
Marine Antibody–Drug Conjugates: Design Strategies and Research Progress

 ,
,
Abstract
:1. Introduction
2. ADCs in Clinical Trial
3. ADCs Design
3.1. Antigen and Indication
3.2. Monoclonal Antibody
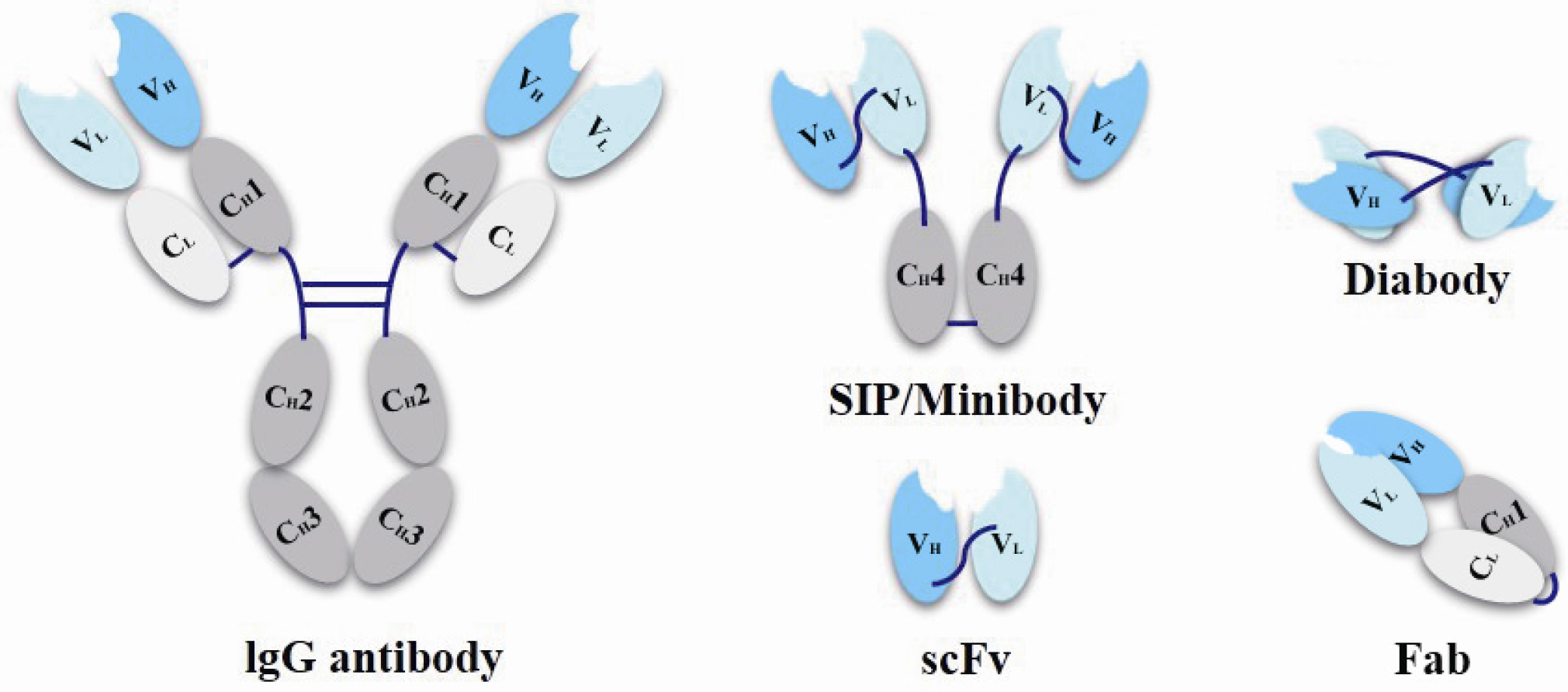
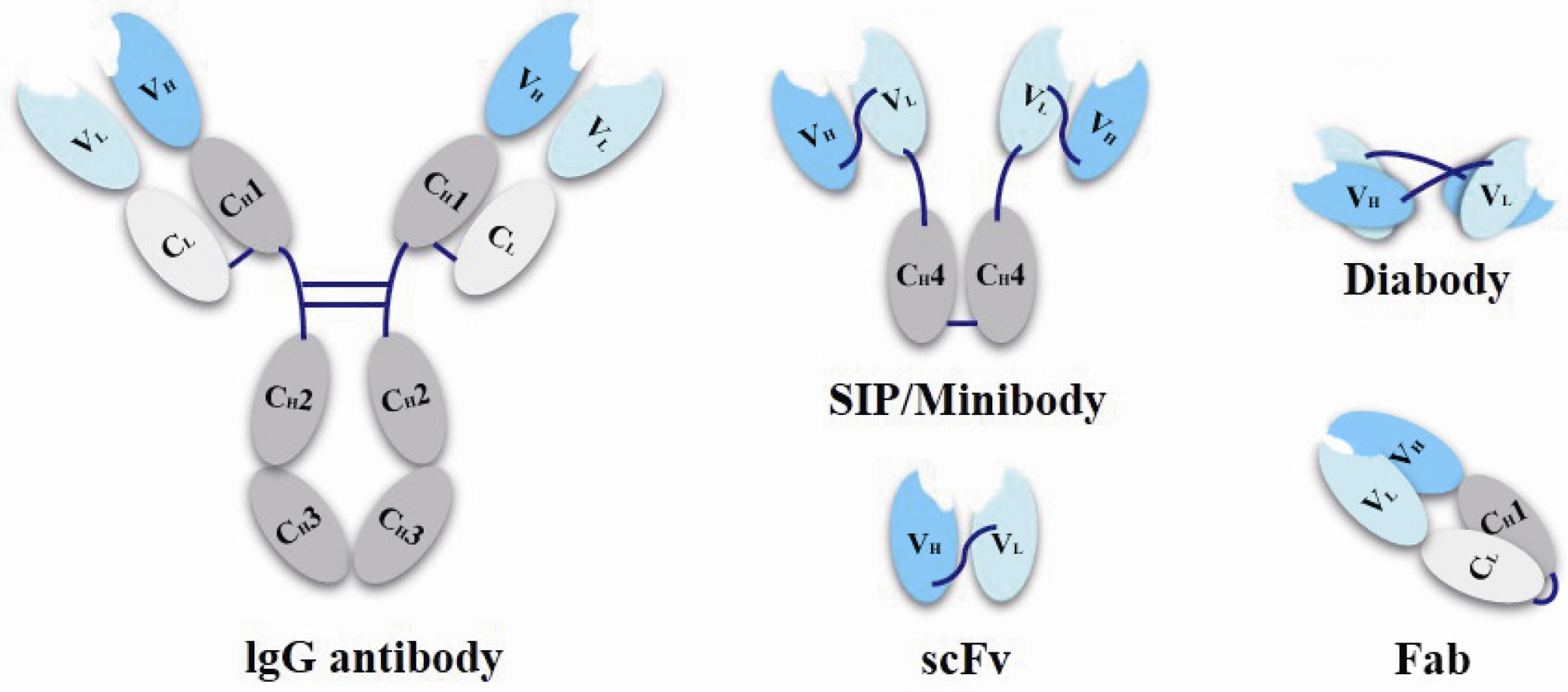
3.2.1. Antibody Structure
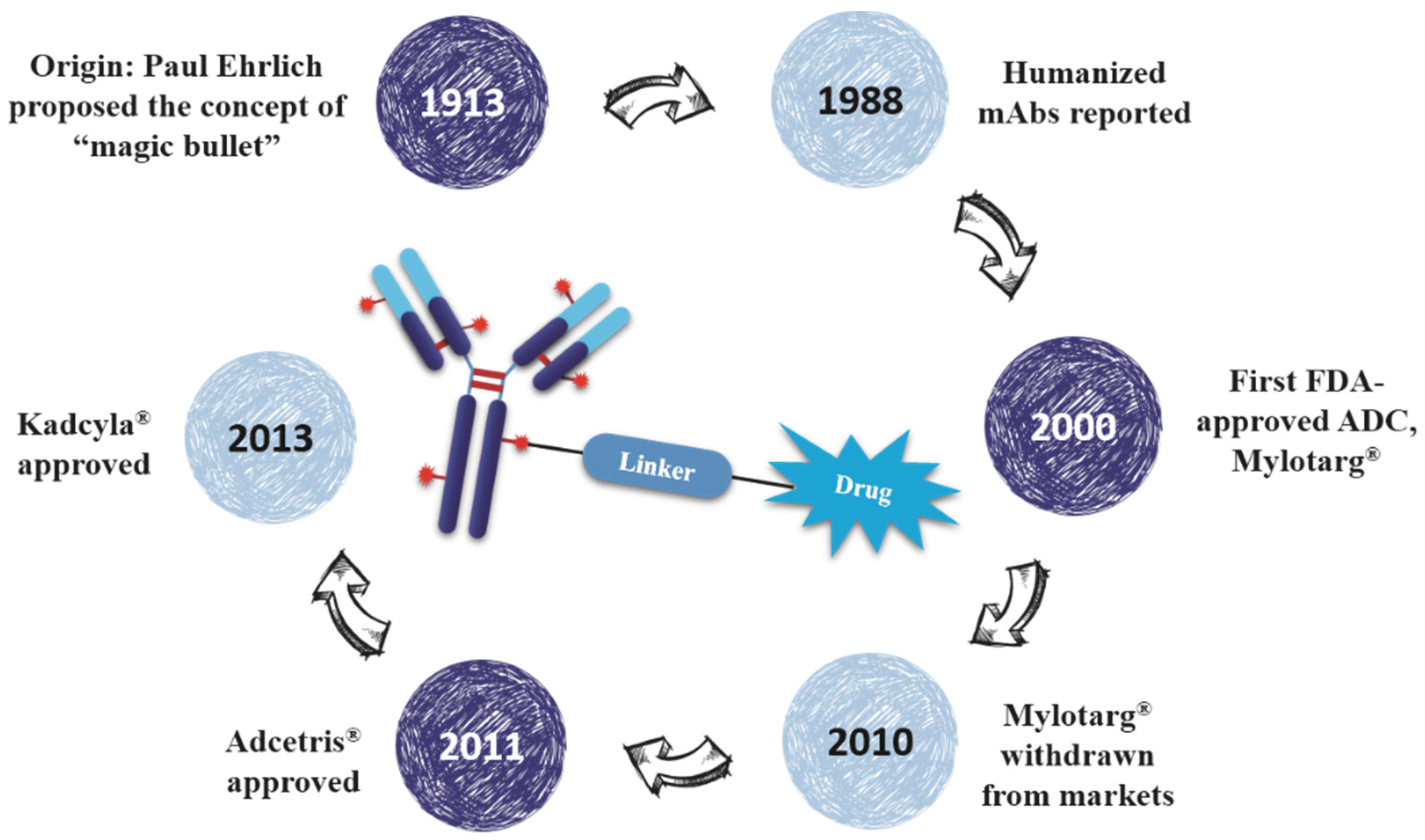
3.2.2. History of Therapeutic MAbs
3.2.3. Novel MAbs
3.3. Site of Conjugation
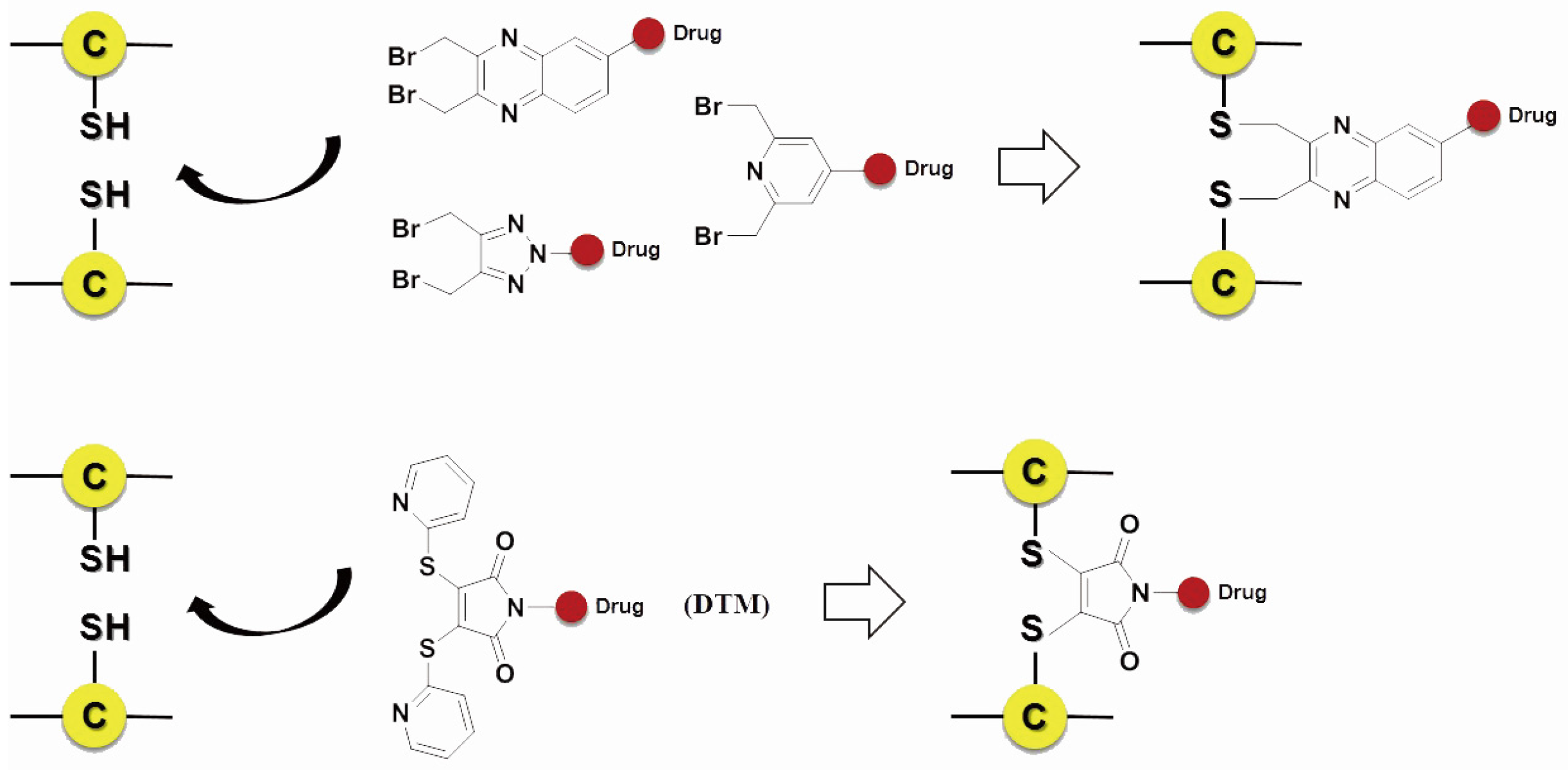
3.3.1. Non-Specific Conjugation
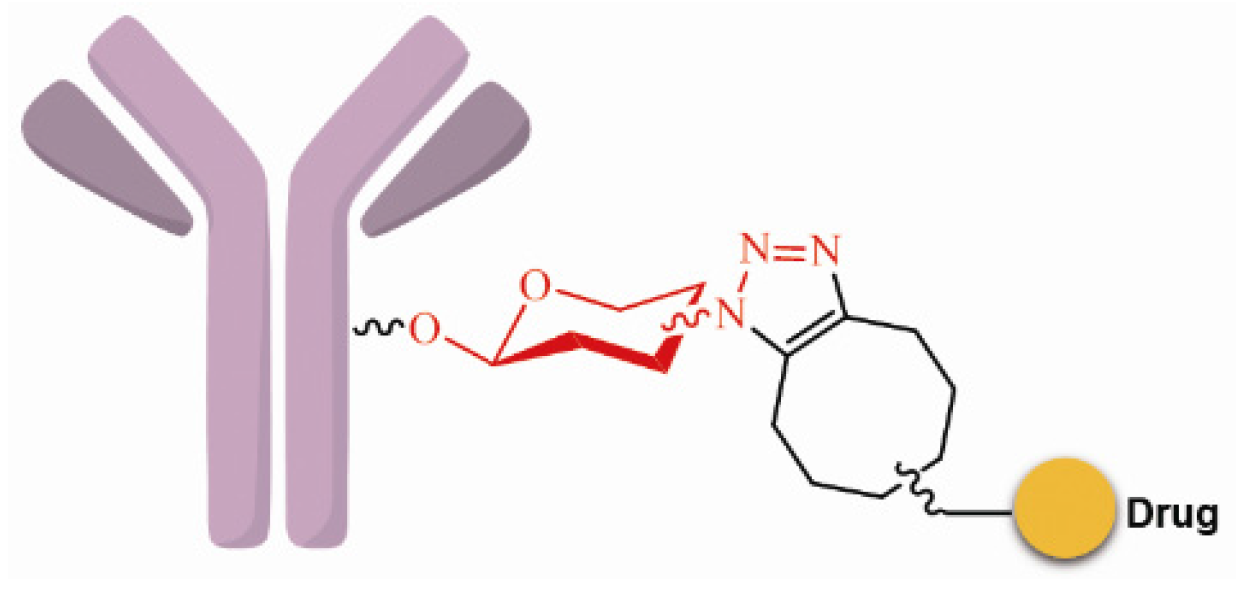
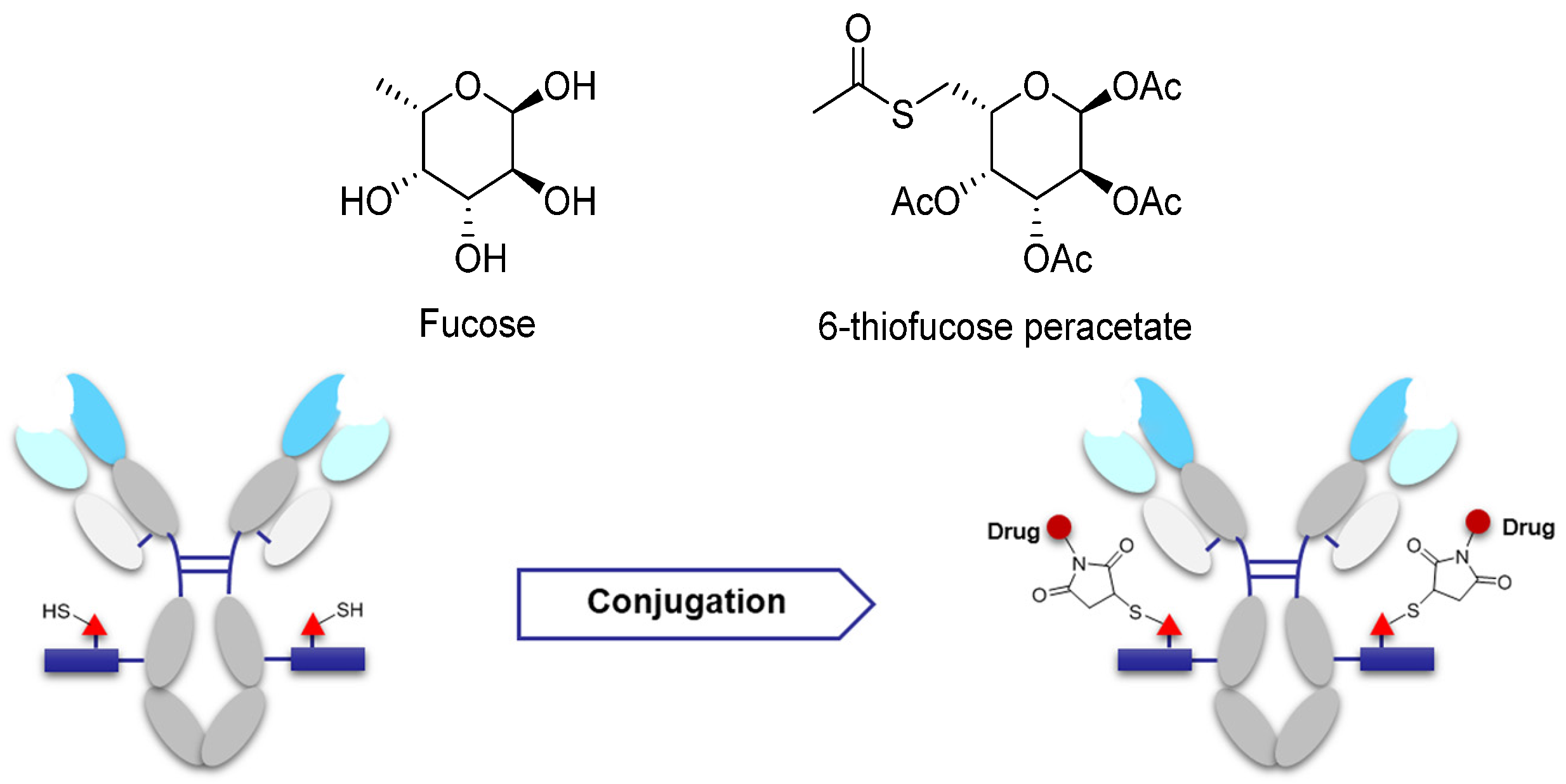
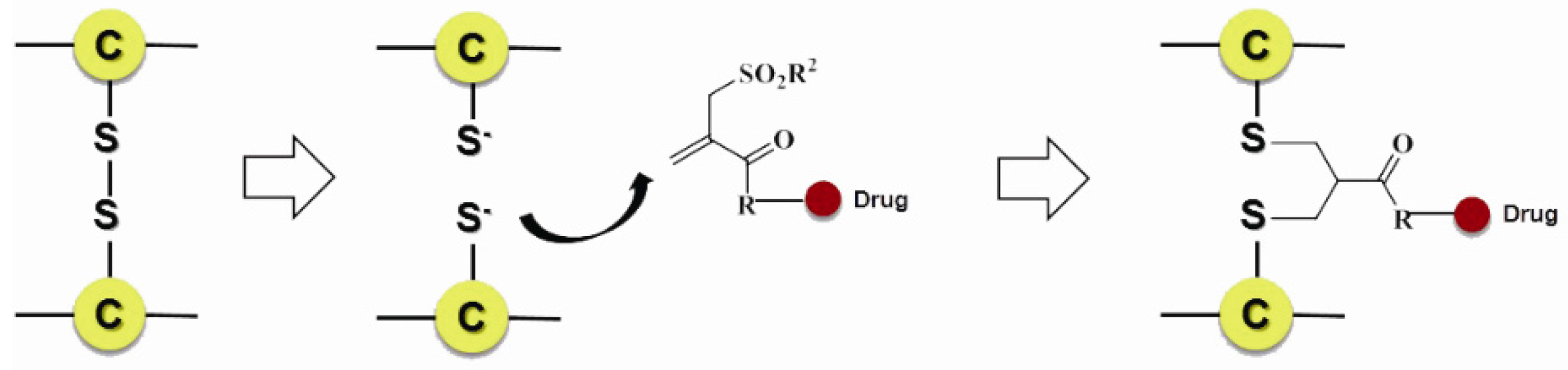
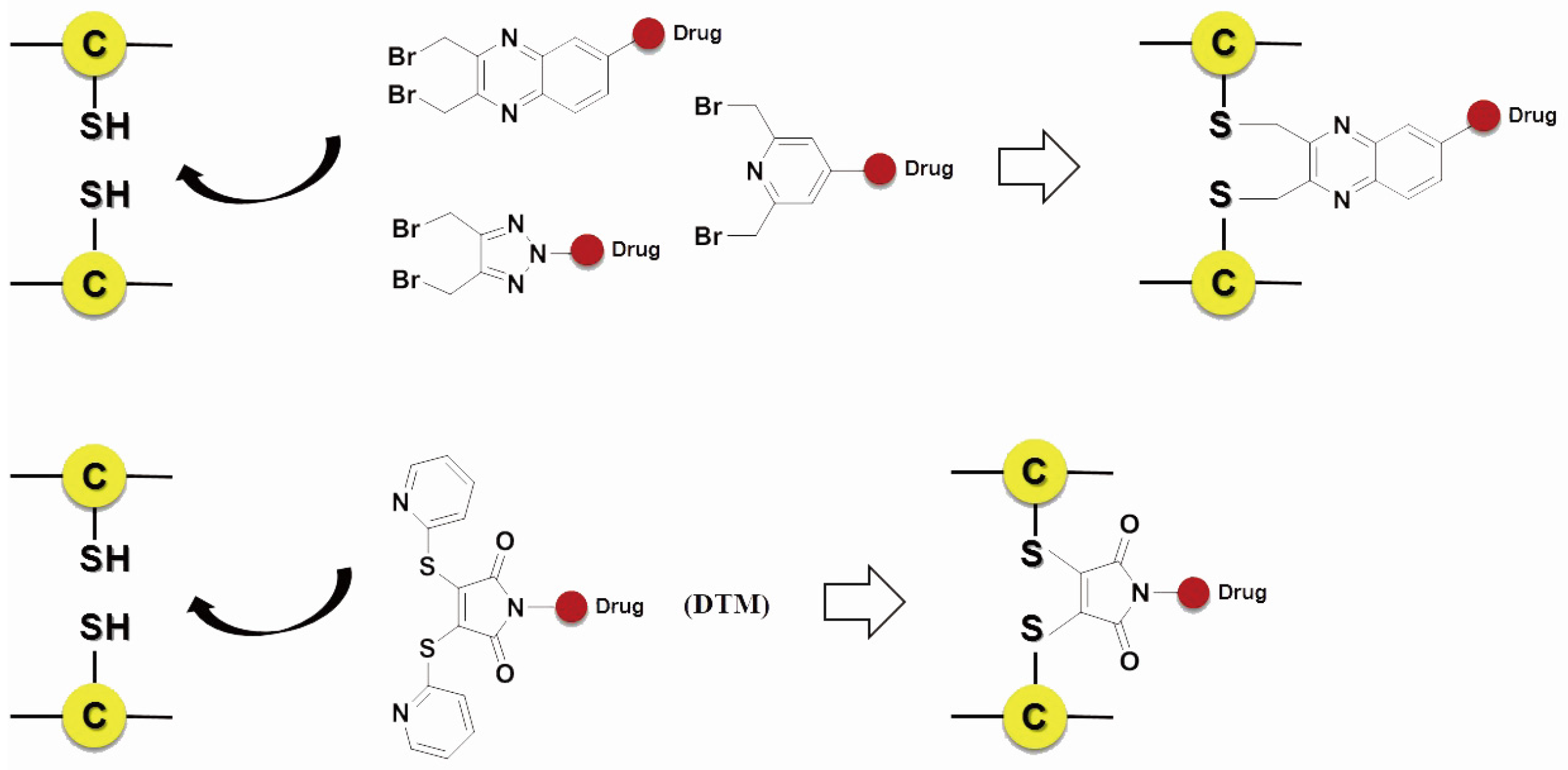
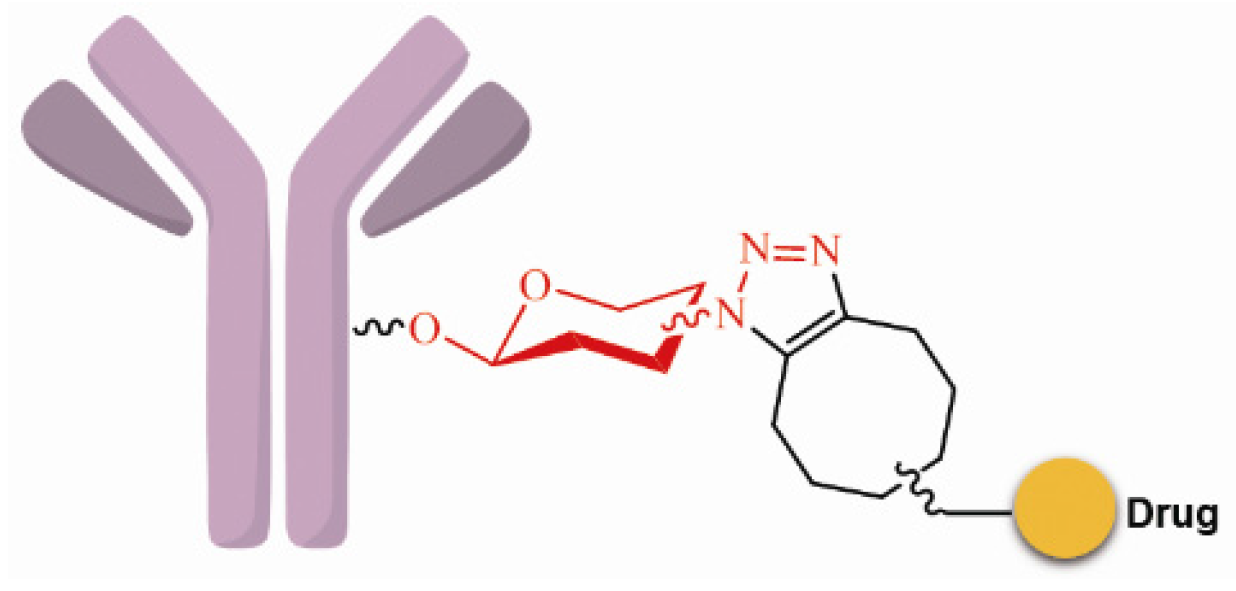
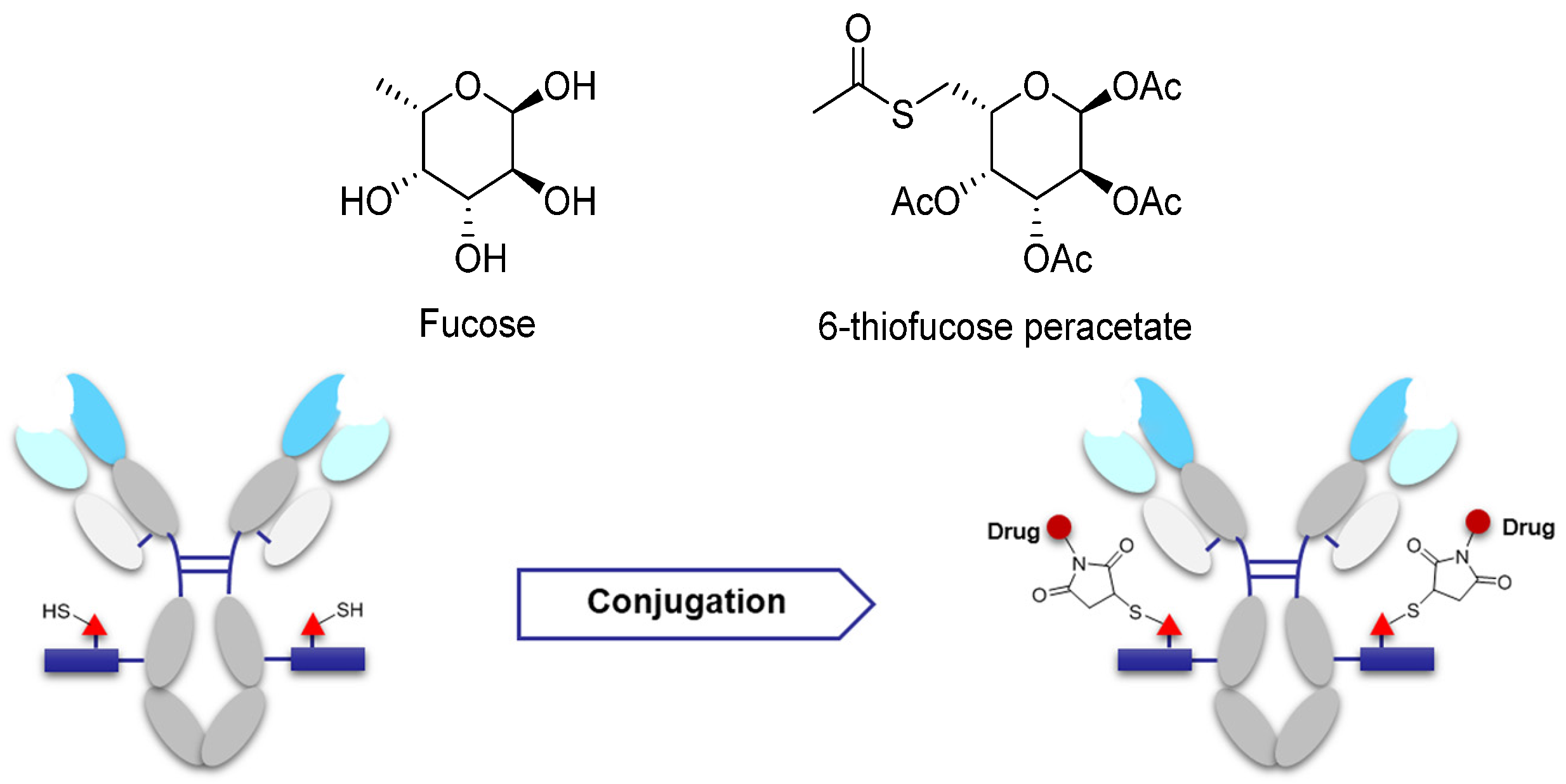
3.3.2. Site-Specific Conjugation
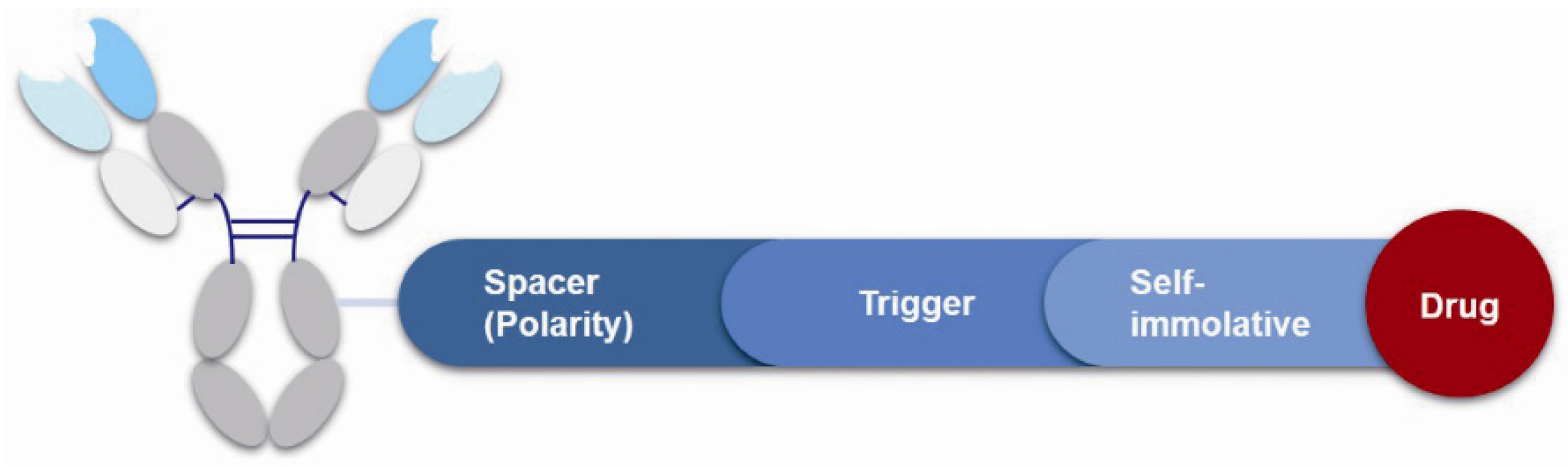
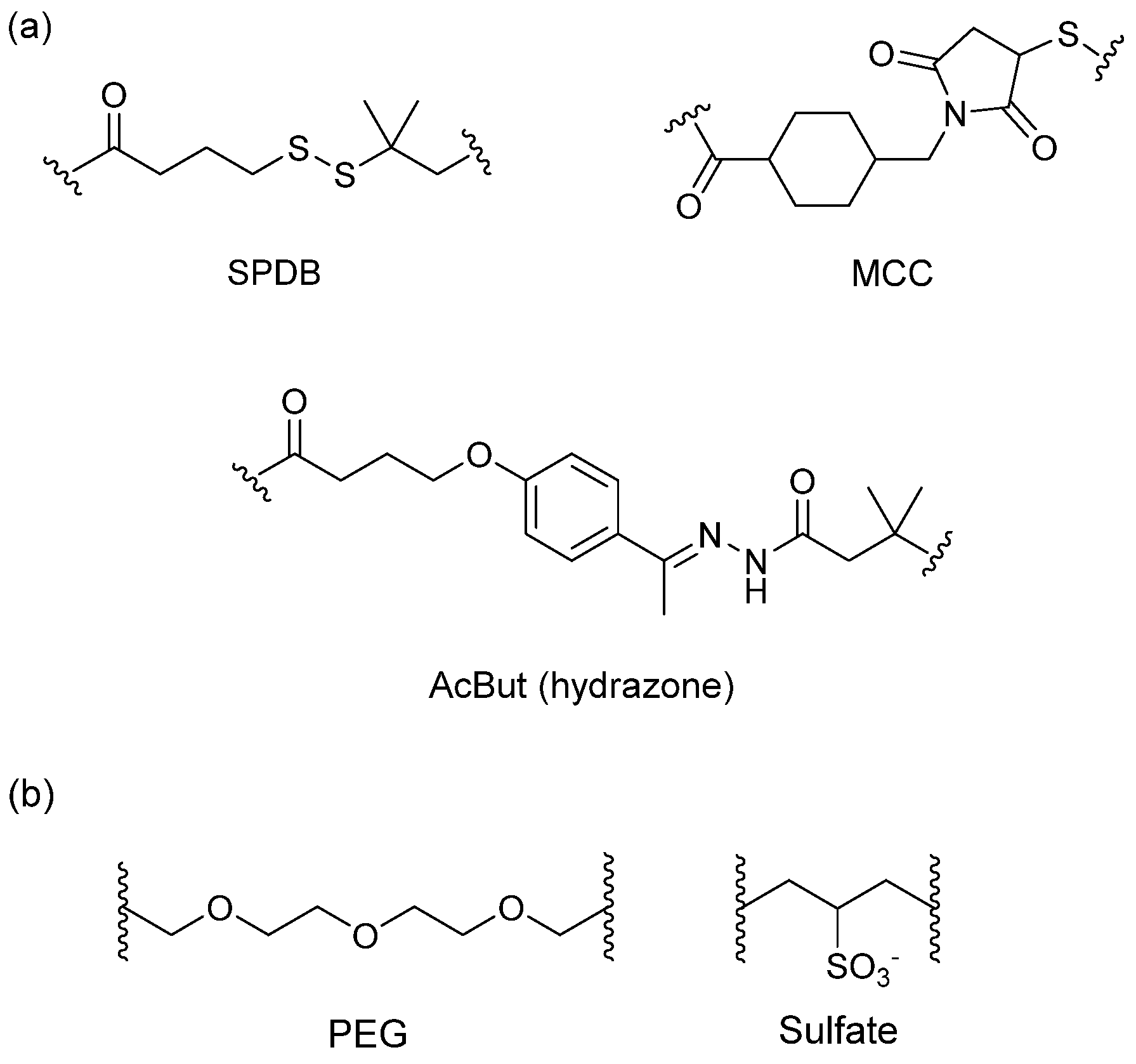
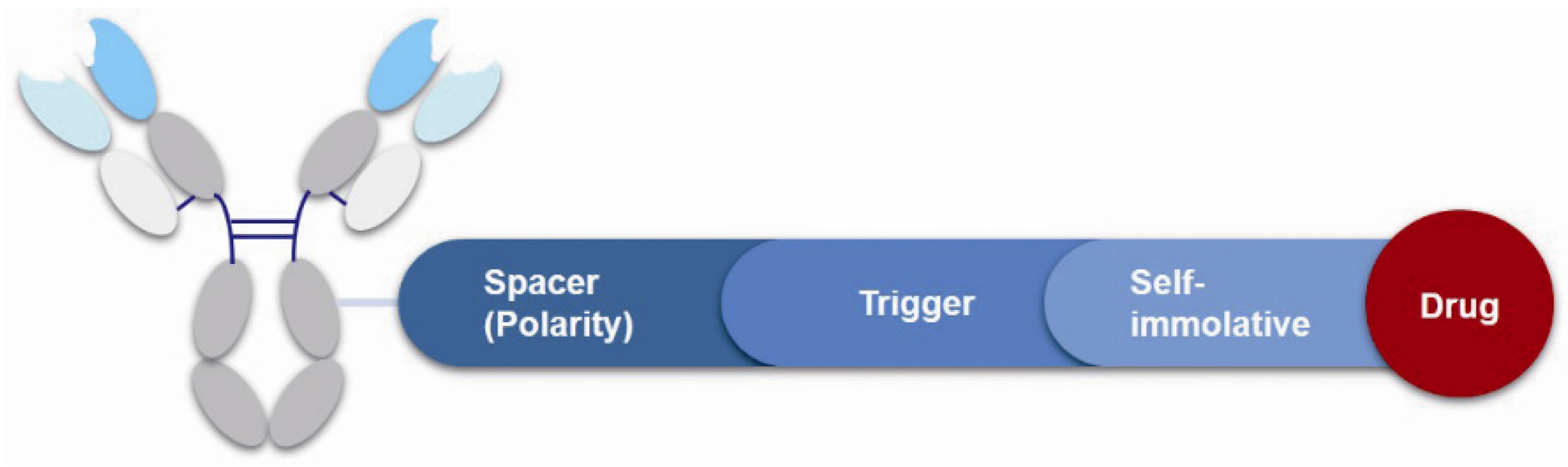
3.4. Linker
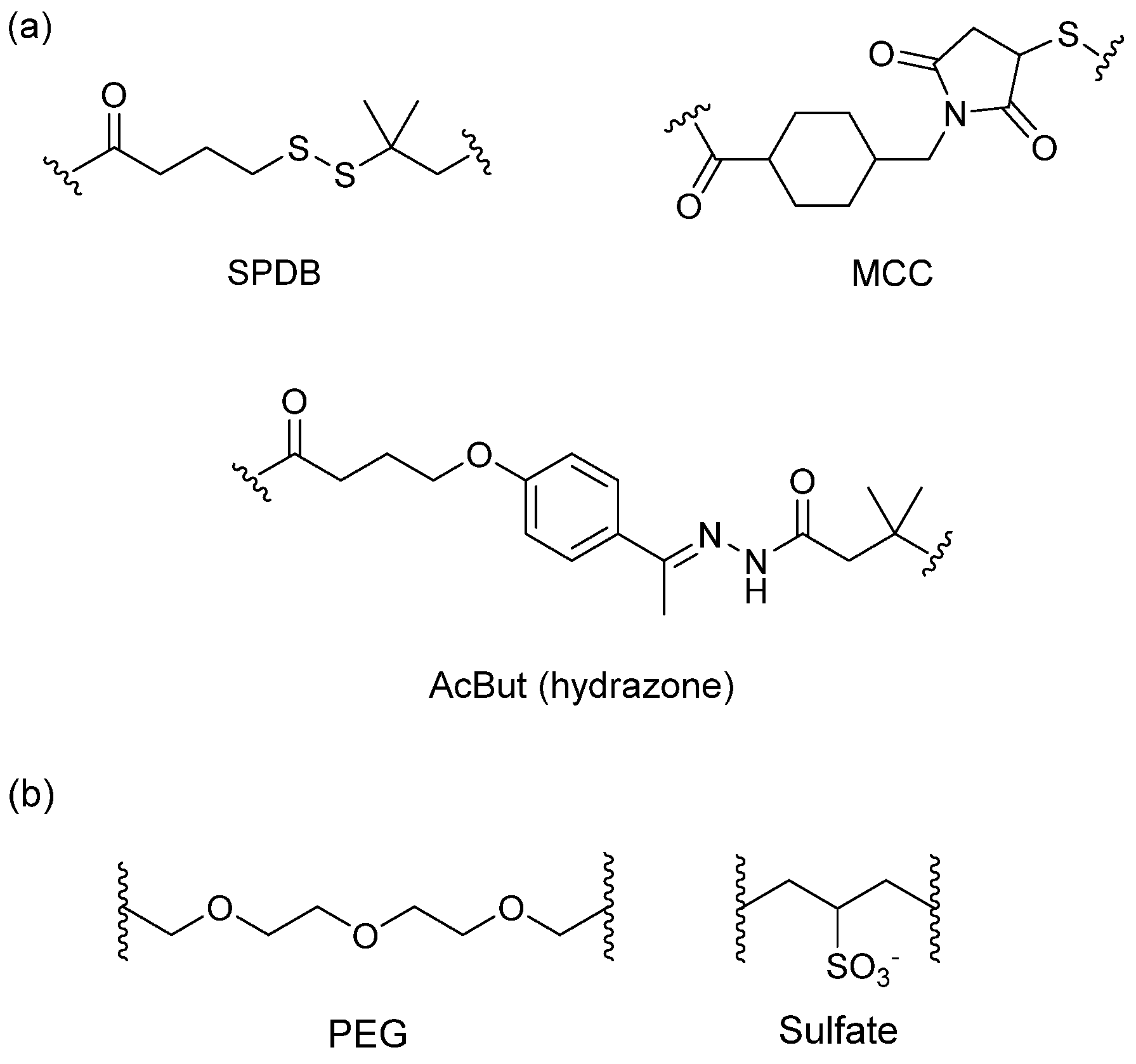
3.4.1. Spacer
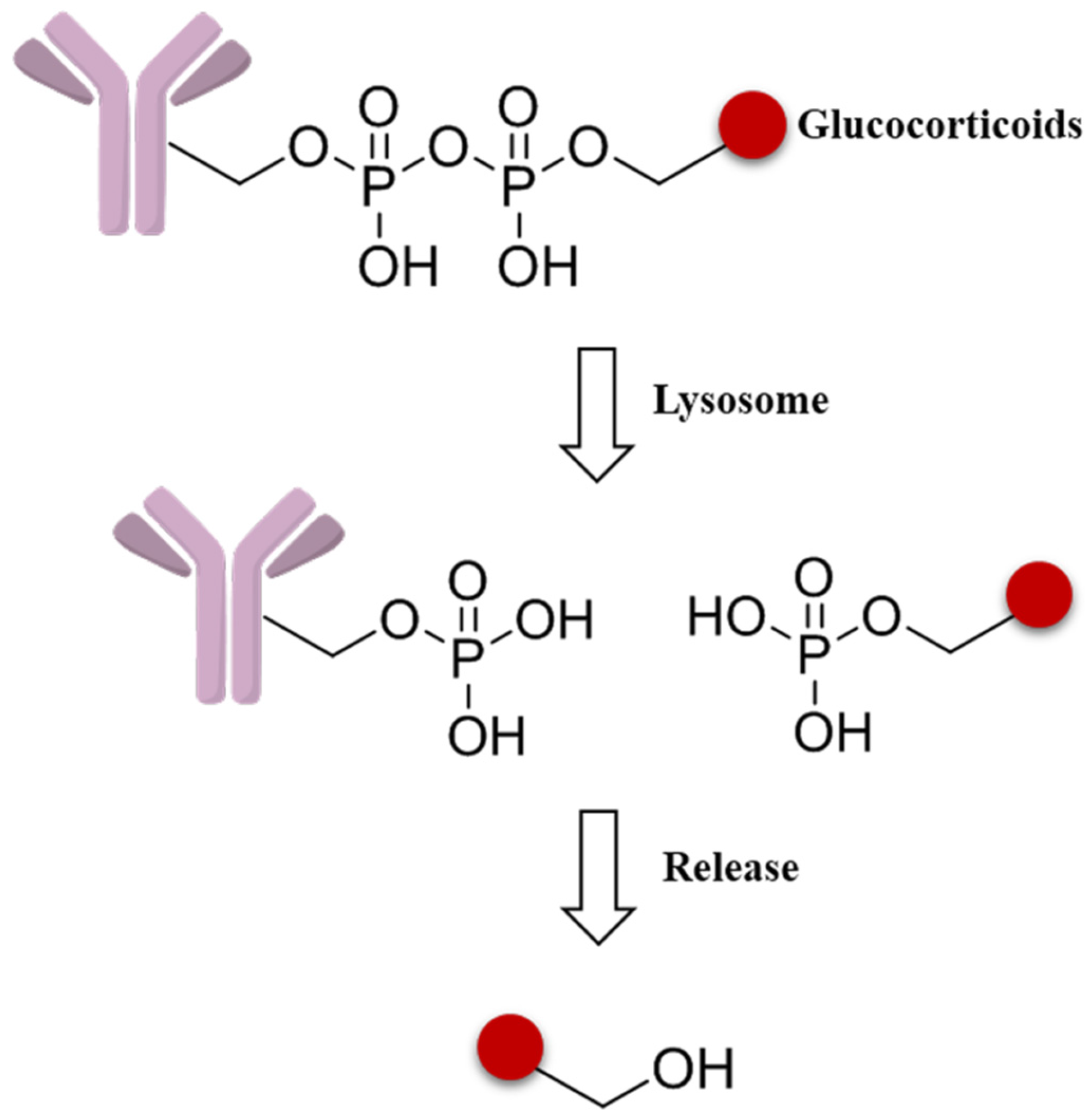
3.4.2. Trigger
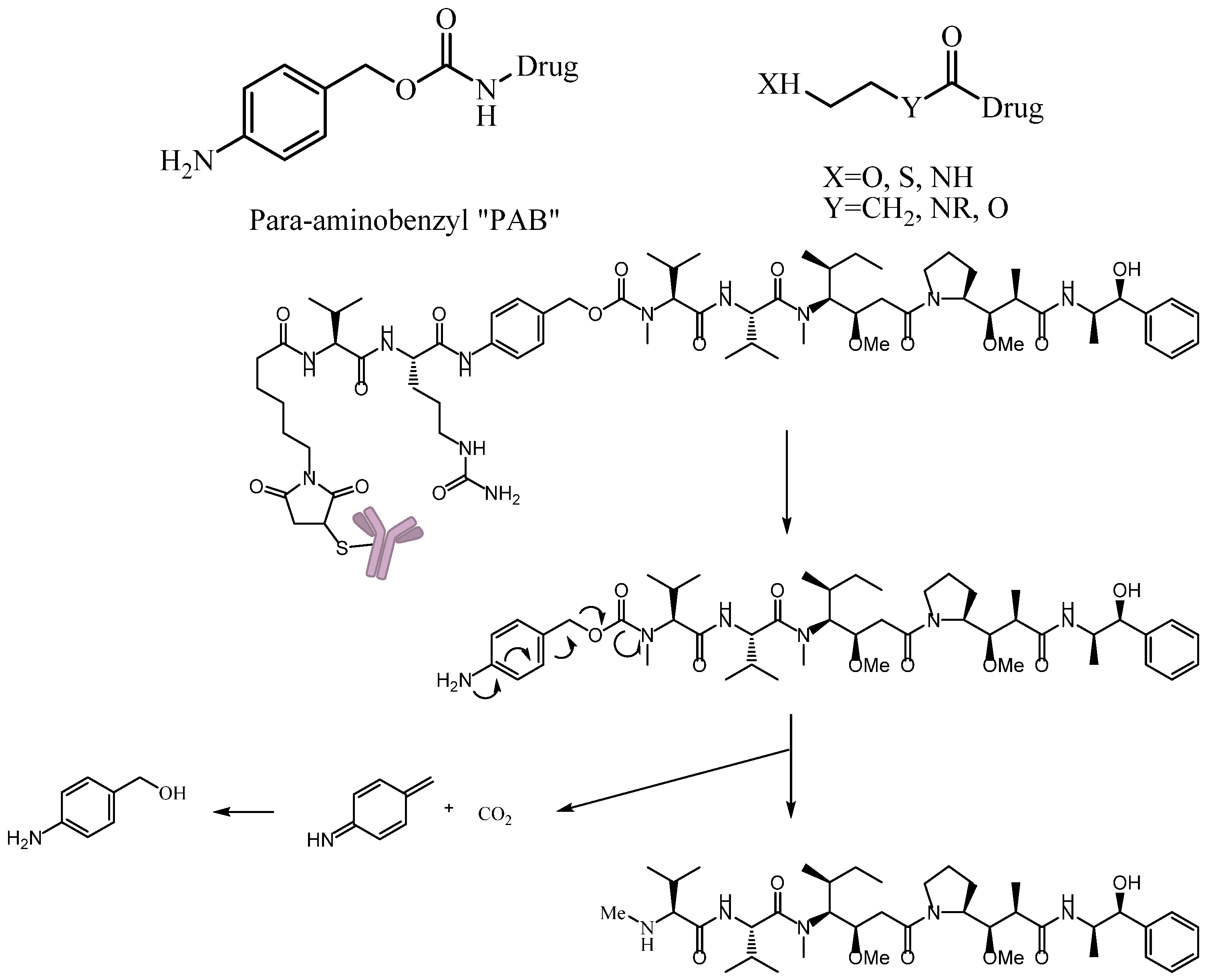
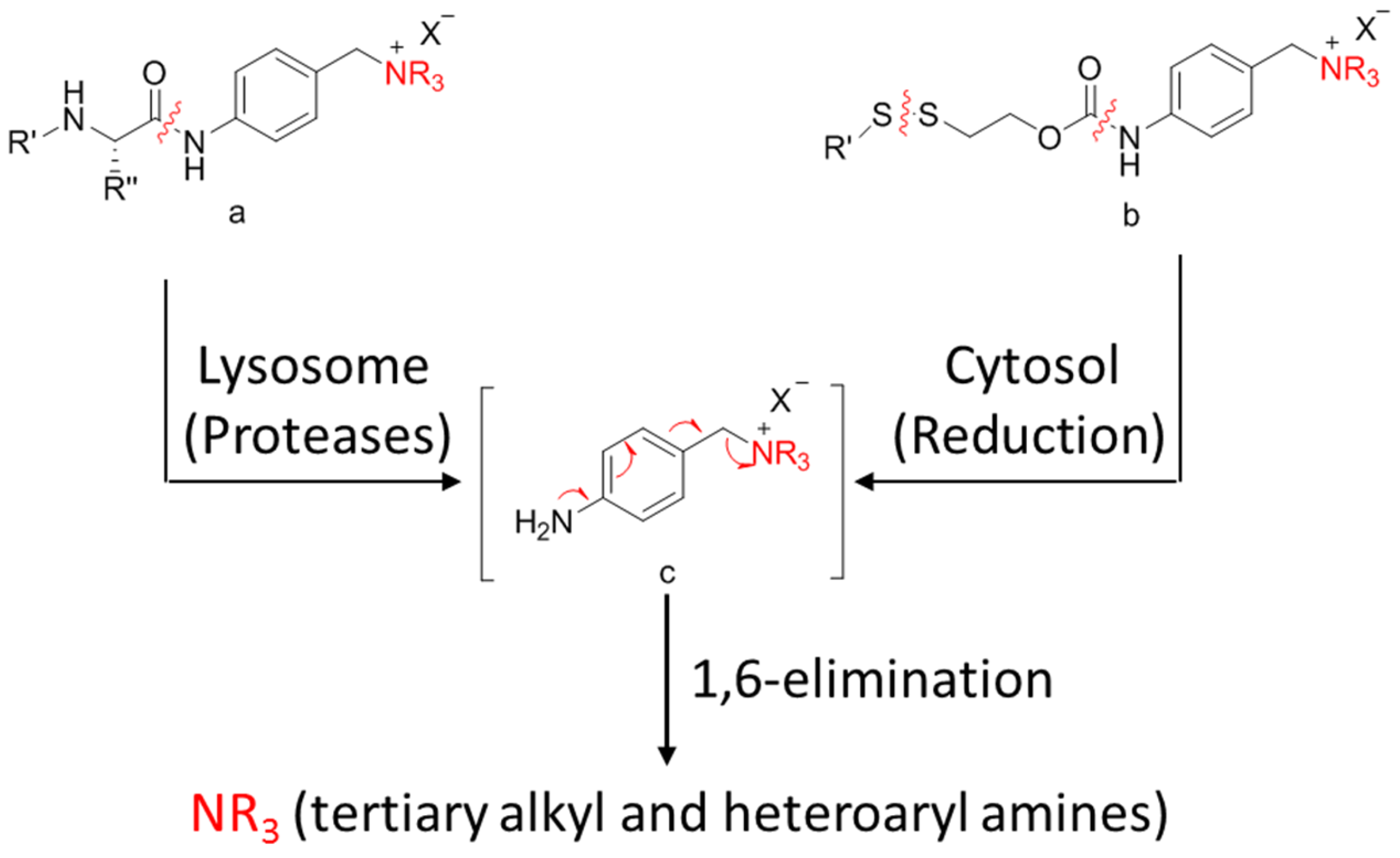
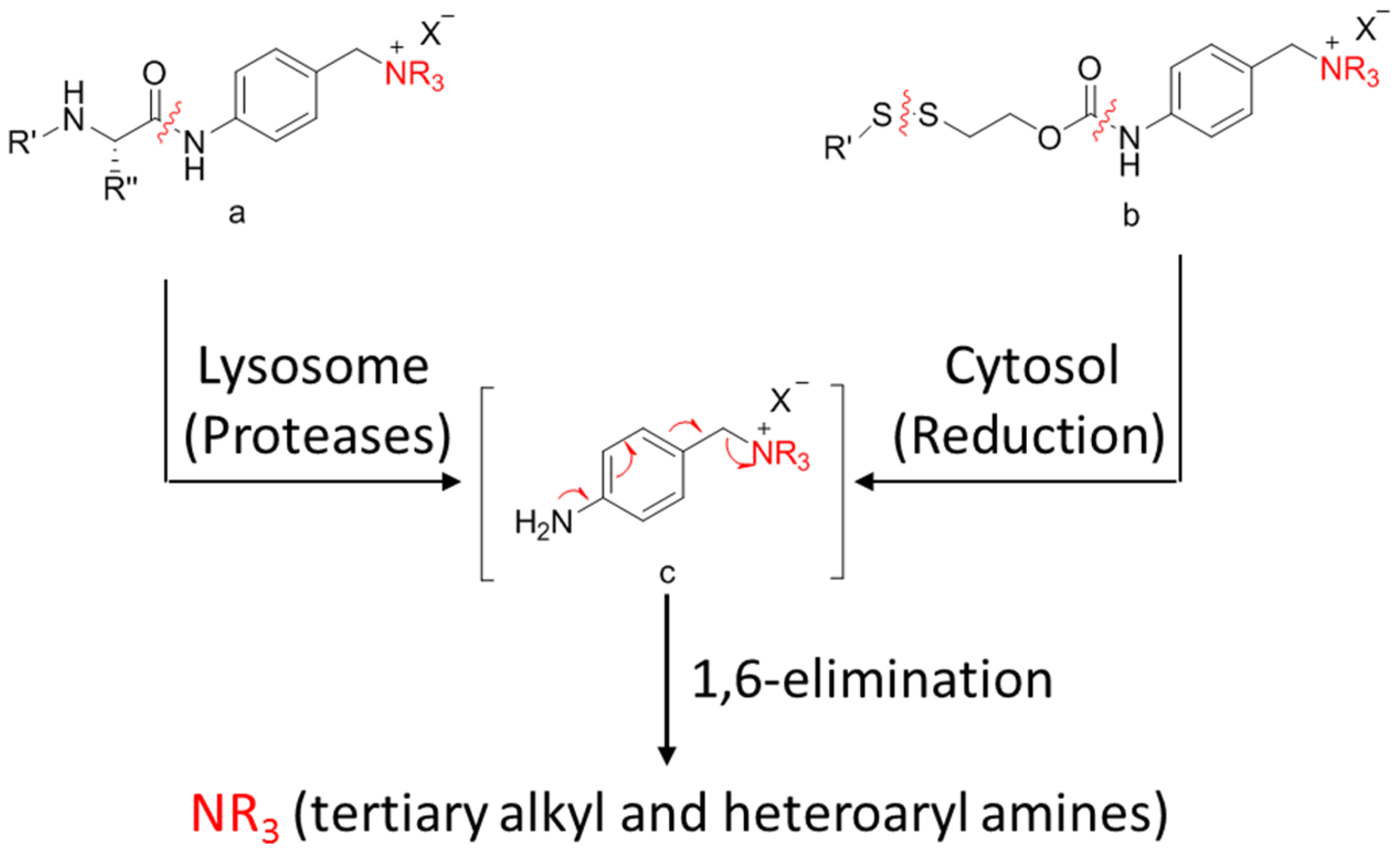
3.4.3. Self-Immolative Spacer
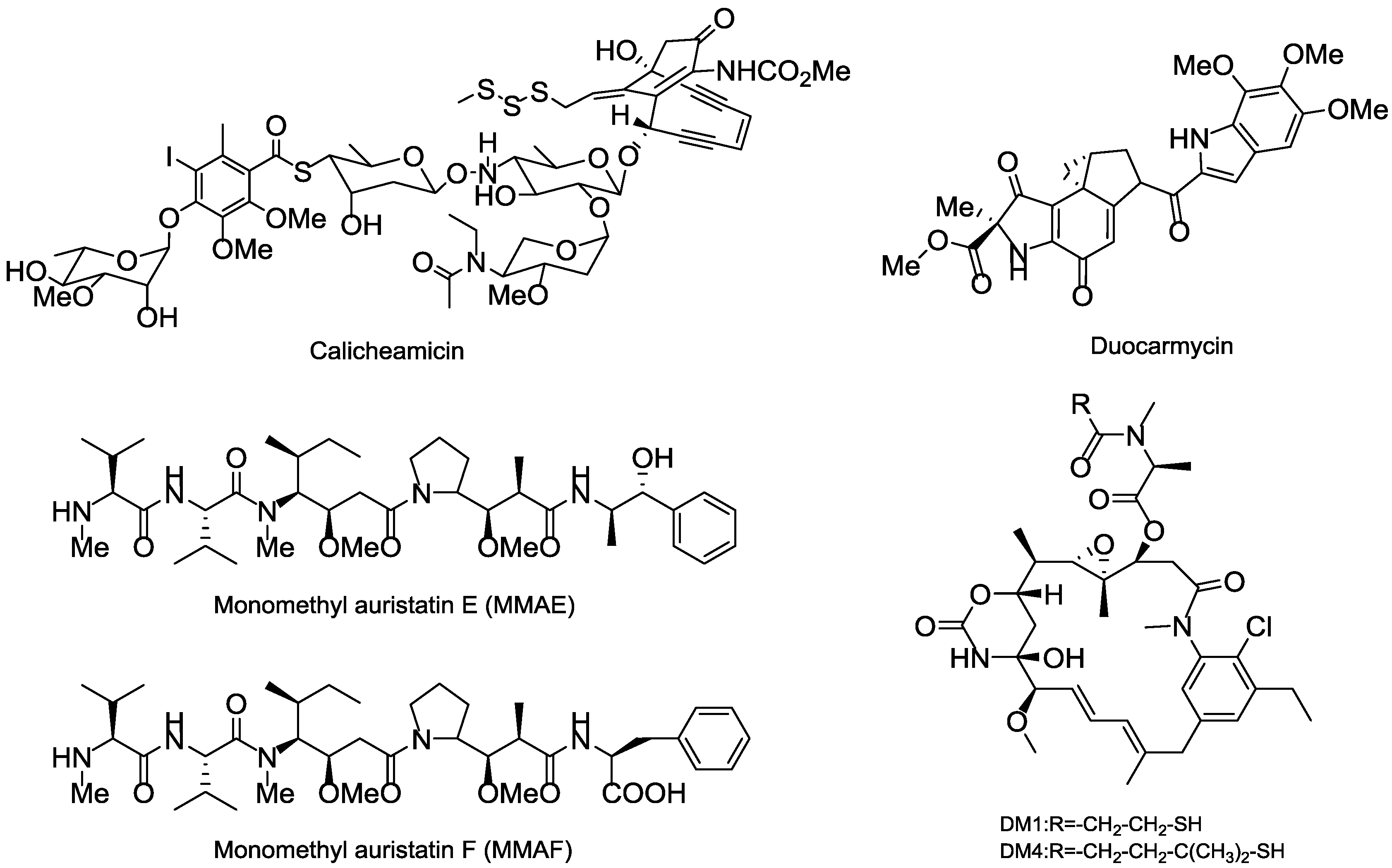
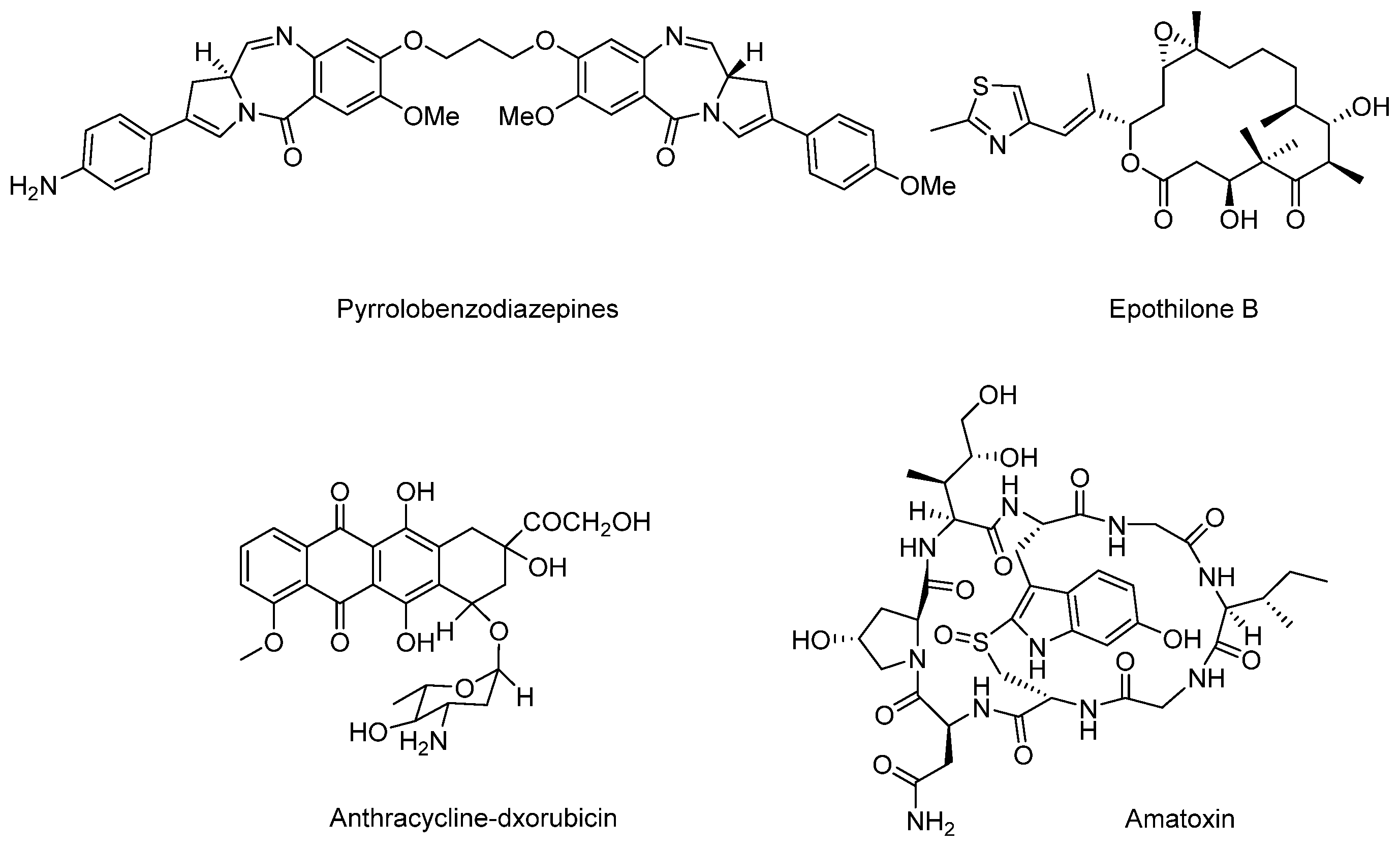
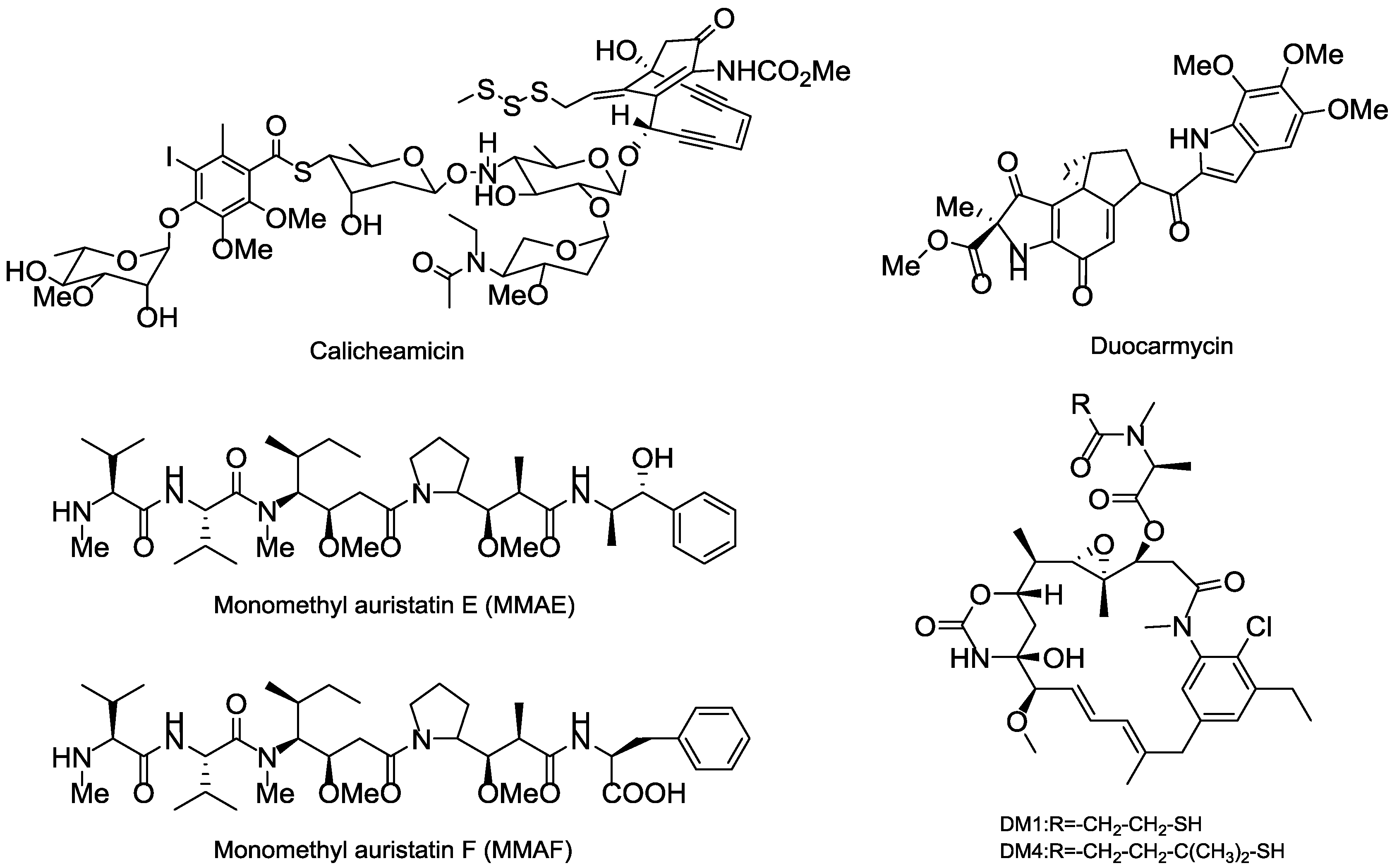
3.5. Cytotoxic Payload
3.5.1. Terrestrial Payloads
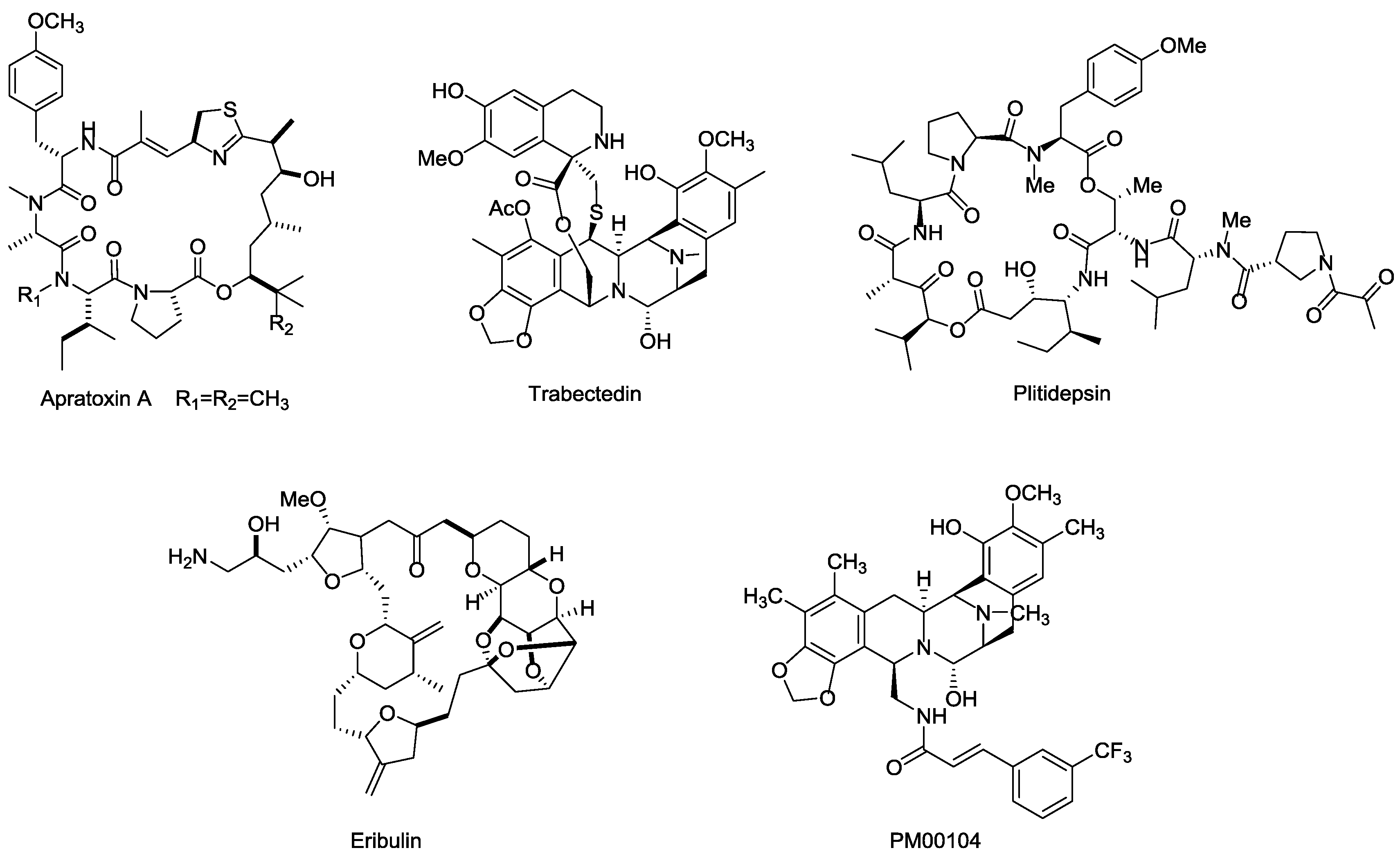
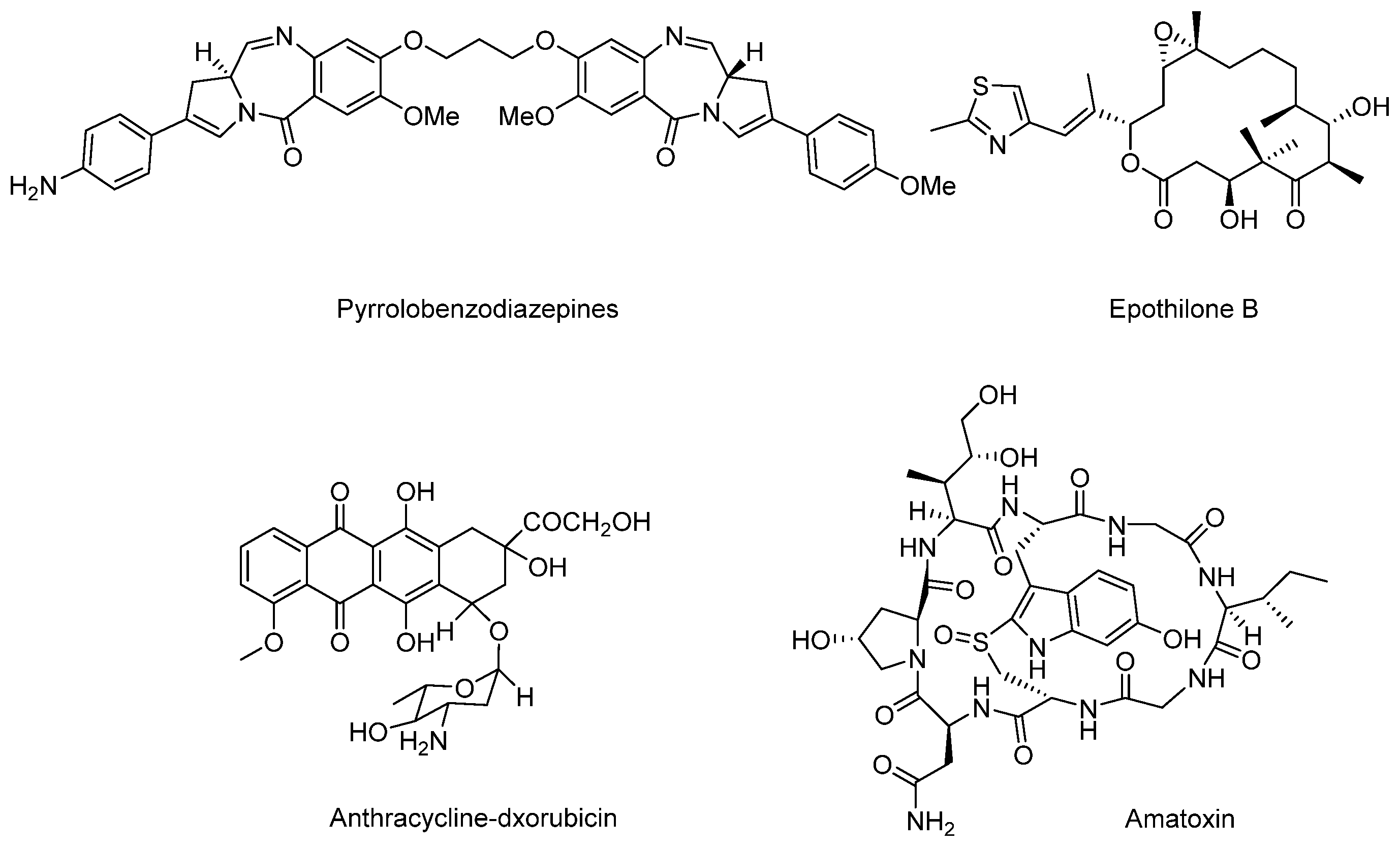
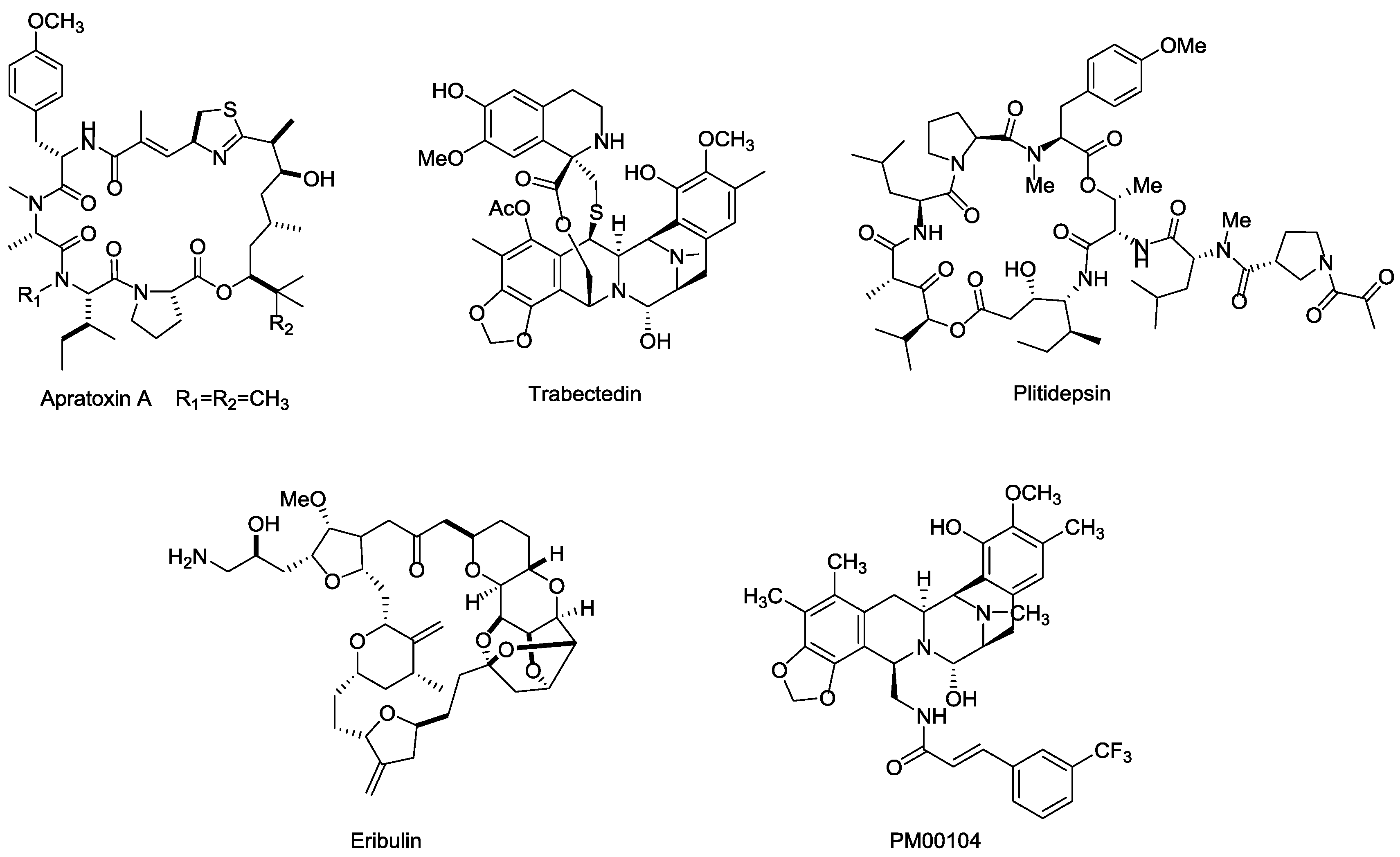
3.5.2. Marine Payloads
3.5.3. Marine Toxins
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AcBut | 4-(4-acetylphenoxy)butanoic acid |
| ADAs | allergic reactions and anti-drug antibodies |
| ADC | antibody–drug conjugate |
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ALCL | anaplastic large cell lymphoma |
| ALL | acute lymphoblastic leukemia |
| AML | acute myelocytic leukemia |
| APL | acute promyelocytic leukemia |
| BCMA | B cell maturation antigen |
| ccRCC | clear cell renal cell carcinoma |
| CDC | complement dependent cytotoxicity |
| Ch | chimeric |
| CLL | chronic lymphocytic leukemia |
| CML | chronic myeloid leukemia |
| CRC | colorectal carcinoma |
| CRPC | castration resistant prostate cancer |
| CTLA4 | cytotoxic T-lymphocyte antigen 4 |
| DARs | Drug: antibody ratios |
| DLBCL | diffuse large B cell lymphoma |
| DM1 | N2′-deacetyl-N2′-(3-mercapto-1-oxopropyl)-maytansine |
| DM4 | N2′-deacetyl-N2′-(4-mercapto-4-methyl-1-oxopentyl)-maytansine |
| DTM | dithiopyridylmaleimide |
| EGFR | epidermal growth factor receptor |
| EpCAM | epithelial cell adhesion molecule |
| ETBR | endothelin B receptor |
| Fab | antigen-binding fragment |
| Fc | crystalline fragment |
| FcRn | human neonatal Fc receptor |
| FDA | Food and Drug Administration |
| FGE | formylglycine-generating enzyme |
| GCC | Guanylyl cyclase C |
| GPNMB | glycoprotein nonmetastatic melanoma protein b |
| HER2 | human epidermalgrowth factor receptor-2 |
| HL | Hodgkin’s lymphoma |
| Hu | human |
| Hz | humanized |
| mAb | monoclonal antibody |
| MC | maleimidocaproyl |
| MCC | maleimidomethyl cyclohexane-1-carboxylate |
| MM | multiple myeloma |
| MMAE | monomethyl auristatin E |
| MMAF | monomethyl auristatin F |
| NHL | non-hodgkin’s lymphoma |
| NSCLC | non-small-cell lung cancer |
| PAB | para-aminobenzyl |
| pAcPhe | p-acetylphenylalanine |
| PBDs | pyrrolobenzodiazepines |
| PEG | polyethylene glycol |
| PK | pharmacokinetic |
| PSMA | prostate specific membrane antigen |
| RCC | renal cell carcinoma |
| SCC | squamous cell carcinoma |
| scFv | single-chain variable fragment |
| SCLC | small-cell lung cancer |
| TF | tissue factor |
| TPBG | trophoblast glycoprotein |
| VEGF | vascular endothelial growth factor |
References
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [PubMed]
- Casi, G.; Neri, D. Antibody-drug conjugates: Basic concepts, examples and future perspectives. J Control. Release 2012, 161, 422–428. [Google Scholar] [PubMed]
- Scotti, C.; Iamele, L.; Vecchia, L. Antibody–drug conjugates: Targeted weapons against cancer. Antibody Technol. J. 2015, 5. [Google Scholar] [CrossRef]
- Hinrichs, M.J.M.; Dixit, R. Antibody drug conjugates: Nonclinical safety considerations. AAPS J. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Sunil, V.; David, M.; Luca, G.; Krop, I.E.; Manfred, W.; José, B.; Mark, P.; Do-Youn, O.; Véronique, D.; Ellie, G. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar]
- Hamilton, G.S. Antibody-drug conjugates for cancer therapy: The technological and regulatory challenges of developing drug-biologic hybrids. Biologicals 2015, 43, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Luisi, D.L.; Pak, R.H. Antibody-drug conjugates: Design, formulation and physicochemical stability. Pharm. Res. 2015, 32, 3541–3571. [Google Scholar] [CrossRef] [PubMed]
- Mccombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Haeuw, J.F.; Wurch, T.; Goetsch, L.; Bailly, C.; Corvaïa, N. The next generation of antibody-drug conjugates comes of age. Discov. Med. 2010, 10, 329–339. [Google Scholar] [PubMed]
- Ritter, A. Antibody-drug conjugates: Looking ahead to an emerging class of biotherapeutic. Pharm. Technol. 2012, 36, 42–47. [Google Scholar]
- Nelson, A.L.; Eugen, D.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S. CD70 as a therapeutic target in human malignancies. Expert Opin. Ther. Targets 2008, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Cardarelli, P.; King, D.; Terrett, J.; Gangwar, S.; Cohen, L.; Pan, C.; Rao, C.; Deshpande, S.; Vangipuram, R.; Passmore, D. Efficacy and safety of a human anti-CD70 antibody-MGBA conjugate. Cancer Res. 2008, 68, 4061. [Google Scholar]
- Haffner, M.C.; Kronberger, I.E.; Ross, J.S.; Sheehan, C.E.; Matthias, Z.; Gilbert, M.; Dietmar, O.; Bettina, Z.; Christian, E.; Yang, X.J. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum. Pathol. 2009, 40, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar] [PubMed]
- Petrylak, D.P.; Vogelzang, N.J.; Chatta, G.S.; Fleming, M.T.; Smith, D.C.; Appleman, L.J.; Arif Hussain, M.M.; Singh, P.; Tagawa, S.T.; Gore, I.; et al. A phase 2 study of prostate specific membrane antigen antibody drug conjugate (PSMA ADC) in patients (pts) with progressive metastatic castration-resistant prostate cancer (mCRPC) following abiraterone and/or enzalutamide (abi/enz). J. Clin. Oncol. 2015, 33, 144. [Google Scholar] [CrossRef]
- Petrylak, D.; Smith, D.C.; Appleman, L.J.; Fleming, M.; Hussain, A.; Dreicer, R.; Sartor, O.; Shore, N.; Vogelzang, N.J.; Youssoufian, H. A phase II trial of prostate-specific membrane antigen antibody drug conjugate (PSMA ADC) in taxane-refractory metastatic castration-resistant prostate cancer (mCRPC). Eur. Urol. Suppl. 2010, 154, 5–6. [Google Scholar]
- Mizuki, T.; Jin-Sung, C.; Hideo, A.; Cruz, P.D.; Kiyoshi, A. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res. 2010, 70, 5778–5787. [Google Scholar]
- Gaboury, H.M.P.L. Glycoprotein nonmetastatic b is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. 2010, 16, 2147–2156. [Google Scholar]
- Kuan, C.T.; Wakiya, K.; Dowell, J.M.; Herndon, J.E.; Reardon, D.A.; Graner, M.W.; Riggins, G.J.; Wikstrand, C.J.; Bigner, D.D. Glycoprotein nonmetastatic melanoma protein b, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin. Cancer Res. 2006, 12, 1970–1982. [Google Scholar] [CrossRef]
- Kam Fai, T.; Michael, J.; Pollack, V.A.; Mccabe, D.A.; Shadish, M.L.; Khramtsov, N.V.; Hackett, C.S.; Shenoy, S.G.; Bing, K.; Boldog, F.L. Cr011, a fully human monoclonal antibody-auristatin e conjugate, for the treatment of melanoma. Clin. Cancer Res. 2006, 12, 1373–1382. [Google Scholar]
- Hamid, O.; Sznol, M.; Pavlick, A.C.; Kluger, H.M.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Davis, T.A.; Crowley, E.; Hwu, P. Frequent dosing and GPNMB expression with CDX-011 (CR011-vcMMAE), an antibody-drug conjugate (ADC), in patients with advanced melanoma. J. Clin. Oncol. 2010, 28, 8525. [Google Scholar]
- Barrow, K.M.; Ward, C.M.; Rutter, J.; Ali, S.; Stern, P.L. Embryonic expression of murine 5T4 oncofoetal antigen is associated with morphogenetic events at implantation and in developing epithelia. Dev. Dyn. 2005, 233, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, E.; Mcgown, A.T.; Rennison, J.; Swindell, R.; Crowther, D.; Starzynska, T.; Stern, P.L. 5T4 oncofetal antigen expression in ovarian carcinoma. Int. J. Gynecol. Cancer 1995, 5, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Starzynska, T.; Marsh, P.J.; Schofield, P.F.; Roberts, S.A.; Myers, K.A.; Stern, P.L. Prognostic significance of 5T4 oncofetal antigen expression in colorectal carcinoma. Br. J. Cancer 1994, 69, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Marc, D.; Geles, K.G.; Follettie, M.T.; Ping, Y.; Michelle, B.; Jonathon, G.; Dijoseph, J.F.; Maha, K.; Shuguang, H.; Veronica, D. Delineation of a cellular hierarchy in lung cancer reveals an oncofetal antigen expressed on tumor-initiating cells. Cancer Res. 2011, 71, 4236–4246. [Google Scholar]
- Sapra, P.; Damelin, M.; Dijoseph, J.; Marquette, K.; Geles, K.G.; Golas, J.; Dougher, M.; Narayanan, B.; Giannakou, A.; Khandke, K. Long-term tumor regression induced by an antibody-drug conjugate that targets 5T4, an oncofetal antigen expressed on tumor-initiating cells. Mol. Cancer Ther. 2013, 12, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Goldmacher, V.S.; Kovtun, Y.V. Antibody-drug conjugates: Using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther. Deliv. 2011, 2, 397–416. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.; Herrmann, R. Overview of monoclonal antibodies in cancer therapy: Present and promise. Crit. Rev. Oncol./Hematol. 2005, 54, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Takamura, I.H.; Nakagawa, T. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008, 68, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Valge-Archer, V.E. Development trends for monoclonal antibody cancer therapeutics. Dressnature Rev. Drug Discov. 2007, 6, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Law, C.; Cerveny, C.; Gordon, K.; Klussman, K.; Mixan, B.; Chace, D.; Meyer, D.; Doronina, S. Efficient elimination of B-lineage lymphomas by anti-CD20-auristatin conjugates. Clin. Cancer Res. 2004, 10, 7842–7851. [Google Scholar] [CrossRef] [PubMed]
- Ober, R.J.; Radu, C.G.; Ghetie, V.; Ward, E.S. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int. Immunol. 2001, 13, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function ☆. Adv. Drug Deliv. Rev. 2006, 58, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Mccafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- René Michael, H.; Cohen, E.H.; Rachel Baribault, K.; Kristin, R.; Sonia, S.; Shannon, H.; Louise, R.; Nicolas, F.; Marc, D.; Henk, P. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 2005, 23, 344–348. [Google Scholar]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.C.; Mashayekh, R.; Wymore, K.; Mccabe, J.G. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994, 368, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.P.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Exp. Cell Res. 2010, 50, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Sliwkowski, M.X.; Ira, M. Antibody therapeutics in cancer. Science 2013, 341, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Rosensweig, C.J.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Isotype and glycoform selection for antibody therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Mark, R. Genentech’s glyco-engineered antibody to succeed rituxan. Nat. Biotechnol. 2014, 32, 6–7. [Google Scholar]
- Mikkel Wandahl, P.; Helle Jane, J.; Klaus, K.; Adam, H.; Charles, P.; John, S.; Rensen, H.; Michael, K. Sym004: A novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010, 70, 588–597. [Google Scholar]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Saga, T.; Neumann, R.D.; Heya, T.; Sato, J.; Kinuya, S.; Le, N.; Paik, C.H.; Weinstein, J.N. Targeting cancer micrometastases with monoclonal antibodies: A binding-site barrier. Proc. Natl. Acad. Sci. USA 1995, 92, 8999–9003. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Philipp, H.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar]
- Borsi, L.; Balza, E.; Bestagno, M.; Castellani, P.; Carnemolla, B.; Biro, A.; Leprini, A.; Sepulveda, J.; Burrone, O.; Neri, D.; et al. Selective targeting of tumoral vasculature: Comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int. J. Cancer 2002, 102, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Berndorff, D.; Borkowski, S.; Sieger, S.; Rother, A.; Friebe, M.; Viti, F.; Hilger, C.S.; Cyr, J.E.; Dinkelborg, L.M. Radioimmunotherapy of solid tumors by targeting extra domain b fibronectin: Identification of the best-suited radioimmunoconjugate. Clin. Cancer Res. 2005, 11, 7053s–7063s. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Schier, R.; Mccall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; de Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010, 70, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 2012, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S. Reduction-alkylation strategies for the modification of specific monoclonal antibody bisulfides. Bioconj. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Panowksi, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar]
- Dere, R.; Yi, J.H.; Lei, C.; Saad, O.M.; Huang, C.; Li, Y.; Baudys, J.; Kaur, S. Pk assays for antibody-drug conjugates: Case study with ado-trastuzumab emtansine. Bioanalysis 2013, 5, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; King, D.J.; Owens, R.J.; Yarranton, G.T.; Millican, A.; Whittle, N.R.; Adair, J.R. Site-specific attachment to recombinant antibodies via introduced surface cysteine residues. Protein Eng. 1990, 3, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and Pk outcomes. Bioconj. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
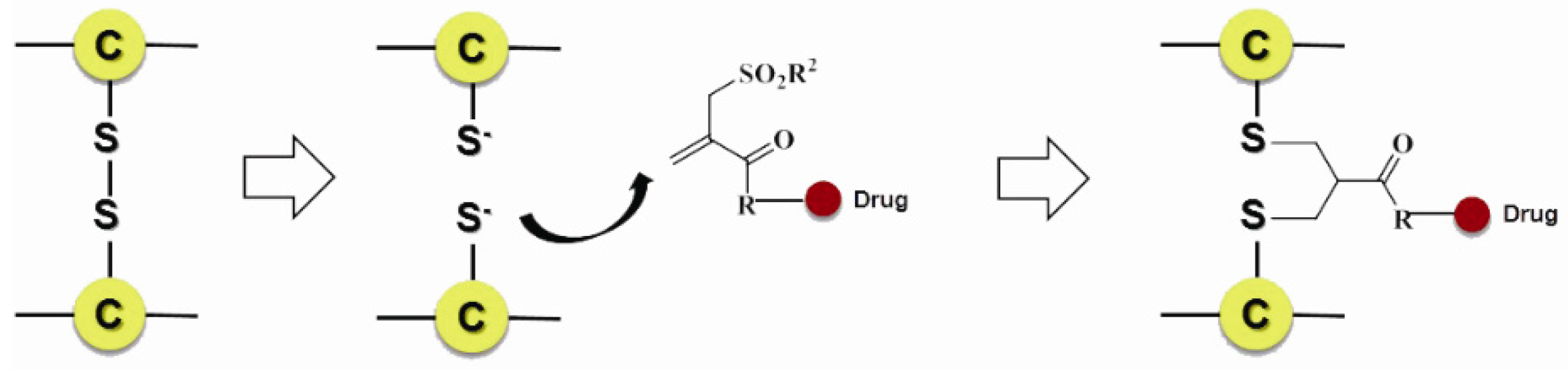
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M. Bridging disulfides for stable and defined antibody drug conjugates. Bioconj. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.T.; Asano, S.; Li, X.; Rader, C.; Barbas, C.F., 3rd. Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconj. Chem. 2014, 25, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native MAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconj. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.L.; Sussman, D. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef] [PubMed]
- Besanceney-Webler, C.; Hao, J.; Wei, W.; Baughn, A.D.; Peng, W. Metabolic labeling of fucosylated glycoproteins in bacteroidales species. Bioorg. Med. Chem. Lett. 2011, 21, 4989–4992. [Google Scholar] [CrossRef] [PubMed]
- Esam, A.S.; Jean-Luc, C.; Christina, L.M.; Sheng-Kai, W.; Chi-Huey, W.; Haltiwanger, R.S. 6-Alkynyl fucose is a bioorthogonal analog for O-fucosylation of epidermal growth factor-like repeats and thrombospondin Type-1 repeats by protein O-fucosyltransferases 1 and 2. Glycobiology 2013, 23, 188–198. [Google Scholar]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current adc linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconj. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
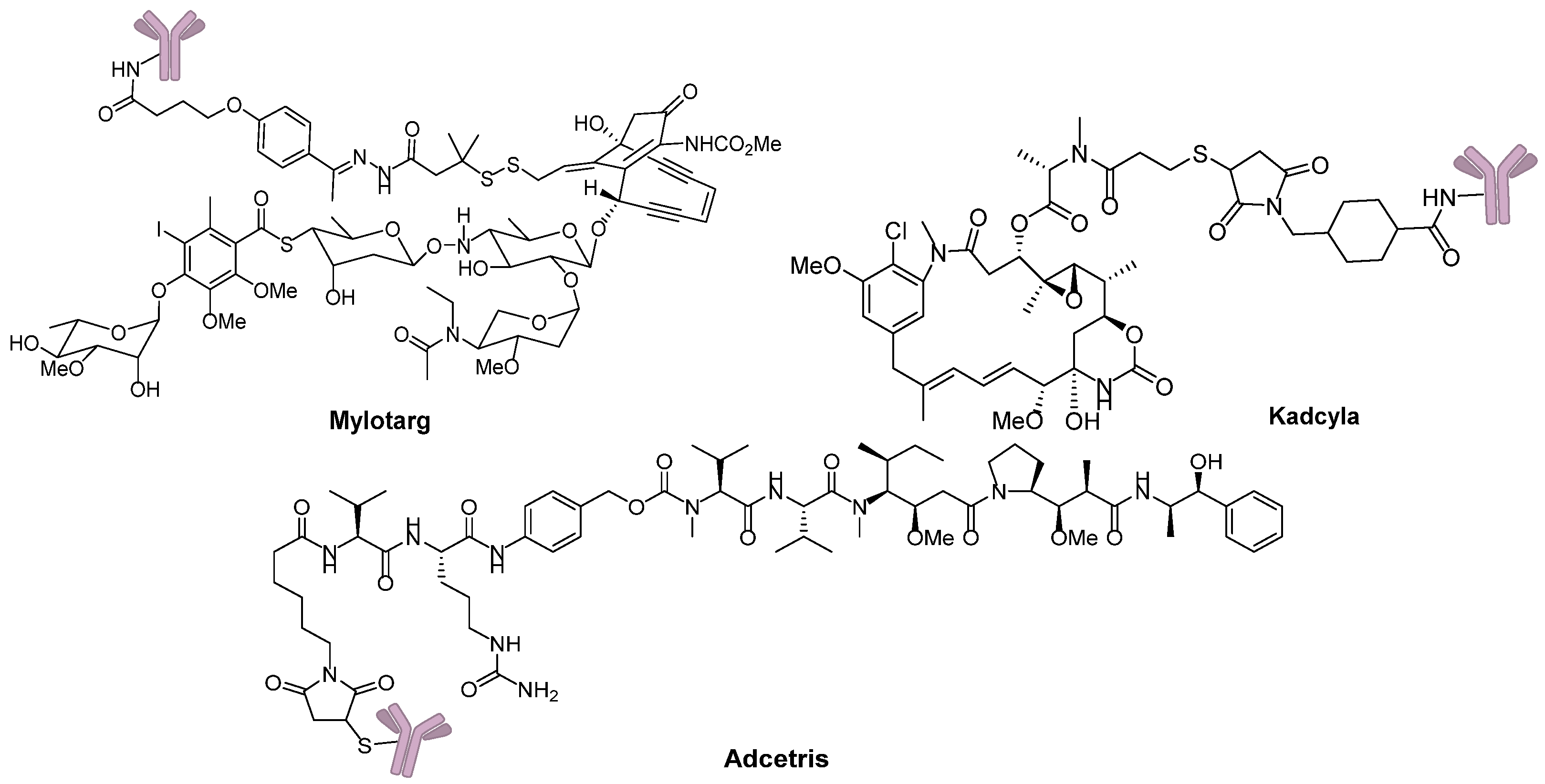
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Nolting, B. Linker technologies for antibody–drug conjugates. Methods Mol. Biol. 2013, 1045, 71–100. [Google Scholar] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Fu, L. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Senter, P.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.; Toki, B.E.; Manikumar, G.; Wani, M.C.; Kroll, D.J.; Jeffrey, S.C. Design, synthesis, and biological evaluation of antibody-drug conjugates comprised of potent camptothecin analogues. Bioconj. Chem. 2009, 20, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A. Structural modifications that alter the p-glycoprotein efflux properties of compounds. J. Med. Chem. 2012, 55, 4877–4895. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Charlene, A.; Gregory, J.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Rajeeva, S.; Yelena, K.; Widdison, W.C. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
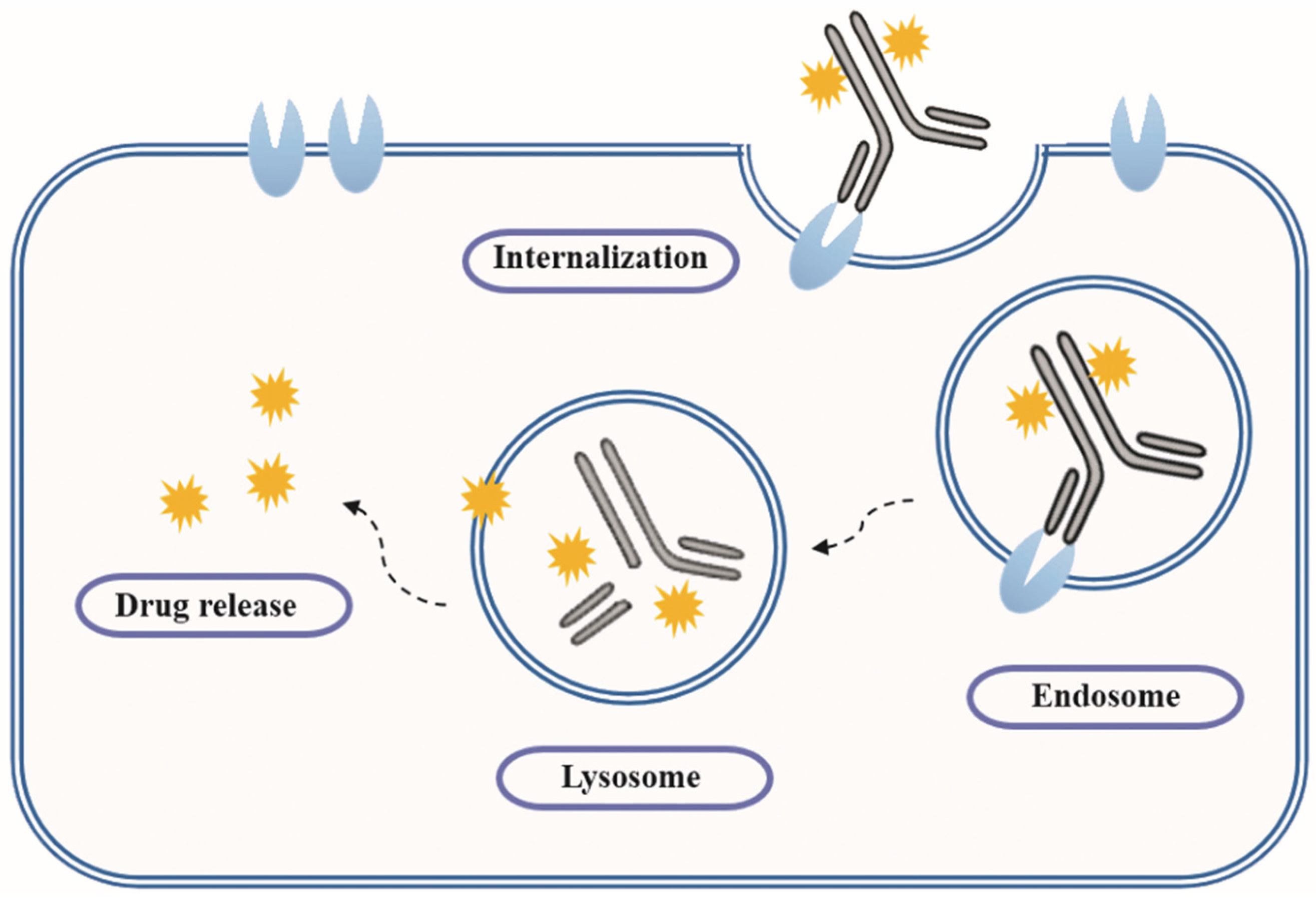
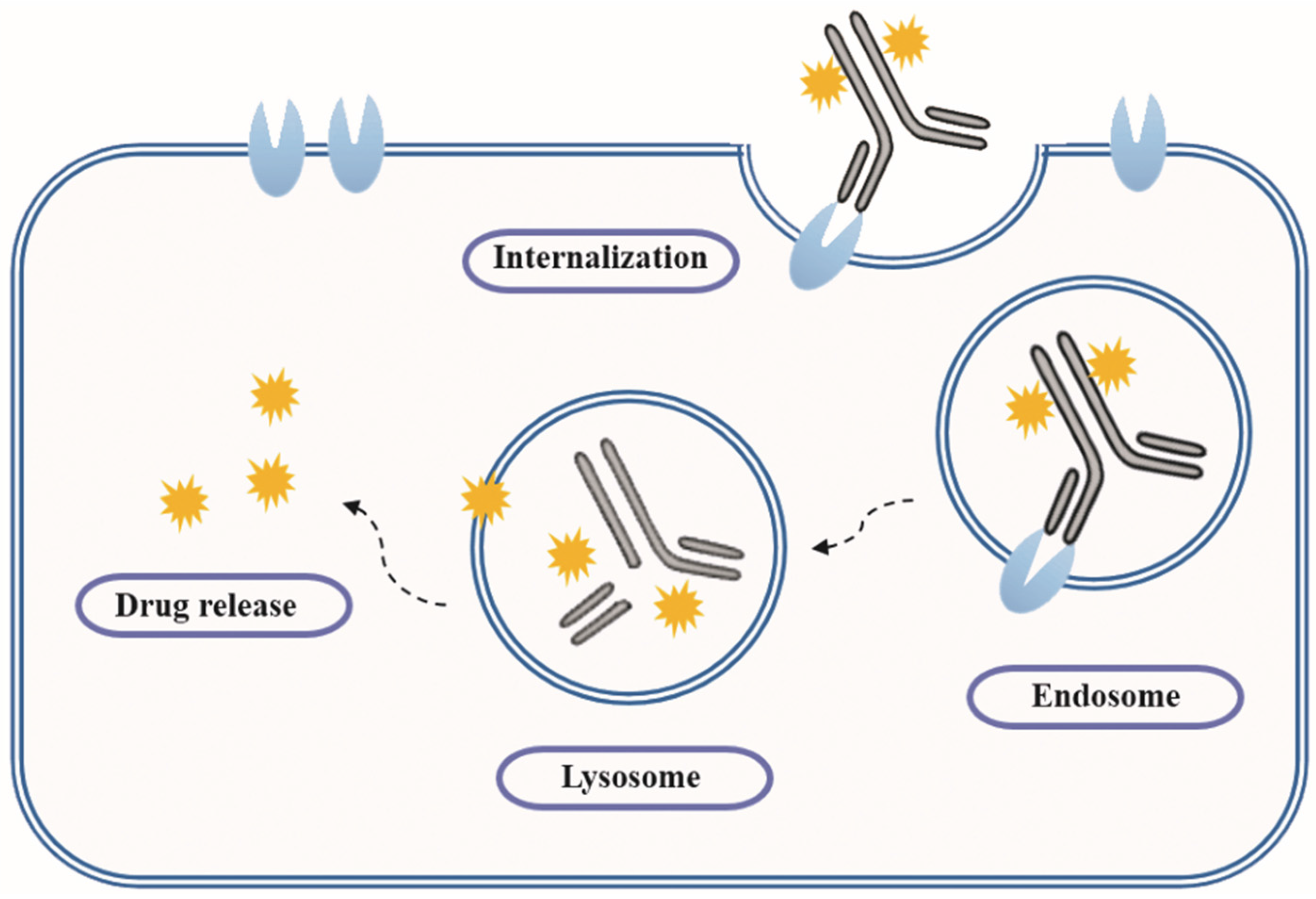
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin b-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconj. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki BETorgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Meyer, B.D.W. Novel peptide linkers for highly potent antibody—Auristatin conjugate. Bioconj. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconj. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. ChemInform 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D. Discovery of pyrophosphate diesters as tunable, soluble and bioorthogonal linkers for site-specific antibody-drug conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, C.A. Self-immolative linkers in polymeric delivery systems. Polym. Chem. 2011, 4, 773–790. [Google Scholar] [CrossRef]
- Toki, B.E.; Cerveny, C.G.; Wahl, A.F.; Senter, P.D. Protease-mediated fragmentation of p-amidobenzyl ethers: A new strategy for the activation of anticancer prodrugs. J. Org. Chem. 2002, 67, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Dewit, M.A.; Gillies, E.R. Design, synthesis, and cyclization of 4-aminobutyric acid derivatives: Potential candidates as self-immolative spacers. Org. Biomol. Chem. 2011, 9, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Staben, L.R.; Koenig, S.G.; Lehar, S.M.; Vandlen, R.; Zhang, D.; Chuh, J.; Yu, S.F.; Ng, C.; Guo, J.; Liu, Y.; et al. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody-drug conjugates. Nat. Chem. 2016, 8, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
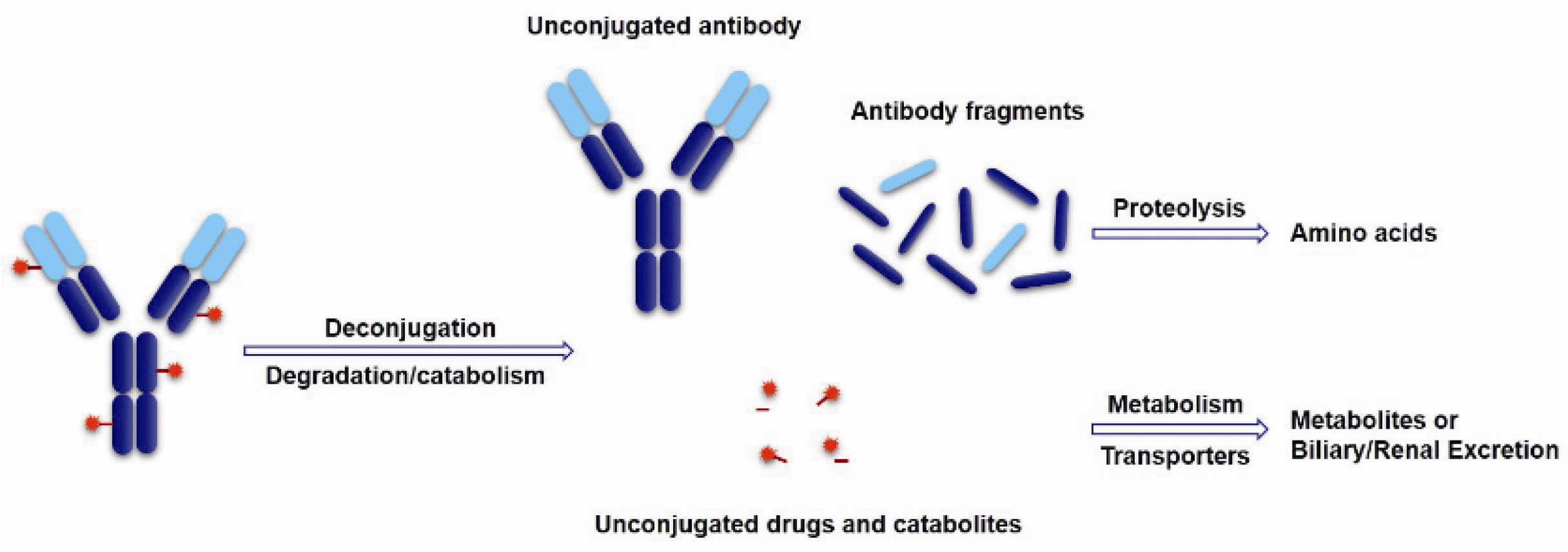
- Lin, K.; Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Koehn, F.E.; Abraham, R.T. The antibody-drug conjugate: An enabling modality for natural product-based cancer therapeutics. Nat. Prod. Rep. 2013, 30, 625–639. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Nicolaou, K.C. The enediyne antibiotics. J. Med. Chem. 1996, 39, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Boger, D.L. Cheminform abstract: The duocarmycins: Synthetic and mechanistic studies. Acc. Chem. Res. 2002, 28, 20–29. [Google Scholar] [CrossRef]
- Lourdes, T.; Alasdair, B.; Indrani, C.; Bilal, S.; Sanjeev, G.; Allen, Z.; Vangipuram, R.; Chetana, R.; Zemin, W.; Chin, P. Novel detection of DNA-alkylated adducts of antibody-drug conjugates with potentially unique preclinical and biomarker applications. Bioanalysis 2013, 5, 1073–1081. [Google Scholar]
- Remillard, S.; Rebhun, L.I.; Howie, G.A.; Kupchan, S.M. Antimitotic activity of the potent tumor inhibitor maytansine. Science 1975, 189, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Yelena, K.; Goldmacher, V.S.; Hongsheng, X.; Steeves, R.M.; Lutz, R.J. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.; Martell, B.A.; Gross, J.L.; Cook, S.B.; Shah, S.A.; Blättler, W.A.; Mckenzie, S.J.; Goldmacher, V.S. Immunoconjugates containing novel maytansinoids: Promising anticancer drugs. Cancer Res. 1992, 52, 127–131. [Google Scholar] [PubMed]
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconj. Chem. 2013, 24, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Zheng, B.; Go, M.A.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M. A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin based adcs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Hallen, H.E.; Hong, L.; Scott-Craig, J.S.; Walton, J.D. Gene family encoding the major toxins of lethal amanita mushrooms. Proc. Natl. Acad. Sci. USA 2007, 104, 19097–19101. [Google Scholar] [CrossRef] [PubMed]
- Hechler, T.; Kulke, M.; Mueller, C.; Pahl, A.; Anderl, J. Amanitin-based antibody-drug conjugates targeting the prostate-specific membrane antigen. Cancer Res. 2014, 74, 664. [Google Scholar] [CrossRef]
- Wakelee, H.A.; Sikic, B.I. Activity of novel cytotoxic agents in lung cancer: Epothilones and topoisomerase I inhibitors. Clin. Lung Cancer 2005, 7, S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Pezzuto, J.M. Natural products as a vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Centre 2015, 25, 41–59. [Google Scholar] [CrossRef] [PubMed]
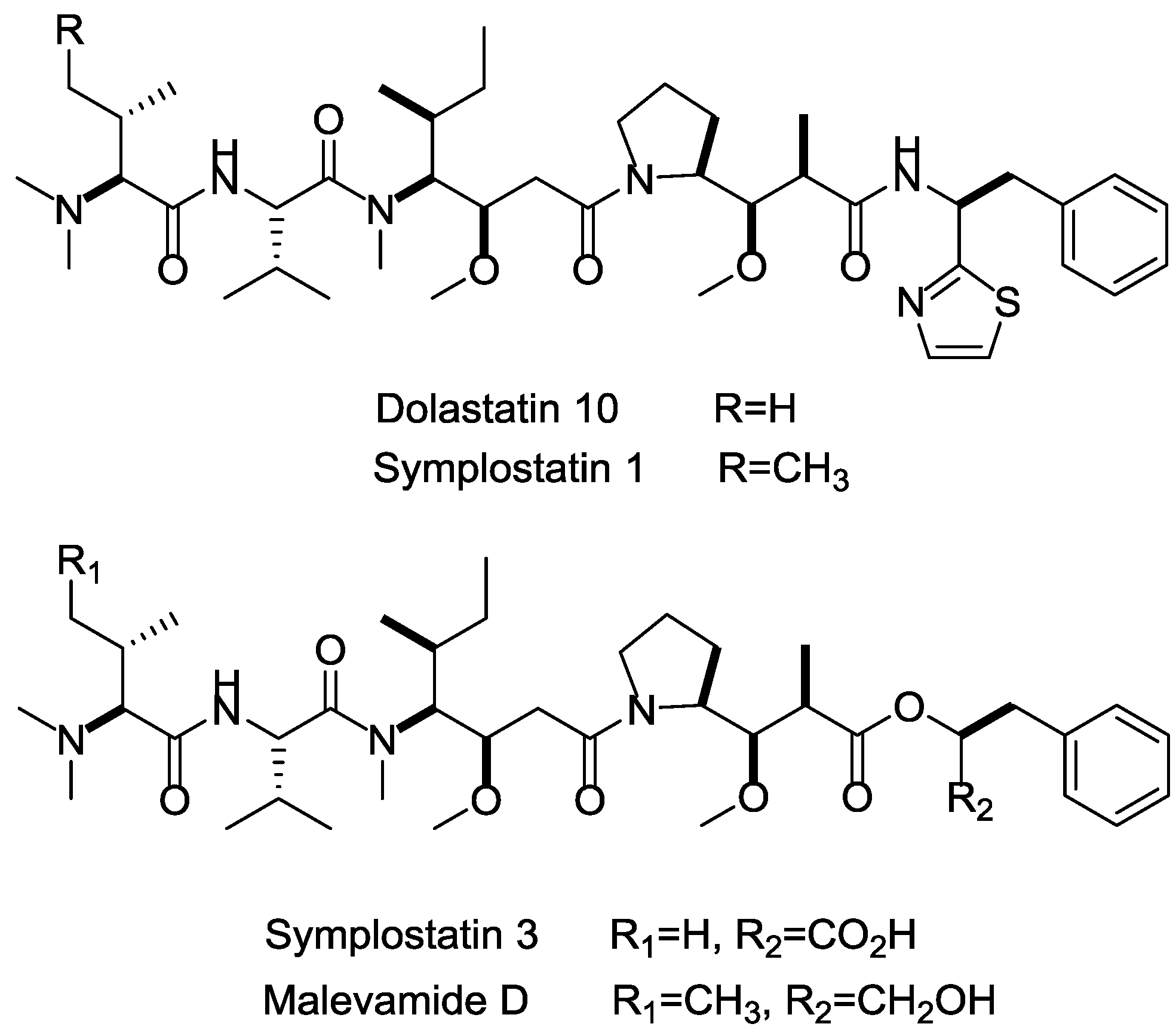
- Tan, L.T. Pharmaceutical agents from filamentous marine cyanobacteria. Drug Discov. Today 2013, 18, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Liua, Z.; Xua, P.; Wub, T.; Zeng, W. Microtubule-targeting anticancer agents from marine natural substance. Anti-Cancer Agents Med. Chem. 2014, 14, 1–9. [Google Scholar] [CrossRef]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Structure-activity studies with chiral isomers and with segments of the antimitotic marine peptide dolastatin 10. Biochem. Pharmacol. 1990, 40, 1859–1864. [Google Scholar] [CrossRef]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Nagle, D.G.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H.; Valeriote, F.A. Symplostatin 1: A dolastatin 10 analogue from the marine cyanobacterium symploca hydnoides. J. Nat. Prod. 1998, 61, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Horgen, F.D.; Kazmierski, E.B.; Westenburg, H.E.; Yoshida, W.Y.; Scheuer, P.J. Malevamide D: Isolation and structure determination of an isodolastatin H analogue from the marine cyanobacterium Symploca hydnoides. J. Nat. Prod. 2002, 65, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin a, a potent novel cytotoxin from the marine cyanobacterium lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. New apratoxins of marine cyanobacterial origin from guam and palau. Bioorg. Med. Chem. 2002, 10, 1973–1978. [Google Scholar] [CrossRef]
- Cooper, E.L.; Yao, D. Diving for drugs: Tunicate anticancer compounds. Drug Discov. Today 2012, 17, 636–648. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Maurizio, D.I.; Paola, A. Trabectedin and plitidepsin: Drugs from the sea that strike the tumor microenvironment. Mar. Drugs 2014, 12, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; Mcintosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Guillem, V.; Garcia, M.; Rivera, F.; Tabernero, J.; Cullell, M.; Lopezmartin, J.A.; Pollard, P.; Dumez, H.; del Muro, X.G. Phase II randomized study of plitidepsin (aplidin), alone or in association with l-carnitine, in patients with unresectable advanced renal cell carcinoma. Mar. Drugs 2009, 7, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Couture, F.; Lorigan, P.; Lüthi, F.; Small, D. Phase II study of weekly plitidepsin as second-line therapy for small cell lung cancer. Lung Cancer 2008, 64, 60–65. [Google Scholar]
- Monaco, R. Novel natural product discovery from marine sponges and their obligate symbiotic organisms. Biorxiv 2014. [Google Scholar] [CrossRef]
- Menis, J.; Twelves, C. Eribulin (halaven): A new, effective treatment for women with heavily pretreated metastatic breast cancer. Breast Cancer Targets Ther. 2011, 3, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.N.; Trivedi, M.V. Eribulin: A novel cytotoxic chemotherapy agent. Ann. Pharmacother. 2012, 46, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Shetty, N.; Gupta, S. Eribulin drug review. South Asian J. Cancer 2014, 3, 57–59. [Google Scholar] [PubMed]
- Chen, R.; Liu, H.; Chen, C. Asymmetric total synthesis of (−)-jorunnamycins A and C and (−)-jorumycin from l-tyrosine. J. Nat. Prod. 2013, 76, 1789–1795. [Google Scholar] [CrossRef]
- Capdevila, J.; Clive, S.; Casado, E.; Michie, C.; Piera, A.; Sicart, E.; Carreras, M.J.; Coronado, C.; Kahatt, C.; Matos-Pita, A.S. A phase i pharmacokinetic study of PM00104 (Zalypsis) administered as a 24-h intravenous infusion every 3 weeks in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Ferrari, S.; Blay, J.Y.; Navid, F.; Lardelli, P.; Alfaro, V.; Siguero, M.; Soman, N.; Chawla, S.P. A phase II multicenter, open-label, clinical and pharmokinetic trial of PM00104 in patients with advanced ewing family of tumors. Investig. New Drugs 2014, 32, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Michela, R.; Roberta, F.; Monique, Z.; Ezia, B.; Luca, P.; Galmarini, C.M.; García-Fernández, L.F.; Carmen, C.; Paola, A.; Eugenio, E. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Langmuir 2013, 12, 3157–3161. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Filamentous tropical marine cyanobacteria: A rich source of natural products for anticancer drug discovery. J. Appl. Phycol. 2010, 22, 659–676. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADCs | Phase | Target | Antibody | Linker | Payload | Indication(s) |
|---|---|---|---|---|---|---|
| Inotuzumab ozogamicin | Phase III | CD22 | Hz IgG4 | Hydrazone | Calicheamicin | NHL, ALL, CML |
| Gemtuzumab ozogamicin | Phase II | CD33 | Hz IgG4 | Hydrazone | Calicheamicin | AML, APL |
| MDX-1203 | Phase I | CD70 | n.d. | Valine-citrulline | Duocarmycin | RCC, NHL |
| Glembatumomab vedotin | Phase II | GPNMB | Hu IgG2 | Valine-citrulline | MMAE | Breast cancer, melanoma |
| Anti-PSMA ADC | Phase I/II | PSMA | Hu IgG1 | Valine-citrulline | MMAE | Prostatic cancer |
| RG-7593/DCDT-2980S | Phase I/II | CD22 | Hz IgG1 | Valine-citrulline | MMAE | NHL, DLBCL |
| RG-7596/DCDS-4501A | Phase I/II | CD79b | Hz IgG1 | Valine-citrulline | MMAE | NHL, DLBCL |
| RG7599/DNIB-0600A | Phase I/II | NaPi2b | Hz IgG1 | n.d. | MMAE | Ovarian cancer, NSCLC |
| MLN0264 | Phase I/II | GCC | n.d. | n.d. | MMAE | Pancreatic cancer, gastric carcinoma |
| ASG-22M6E | Phase I | Nectin-4 | Hu IgG1 | Valine-citrulline | MMAE | Solid tumors |
| ASG-5ME | Phase I | SLC44A4 | Hu IgG2 | Valine-citrulline | MMAE | Pancreatic cancer, prostatic cancer |
| AGS-67E | Phase I | CD37 | Hu IgG2 | Valine-citrulline | MMAE | Lymphoma, AML |
| BAY-79-4620 | Phase I | CA-IX | Hu IgG1 | Valine-citrulline | MMAE | Solid tumors |
| RG7458/DMUC-5754A | Phase I | MUC16 | IgG1 | n.d. | MMAE | Ovarian cancer, pancreatic cancer |
| RG7636 | Phase I | ETBR | n.d. | n.d. | MMAE | Melanoma |
| AGS-15ME | Phase I | SLITRK6 | Hu IgG2 | Cleavable linker | MMAE | Urothelial neoplasms |
| HuMax®-TF | Phase I | TF | Hu IgG1 | Valine-citrulline | MMAE | Solid tumors |
| SGN-LIV1A | Phase I | LIV-1 | Hz IgG1 | Valine-citrulline | MMAE | Breast cancer |
| AGS-16C3F | Phase I/II | ENPP3 | Hu IgG2 | MC | MMAF | RCC |
| SGN-CD19A | Phase I/II | CD19 | Hz IgG1 | MC | MMAF | Lymphoma, DLBCL |
| ABT-414 | Phase I/II | EGFR | Hu IgG1 | n.d. | MMAF | Solid tumors, glioma, SCC |
| GSK-2857916 | Phase I | BCMA | Hz IgG1 | MC | MMAF | MM |
| AGS-16M8F | Phase I | ENPP3 | Hu IgG2 | MC | MMAF | RCC |
| A1-mc-MMAF | Phase I | 5T4 | Hz IgG1 | MC | MMAF | Solid tumors |
| IMGN-529 | Phase I/II | CD37 | IgG1 | Thioether | DM1 | NHL, CLL, DLBLC |
| Lorvotuzumab mertansine | Phase I/II | CD56 | Hz IgG1 | SPP | DM1 | SCLC, MM |
| AMG-172 | Phase I | CD27L | Hu IgG1 | MCC | DM1 | ccRCC |
| IMGN-289 | Phase I | EGFR | Hz IgG | SMCC | DM1 | Solid tumors |
| AMG-595 | Phase I | EGFRvIII | n.d. | SMCC | DM1 | Glioma |
| SAR-3419 | Phase II | CD19 | Hz IgG1 | SPDB | DM4 | NHL, DLBCL |
| BT-062 | Phase II | CD138 | Ch IgG4 | SPDB | DM4 | MM |
| BAY-94-9343 | Phase I/II | Mesotherin | Hu IgG1 | SPDB | DM4 | Solid tumors |
| IMGN-853 | Phase I/II | FOLR1 | IgG1 | n.d. | DM4 | Solid tumors |
| SAR-566658 | Phase I | CA6 | Hu IgG1 | SPDB | DM4 | Solid tumors |
| IMGN-388 | Phase I | Integrinαvβ3 | Hu IgG1 | SPDB | DM4 | Solid tumors |
| BIIB-015 | Phase I | Cripto | Hz IgG1 | SPDB | DM4 | Solid tumors |
| Labetuzumab-SN-38 | Phase I/II | CD66e | Hz IgG1 | Phenylalanine-lysine | SN38 | CRC |
| IMMU-132 | Phase I/II | TROP-2 | Hz IgG1 | CL2A | SN38 | Epithelial carcinomas, solid tumors |
| SGN-CD33A | Phase I/II | CD33 | Hz IgG1 | Valine-alanine | PBDs | AML, APL |
| SGN-CD70A | Phase I | CD70 | n.d. | Valine-alanine | PBDs | RCC, lymphoma |
| SC16LD6.5 | Phase I/II | Fyn3 | SC16 | n.d. | D6.5 | SCLC |
| Milatuzumab doxorubicin | Phase I/II | CD74 | Hz IgG1 | Hydrazone | Doxorubicins | MM |
| SYD985 | Phase I | HER2 | Hz IgG | n.d. | n.d. | Solid tumors |
| IGN523 | Phase I | CD98 | Hz IgG | n.d. | n.d. | AML |
| Indications | Antigen Targets |
|---|---|
| NHL | CD19, CD20, CD21, CD22, CD37, CD70, CD72, CD79a/b, CD180 |
| HL | CD30 |
| AML | CD33, CD98 |
| MM | CD56, CD74, CD138, ETBR |
| Lung cancer | CD24, CD56, CD326, Cripto, FAP, Mesothelin, GD2, 5T4, NaPi2b, FOLR1, Integrinαvβ3, Fyn3 |
| CRC | CD74, CD174, CD227, CD326, CRIPTO, FAP, ED-B, CD66e |
| Pancreatic cancer | CD74, CD227, nectin-4, CA19-9, MUC-4, MUC16, alpha v beta6, GCC |
| Breast cancer | CD174, GPNMB, CRIPTO, nectin-4, LIV1A |
| Ovarian cancer | MUC16, TIM-1, Mesothelin, NaPi2b |
| Melanoma | GD2, GPNMB, ED-B, PMEL 17, ETBR |
| Prostate cancer | PSMA, STEAP-1, TENB2 |
| Renal cancer | CAIX, TIM-1, CD27L, CD70, ENPP3 |
| Mesothelioma | Mesothelin |
| Urothelial cancer | SLITRK6 |
| Glioma | EGFRvIII |
| Generic Name | Trade Name | Target | Antibody | Indication(s) | Year of First Approval |
|---|---|---|---|---|---|
| Edrecolomab | Panorex | EpCAM | Murine IgG2 | CRC | 1995 |
| Ibritumomab tiuxetan | Zevalin | CD23 | Murine IgG1 | NHL | 2002 |
| 131I-labeled tositumumab | Bexxar | CD20 | Murine IgG2 | NHL | 2003 |
| Rituximab | Rituxan | CD20 | Chimeric IgG1 | B-NHL | 1997 |
| 131I-labeled ch-TNT | n.d. | n.d. | Chimeric IgG1 | Lung cancer | 2003 |
| Cetuximab | Erbitux | EGFR | Chimeric IgG1 | CRC | 2004 |
| Brentuximab | Adcetris | CD30 | Chimeric IgG1 | ALCL, HL | 2011 |
| Trastuzumab | Herceptin | HER2 | Humanized IgG1 | Breast cancer | 1998 |
| Gentuzumab ozogamcin | Mylotarg | CD33 | Humanized IgG4 | Leukemia | 2000 |
| Alemtuzumab | Campath | CD52 | Humanized IgG1 | B-CLL | 2001 |
| Bevacizumab | Avastin | VEGF | Humanized IgG1 | CRC, lung, breast cancer | 2004 |
| Nimotuzumab | TheraCIM | EGFR | Humanized IgG1 | Epithelial cancer | 2005 |
| Pertuzumab | Perjeta | HER2 | Humanized IgG1 | Breast cancer | 2012 |
| Panitumumab | Vectibix | EGFR | Human IgG2 | CRC | 2007 |
| Ofatumumab | Arzerra | CD20 | Human IgG1 | CLL | 2009 |
| Ipilimumab | Yervoy | CTLA4 | Human IgG1 | Melanoma | 2011 |
| Cleavable Linker | Non-Cleavable Linker |
|---|---|
 |  |
 |  |
 |  |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-J.; Li, Y.-Y.; Liu, X.-Y.; Lu, X.-L.; Cao, X.; Jiao, B.-H. Marine Antibody–Drug Conjugates: Design Strategies and Research Progress. Mar. Drugs 2017, 15, 18. https://doi.org/10.3390/md15010018
Wang Y-J, Li Y-Y, Liu X-Y, Lu X-L, Cao X, Jiao B-H. Marine Antibody–Drug Conjugates: Design Strategies and Research Progress. Marine Drugs. 2017; 15(1):18. https://doi.org/10.3390/md15010018
Chicago/Turabian StyleWang, Yu-Jie, Yu-Yan Li, Xiao-Yu Liu, Xiao-Ling Lu, Xin Cao, and Bing-Hua Jiao. 2017. "Marine Antibody–Drug Conjugates: Design Strategies and Research Progress" Marine Drugs 15, no. 1: 18. https://doi.org/10.3390/md15010018