
Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in Vitro

 , ,
, ,
Abstract
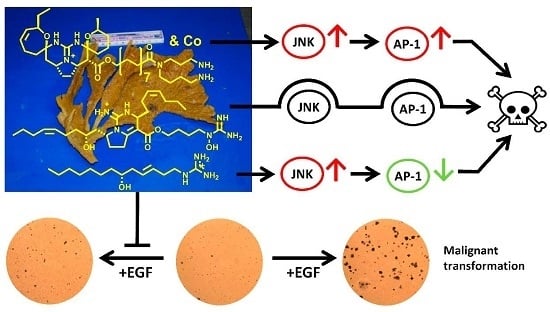
:
1. Introduction
2. Results and Discussion
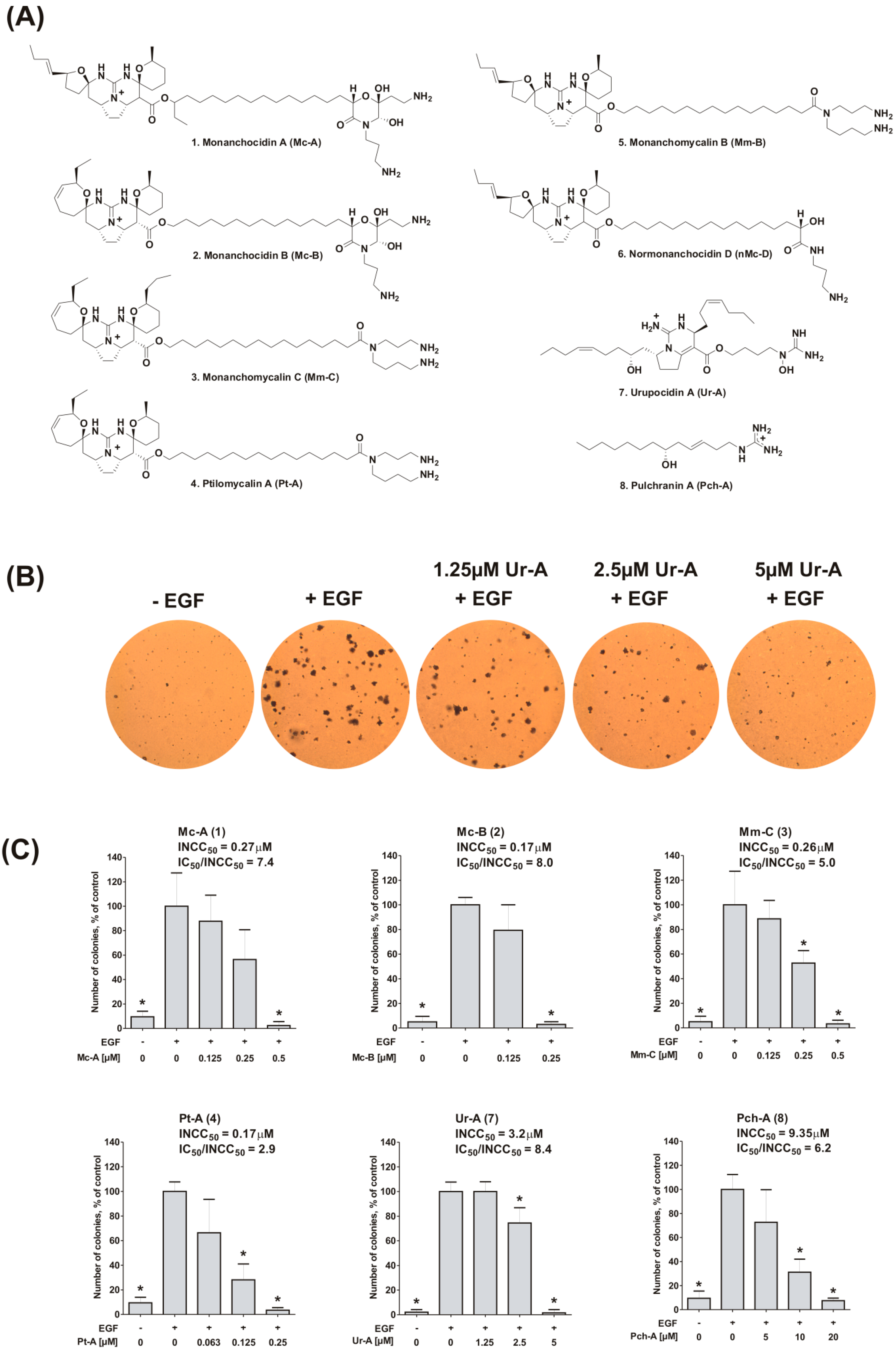
2.1. Inhibition of EGF-Induced Malignant Transformation of Murine Epithelial Cells
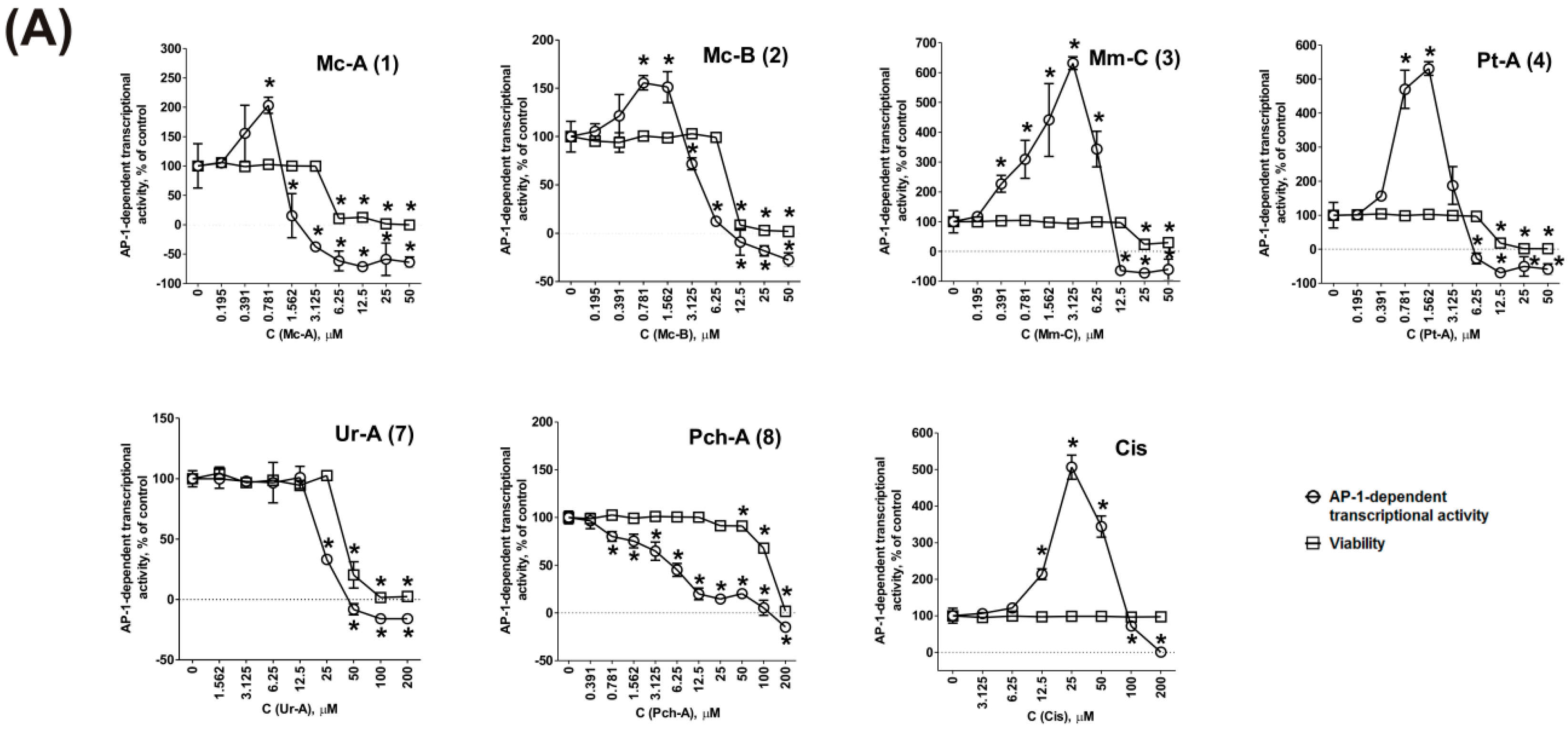
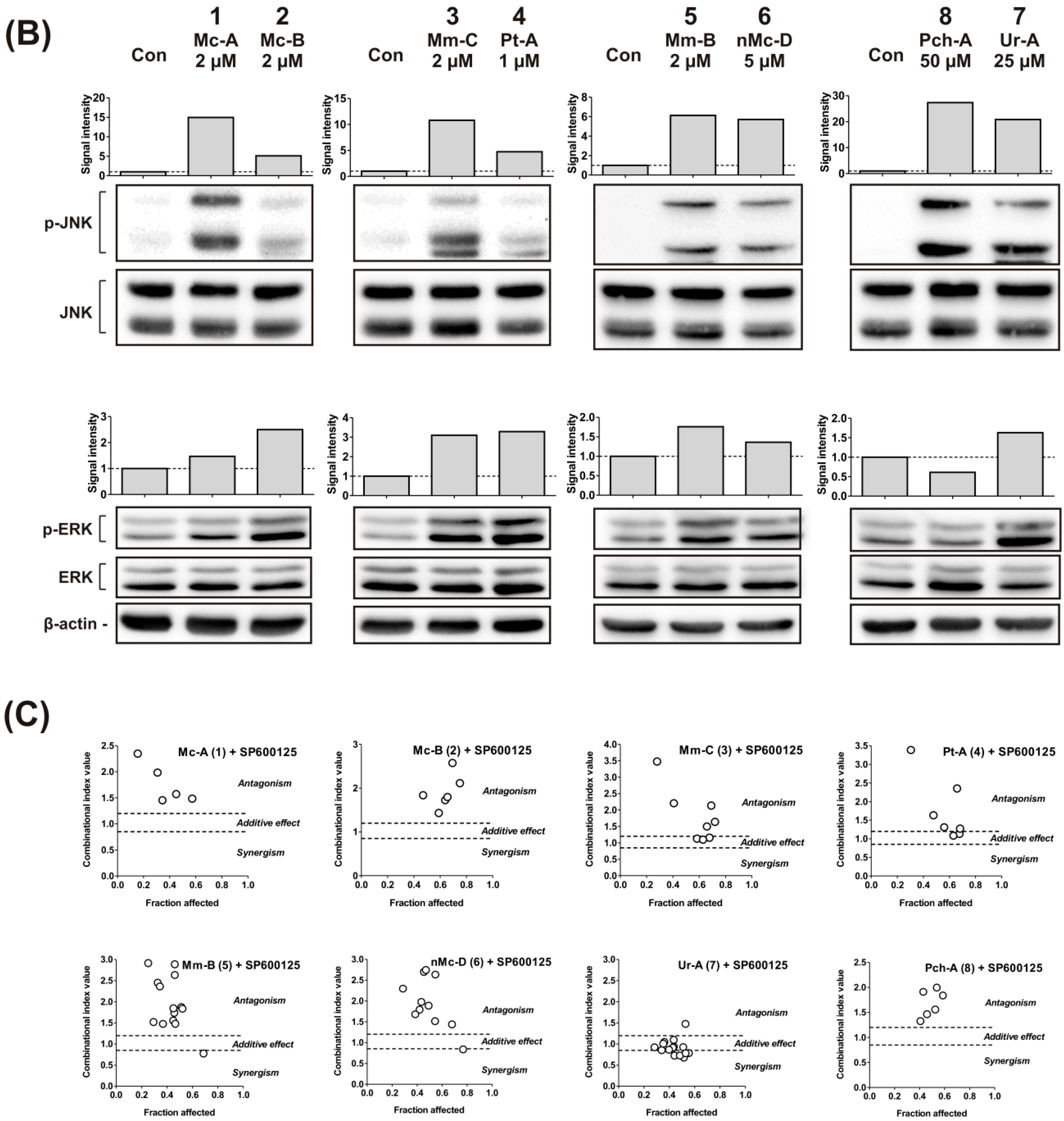
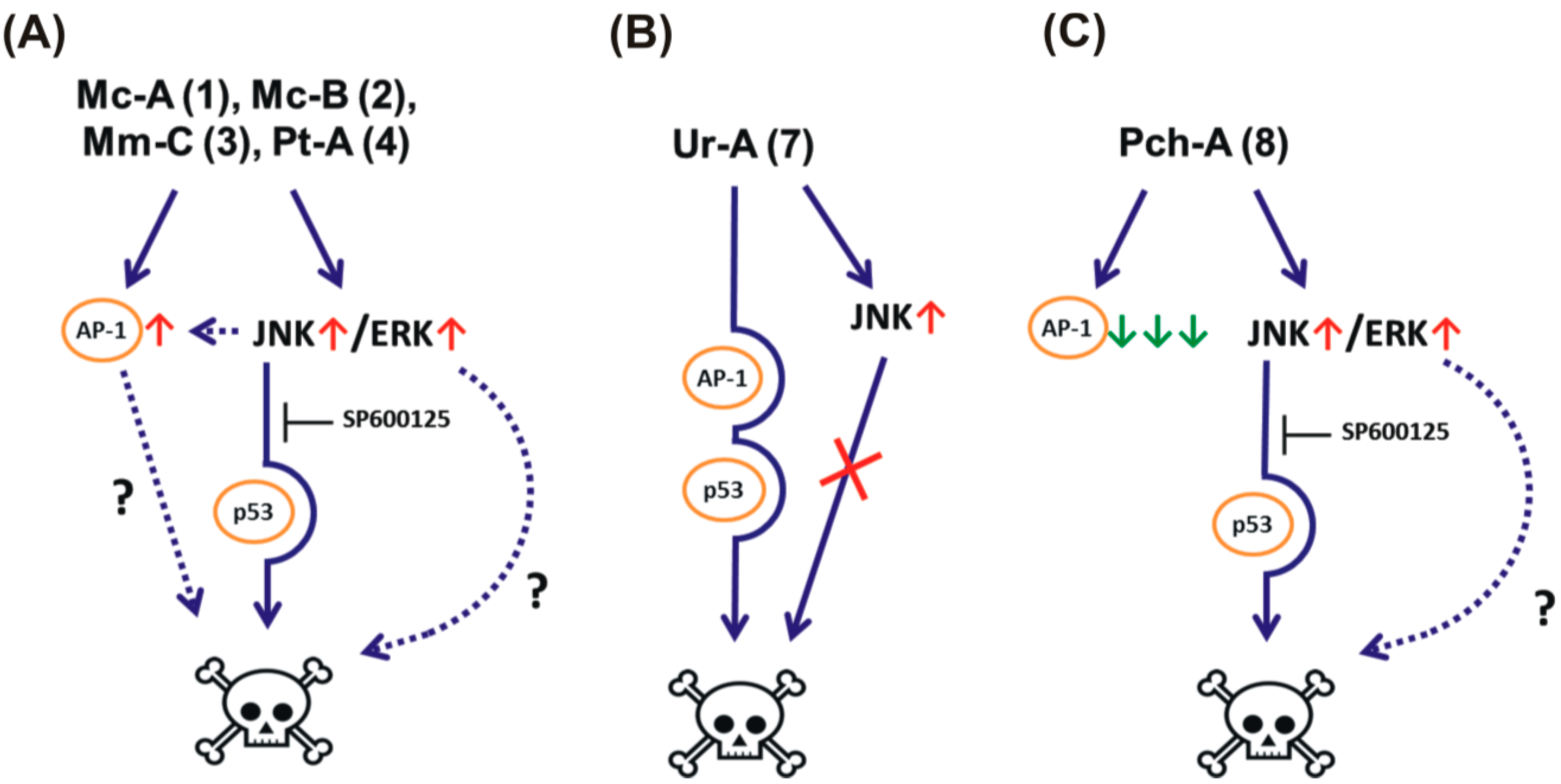
2.2. Effect on MAPK/AP-1 Signaling
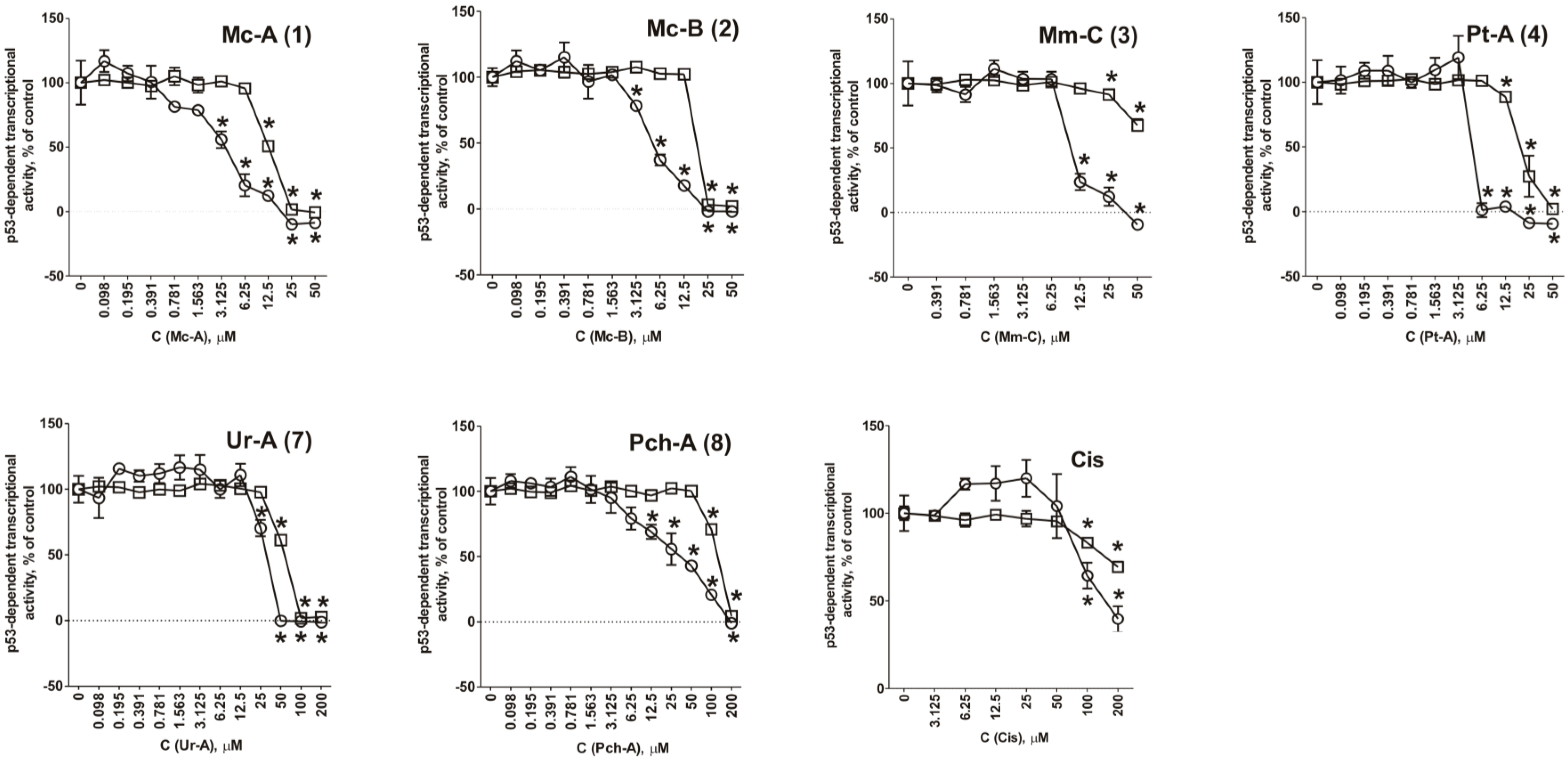
2.3. Role of p53 in Cytotoxic Effect of the Guanidine Alkaloids
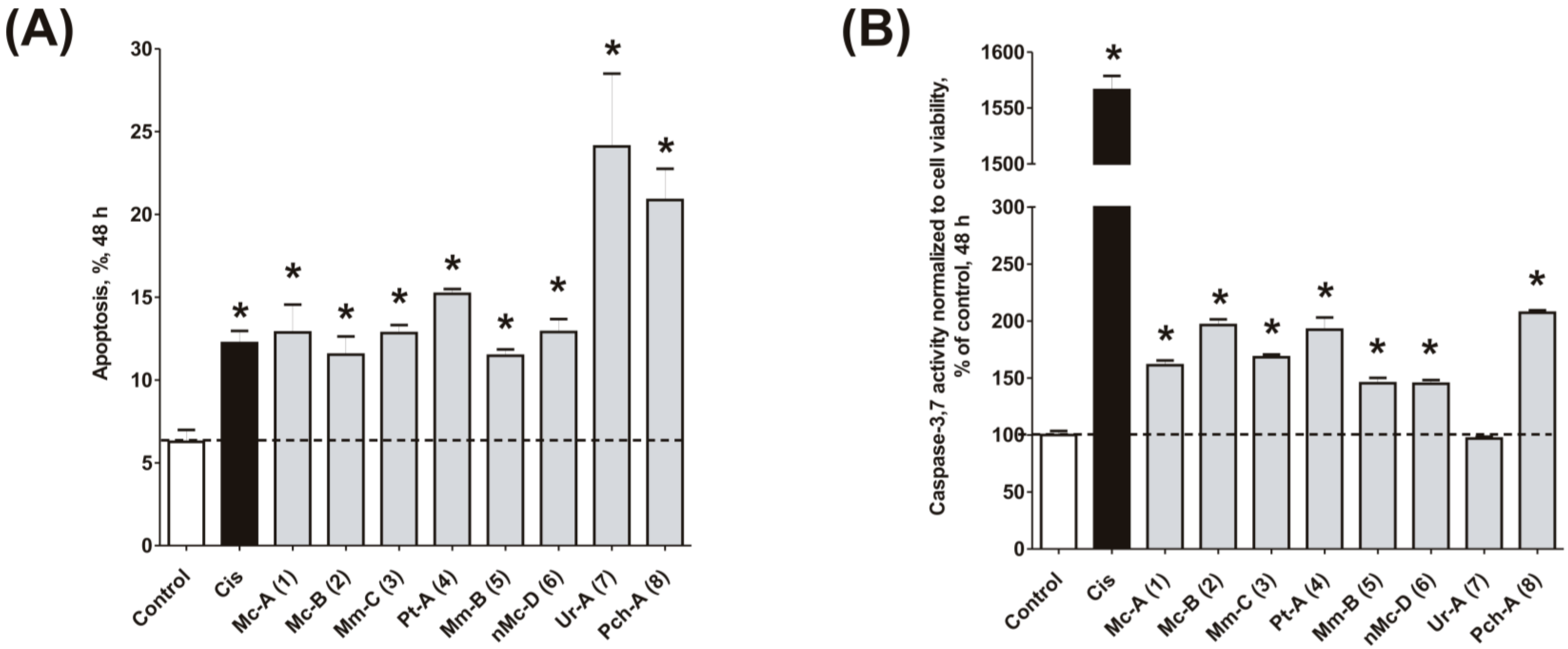
2.4. Induction of Programmed Cell Death and Cell Cycle Arrest in Cancer Cells
3. Materials and Methods
3.1. Reagents and Antibodies
3.2. Cell Culture
3.3. In Vitro MTT-Based Drug Sensitivity Assay
3.4. Anchorage-Independent Neoplastic Transformation (Colony Growth Assay)
3.5. Determination of the Effect of Сompounds on the Basal Transcriptional Activity of AP-1 and p53
3.6. Cell Cycle and DNA Fragmentation Analysis
3.7. Examination of Antagonistic Effects of JNK1/2 Inhibitor SP600125 on the Cytotoxicity of the Tested Compounds
3.8. Western Blotting
3.9. Caspase-3/7 Activity Assay
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Trindade-Silva, A.E.; Romminger, S.; Morais, R.P.; Bandeira, K.; Mizuno, C.M. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2010, 27, 1871–1907. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Kelly, M.; Bowling, J.; Sims, J.; Waters, A.; Hamann, M. Advancement into the arctic region for bioactive sponge secondary metabolites. Mar. Drugs 2011, 9, 2423–2437. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Trindade-Silva, A.E.; Santos, M.F.C. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2012, 29, 1382–1406. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Kossuga, M.H. Natural guanidine derivatives. Nat. Prod. Rep. 2005, 22, 516–550. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S. Natural guanidine derivatives. Nat. Prod. Rep. 2002, 19, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Hamann, M.T.; Gul, W.; Hammond, N.L.; Yousaf, M.; Bowling, J.J.; Schinazi, R.F.; Wirtz, S.S.; Andrews, G.D.C.; Cuevas, C. Modification at the C9 position of the marine natural product isoaaptamine and the impact on HIV-1, mycobacterial, and tumor cell activity. Bioorgan. Med. Chem. 2006, 14, 8495–8505. [Google Scholar]
- Guzii, A.G.; Makarieva, T.N.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Dyshlovoy, S.A.; Krasokhin, V.B.; Stonik, V.A. Monanchocidin: A new apoptosis-inducing polycyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2010, 12, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Kuzmich, A.S.; Lee, H.S.; Stonik, V.A. Monanchomycalins A and B, unusual guanidine alkaloids from the sponge Monanchora pulchra. Tetrahedron Lett. 2012, 53, 4228–4231. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.S.; Stonik, V.A. Monanchocidins B–E: Polycyclic guanidine alkaloids with potent antileukemic activities from the sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Guzii, A.G.; Makarieva, T.N.; Korolkova, Y.V.; Andreev, Y.A.; Mosharova, I.V.; Tabakmaher, K.M.; Denisenko, V.A.; Dmitrenok, P.S.; Ogurtsova, E.K.; Antonov, A.S.; et al. Pulchranin A, isolated from the far-eastern marine sponge, Monanchora pulchra: The first marine non-peptide inhibitor of TRPV-1 channels. Tetrahedron Lett. 2013, 54, 1247–1250. [Google Scholar] [CrossRef]
- Bensemhoun, J.; Bombarda, I.; Aknin, M.; Vacelet, J.; Gaydou, E.M. Ptilomycalin D, a polycyclic guanidine alkaloid from the marine sponge Monanchora dianchora. J. Nat. Prod. 2007, 70, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Gallimore, W.A.; Kelly, M.; Scheuer, P.J. Alkaloids from the sponge Monanchora unguifera. J. Nat. Prod. 2005, 68, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Hauschild, J.; Amann, K.; Tabakmakher, K.M.; Venz, S.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; Fedorov, S.N.; et al. Marine alkaloid monanchocidin A overcomes drug resistance by induction of autophagy and lysosomal membrane permeabilization. Oncotarget 2015, 6, 17328–17341. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Venz, S.; Hauschild, J.; Tabakmakher, K.M.; Otte, K.; Madanchi, R.; Walther, R.; Guzii, A.G.; Makarieva, T.N.; Shubina, L.K.; et al. Anti-migratory activity of marine alkaloid monanchocidin A—proteomics-based discovery and confirmation. Proteomics 2016, 16, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- Tabakmakher, K.M.; Denisenko, V.A.; Guzii, A.G.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Lee, H.S.; Makarieva, T.N. Monanchomycalin C, a new pentacyclic guanidine alkaloid from the far-eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2013, 8, 1399–1402. [Google Scholar] [PubMed]
- Black, G.P.; Coles, S.J.; Hizi, A.; Howard-Jones, A.G.; Hursthouse, M.B.; McGown, A.T.; Loya, S.; Moore, C.G.; Murphy, P.J.; Smith, N.K.; et al. Synthesis and biological activity of analogues of ptilomycalin A. Tetrahedron Lett. 2001, 42, 3377–3381. [Google Scholar] [CrossRef]
- Hua, H.M.; Peng, J.; Dunbar, D.C.; Schinazi, R.F.; de Castro Andrews, A.G.; Cuevas, C.; Garcia-Fernandez, L.F.; Kelly, M.; Hamann, M.T. Batzelladine alkaloids from the caribbean sponge Monanchora unguifera and the significant activities against HIV-1 and aids opportunistic infectious pathogens. Tetrahedron 2007, 63, 11179–11188. [Google Scholar] [CrossRef]
- Laville, R.; Thomas, O.P.; Berrué, F.; Marquez, D.; Vacelet, J.; Amade, P. Bioactive guanidine alkaloids from two caribbean marine sponges. J. Nat. Prod. 2009, 72, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Ogurtsova, E.K.; Denisenko, V.A.; Dmitrenok, P.S.; Tabakmakher, K.M.; Guzii, A.G.; Pislyagin, E.A.; Es’kov, A.A.; Kozhemyako, V.B.; Aminin, D.L.; et al. Urupocidin A: A new, inducing inos expression bicyclic guanidine alkaloid from the marine sponge Monanchora pulchra. Org. Lett. 2014, 16, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Tabakmakher, K.M.; Makarieva, T.N.; Denisenko, V.A.; Guzii, A.G.; Dmitrenok, P.S.; Kuzmich, A.S.; Stonik, V.A. Normonanchocidins A, B and D, new pentacyclic guanidine alkaloids from the far-eastern marine sponge Monanchora pulchra. Nat. Prod. Commun. 2015, 10, 913–916. [Google Scholar] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid differentiation in K562 chronic myelogenous cells induced by crambescidin 800, a pentacyclic guanidine alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Aron, Z.D.; Pietraszkiewicz, H.; Overman, L.E.; Valeriote, F.; Cuevas, C. Synthesis and anticancer activity of side chain analogs of the crambescidin alkaloids. Bioorg. Med. Chem. Lett. 2004, 14, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Ternon, E.; Lopez-Alonso, H.; Thomas, O.P.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Crambescidin-816 acts as a fungicidal with more potency than crambescidin-800 and-830, inducing cell cycle arrest, increased cell size and apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic guanidine alkaloids from the marine sponge Crambe crambe and Ca++ channel blocker activity of crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Vale, C.; Bondu, S.; Thomas, O.P.; Vieytes, M.R.; Botana, L.M. Differential effects of crambescins and crambescidin 816 in voltage-gated sodium, potassium and calcium channels in neurons. Chem. Res. Toxicol. 2013, 26, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; Lopez-Alonso, H.; Roel, M.; Vieytes, M.R.; Thomas, O.; Ternon, E.; Vega, F.V.; Botana, L.M. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharmacol. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.; Ermakova, S.; Zvyagintseva, T.; Stonik, V. Anticancer and cancer preventive properties of marine polysaccharides: Some results and prospects. Mar. Drugs 2013, 11, 4876–4901. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.; Fedorov, S. Marine low molecular weight natural products as potential cancer preventive compounds. Mar. Drugs 2014, 12, 636–671. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Bode, A.M.; Stonik, V.A.; Gorshkova, I.A.; Schmid, P.C.; Radchenko, O.S.; Berdyshev, E.V.; Dong, Z.G. Marine alkaloid polycarpine and its synthetic derivative dimethylpolyearpine induce apoptosis in JB6 cells through p53-and caspase 3-dependent pathways. Pharm. Res. 2004, 21, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Makarieva, T.N.; Guzii, A.G.; Shubina, L.K.; Kwak, J.Y.; Stonik, V.A. Marine two-headed sphingolipid-like compound rhizochalin inhibits EGF-induced transformation of JB6 P+ Cl41 cells. Lipids 2009, 44, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Radchenko, O.S.; Shubina, L.K.; Balaneva, N.N.; Bode, A.M.; Stonik, V.A.; Dong, Z. Evaluation of cancer-preventive activity and structure-activity relationships of 3-demethylubiquinone Q2, isolated from the ascidian aplidium glabrum, and its synthetic analogs. Pharm. Res. 2006, 23, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Shubina, L.K.; Bode, A.M.; Stonik, V.A.; Dong, Z.G. Dactylone inhibits epidermal growth factor-induced transformation and phenotype expression of human cancer cells and induces G1/S arrest and apoptosis. Cancer Res. 2007, 67, 5914–5920. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Kalinovsky, A.I.; Shubina, L.K.; Bokemeyer, C.; Stonik, V.A.; Honecker, F. Mycalamide A shows cytotoxic properties and prevents EGF-induced neoplastic transformation through inhibition of nuclear factors. Mar. Drugs 2012, 10, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Fedorov, S.N.; Shubina, L.K.; Kuzmich, A.S.; Bokemeyer, C.; Keller-von Amsberg, G.; Honecker, F. Aaptamines from the marine sponge Aaptos sp. Display anticancer activities in human cancer cell lines and modulate AP-1-, NF-kappa B-, and p53-dependent transcriptional activity in mouse JB6 Cl41 cells. Biomed. Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.; Dyshlovoy, S.; Monastyrnaya, M.; Shubina, L.; Leychenko, E.; Kozlovskaya, E.; Jin, J.O.; Kwak, J.Y.; Bode, A.M.; Dong, Z.G.; et al. The anticancer effects of actinoporin rtx-a from the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 2010, 55, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Menchinskaya, E.S.; Venz, S.; Rast, S.; Amann, K.; Hauschild, J.; Otte, K.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; et al. The marine triterpene glycoside frondoside A exhibits activity in vitro and in vivo in prostate cancer. Int. J. Cancer 2016, 138, 2450–2465. [Google Scholar] [CrossRef] [PubMed]
- Colburn, N.H.; Gindhart, T.D. Specific binding of transforming growth factor correlates with promotion of anchorage independence in EGF receptorless mouse JB6 cells. Biochem. Biophys. Res. Commun. 1981, 102, 799–807. [Google Scholar] [CrossRef]
- Colburn, N.H.; Former, B.F.; Nelson, K.A.; Yuspa, S.H. Tumour promoter induces anchorage independence irreversibly. Nature 1979, 281, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Colburn, N.H.; Koehler, B.A.; Nelson, K.J. A cell culture assay for tumor-promoter-dependent progression toward neoplastic phenotype: Detection of tumor promoters and promotion inhibitors. Teratog. Carcinog. Mutagen. 1980, 1, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Ma, W.Y.; Young, M.R.; Colburn, N.; Dong, Z.G. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc. Natl. Acad. Sci. USA 1998, 95, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.G.; Birrer, M.J.; Watts, R.G.; Matrisian, L.M.; Colburn, N.H. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. USA 1994, 91, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Ma, W.Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z.G. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA 1997, 94, 5826–5830. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.C.; Nair, R.; Tulsian, P.; Camalier, C.E.; Hegamyer, G.A.; Young, M.R.; Colburn, N.H. Transformation nonresponsive cells owe their resistance to lack of p65/nuclear factor-kappa B activation. Cancer Res. 2001, 61, 4160–4168. [Google Scholar] [PubMed]
- Li, J.J.; Westergaard, C.; Ghosh, P.; Colburn, N.H. Inhibitors of both nuclear factor-kappa beta and activator protein-1 activation block the neoplastic transformation response. Cancer Res. 1997, 57, 3569–3576. [Google Scholar] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.N.; Shubina, L.K.; Kicha, A.A.; Ivanchina, N.V.; Kwak, J.Y.; Jin, J.O.; Bode, A.M.; Dong, Z.G.; Stonik, V.A. Proapoptotic and anticarcinogenic activities of leviusculoside G from the starfish Henricia leviuscula and probable molecular mechanism. Nat. Prod. Commun. 2008, 3, 1575–1580. [Google Scholar]
- Foletta, V.C.; Segal, D.H.; Cohen, D.R. Transcriptional regulation in the immune system: All roads lead to AP-1. J. Leukoc. Biol. 1998, 63, 139–152. [Google Scholar] [PubMed]
- Ameyar, M.; Wisniewska, M.; Weitzman, J.B. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Berry, A.; Goodwin, M.; Moran, C.L.; Chambers, T.C. AP-1 activation and altered AP-1 composition in association with increased phosphorylation and expression of specific Jun and Fos family proteins induced by vinblastine in KB-3 cells. Biochem. Pharmacol. 2001, 62, 581–591. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.E.; Birrer, M.J.; Chambers, T.C. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001, 61, 4450–4458. [Google Scholar] [PubMed]
- Gopalakrishnan, A.; Xu, C.J.; Nair, S.S.; Chen, C.; Hebbar, V.; Kong, A.N. Modulation of activator protein-1 (AP-1) and MAPK pathway by flavonoids in human prostate cancer PC3 cells. Arch. Pharm. Res. 2006, 29, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Bahn, J.H.; Whitlock, N.C.; Baek, S.J. Activating transcription factor 2 (ATF2) controls tolfenamic acid-induced atf3 expression via map kinase pathways. Oncogene 2010, 29, 5182–5192. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3-and 10-bromofascaplysins is mediated by caspase-8,-9,-3-dependent apoptosis. Bioorgan. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef] [PubMed]
- Shubina, L.K.; Fedorov, S.N.; Radchenko, O.S.; Balaneva, N.N.; Kolesnikova, S.A.; Dmitrenok, P.S.; Bode, A.; Dong, Z.; Stonik, V.A. Desmethylubiquinone Q2 from the far-eastern ascidian Aplidium glabrum: Structure and synthesis. Tetrahedron Lett. 2005, 46, 559–562. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA damaging agents induce expression of fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Yu, Q. Restoring p53-mediated apoptosis in cancer cells: New opportunities for cancer therapy. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2006, 9, 19–25. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.C.; Becerril, C.; Ljungman, M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 2001, 20, 6805–6808. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, E.G.; Voutsinas, G.E.; Karkoulis, P.K.; Aravantinos, G.; Margaritis, L.H.; Stravopodis, D.J. Human bladder cancer cells undergo cisplatin-induced apoptosis that is associated with p53-dependent and p53-independent responses. Int. J. Oncol. 2009, 35, 401–416. [Google Scholar] [PubMed]
- Zamble, D.B.; Jacks, T.; Lippard, S.J. P53-dependent and -independent responses to cisplatin in mouse testicular teratocarcinoma cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2014, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Venz, S.; Shubina, L.K.; Fedorov, S.N.; Walther, R.; Jacobsen, C.; Stonik, V.A.; Bokemeyer, C.; Balabanov, S.; Honecker, F. Activity of aaptamine and two derivatives, demethyloxyaaptamine and isoaaptamine, in cisplatin-resistant germ cell cancer. J. Proteom. 2014, 96, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Naeth, I.; Venz, S.; Preukschas, M.; Sievert, H.; Jacobsen, C.; Shubina, L.K.; Gesell Salazar, M.; Scharf, C.; Walther, R.; et al. Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with antiproliferative activity. J. Proteome Res. 2012, 11, 2316–2330. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Cytotoxic Activity, IC50 [µМ], 48 h | |
|---|---|---|---|
| HeLa Cells | JB6 P+ Cl41 Cells | ||
| 1 | Monanchocidin A (Mc-A) | 1.39 | 2.01 |
| 2 | Monanchocidin B (Mc-B) | 0.58 | 1.36 |
| 3 | Monanchomycalin C (Mm-C) | 1.84 | 1.31 |
| 4 | Ptilomycalin A (Pt-A) | 1.1 | 0.5 |
| 5 | Monanchomycalin B (Mm-B) | 1.5 | 1.72 |
| 6 | Normonanchocidin D (nMc-D) | 2.1 | 5.2 |
| 7 | Urupocidin A (Ur-A) | 28.7 | 27 |
| 8 | Pulchranin A (Pch-A) | 51 | 58 |
| Cisplatin | 4.75 | 30.2 | |
| - | Control | 1 (Mc-A) | 2 (Mc-B) | 3 (Mm-C) | 4 (Pt-A) | 5 (Mm-B) | 6 (nMc-D) | 7 (Ur-A) | 8 (Pch-A) | Cis |
|---|---|---|---|---|---|---|---|---|---|---|
| G1-phase | 69.5 ± 1 | 62 ± 0.7 | 63.8 ± 1.5 | 65.6 ± 1.8 | 65.4 ± 0.4 | 66.3 ± 0.9 | 71.4 ± 0.1 | 65.3 ± 3 | 73 ± 1.5 | 7 ± 0.6 |
| S-phase | 13.8 ± 0.4 | 17 ± 0.7 * | 21 ± 0.1 * | 21.5 ± 1.4 * | 22.6 ± 2 * | 23.7 ± 1.2 * | 17.6 ± 0.2 * | 10.6 ± 0.8 | 11.2 ± 1.5 | 15.6 ± 3.5 |
| G2/M-phase | 16.3 ± 0.7 | 21.4 ± 1.3 * | 14.9 ± 0.8 | 12.1 ± 2.1 | 12.2 ± 1.5 | 10.3 ± 1.1 | 10.5 ± 0.8 | 23.7 ± 0.5 * | 15.7 ± 0.8 | 77.6 ± 3.7 * |
| Cell cycle arrest | S, G2/M | S | S | S | S | S | G2/M | no | G2/M | |
| Raw data |  |  |  |  |  |  |  |  |  |  |
| Effect | 1 (Mc-A) | 2 (Mc-B) | 3 (Mm-C) | 4 (Pt-A) | 5 (Mm-B) | 6 (nMc-D) | 7 (Ur-A) | 8 (Pch-A) | Cis |
|---|---|---|---|---|---|---|---|---|---|
| Effect on DNA fragmentation at IC50 | ↑↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑↑ | ↑↑↑ | ↑↑ |
| Effect on caspase-3/7 activity at IC50 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | no | ↑↑ | ↑↑↑ |
| Cell cycle arrest | S, G2/M | S | S | S | S | S | G2/M | no | G2/M |
| Effect on AP-1-transcriptional activity | ↑ | ↑ | ↑ | ↑ | - | - | no | ↓ | ↑ |
| Effect on ERK phosphorylation | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | no | - |
| Effect on JNK phosphorylation | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | - |
| Effect of SP600125 (JNK inhibitor) on the drug cytotoxic activity | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | no | ↓ | - |
| Activation of p53-transcriptional activity | no | no | no | no | no | no | no | no | no |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in Vitro. Mar. Drugs 2016, 14, 133. https://doi.org/10.3390/md14070133
Dyshlovoy SA, Tabakmakher KM, Hauschild J, Shchekaleva RK, Otte K, Guzii AG, Makarieva TN, Kudryashova EK, Fedorov SN, Shubina LK, et al. Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in Vitro. Marine Drugs. 2016; 14(7):133. https://doi.org/10.3390/md14070133
Chicago/Turabian StyleDyshlovoy, Sergey A., Kseniya M. Tabakmakher, Jessica Hauschild, Regina K. Shchekaleva, Katharina Otte, Alla G. Guzii, Tatyana N. Makarieva, Ekaterina K. Kudryashova, Sergey N. Fedorov, Larisa K. Shubina, and et al. 2016. "Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation in Vitro" Marine Drugs 14, no. 7: 133. https://doi.org/10.3390/md14070133