methylpropionate | [255] |
| Myriaporone 4 | T | 27 | Methyl-(S)-(+)-3-hydroxy-2-methylpropionate | [255] |
| Nakiterpiosin | T | 23 | 3-bromo-2-methylbenzenecarboxylic acid | [256] |
| Neoamphimedine | T | 10 | 2,5-Dimethoxy-3-nitrobenzoate | [257] |
| Neopetrosiamides A and B | T | 4 | Resin-bound linear peptide | [258] |
| Netamine M | N/A | N/A | N/A | N/A |
| Ningalin B | T | 8 | 6-Bromoveratraldehyde | [259] |
| Onnamide A | T | 5 | 5-iodopentadienoic acid | [260] |
| Pachastrissamine (jaspine B) | T | 8 | 6-heptenal | [261] |
| Palau’amine | T | 28 | 3-cyclohexene-1-carboxylic acid | [262] |
| Pateamine A | T | 29 | Dimethyl l-malate and S-methyl 3-hydroxy-2-methylpropionate | [263] |
| Peloruside A | T | 22 | 3-Methyl-1-butyne | [264] |
| Petrosaspongiolide M | N/A | N/A | N/A | N/A |
| Philinopside A and E | N/A | N/A | N/A | N/A |
| PM050489 | T | 35 | 1,3-Propanediol | [265] |
| PM060184 | T | 33 | 1,3-Propanediol | [265] |
| Psammaplin A | T | 5 | 3-bromo-4-hydroxybenzaldahyde | [266] |
| Psammaplysene A | T | 5 | 4-Iodophenol | [267] |
| Renieramycin M | T | 21 | N-Trityl-l-serine methyl ester | [268] |
| Ritterazine B | N/A | N/A | N/A | N/A |
| Salicylihalamide A | T | 16 | ethylene glycol | [269] |
| Sarcodictyin A | T | 25 | (+)-Carvone | [270] |
| Sceptrin | T | 25 | l-Glutamic acid | [271] |
| Secobatzellines A and B | T | 12 | 2,4,5-trimethoxy-benzaldehyde | [272] |
| Simplextone C | N/A | N/A | N/A | N/A |
| Sipholenol A | N/A | N/A | N/A | N/A |
| Sodwanone V | N/A | N/A | N/A | N/A |
| Spisulosine (ES-285) | T | 9 | (S)-Garner’s aldehyde. | [273] |
| Spongiacidin C | N/A | N/A | N/A | N/A |
| Spongistatin 1 (Altohyrtin A) | T | 27 | (S)-glycidol | [274] |
| Stelletin A | N/A | N/A | N/A | N/A |
| Strongylophorine 8 and 26 | N/A | N/A | N/A | N/A |
| Subereamolline A | T | 10 | 2-Hydroxy-4-methoxybenzaldehyde | [275] |
| Thorectidaeolide A | N/A | N/A | N/A | N/A |
| Trabectedin | T | 28 | 1,3-propanodiol | [276] |
| Variolin B | T | 8 | 4-chloro-2-methylthiopyrimidine | [277] |
| Vitilevuamide | N/A | N/A | N/A | N/A |
| Waixenicin A | N/A | N/A | N/A | N/A |
| Zampanolide | T | 20 | d-(−)-Aspartic acid | [278] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, N.G.M.; Dasari, R.; Chandra, S.; Kiss, R.; Kornienko, A. Marine Invertebrate Metabolites with Anticancer Activities: Solutions to the “Supply Problem”. Mar. Drugs 2016, 14, 98. https://doi.org/10.3390/md14050098
Gomes NGM, Dasari R, Chandra S, Kiss R, Kornienko A. Marine Invertebrate Metabolites with Anticancer Activities: Solutions to the “Supply Problem”. Marine Drugs. 2016; 14(5):98. https://doi.org/10.3390/md14050098
Chicago/Turabian StyleGomes, Nelson G. M., Ramesh Dasari, Sunena Chandra, Robert Kiss, and Alexander Kornienko. 2016. "Marine Invertebrate Metabolites with Anticancer Activities: Solutions to the “Supply Problem”" Marine Drugs 14, no. 5: 98. https://doi.org/10.3390/md14050098