The Oxepane Motif in Marine Drugs


1
GIR MIOMeT, IU CINQUIMA/Inorganic Chemistry, University of Valladolid, Campus Miguel Delibes, 47011 Valladolid, Spain
2
Department of Organic Chemistry, University of Valladolid, Campus Miguel Delibes, 47011 Valladolid, Spain
*
Author to whom correspondence should be addressed.
Mar. Drugs 2017, 15(11), 361; https://doi.org/10.3390/md15110361
Submission received: 29 September 2017
/
Revised: 3 November 2017
/
Accepted: 8 November 2017
/
Published: 15 November 2017
(This article belongs to the Special Issue Bioactive Marine Natural Compounds with Heterocyclic Motifs in their Structure)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Oceans have shown to be a remarkable source of natural products. The biological properties of many of these compounds have helped to produce great advances in medicinal chemistry. Within them, marine natural products containing an oxepanyl ring are present in a great variety of algae, sponges, fungus and corals and show very important biological activities, many of them possessing remarkable cytotoxic properties against a wide range of cancer cell lines. Their rich chemical structures have attracted the attention of many researchers who have reported interesting synthetic approaches to these targets. This review covers the most prominent examples of these types of compounds, focusing the discussion on the isolation, structure determination, medicinal properties and total synthesis of these products.
1. Introduction
More than 70% of Earth surface is covered by water, 96.5% of which is found in the oceans. This means that the planet’s largest habitat is the ocean, which is the ecosystem where the major part of animals and plants of Earth lives. This includes microscopic algae, marine plants with roots, sponges, corals and all types of fishes, within others. These marine organisms are a great source of natural products with important biological activities.
The continuous search for compounds with pharmacological properties is one of the main aims of scientists. Natural products have always provided a great contribution to medicine since the first discovery of drugs with positive impact in human health. For thousands of years the common sources of natural products with potential biological activities have been microorganisms and land plants. The search for natural products in the sea is much recent, since a parallel development of the appropriate technology was needed. However, since the 70’s the number of isolated marine natural products with outstanding biological properties has grown enormously [1].
A group of marine natural products that attract special interest is polyfunctionalized cyclic ethers. Within them, natural products containing an oxepanyl moiety have been frequently found in sponges, corals, or different marine fungus, within others. Their interesting biological properties include anticancer, antibacterial or antifungal activities. From a structural point of view, many of them are terpenes presenting a great diversity of rings and chains bonded to the oxepanyl ring.
Their challenging structure, together with their promising medicinal properties, has prompted many researchers to try to develop synthetic methodologies to access this class of marine drugs.
In this review, we try to cover marine drugs that contain a single oxepanyl ring in their structure. However, the coverage will not be comprehensive, since we intend to provide a general overview of the interesting biological, structural and synthetic possibilities of this class of metabolites. Within others, we have omitted the family of lauroxanes, since it has already been reviewed by Fujiwara [2]. We also omitted classical examples of marine polyether biotoxins, such as brevetoxins, ciguatoxins, gambierol, etc., which have been addressed by Nicolau [3]. Some of them, along with abudinol B, brevenal, armatol A, enshuol and others, have been recently reviewed by Jamison [4]. Toxicols, adociasulfates and the latest example of halicloic acids A and B are also interesting substances and appear in a recent review [5]. Of course, we could mention many other families and examples of marine natural products that bear the oxepane moiety (spiroxins, phomactins, clavulazols, etc.) but it is out of the scope of our work. On the other hand, some isolated examples of the compounds described here are also present in other reviews devoted to marine triterpenes [6] or marine natural products [7].
This review is roughly organized in five sections, according to the structure and biogenetic origin of the compounds. For some of the families, only structure determination, isolation and biological activity have been reported. For the rest of the products some of the most relevant synthetic approaches reported till date are described.
2. Halogenated Sesquiterpenoids: Aplysistatin and Palisadins
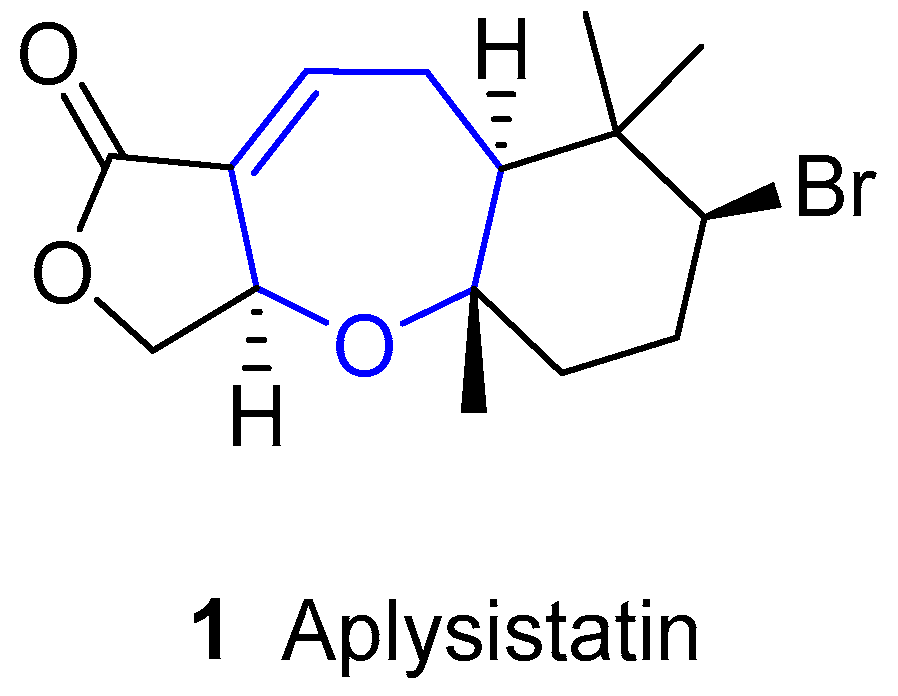
Aplysistatin was first isolated from the South Pacific Ocean sea hare Aplysia angasi in 1977 [8]. In his paper, Pettit pointed out that this compound could be derived from its diet, since it is known that sea hares usually graze algae and other similar metabolites had already been isolated from algae [9,10,11,12]. This hypothesis was confirmed in 1980, when aplysistatin was collected from the alga Laurencia cf. palisada Yamada in 1980 [13]. Since then, it has been identified in many other Laurencia species, namely Laurencia filiformis [14], Laurencia implicata [15], Laurencia flexilis [16], Laurencia karlae [17], Laurencia luzonensis [18], Laurencia saitoi [19], Laurencia similis [20] and Laurencia snackeyi [21].
This sesquiterpene bears a unique structure, as shown in Figure 1. Its absolute configuration was established by X-ray diffraction [8,22], showing a trans-anti stereochemistry for the fused rings.
As for its biological activity, it was reported to inhibit progression of murine lymphocytic leukemia P-388 (T/C 175 at 400 mg/kg) [8] and a broad range of other cultured tumor cells. It also showed antimalarial activity [23], anti-inflammatory activity and ability to suppress the expressions of iNOS and COX-2 enzymes [24].
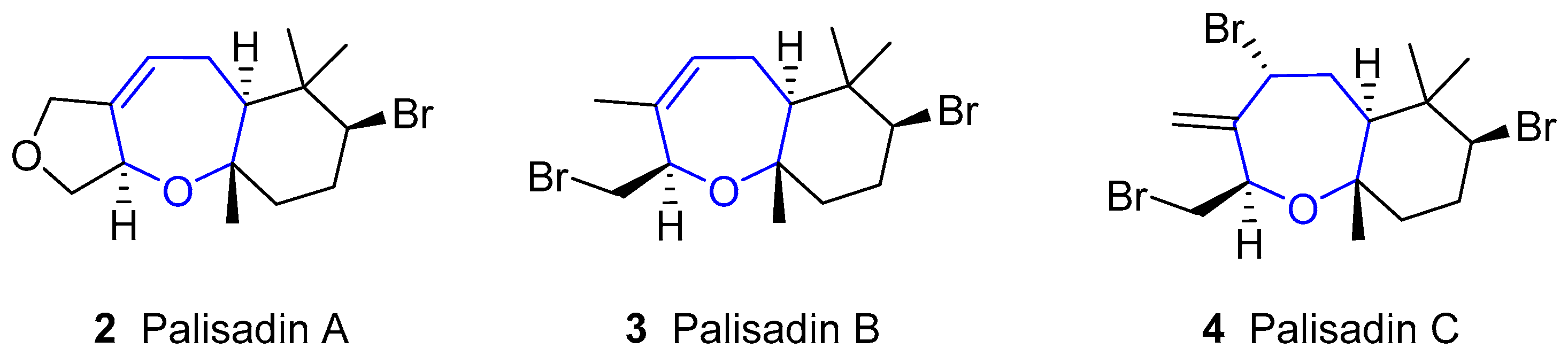
In 1980, Fenical reported the isolation of palisadin A, palisadin B and other three related compounds from Laurencia cf. palisada [13]. Later, palisadin C [16], along with other substituted palisadins, has been isolated (structures of this family are shown in Figure 2). They have a similar structure to aplysistatin, although lacking the lactone ring, and are also present in many algae of the Laurencia species.
5β-hydroxipalisadin B (Figure 3) showed effective anti-inflammatory properties, reducing stress-induced reactive oxygen species formation, and inhibiting the lipopolysaccharide-induced NO production in zebrafish embryos [25].
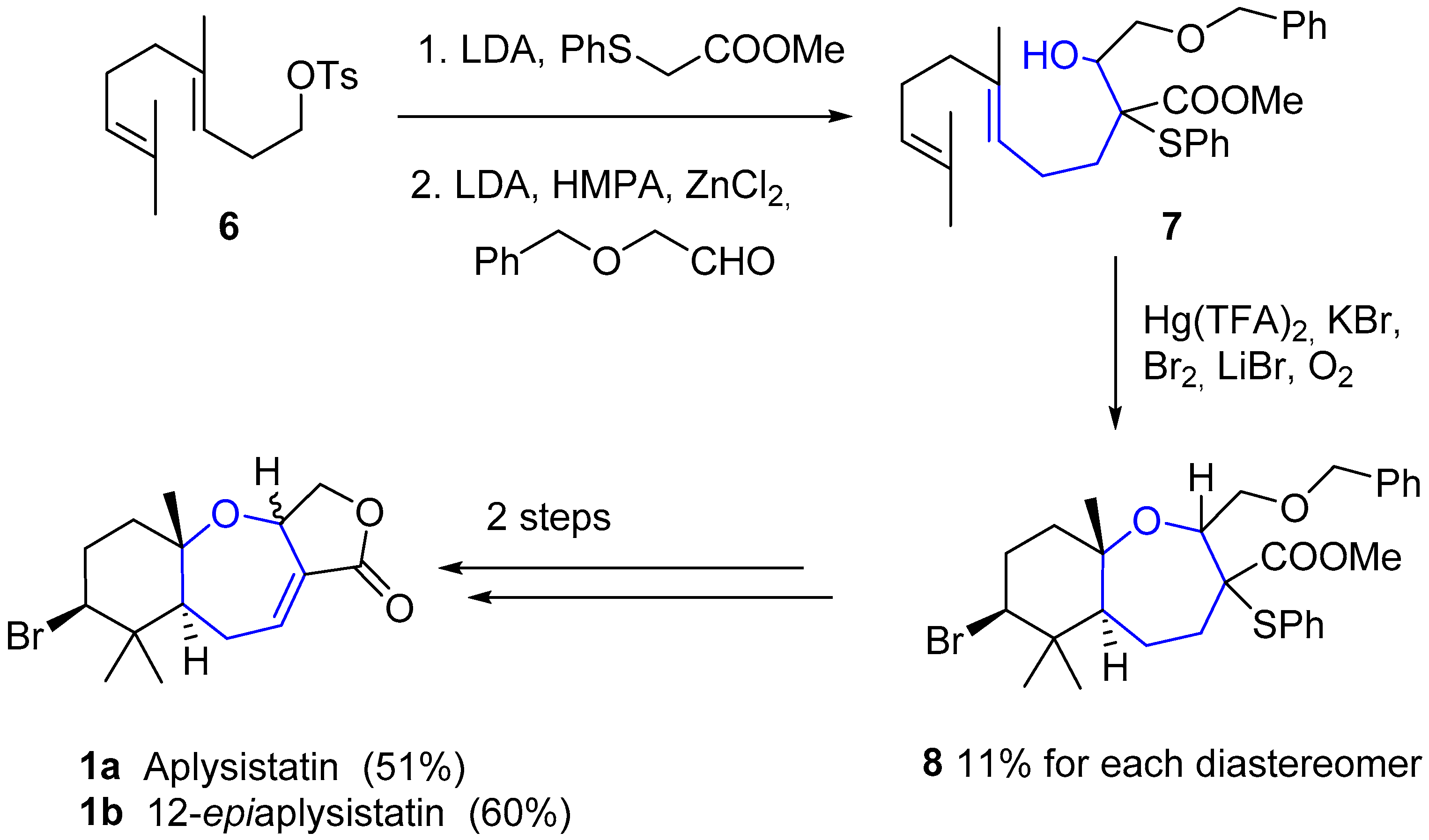
The unusual, but not exceptionally complex, structure of these compounds has attracted the attention of synthetic chemists. In 1979, just two years after its isolation, Hoye and coworkers reported the first total synthesis of aplysistatin [26]. Diene 7 was obtained in two steps from the p-toluenesulfonate ester of homogeraniol 6, by alkylation with the enolate anion of 2-phenyltioacetate and subsequent aldol condensation. An additional key Hg(TFA)2/bromine-mediated cyclization provided a mixture of diastereomeric oxepins 8, which in two steps (oxidative elimination and final debenzylation-lactonization) provided aplysistatin (1a) and 12-epi-aplysistatin (1b) (see Scheme 1). In the next few years some other total syntheses of 1 were reported which did not imply any improvements in either yields or stereoselectivities [27,28,29,30,31,32].
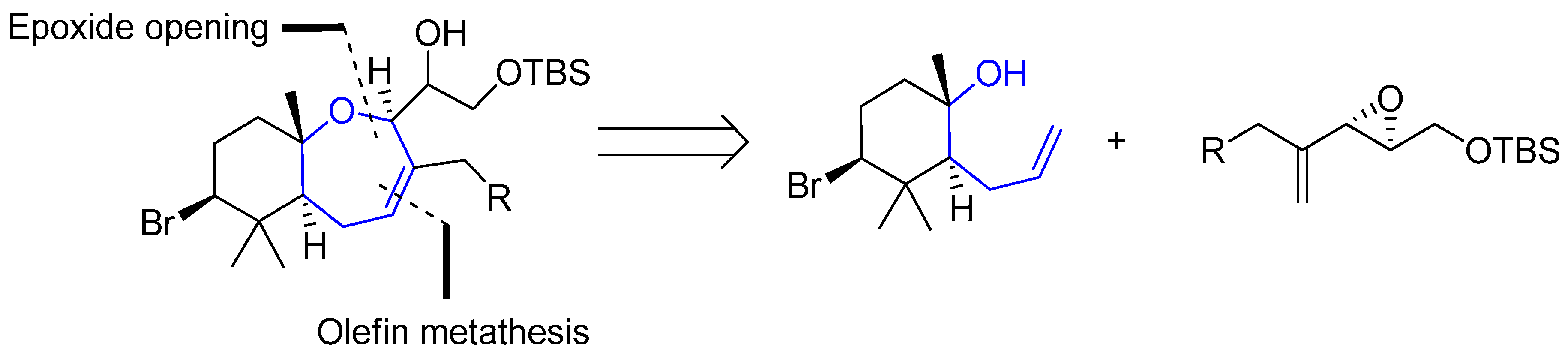
Regarding palisadins, Yamashita’s group performed the first total synthesis of (+)-palisadin A and (+)-hydroxypalisadin B [33], also relying on a Hg(TFA)2/bromine-mediated cyclization as the key step. In 2004 Couladouros’ group published a general approach to trans-fused oxepene-cyclogeranyl systems, and applied it to the synthesis of aplysistatin, palisadins A and B and 12-hydroxypalisadin B [34]. Their strategy followed an alternative route based on the formation of the oxepanyl derivative by addition of a tertiary alcohol to a 1,2-disubstituted epoxide and subsequent ring closing metathesis Scheme 2.
This oxepene intermediate provided easy access to the desired natural products in three or four additional steps, as depicted in Scheme 3.
3. Marine Diterpenes: Oxepin Lobatrienol
Lobanes are a family of diterpenes from genus Lobophytum, also known as “devil’s hand corals”. Their habitat covers the shallow waters of Indo-Pacific coasts [35,36], although these compounds have been also isolated from Eunicea fusca [37,38], Sinularia [39,40,41] and Sarcophyton [42] species. Their structure is based on the main scaffold of β-elemene and one of the members of this family, the one called oxepin lobatrienol, possess an oxepane-like moiety (see Figure 4). In such molecule, an oxepanol moiety has substituted a prop-1-en-2-yl unit in C4 [43]. Despite their remarkable bioactivity, few efforts have been devoted to the synthesis of this kind of compounds [44,45,46,47].
All members of lobane family are lethal against Cladosporium cucumerinum, a pathogen fungus responsible for scab disease that affects cucumbers, and against genus Artemia species (commonly known as brine shrimps). Apparently, the nature of the isoprenyl fragment attached to the basic structure of β-elemene determines which of the two properties predominates. It seems that when the activity against the fungus is increased, the activity against brine shrimps is decreased, and vice versa. These findings suggest a non-generic toxicity and a specific mode of action is yet to be discovered. Interestingly, oxepin lobatrienol possess intermediate activity and it performs well in both tasks, demonstrating, once more, the importance of oxepane-like structures.
4. Marine Triterpenes
This huge group consists of a set of triterpenes bearing oxepane moieties [48,49]. It is believed that their biosynthesis comes from the metabolism of very simple squalene building blocks. They have been named according to their origin, either from the species or the place where they were obtained. Bearing in mind the complexity of the classification, we decided to divide it into 5 categories.
4.1. Sipholenols Family
The first compound of this group (sipholenol A) was isolated by Shmueli, Kashman and coworkers [50] from the colonial tube-sponge Siphonochalina siphonella, a species whose habitat is the Red Sea. Just two years later, up to nine molecules were already known [51,52]. Less attention was paid in the 90s (mainly due to the discovery of similar families such as sodwanones) until the beginning of the 21st century, when interest for these species was renewed. Nowadays, the number of compounds belonging to sipholenols has increased up to thirty [53,54,55].
From the structural point of view, the molecules contain a hydroazulene fragment (with variable insaturations) linked through an ethylene bridge to a trans-decahydrobenzoxepin, as seen in Figure 5. It must be noted that this is the most common scaffold, but not present in all of them. There are some differences in the last set of discovered molecules, e.g., the rearrangement of azulene fragment or the substitution of an oxepane by a tetrahydropyran.
These molecules have shown good anti-cancer properties [56]. In more detail, they are able to reverse P-glycoprotein-mediated multi drug resistance in cancer cells by inhibiting the drug efflux from this protein, along with other effects [57,58,59,60]. Currently, their antiproliferative effects towards human hepatic and colorectal cancer cells are being studied [61].
Moreover, this family can be used as antifouling agent owing to its ability to disrupt settlement of barnacle larvae.
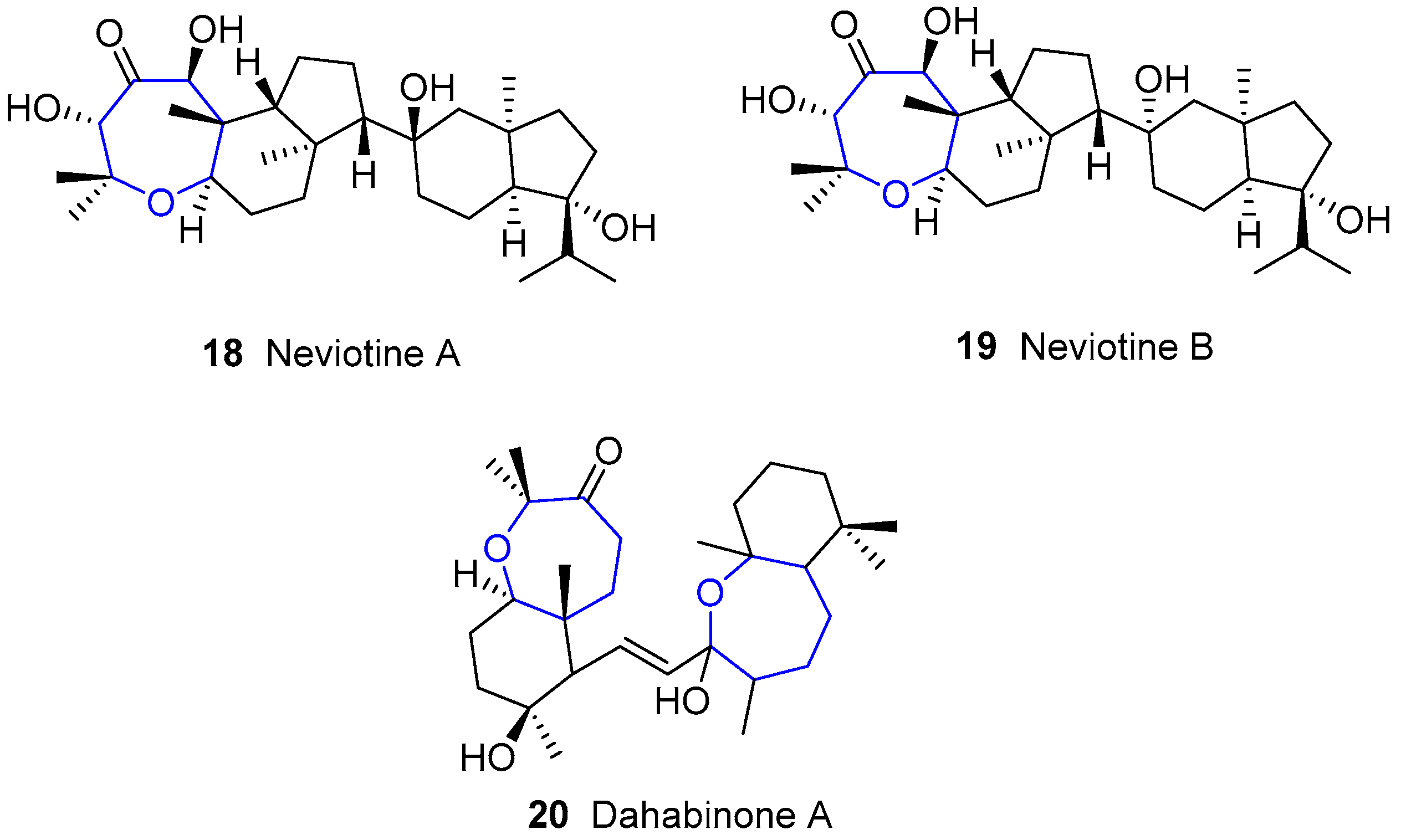
4.2. Neviotanes and Dahabanes
Along with sipholenols, Kashman and collaborators also reported the isolation of Neviotines A and B with Dahabinone A from the same sponge Siphonochalina siphonella. Such names have their origin in the place from which these species were extracted: Nevi’ot (Israel) and Dahlak islands (Erithrea), respectively [53,62]. The structure of the latter compound is very similar to that from sipholenols family, bearing a second oxepane unit fused to a cyclohexane instead of the azulene moiety. Unlike sipholenols and dahabanes, neviotanes possess a pentacyclic core in which there are two groups connecting though a single bond (Figure 6).
In addition, new compounds of this family have been reported recently [63].
4.3. Sodwanones
This class of compounds was first isolated from the Indo-Pacific fan sponge Axinella weltneri, collected during the summer of 1992 in Sodwana Bay, South Africa [64,65]. Since then, they have been found in other species, namely Ptilocaulis spiculifer [66], and Axinella cf. bidderi [67], and so far they form a family of more than 20 compounds [68,69,70,71,72]. They are examples of the rare marine triterpenes, which have a common structure showing an oxepane-cycloalkane moiety. As far as we know, no attempts of synthesis of any of the sodwanones have been done.
Many of the sodwanones have shown interesting properties. Sodwanone A (Figure 7) was active against several cell lines, such as a lung carcinoma cell line [67], three esophageal cancer cell lines [73], an ovarian cancer one [71] and some others. Sodwanones G, H and I were found to be toxic against several cancer cell lines [69]. Sodwanone M was reported to be cytotoxic to P-388 murine leukemia cells [68]. Sodwanone S was moderately active against several lines [71] and some other members of the sodwanone family inhibited hypoxia-induced HIF-1 activation in breast and prostate tumor cells [72].
4.4. Shaagrockols
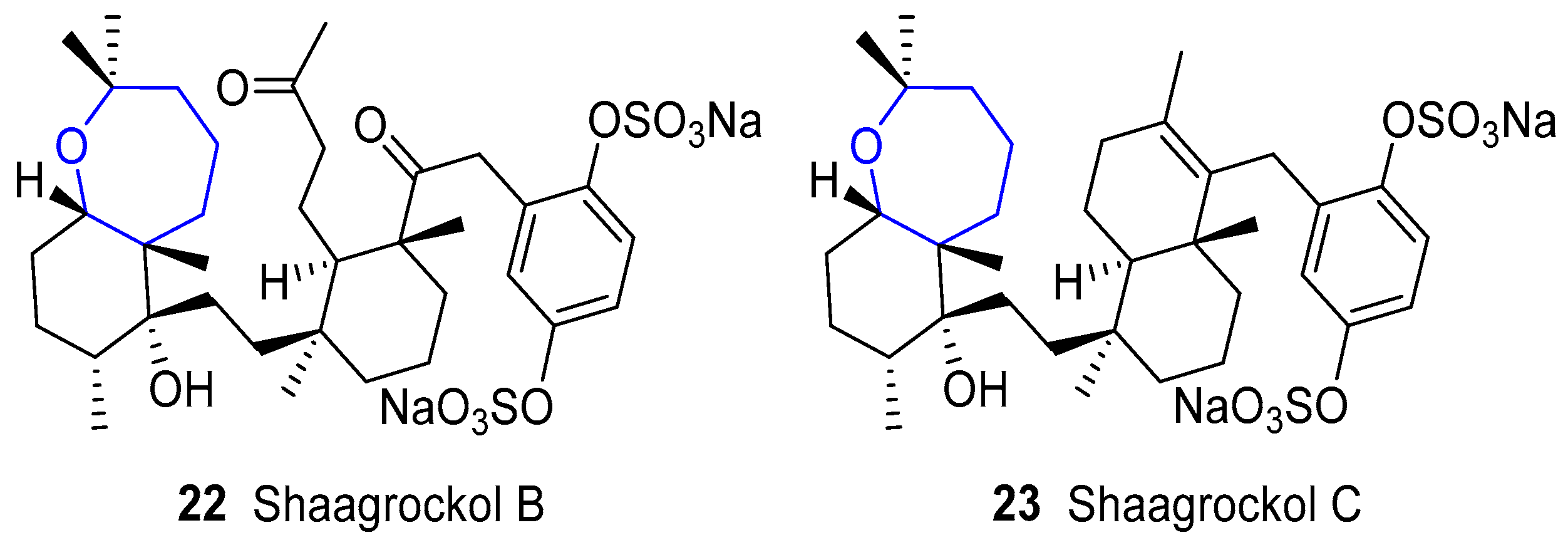
In parallel to the discoveries of sodwanones, two new compounds were reported by Kashman’s group in 1992 called Shaagrockols B and C from the Red Sea sponge Toxiclona toxius [74,75]. Their name comes from the place where the sponge was collected: Shaag rock, in the entrance to the Gulf of Suez. Structurally, they are very closely related to sodwanones and the main difference arises from the additional tetrahydroquinone fragment that bears sulfonate groups [76,77], as depicted in Figure 8. They have shown antifungal activity against Candida albicans and inhibition of Human Immunodeficiency Virus Type 1 Reverse Transcriptase (HIV-1 RT).
4.5. Raspacionins
This family was firstly discovered by Puliti, Madaio and collaborators between 1991 and 1992. Compounds were isolated from red encrusting sponge Raspaciona aculeata [80,81,82,83]. From the structural point of view, these quasi-symmetrical molecules resemble dahabinone as they contain two oxepane subunits (Figure 9). After such initial discoveries, other minor secondary metabolites were found [84,85].
5. Meroterpenoids: Austalides
This family consists of a 28-membered group of molecules whose origin comes from Penicillium and Aspergillus marine-derived genera. First discovered in 1981 by Vleggaar and coworkers and new compounds are nowadays being reported [86,87,88,89,90,91,92]. They are meroterpenoids possessing 4 to 6 carbo and heterocycles, from which 12 of them bear an oxepane-like structure. Their basic structure is based on a pentacyclic 5/6/6/6/7 system, as shown in Figure 10. It is known that their biosynthesis has its origin in a farnesyl phthalide derivative, which, upon cyclization and oxidative modifications, furnishes this family [93,94,95]. Many beneficial effects have been found, such as anticancer, antibacterial, antiviral and antifouling properties; most of them due to specific inhibition of crucial target proteins (α-glucosidase, AP-1 transcription factor, endo-1,3-β-d-glucanase, etc.).
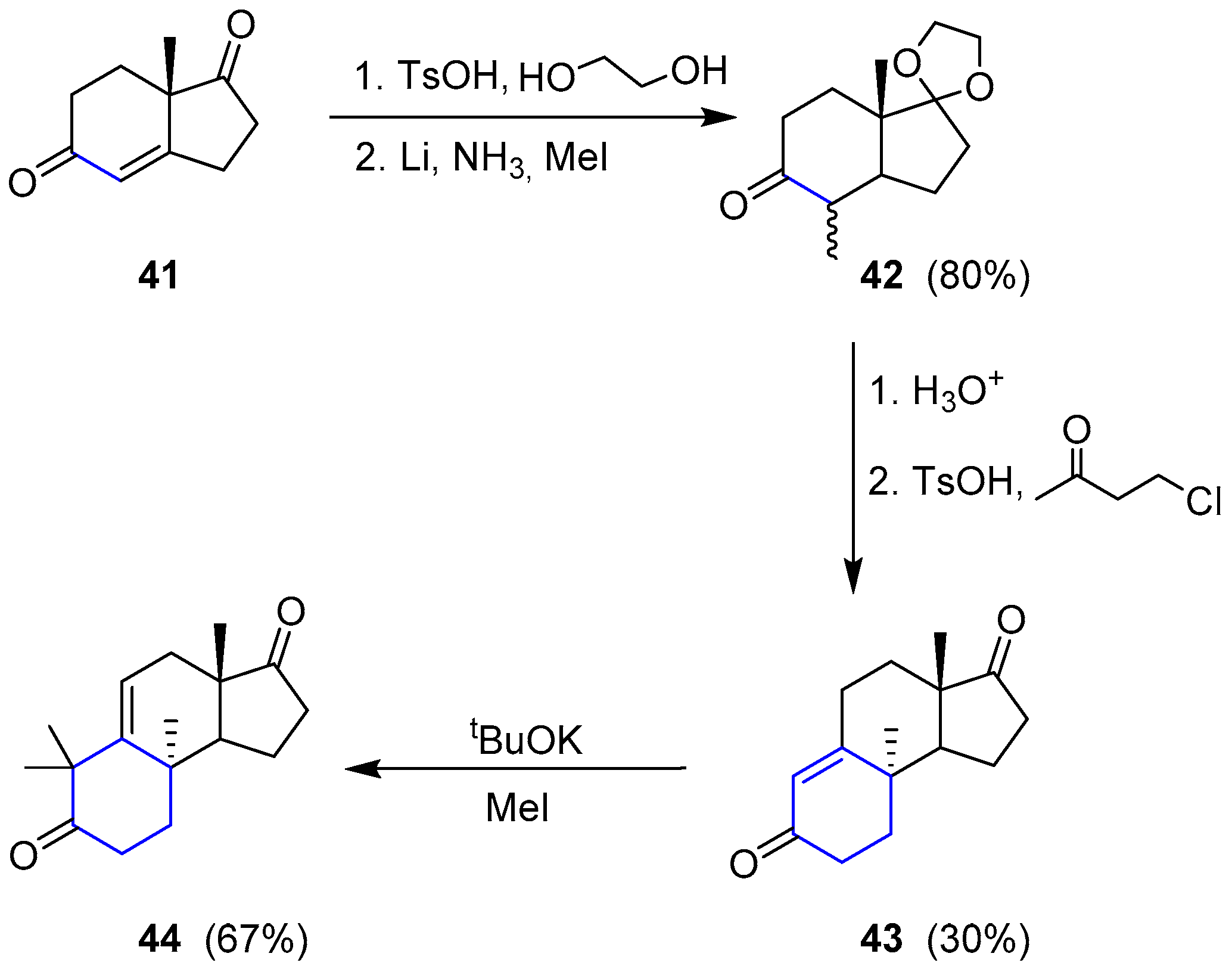
The only attempt regarding the total synthesis of a member of this family was reported by Paquette and coworkers in 1994 [96,97]. The selected molecule was (−)-Austalide B. They envisaged a retrosynthetic analysis by splitting the preparation into two sections. The so-called western sector was considered the most challenging difficulty. To access compound 38, lactone 39 seemed to be the best precursor, which could derive from a ring expansion of a tetrahydrofuran (40) through a Baeyer-Villiger oxidation. Such molecule could be obtained from very simple diketone 41 after a Robinson annulation and subsequent ring expansion. On the other hand, the phthalide moiety (eastern sector) could be easily connected through a sequence of conventional proceedings to dihydropyran (38) (Scheme 4).
Starting by readily available 41, the saturated ketone was regioselectively protected and subjected to metal reduction in the presence of methyl iodide to give 42. Robinson annulation did not yield the expected cyclization, so the reaction with 4-chloro-2-butanone under acidic conditions was used instead to obtain 43. This compound was dimethylated to furnish 44 and complete the first stage of the western section synthesis (Scheme 5).
Then, compound 44 was oxidized with osmium tetroxide to give a diol whose secondary alcohol was selectively protected as SEM ether 45. Under Baeyer-Villiger conditions only the cyclohexanone moiety underwent the homologation process resulting in 46. The second Bayer-Villiger oxidation could be accomplished upon formation of the corresponding ortho lactone 47, thus obtaining the expected complete western section 48. The process is summarized in Scheme 6.
The synthesis of the eastern section (Scheme 7) turned out to be much harder than initially expected. Finally, after two unsuccessfully attempts, Paquette and coworkers were able to achieve desired Austalide B and the procedure is described as follows. Lactone 48 was subjected to C-alkylation (with cyanoformate) and subsequent O-triflation to give 49. This triflate was coupled with previously prepared stannane 50 under standard Stille conditions to obtain compound 51. Then, intramolecular cyclization and further benzannulation followed by methylation gave rise to 52, whose main scaffold was very close to the natural desired product. Finally, deprotection of SEM group in the western section and inversion at C6 in a 3-step sequence ultimately yielded (−)-Austalide B (28).
6. Alkaloids
6.1. Bromotyrosine Alkaloids: Psammaplysins and Ceratinamides
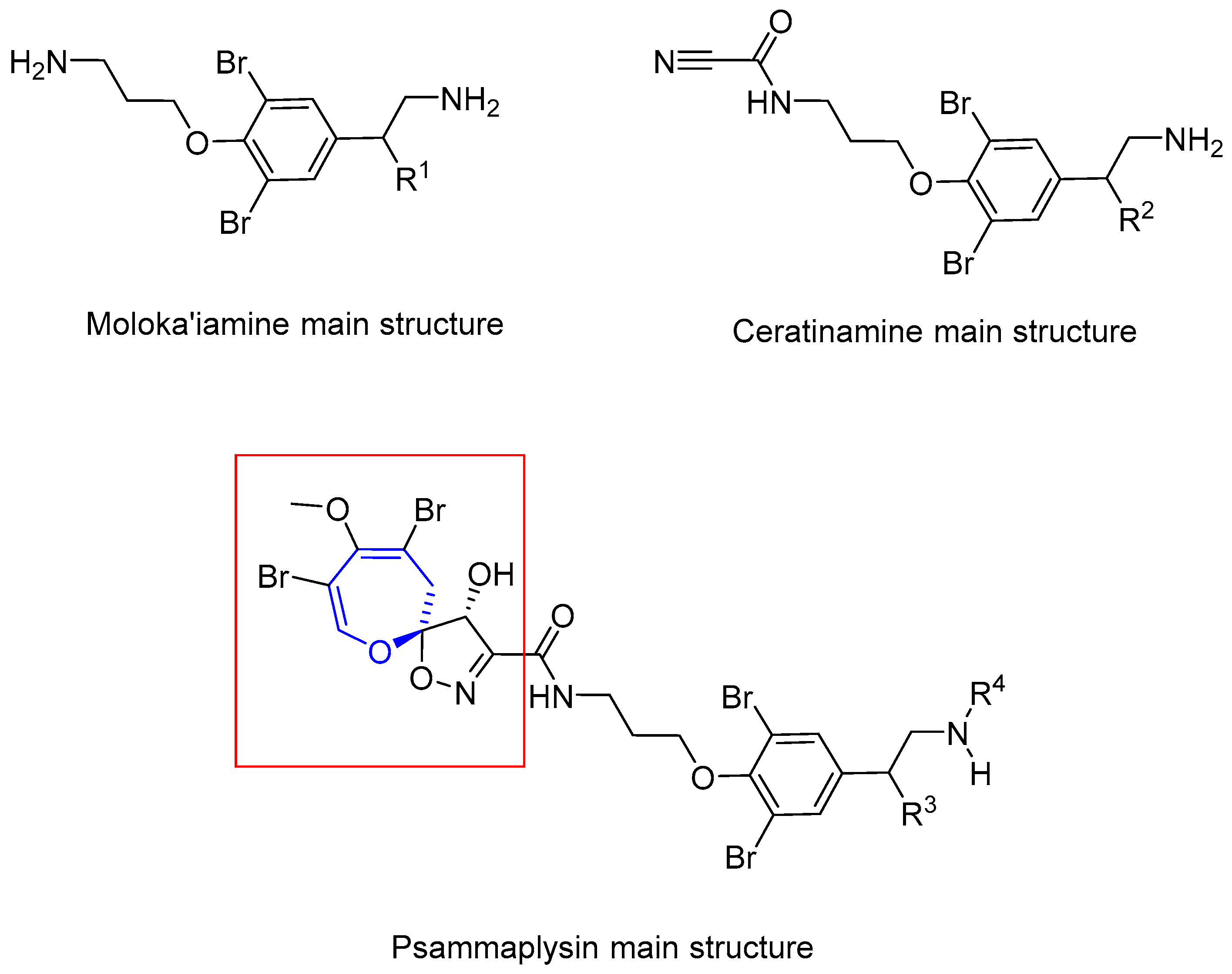
In 1983, Kashman and coworkers reported the isolation of two new compounds from sponge Psammaplysilla purpurea [98]. They showed antibiotic properties, being active against gram-positive bacteria and E. coli. Initially, their structures were wrongly assigned as spirocyclohexadienyloxazoline derivatives. Two years later, Scheuer and Clardy elucidated the correct structure (Figure 11), based on a deeper study of the NMR spectra and single-crystal X-ray diffraction [99]. The absolute configuration for psammaplysin A was assigned as (6S*,7S*), but recently it has been corrected to (6R,7R) by Garson and Kurtán [100].
In the nineties, some other members of the family were reported. Psammaplysin C was isolated from Psammaplysilla purpurea by Ireland and coworkers in 1992 [101]. It has identical structure to psammaplysin B, except for the amine substitution. Later on, Scheuer and coworkers reported the new psammaplysins D and E from an unknown species of Aplysinella sponge, collected at Pingelap Atoll, Micronesia [102]. The former has an amide group instead of the terminal amine and the latter has an unprecedented cyclopentenedione ring. In 1996, Fusetani’s group isolated ceratinamides A and B, named after the sponge they were isolated from, Pseudoceratina purpurea [103]. These new compounds, along with Psammaplysins A and E showed antifouling activity against Balanus amphitrite. Finally, psammaplysin F was reported by Schmitz and coworkers, from a sponge believed to be from the Aplisynella genus [104]. The structures of these compounds are shown in Figure 12.
In the 2000s no new members of this family were reported. Bewley and coworkers reported that psammaplysins A and B inhibit mycothiol-S-conjugate amidase from Mycobacterium tuberculosis and Mycobacterium smegmatis, also inhibiting growth of the latter [105,106].
In 2010, Quinn and coworkers isolated psammaplysin G from an Australian sponge of Hyatella species [107]. It is the first example of this family that bears a terminal N-methylurea moiety. Both psammaplysin F and G showed antimalarial activity. Psammaplysin G was very active against a chloroquine-resistant (Dd2) strain of Plasmodium falciparum at 40 µM but was not cytotoxic at all to HEK293 (a human embryonic kidney cell line). Psammaplysin F obtained better values (1.4 and 0.87 µM) against Dd2 and 3D7 (chloroquine-sensitive) strains, respectively. In 2011, the same group reported the isolation of psammaplysin H, being also active against P. falciparum, and quite selective (>97-fold) [108]. Ramsey and McAlpine reported antibiotic activity for psammaplysins F and H against Gram-positive bacteria. Psammaplysin F produced an unequal chromosome partitioning between daughter cells, placing it as a possible new lead antibiotic.
In the past few years, many other compounds have appeared. In 2012, Wright reported psammaplysins I and J from a Suberea species sponge [109], but did not assess their biological activity. In the same year, Garson found 21 new derivatives from the sponge Aplysinella strongylata [110]. Five of them had different chains attached to C-16, and the rest had terminal fatty acid chains. 19-Hydroxypsammaplysin E inhibited growth of P. falciparum (IC50 = 6.4 µM). Another four compounds of this kind were isolated by Lee and coworkers from a sponge of the genus Suberea in 2013 [111]. Most of them had a significant activity against several cancer cell lines. Lee also assessed some moloka’iamines and ceratinamides from the same sponge, and they exhibited no activity up to 70 µM, thus concluding that the spirooxepinisoxazoline moiety is crucial for their anticancer activity. Comparison of the structures can be seen in Figure 13.
To the best of our knowledge, no total synthesis of any of the compounds of this family has been reported.
6.2. Guanidinium Alkaloids
The family of guanidinium alkaloids containing oxepane rings is diverse since it contains over 15 members. Fortunately, they share some structural similarities [112]. A triazaperhydroacenaphthalene skeleton directly connected to an oxepane and a tetrahydropyran, giving rise to the so-called pentacyclic “vessel unit”, is common in all molecules. Then, a very long chain is linked to this scaffold containing a hydrocarbon fatty acid functionalized as amide with a spermidine moiety furnishing the “anchor unit”. The reason why these two parts are called like this are due to the likeness of the molecule to a macroscopic ship trailing an anchor [113]. These structures are summarized in Figure 14.
All of them have been primarily isolated from marine sponges. The first member was discovered by Kashman’s group in 1989 from Hemimycale sp. of the Red Sea and the Caribbean Ptilocaulis spiculifer [114]. Since then, a plethora of new molecules have been reported, and new names were coined, such as monanchomycalins, crambescidin, neofolitispates, etc. All of them based on the marine species from which the compound is obtained [113,114,115,116,117,118,119,120].
Given that the guanidine motif is present in arginine aminoacid and guanine nucleobase, it is not surprising that this functional group appears in countless natural products, and it is even less surprising that it possesses outstanding beneficial properties. It has been demonstrated that this family show antiviral [122], fungicidal [123] and anticancer effects [124,125,126,127,128]. The mechanisms of action for this latter activity has been studied and now it is known that they are excellent Ca2+-channel blockers and good inhibitors of enzymes Na+, K+ or Ca2+-ATP-ase [129,130]. There are other applications; for instance, Crambescidin 800 has revealed exceptional protection of cell lines under oxidative stress, typically present in neuronal degenerative diseases [131].
Concerning the synthesis of the members in this family, fantastic efforts towards the development of chemical tools to prepare these intriguing structures have been addressed [121,132,133,134,135,136,137,138,139,140]. Since there are several in-depth works regarding the total synthesis of many compounds (or their fragments) in the family [141,142,143,144,145], addressing all of them would be out of the scope of this review. Therefore, we will cover herein, as an example, the preparation of the simple Crambescidin 359 which structure coincides with the main core “vessel unit”.
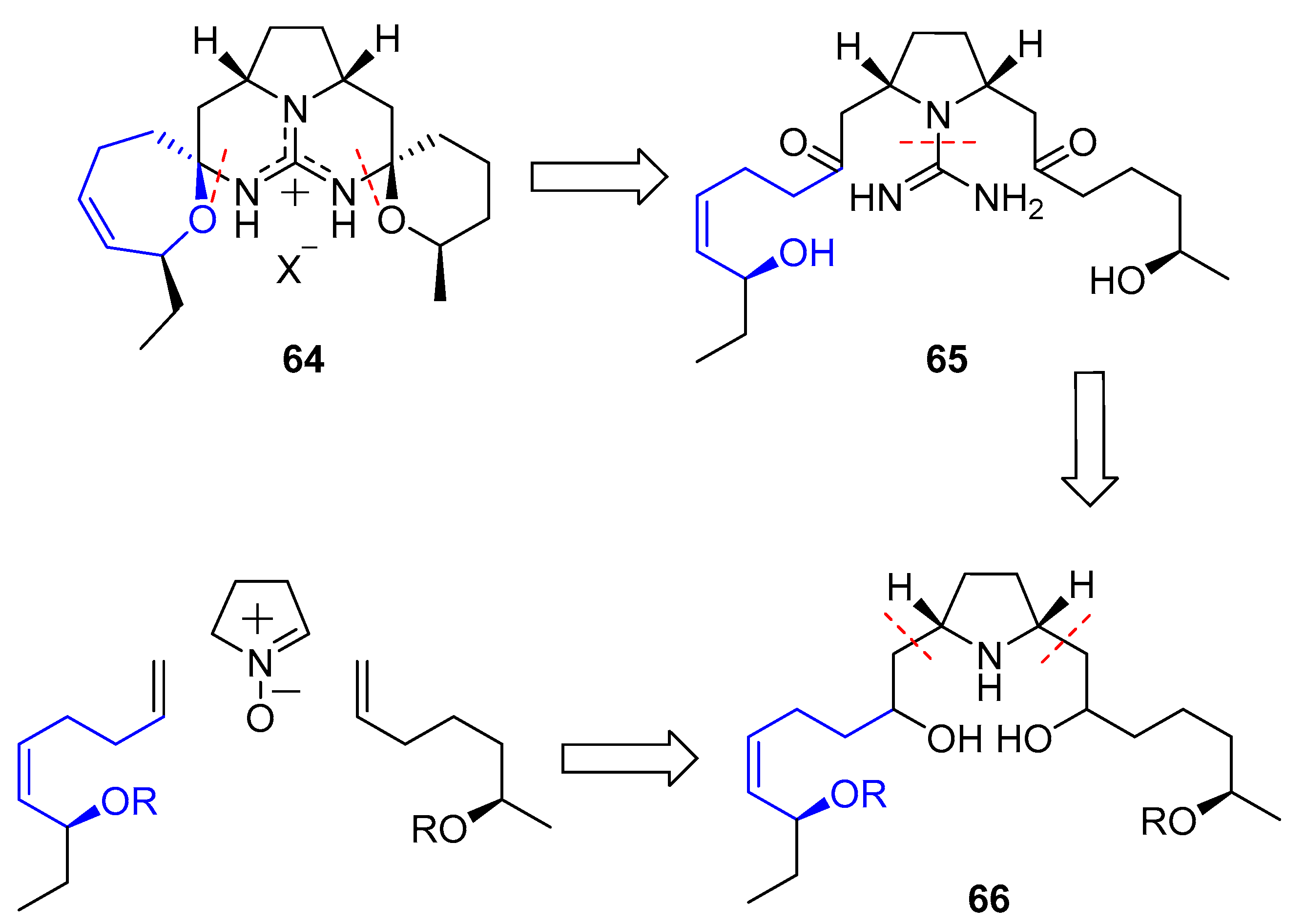
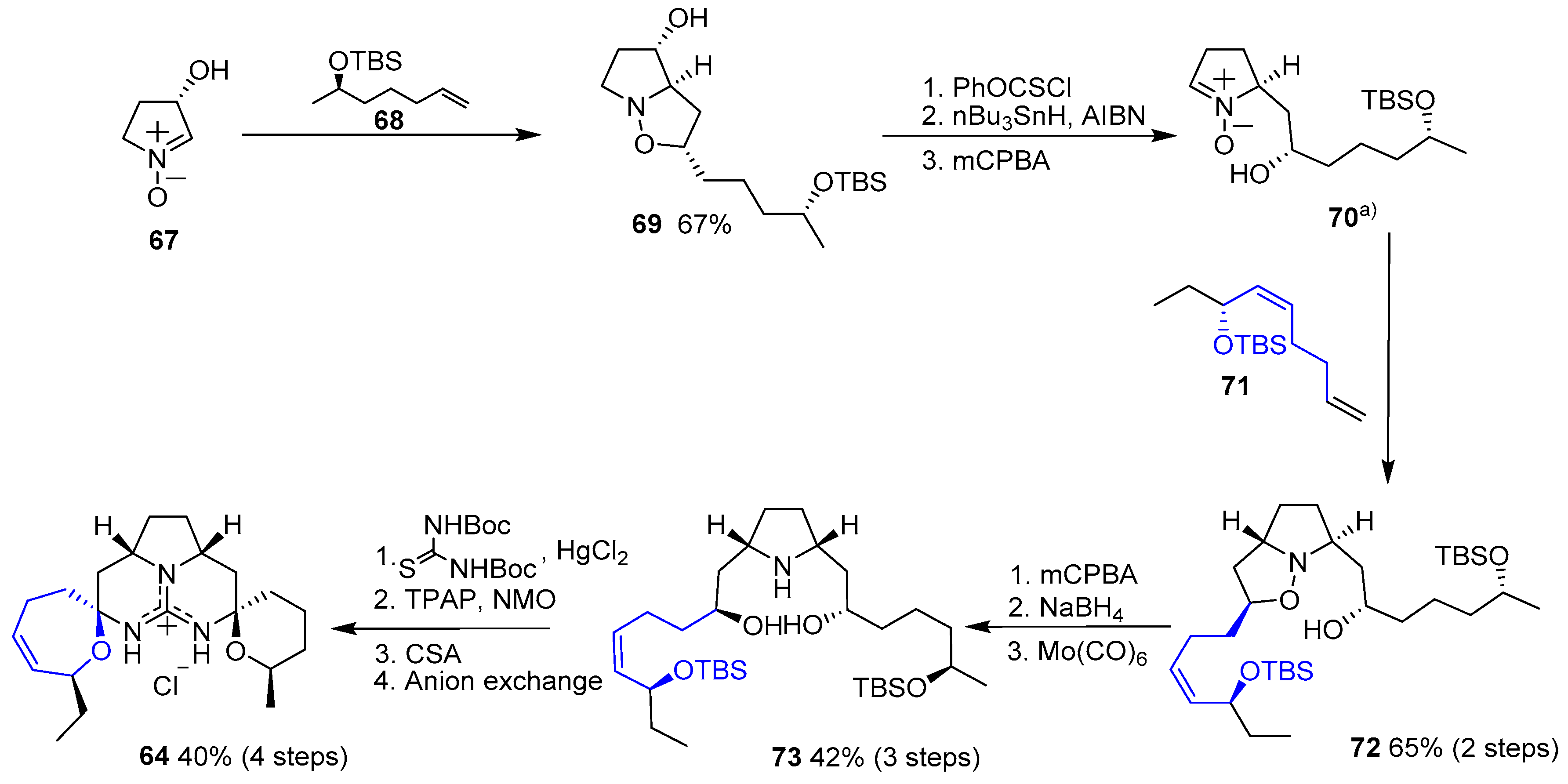
In 2002, Nagasawa and coworkers published the first total synthesis of this natural product [146]. They conceived a double 1,3-dipolar cycloaddition between a commercially available nitrone and two distinct terminal olefins which would have been previously prepared. Resulting pyrrolidine 66 would easily yield guanidine 65 and, finally, a double intramolecular condensation would furnish the desired compound (64), as shown in Scheme 8.
1,3-Dipolar cycloaddition of nitrone 67 with alkene 68 yielded isoxazolidine 69. Then, removal of the hydroxyl group, followed by treatment with mCPBA provided free nitrone 70. A second cycloaddition with olefin 71 furnished compound 72, which was readily transformed in pyrrolidine 73 by oxidation with mCPBA and subsequent reduction. Reaction with bis-N-Boc thiourea, followed by oxidation with TPAP/NMO system, deprotection with mild camphorsulfonic acid (CSA) and final N,O-acetalization gave 64 (Scheme 9).
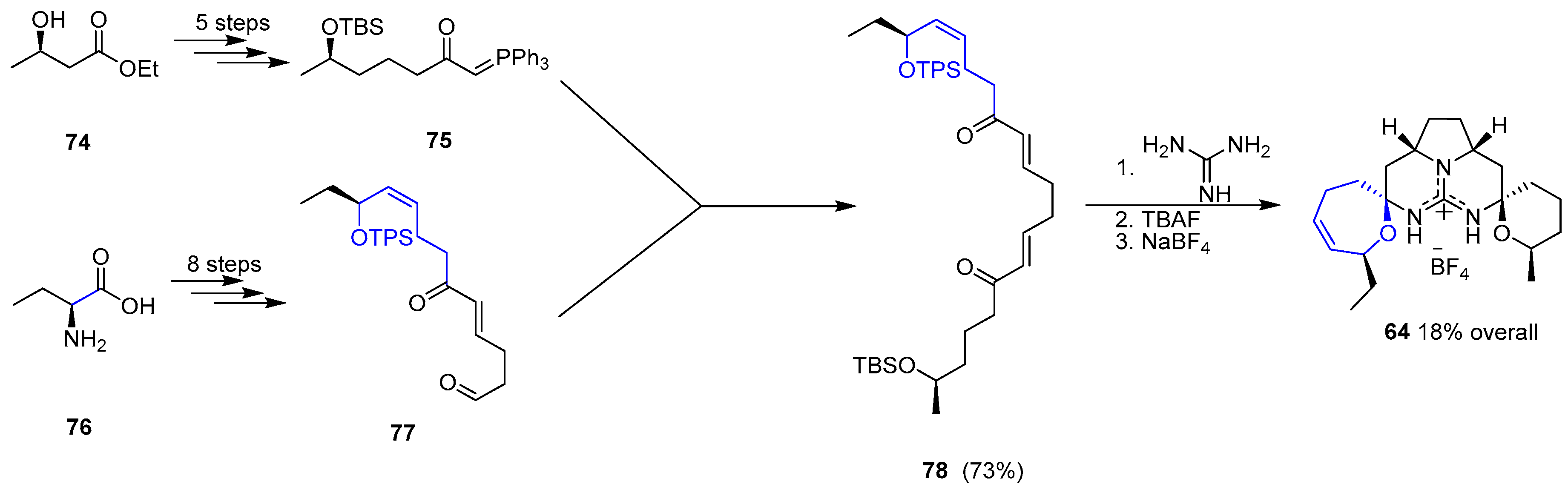
One year later Murphy and coworkers reported an alternative synthetic approach to 64 [147]. The strategy relied on the preparation of a suitable bis-enone 78 ready to couple with a guanidine moiety to provide the pentacyclic unit of 64 in a one-step process (Scheme 10). Hence, a convergent synthesis of two different blocks (75 and 77) gave access to desired bis-enone 78 in good overall yield (14 steps). Condensation with guanidine followed by deprotection of two silyl groups (TBS and TPS) furnished Crambescidin 359 (64) bearing BF4 anion upon treatment with NaBF4.
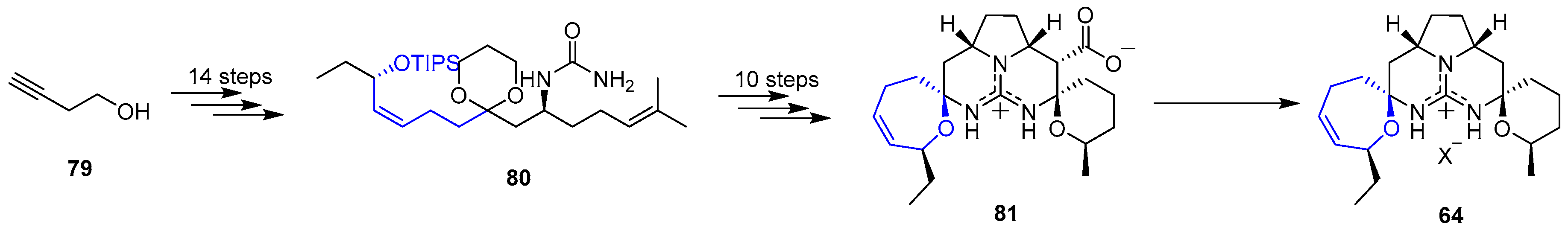
On the course of synthetic studies towards guanidinium alkaloids, Overman’s group published a different stepwise method [148] that started with commercially available 3-butynol (79) whose chain was elongated with the incorporation of a guanidine group to get 80. Then, a set of 10 steps allowed the synthesis of guanidinium carboxylate 81. This compound was an extraordinary precursor for the preparation of other crambescidinds (and more members in guanidinium alkaloids family) and, due to that reason, it was extensively studied. Interestingly, it was found that, upon standing 81 in several buffers, decarboxylation mildly occurred yielding Crambescidin 359 (64). This approach is summarized in Scheme 11.
6.3. Oxepinamides Family
This large group has been isolated from a set of marine fungus species associated to very distinct organisms. They possess interesting beneficial properties; however, they are especially important due to their anti-cancer activity [149]. They all bear an oxepin fused to a pyrimidinone moiety, as the main structure, and a six- or seven-membered lactam. In the first case, the compounds are also known as diketopiperazines, which correspond to another vast family of species (marine or not), for which oxepane-derived molecules will be addressed later on.
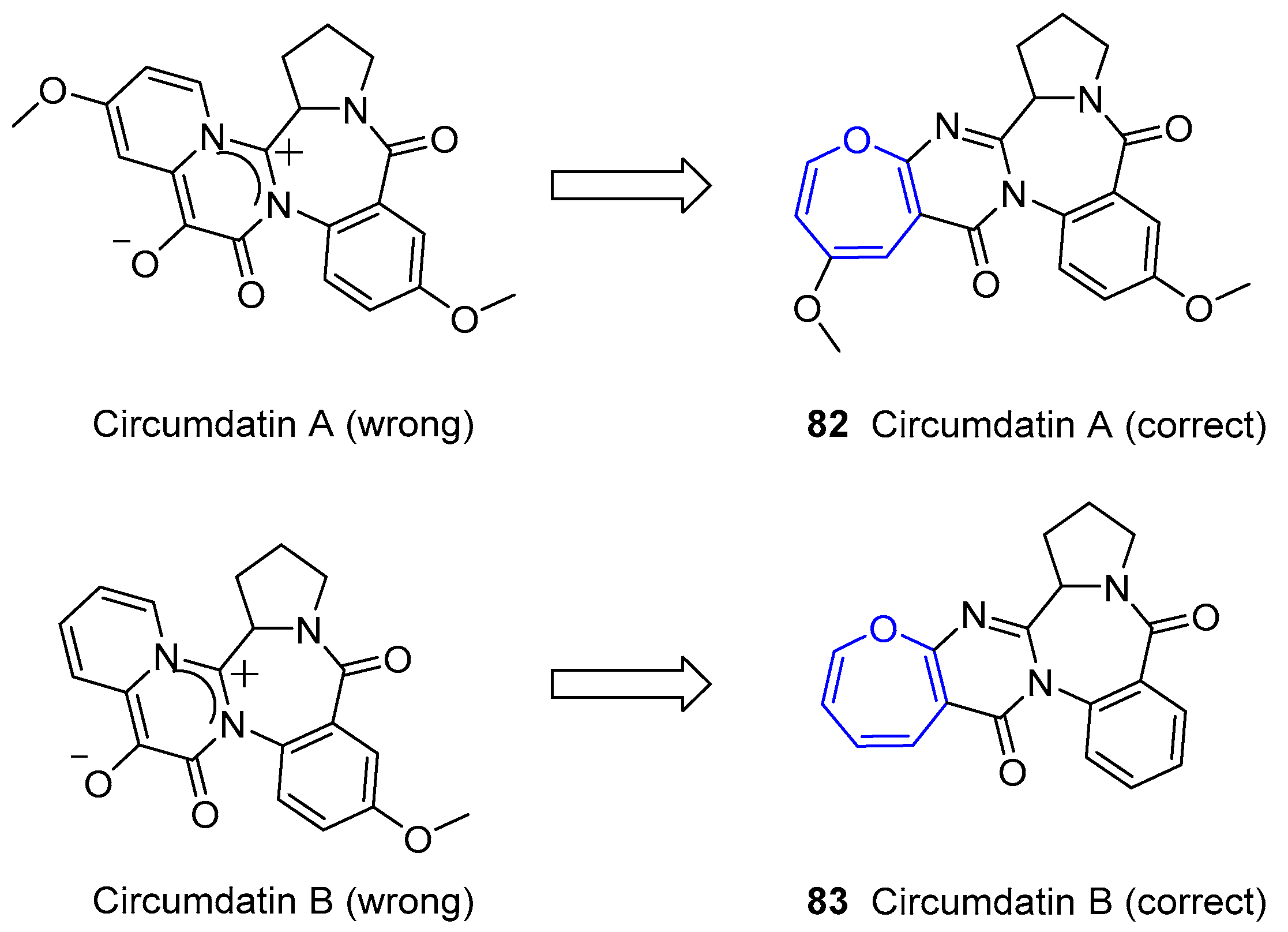
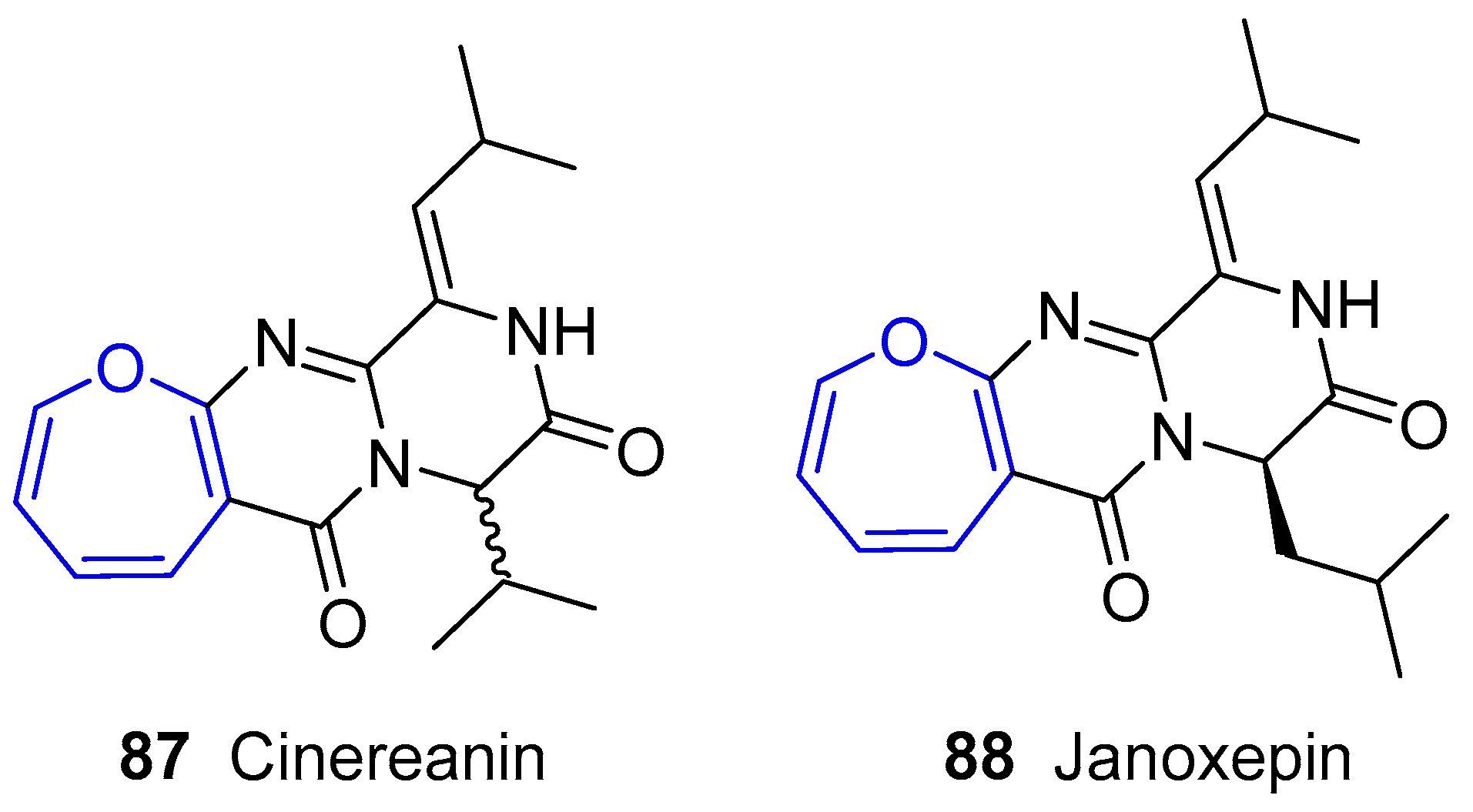
The first example of this family, called Cinereanin, was isolated by Springer, Arison, Roberts and collaborators from Botrytis cinereal sunflower seeds in 1988 and showed to have plant growth regulating properties [150]. Its structure remained unknown for years until Christophersen’s group reported the isolation of three benzodiazepine alkaloids (called Circumdatins A, B and C) from a culture of the fungus Aspergillus ochraceus [151]. Although it is known that this is a common soil fungus, it has been demonstrated its adaptation to other niches, such as marine ecosystem. Interestingly, the initial zwitterionic proposed structures of two of them (A and B) were wrongly assigned according to recorded NMR data. In fact, no oxepane-like group was present. This misled the scientific community for nine years until Kusumi and coworkers could isolate a set of alkaloids from fungus Aspergillus ostianus and grow single-crystals of Circumdatins A and B suitable for X-ray analysis [152], which yielded the final corrected structure as shown in Figure 15. These compounds were also detected and confirmed by Alfonso, Botana and collaborators from original fungus Aspergillus ochraceus [153].
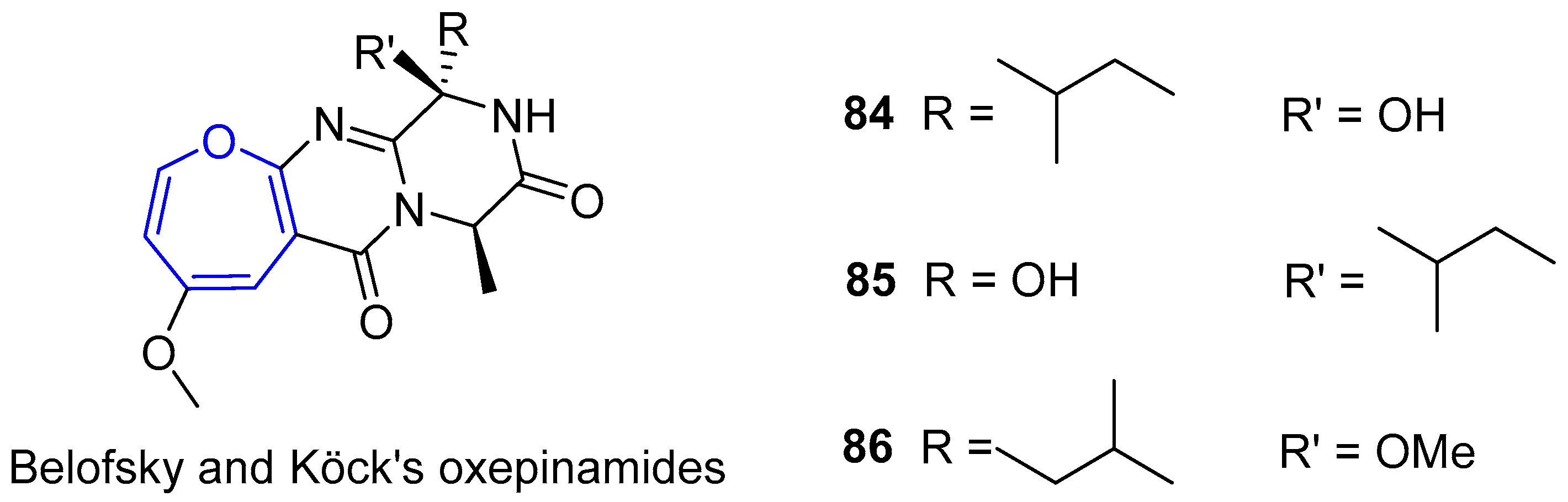
In parallel to Circumdatins discoveries, Belofsky, Köck’s group was able to isolate several bioactive metabolites from a fungus of the genus Acremonium, which were collected from the surface of Caribbean tunicate Ecteinascidia turbinata [154]. Three of them, generally called Oxepinamides A–C, contained oxepin derivatives with a structure very similar to the first one described by Springer, Arison, Roberts and collaborators, as depicted in Figure 16. It is worth noting that Oxepinamide A was capable of inhibiting ear edema induced in mice by using resiniferatoxin.
Later on, Sprogøe’s group reported two new compounds extracted from fungus Aspergillus janus [155]. One of them was called Janoxepin and had a very close structure to Cinereanin (Figure 17). It turned out to be active against the malaria parasite Plasmodium falciparum 3D7.
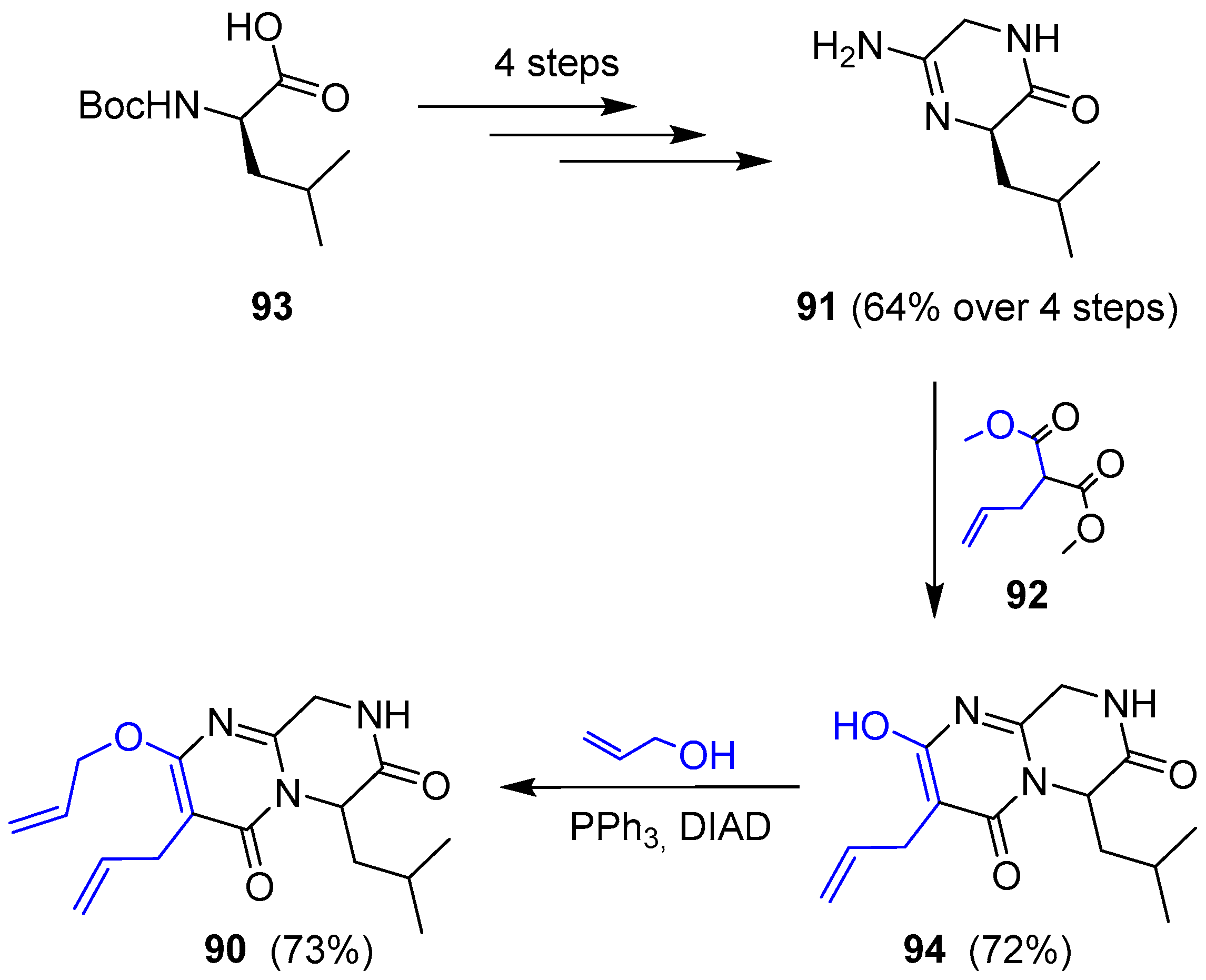
Janoxepin is the only compound of this family of oxepinamides from fungus species that has been synthesized. In 2012 Taylor’s group reported its total synthesis [156], according to the following retrosynthetic analysis: the key step is a ring closing metathesis (RCM) of a diallylated pyrimidinone 90, which should be easily prepared from guanidine 91 and dimethyl allylmalonate (Scheme 12).
Thus, amidine 91 was obtained in good yield in four consecutive steps (coupling with aminoacetonitrile, Boc deprotection, oxime formation-hydrogenation and final cyclization) starting from commercially available N-Boc-d-leucine. Further condensation with malonate 92 gave pyrimidone 94 along with unwanted total racemization (only 5% ee was detected). This molecule was turned into compound 90 by Mitsunobu reaction with allylic alcohol (Scheme 13).
Olefin metathesis was readily performed using second-generation Grubb’s catalyst, prior protection of 90 as imidate 95. Resulting compound 95 underwent the incorporation of the side chain through an aldol reaction with iso-butyraldehyde, followed by a chlorination and dehydrochlorination sequence furnishing dihydro-oxepin 96 (Scheme 14).
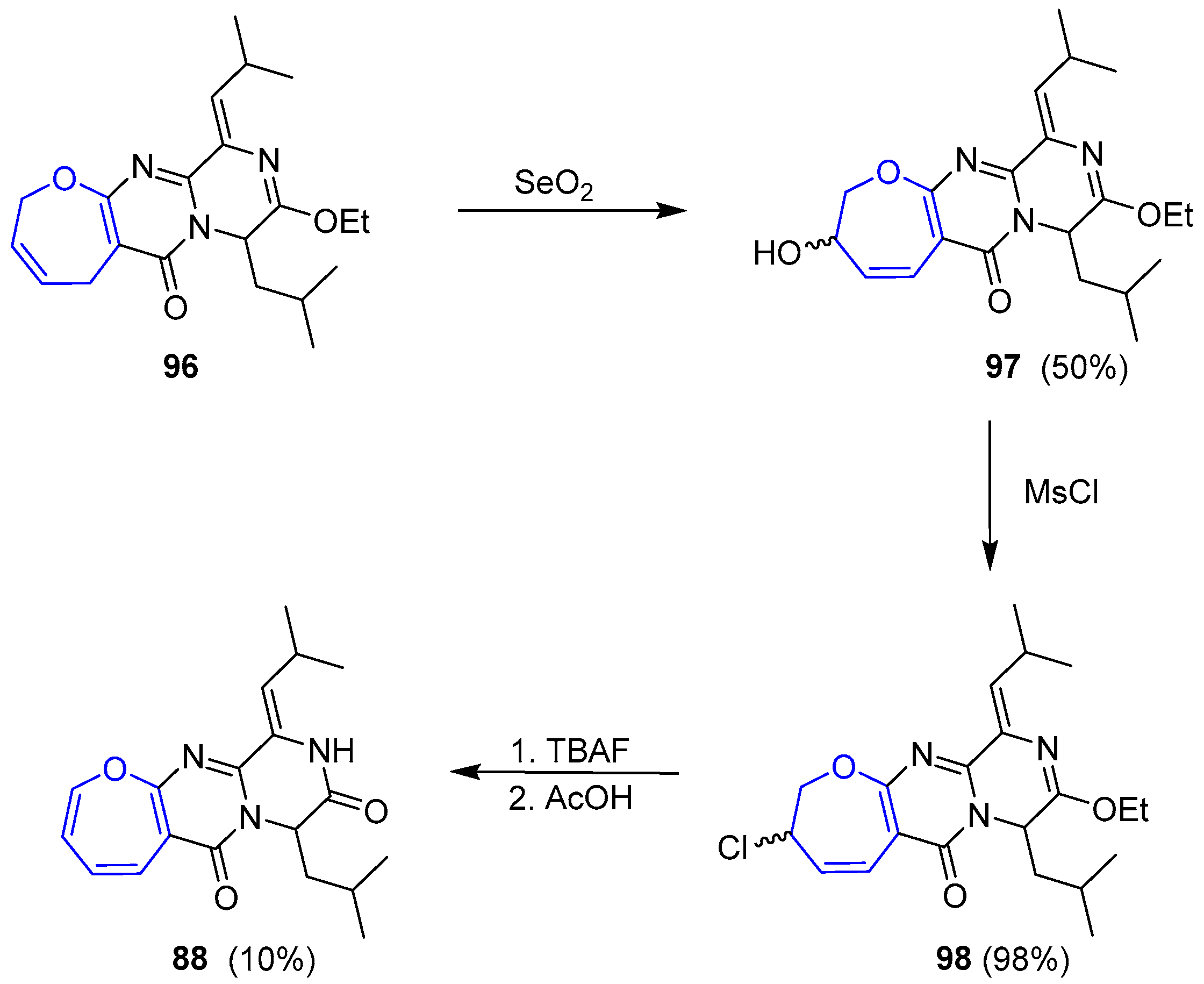
Formation of desired oxepin turned out to be extremely troublesome and, after many attempts, the only feasible procedure consisted of an allylic oxidation with SeO2 to give an alcohol 97 that was readily substituted by a chloride 98. Dehydrohalogenation was achieved but yields could never be optimized and only 10% of 98 was obtained. Nevertheless, (±)-Janoxepin (88) could be finally prepared after a simple deprotection step (Scheme 15).
Ahn, Oh and collaborators described, in 2011, four compounds from an ethyl acetate fraction of the marine-derived fungus Aspergillus sp. SF-5044 collected from Dadaepo Beach, Busan, Korea [157]. Two of them were oxepinamides, called Protuboxepins A and B, whose structure is shown in Figure 18. The reader should rapidly realize that these piperazines are structurally similar to Cinereanin and Janoxepin. Interestingly, both compounds contain the rare d-phenylalanine amino acid fragment. However, their anti-cancer activities towards several carcinoma cell lines were moderate. Nonetheless, Kihm and Ahn’s group reported the mechanism of tumor cell growth inhibition by Protuboxepin A [158]. They demonstrated that, due to an α,β-tubulin binding, microtubule dynamics were modified in such an extent that chromosomes were misaligned in metaphase, finally producing cell apoptosis.
In the last few years, up to six new rare oxepinamides have emerged from different fungus species [159,160,161,162]. They resemble structures already described here and are depicted in Figure 19. Apart from their structural interest, they have showed new bioactive abilities, such as antibacterial and antifungal properties.
6.4. Diketopiperazine Sulfides
As commented above, diketopiperazine structures bearing oxepane-like moieties are present in other families. One interesting family corresponds to disulfide (trisulfide or tetrasulfide) derivatives. Due to the presence of such functional group, all these compounds exhibit outstanding biomedical applications (anticancer, antifungal, antiviral, etc.) [163,164,165,166,167,168,169,170,171].
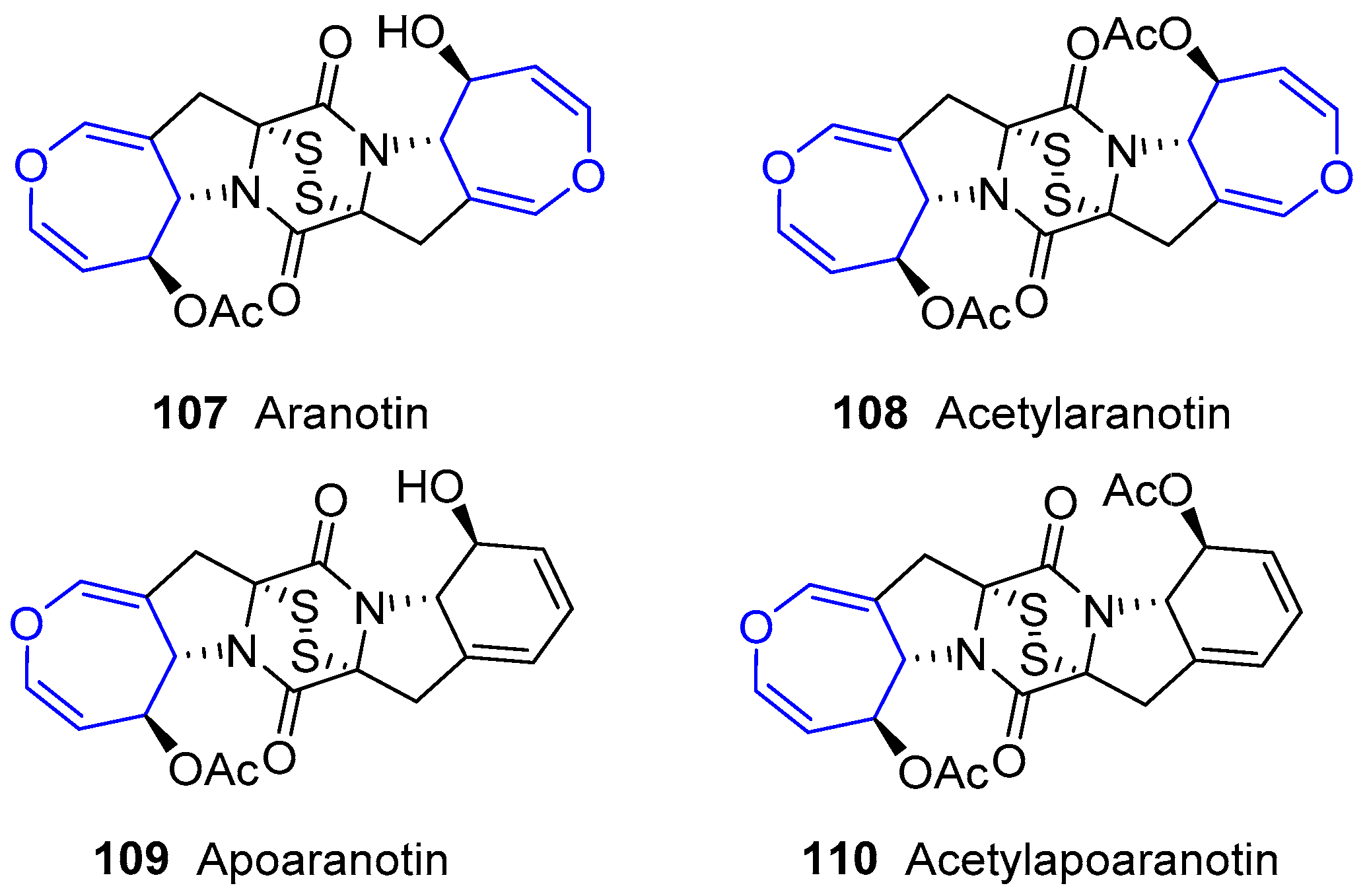
The first members in this family were obtained from fungus Arachniotus aureus in 1968, and were named Aranotin and Apoaranotin. Along with their acetylated derivatives, they were considered metabolites from such species and very related to formerly known sulfur-containing diketopiperazines, e.g., Gliotoxin and Sporidesmin (Figure 20) [172,173,174,175,176]. Structural characteristics are a central diketopiperazine scaffold, a disulfur bridge and a functionalized oxepin-fused pyrrolidine. Their biosynthesis pathway has been recently unraveled by Wang and collaborators using genome-based deletion analysis [177]. These compounds are present in other classes of fungus, such as Aspergillus sp. KMD 901, and showed excellent apoptosis-inducing effects towards colon cancer cell lines [178].
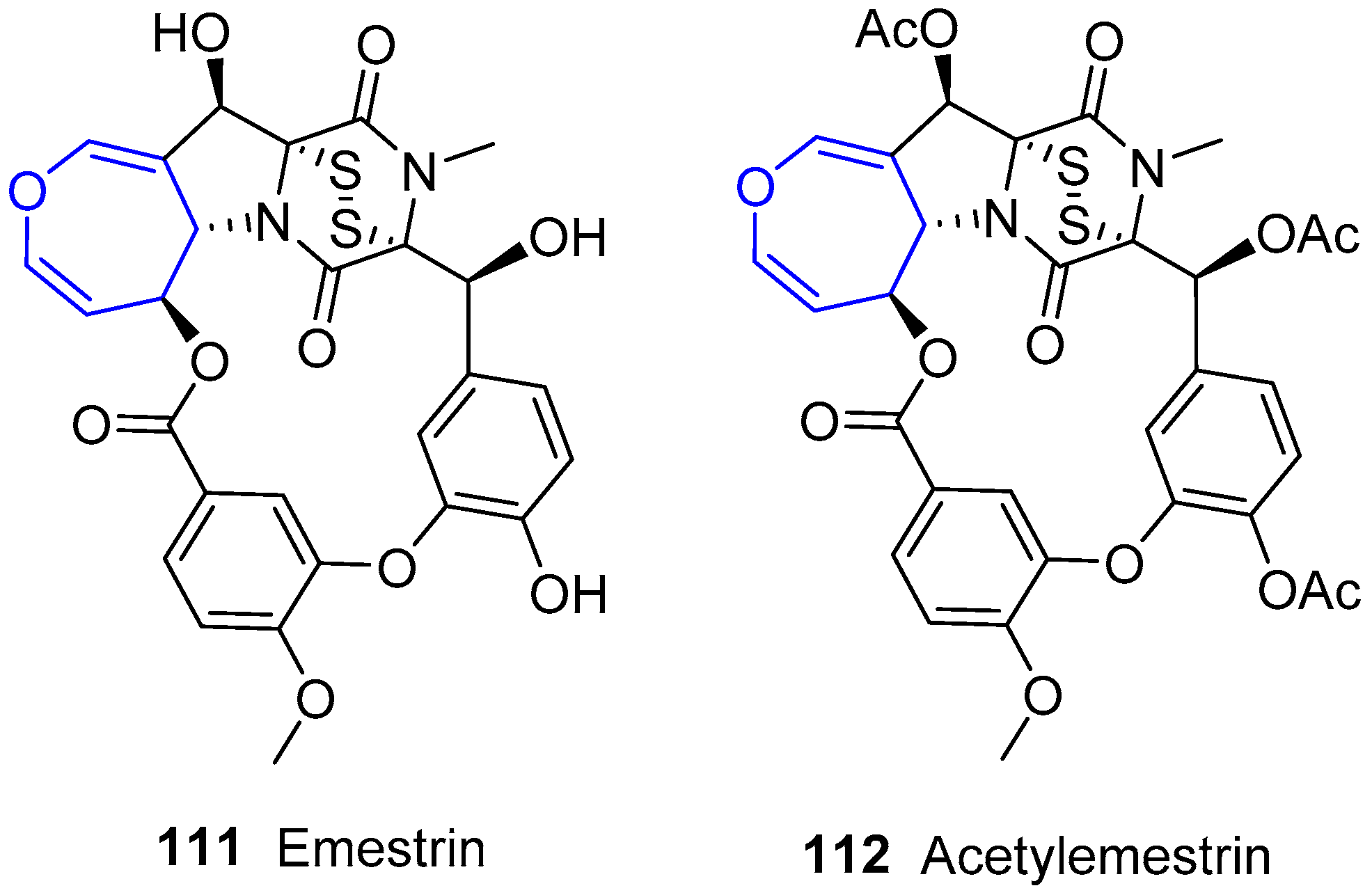
Another molecule of this family was obtained by Kawai’s group from Emericella striata, a thermotolerant fungus collected in Nepal [179,180,181]. It was called Emestrin and was reported as its acetylated form (Figure 21). Although not strictly marine, this compound belongs to the family due to its similarity to the previous structures and it is worth addressing. The previously described main scaffold is maintained but, in this case, diketopiperazine and oxepin moieties are additionally connected through a diphenyl ether chain giving rise to a macrocyclic lactone. Years later, the same group found structurally related molecules from Emericella foveolata [182].
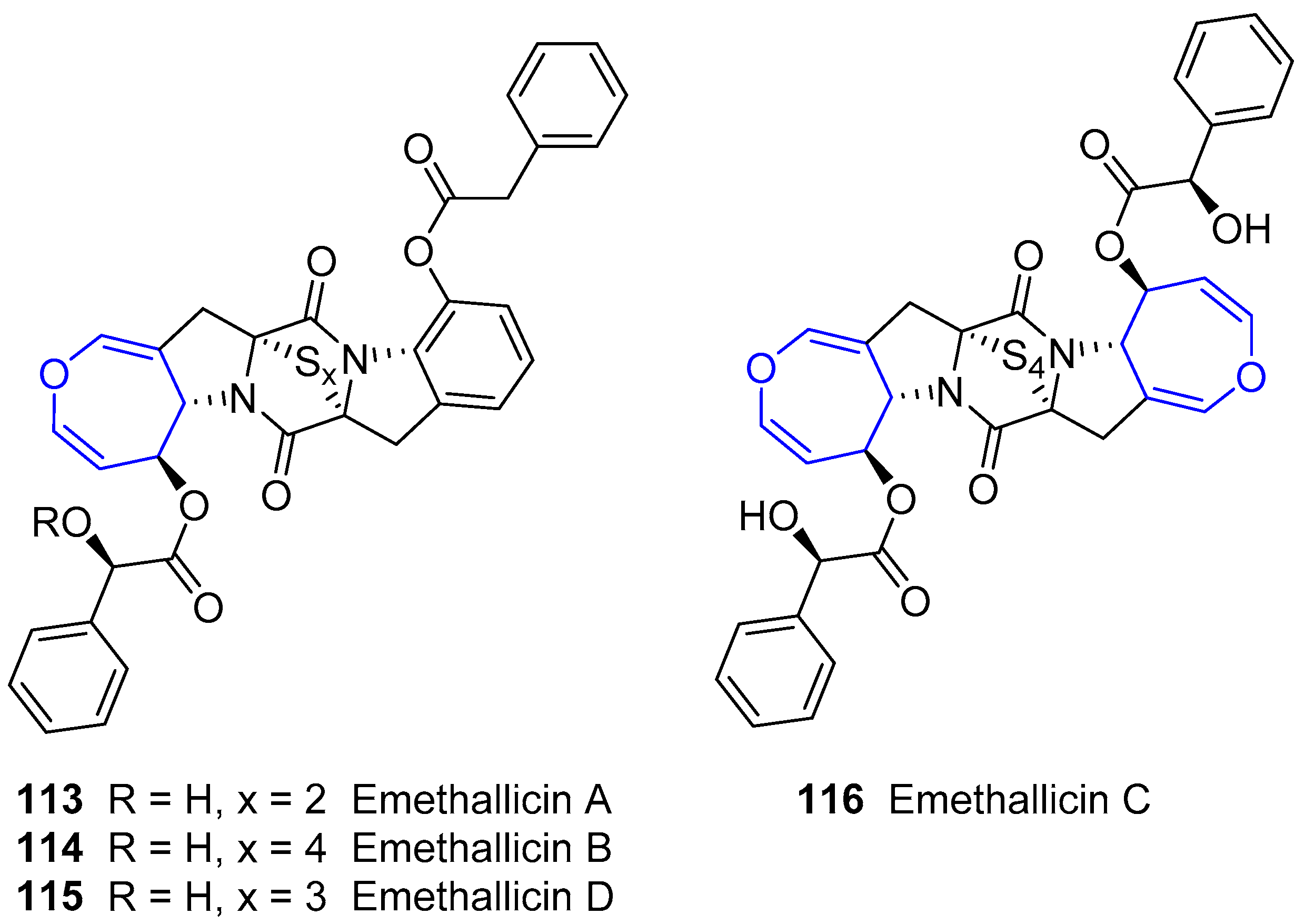
The same researchers reported the isolation of a small group of diketopiperazines arising from Emericella heterothallica extracts which were called Emethallicins [183,184]. Their structures resemble those from Aranotin, having a different acetylating group and a variable number of sulfur atoms (Figure 22). This feature was also observed for already described Emestrin derivatives. They have shown antiallergic properties owing to the observation of histamine release inhibition from mast cells and towards 5-lipoxygenase.
Interestingly, the synthesis of the intriguing structures of this family has attracted the attention of several authors. Thus, many synthetic approaches towards the partial synthesis of these compounds or analogues have been carried out and published [185,186,187,188,189,190,191,192]. In 2012, Reisman and coworkers reported the first total synthesis of an oxepin-containing diketopiperazine disulfide, (−)-Acetylaranotin, over 40 years after its discovery [193]. Due to the C2-symmetric nature of this compound, the strategy to synthesize this compound was based on the obtention of an oxepin fused to proline 118 which, after dimerization product 117 and a final step of sulfenylation, could provide (−)-Acetylaranotin. Proline oxepin 118 was foreseen to be prepared by a metal vinylidene-mediated 7-endo cycloisomerization from an alkynal precursor (119) that could be obtained from proline 120. This molecule can be synthesized through an asymmetric 1,3-dipolar cycloaddition of an acrylate and the azomethine ylide resulting from the coupling of a glycinate and cinnamaldehyde (Scheme 16).
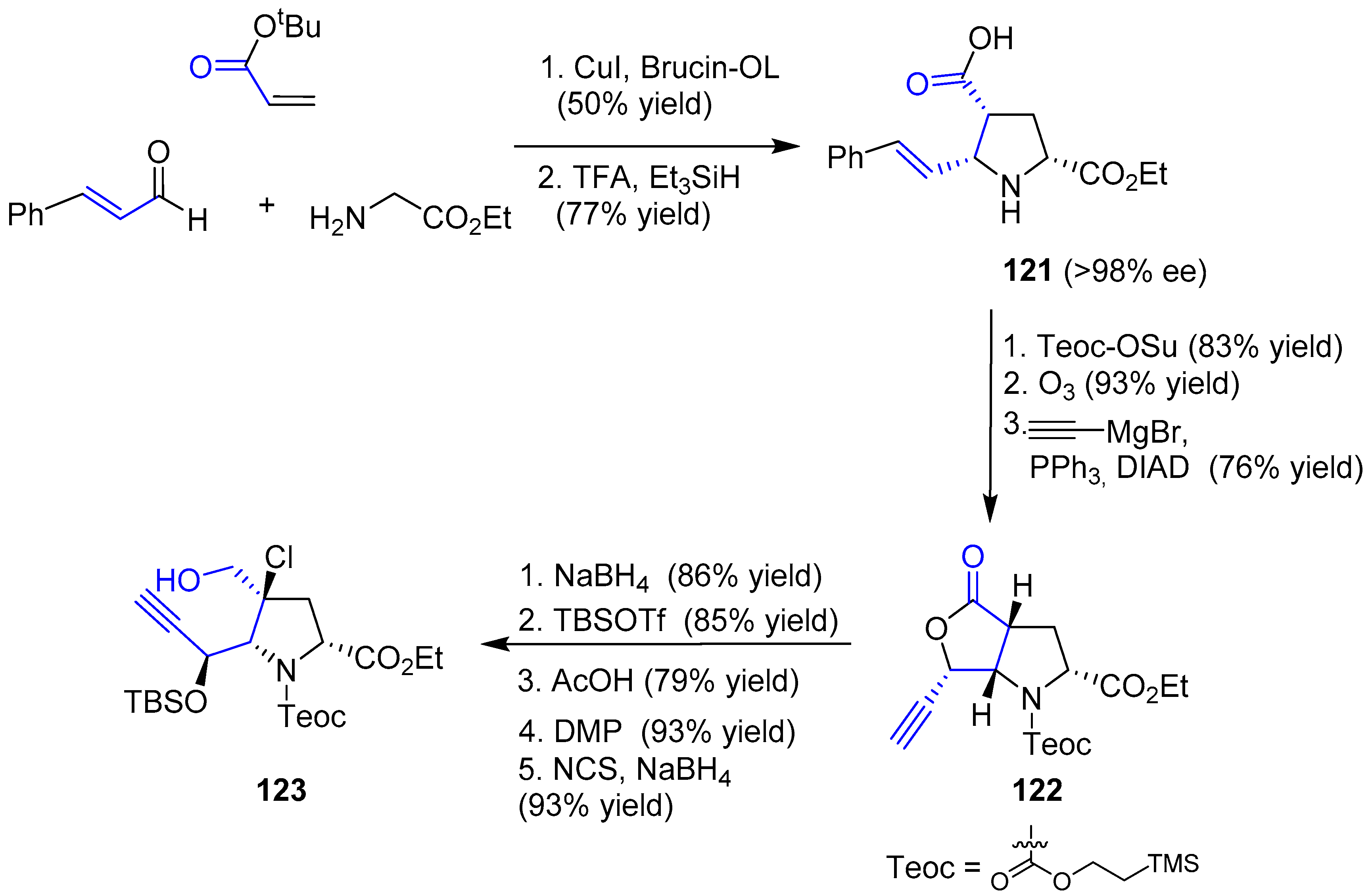
The enantioselective synthesis of pyrrolidine 121 was achieved by an asymmetric 1,3-dipolar cycloaddition mediated by catalytic copper iodide and Brucin-OL as the chiral ligand, followed by hydrosilane-reduction and deprotection of tBu group. Then, four consecutive steps, such as Teoc-protection, ozonolysis, addition of ethynyl Grignard reagent and final Mitsunobu reaction, gave rise to chiral alkynyl lactone 122. Finally, reduction with sodium borohydride, protection of the resulting alcohol and a sequence of Dess-Martin oxidation, chlorination of the α carbon and aldehyde reduction furnished the desired alkynol 123. The whole first stage is summarized in Scheme 17.
The second stage consisted of the construction of oxepin structure, dimerization and final sulfenylation. The use of catalytic [RhCl(COD)]2 in the presence of tris(p-fluorophenyl)phosphine yielded the expected oxepin 124, which was subjected to two different procedures. In both, a step of dehydrohalogenation and deprotection of orthogonal functional groups (carbamate into amine and ester into acid) resulted in two complementary peptides that reacted in the presence of BOP-Cl, a common reagent to activate carboxyl groups for peptide coupling, to get 125. Treatment with TBAF allowed the second amide formation (126). Surprisingly, epimerization of both stereogenic carbons in diketopiperazine skeleton occurred, but the mechanism was unclear. Nevertheless, episulfide formation was the last difficulty to be overcome. Treatment with NaHDMS and S8 furnished a tetrasulfide (127) with the appropriate stereochemistry which, after diacetylation of two hydroxyl groups, reduction with propanedithiol and further mild oxidation of the resulting dithiol gave expected (−)-Acetylaranotin (Scheme 18). The whole process required 18 steps and the key step relied on a Rh-catalyzed 7-endo cyclization to obtain the monomeric oxepin 124.
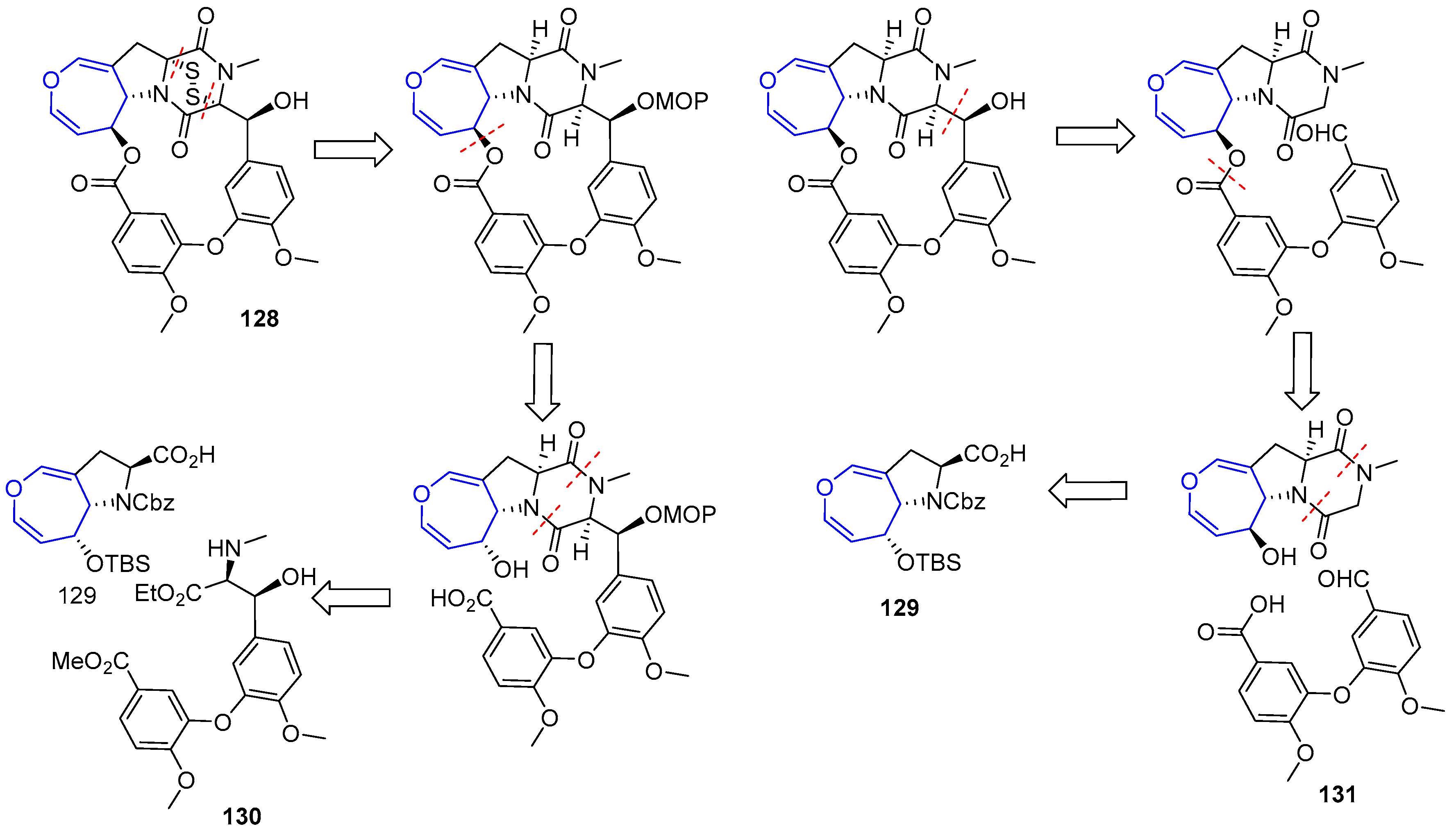
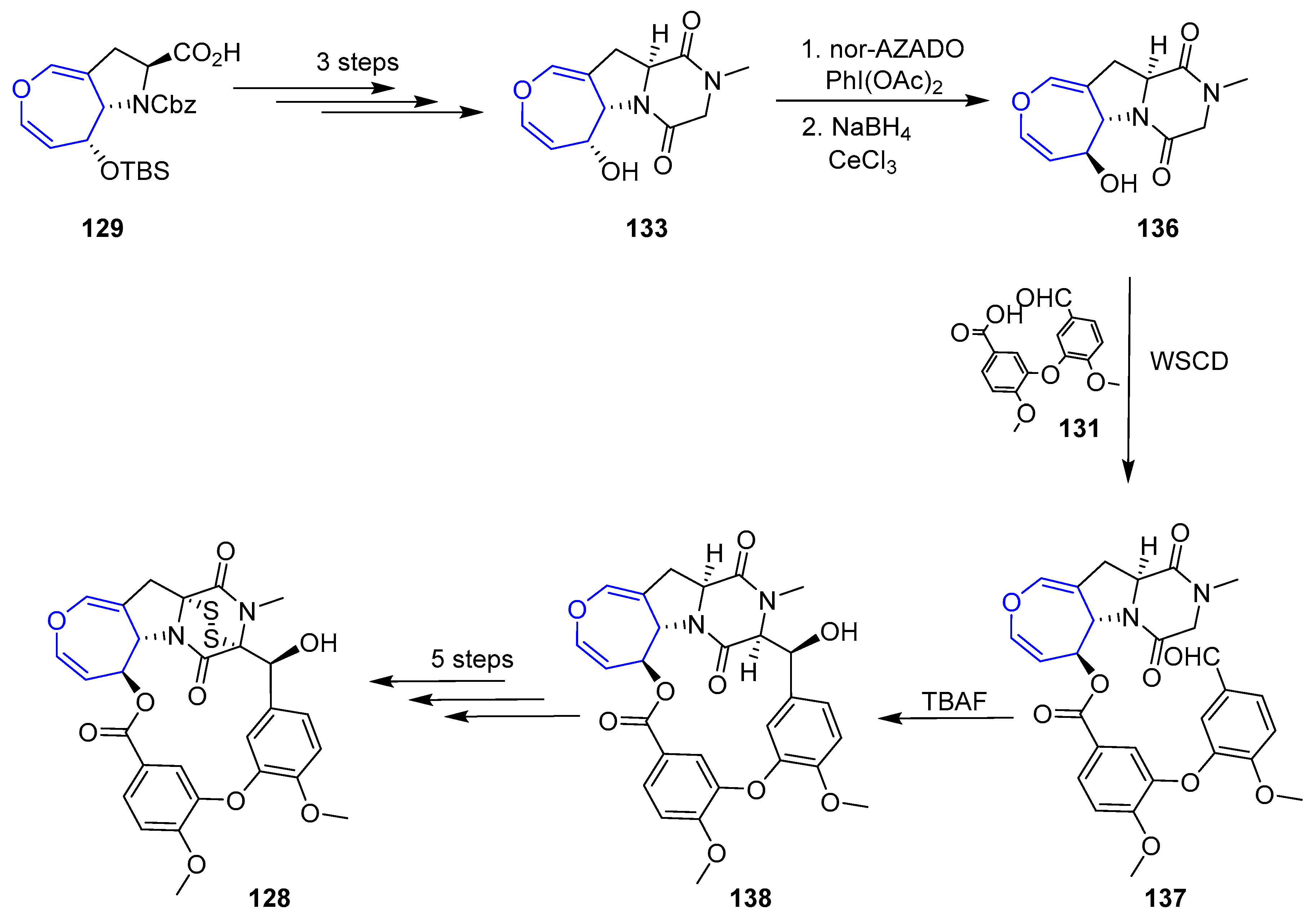
In recent years, other diketopiperazin sulfides total syntheses have been disclosed based on the contribution made by Reisman [193]. Compound (+)-MPC1001B (128), a molecule closely related to Emestrin, was prepared in 2004 [194,195]. The key step of the process involved the elusive 15-membered macrolactone formation and two plausible retrosynthetic analyses were devised. In both, sulfenylation would take place in the final step and oxepin proline moiety (129) was considered as the key intermediate from which macrocycle would be built. The main difference relied on the way the macroring would be closed. In the first one, a Mitsunobu reaction through an alcohol directly linked to oxepin fragment is proposed, whereas in the second one an aldol reaction in diketopiperazin moiety would be carried out (Scheme 19).
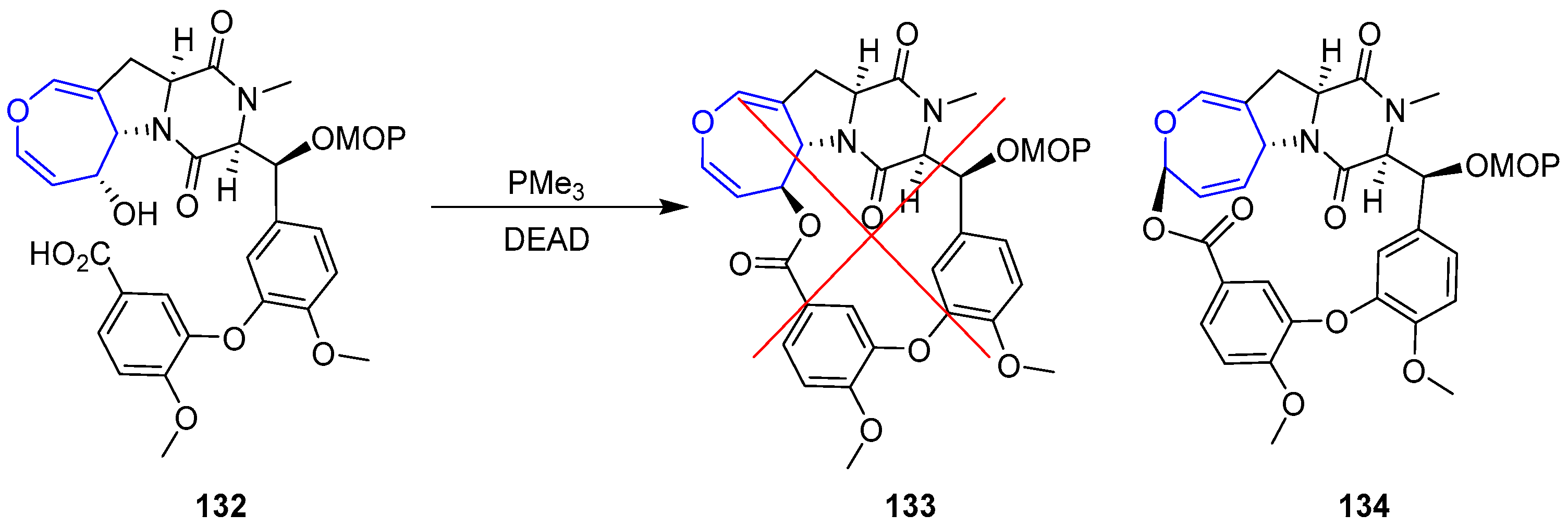
Unfortunately, macrocyclization of 132 under Mitsunobu conditions failed to provide the desired lactone 133, instead regioisomer 134 was isolated in high yields (Scheme 20).
From easily accessible proline 129, a key precursor of the second strategy, was readily obtained diketopiperazine 133. Inversion of the configuration at C-6 (via a mild oxidation and Luche reduction), followed by condensation with anisic acid derivative 131 using WSCD provided ester 137. To the delight of the authors, now the crucial formation of the macrolactone (138) formation could be easily achieved in the presence of TBAF. From that point, sulfenylation was carried out using a modification of Reisman’s protocol (with a novel trityl trisulfide reagent) although the secondary alcohol present in the structure had to be protected in the process, increasing the expected number of steps. Nevertheless, elusive diketopiperazin disulfide (+)-MPC1001B could be obtained for the first time (Scheme 21).
Simultaneously, Reisman’s group also reported the total synthesis of (−)-Acetylapoaranotin using a very close methodology [196]. Owing to the nonsymmetric nature of this compound, the procedure had to be a step-wise preparation of two proline key intermediates which were readily coupled and finally sulfenylated.
In summary, marine drugs containing 7-membered oxacycles are abundant in nature. They show a rich variety of structures whose intriguing features have attracted the attention of many synthetic chemists. Moreover, their relevant biological properties include antimalarial, antifungal, antibacterial activities and, most remarkably, high cytotoxicity against a wide number of cancer cells. In this review, we have tried to show an overview of these types of compounds, focusing on their isolation, structure determination, biological properties and synthetic approaches.
Acknowledgments
We gratefully acknowledge financial support from the Spanish Ministry of Economy (CTQ2016-75253-P). C.D.-P. acknowledges a predoctoral Grant (Q4718001C), funded by the European Social Fund and the “Junta de Castilla y León”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blunt, J.W.; Copp, B.R.; Hu, W.-P.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2008, 25, 35–94. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K. Total Synthesis of Medium-Ring Ethers from Laurencia Red Algae. In Marine Natural Products; Kiyota, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 97–148. [Google Scholar]
- Nicolaou, K.C.; Frederick, M.O.; Aversa, R.J. The Continuing Saga of the Marine Polyether Biotoxins. Angew. Chem. Int. Ed. 2008, 47, 7182–7225. [Google Scholar] [CrossRef] [PubMed]
- Vilotijevic, I.; Jamison, F.T. Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades. Mar. Drugs 2010, 8, 763–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.T. Biogenetic Relationships of Bioactive Sponge Merotriterpenoids. Mar. Drugs 2017, 15, 285. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.J.; Souto, M.L.; Norte, M. Marine polyether triterpenes. Nat. Prod. Rep. 2000, 17, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 1994, 11, 355–394. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Herald, C.L.; Allen, M.S.; Von Dreele, R.B.; Vanell, L.D.; Kao, J.P.Y.; Blake, W. Antineoplastic agents. 48. The isolation and structure of aplysistatin. J. Am. Chem. Soc. 1977, 99, 262–263. [Google Scholar] [CrossRef] [PubMed]
- Palaniveloo, K.; Vairappan, C.S. Chemical relationship between red algae genus Laurencia and sea hare (Aplysia dactylomela Rang) in the North Borneo Island. J. Appl. Phycol. 2014, 26, 1199–1205. [Google Scholar] [CrossRef]
- De Nys, R.; Steinberg, P.D.; Rogers, C.N.; Charlton, T.S.; Duncan, M.W. Quantitative variation of secondary metabolites in the sea hare Aplysia parvula and its host plant, Delisea pulchra. Mar. Ecol. Prog. Ser. 1996, 130, 135–146. [Google Scholar] [CrossRef]
- Rogers, C.N.; De Nys, R.; Charlton, T.S.; Steinberg, P.D. Dynamics of Algal Secondary Metabolites in Two Species of Sea Hare. J. Chem. Ecol. 2000, 26, 721–744. [Google Scholar] [CrossRef]
- Pennings, S.C.; Paul, V.J. Sequestration of dietary secondary metabolites by three species of sea hares: Location, specificity and dynamics. Mar. Biol. 1993, 117, 535–546. [Google Scholar] [CrossRef]
- Paul, V.J.; Fenical, W. Palisadins A, B and related monocyclofarnesol-derived sesquiterpenoids from the red marine alga Laurencia cf. palisada. Tetrahedron Lett. 1980, 21, 2787–2790. [Google Scholar] [CrossRef]
- Capon, R.; Ghisalberti, E.L.; Jefferies, P.R.; Skelton, B.W.; White, A.H. Sesquiterpene metabolites from laurencia filiformis. Tetrahedron 1981, 37, 1613–1621. [Google Scholar] [CrossRef]
- Wright, A.D.; König, G.M.; Sticher, O. New Sesquiterpenes and C15 Acetogenins from the Marine Red Alga Laurencia implicata. J. Nat. Prod. 1991, 54, 1025–1033. [Google Scholar] [CrossRef]
- De Nys, R.; Wright, A.D.; König, G.M.; Sticher, O.; Alino, P.M. Five New Sesquiterpenes from the Red Alga Laurencia flexilis. J. Nat. Prod. 1993, 56, 877–883. [Google Scholar] [CrossRef]
- Su, J.-Y.; Zhong, Y.-L.; Zeng, L.-M.; Wu, H.-M.; Ma, K. Terpenoids from Laurencia karlae. Phytochemistry 1995, 40, 195–197. [Google Scholar] [CrossRef]
- Kuniyoshi, M.; Marma, M.S.; Higa, T.; Bernardinelli, G.; Jefford, C.W. New Bromoterpenes from the Red Alga Laurencia luzonensis. J. Nat. Prod. 2001, 64, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yuan, Z.-H.; Li, J.; Guo, S.-J.; Deng, L.-P.; Han, L.-J.; Zhu, X.-B.; Shi, D.-Y. Sesquiterpenes from the Marine Red Alga Laurencia saitoi. Helv. Chim. Acta 2009, 92, 1291–1297. [Google Scholar] [CrossRef]
- Su, H.; Shi, D.-Y.; Li, J.; Guo, S.-J.; Li, L.-L.; Yuan, Z.-H.; Zhu, X.-B. Sesquiterpenes from Laurencia similis. Molecules 2009, 14, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Lee Tan, K.; Matsunaga, S.; Vairappan, C.S. Halogenated chamigranes of red alga Laurencia snackeyi (Weber-van Bosse) Masuda from Sulu-Sulawesi Sea. Biochem. Syst. Ecol. 2011, 39, 213–215. [Google Scholar] [CrossRef]
- Von Dreele, R.B.; Kao, J.P.Y. The structure of the sesquiterpene aplysistatin. Acta Crystallogr. B 1980, 36, 2695–2698. [Google Scholar] [CrossRef]
- König, G.M.; Wright, A.D.; Sticher, O.; Angerhofer, C.K.; Pezzuto, J.M. Biological Activities of Selected Marine Natural Products. Planta Med. 1994, 60, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Vairappan, C.S.; Kamada, T.; Lee, W.-W.; Jeon, Y.-J. Anti-inflammatory activity of halogenated secondary metabolites of Laurencia snackeyi (Weber-van Bosse) Masuda in LPS-stimulated RAW 264.7 macrophages. J. Appl. Phycol. 2013, 25, 1805–1813. [Google Scholar] [CrossRef]
- Wijesinghe, W.A.; Kim, E.A.; Kang, M.C.; Lee, W.W.; Lee, H.S.; Vairappan, C.S.; Jeon, Y.J. Assessment of anti-inflammatory effect of 5beta-hydroxypalisadin B isolated from red seaweed Laurencia snackeyi in zebrafish embryo in vivo model. Environ. Toxicol. Pharmacol. 2014, 37, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Kurth, M.J. Total synthesis of dl-aplysistatin. J. Am. Chem. Soc. 1979, 101, 5065–5067. [Google Scholar] [CrossRef]
- Hoye, T.R.; Caruso, A.J.; Dellaria, J.F.; Kurth, M.J. Two syntheses of dl-aplysistatin. J. Am. Chem. Soc. 1982, 104, 6704–6709. [Google Scholar] [CrossRef]
- Shieh, H.-M.; Prestwich, G.D. Chiral, biomimetic total synthesis of (−)-aplysistatin. Tetrahedron Lett. 1982, 23, 4643–4646. [Google Scholar] [CrossRef]
- White, J.D.; Nishiguchi, T.; Skeean, R.W. Stereoselective, biogenetically patterned synthesis of (.+-.)-aplysistatin. J. Am. Chem. Soc. 1982, 104, 3923–3928. [Google Scholar] [CrossRef]
- Gosselin, P.; Rouessac, F. Polycyclisations cationiques de polyenes via leurs bromohydrines—II synthese de la (±) aplysistatine. Tetrahedron Lett. 1983, 24, 5515–5518. [Google Scholar] [CrossRef]
- Kraus, G.A.; Gottschalk, P. A direct synthesis of .beta.-hydroxybutyrolactones. Total synthesis of dendrolasin and formal total synthesis of aplysistatin. J. Org. Chem. 1983, 48, 5356–5357. [Google Scholar] [CrossRef]
- Tanaka, A.; Otsuka, S.; Yamashita, K. Biomimetic Total Synthesis of (−)-Aplysistatin. Agric. Biol. Chem. 1984, 48, 2535–2540. [Google Scholar] [CrossRef]
- Tanaka, A.; Suzuki, M.; Yamashita, K. Total Synthesis of (+)-Palisadin A and (+)-12-Hydroxypalisadin B. Agric. Biol. Chem. 1986, 50, 1069–1071. [Google Scholar] [CrossRef]
- Couladouros, E.A.; Vidali, V.P. Novel Stereocontrolled Approach to syn- and anti-Oxepene-Cyclogeranyl trans-Fused Polycyclic Systems: Asymmetric Total Synthesis of (−)-Aplysistatin, (+)-Palisadin A, (+)-Palisadin B, (+)-12-Hydroxy-Palisadin B, and the AB Ring System of Adociasulfate-2 and Toxicol A. Chem. Eur. J. 2004, 10, 3822–3835. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.; Wells, R. Isolation of Some Novel Diterpenes from a Soft Coral of the Genus Lobophytum. Aust. J. Chem. 1979, 32, 1345–1351. [Google Scholar] [CrossRef]
- Raju, B.L.; Subbaraju, G.V.; Rao, C.B.; Trimurtulu, G. Two New Oxygenated Lobanes from a Soft Coral of Lobophytum Species of the Andaman and Nicobar Coasts. J. Nat. Prod. 1993, 56, 961–966. [Google Scholar] [CrossRef]
- Shin, J.; Fenical, W. Fuscosides A-D: Anti-inflammatory diterpenoid glycosides of new structural classes from the caribbean gorgonian Eunicea fusca. J. Org. Chem. 1991, 56, 3153–3158. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Berrue, F.; Kerr, R.G. Eunicidiol, an Anti-inflammatory Dilophol Diterpene from Eunicea fusca. J. Nat. Prod. 2012, 75, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Hamada, T.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Structures and Absolute Configuration of New Lobane Diterpenoids from the Okinawan Soft Coral Sinularia flexibilis. Chem. Lett. 1992, 21, 33–36. [Google Scholar] [CrossRef]
- Kusumi, T.; Hamada, T.; Ishitsuka, M.O.; Ohtani, I.; Kakisawa, H. Elucidation of the relative and absolute stereochemistry of lobatriene, a marine diterpene, by a modified Mosher method. J. Org. Chem. 1992, 57, 1033–1035. [Google Scholar] [CrossRef]
- Chai, M.-C.; Wang, S.-K.; Dai, C.-F.; Duh, C.-Y. A Cytotoxic Lobane Diterpene from the Formosan Soft Coral Sinularia inelegans. J. Nat. Prod. 2000, 63, 843–844. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, I.; Jhaumeer-Laulloo, S.B.; Bontemps, N.; Banaigs, B.; Aknin, M. New Lobane and Cembrane Diterpenes from Two Comorian Soft Corals. Mar. Drugs 2010, 8, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Edrada, R.A.; Proksch, P.; Wray, V.; Witte, L.; van Ofwegen, L. Four New Bioactive Lobane Diterpenes of the Soft Coral Lobophytum pauciflorum from Mindoro, Philippines. J. Nat. Prod. 1998, 61, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, H.; Yamabe, O.; Kato, M. Synthetic study of marine lobane diterpenes: Efficient synthesis of (+)-fuscol. J. Chem. Soc. Perkin Trans. 1 1998, 217–222. [Google Scholar] [CrossRef]
- Kato, M.; Kosugi, H.; Ichiyanagi, T.; Yamabe, O. Synthetic study of marine lobane diterpenes. Enantioselective syntheses of lobatrienolide and lobatrientriol from (+)-nopinone. J. Chem. Soc. Perkin Trans. 1 1999, 783–788. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Kerr, R.G. Semisynthesis of fuscoside B analogues and eunicosides, and analysis of anti-inflammatory activity. Tetrahedron 2011, 67, 3053–3061. [Google Scholar] [CrossRef]
- Edrada, R.A.; Wray, V.; Handayani, D.; Schupp, P.; Balbin-Oliveros, M.; Proksch, P. Structure-activity relationships of bioactive metabolites from some Indo-Pacific marine invertebrates. Stud. Nat. Prod. Chem. 2000, 21, 251–292. [Google Scholar] [CrossRef]
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from Marine Invertebrates: Structures, Occurrence, and Ecological Implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, M.; Mohamed, T.; Alhammady, M.; Shaheen, A.; Reda, E.; Elshamy, A.; Aziz, M.; Paré, P. Molecular Architecture and Biomedical Leads of Terpenes from Red Sea Marine Invertebrates. Mar. Drugs 2015, 13, 3154–3181. [Google Scholar] [CrossRef] [PubMed]
- Shmueli, U.; Carmely, S.; Groweiss, A.; Kashman, Y. Sipholenol and sipholenone, two new triterpenes from the marine sponge siphonochalina siphonella (levi). Tetrahedron Lett. 1981, 22, 709–712. [Google Scholar] [CrossRef]
- Carmely, S.; Kashman, Y. The sipholanes, a novel group of triterpenes from the marine sponge Siphonochalina siphonella. J. Org. Chem. 1983, 48, 3517–3525. [Google Scholar] [CrossRef]
- Carmely, S.; Loya, Y.; Kashman, Y. Siphenellinol, a new triterpene from the marine sponge siphonochalinasiphonella. Tetrahedron Lett. 1983, 24, 3673–3676. [Google Scholar] [CrossRef]
- Kashman, Y.; Yosief, T.; Carmeli, S. New Triterpenoids from the Red Sea Sponge Siphonochalina siphonella. J. Nat. Prod. 2001, 64, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Abraham, I.; Carvalho, P.; Kuang, Y.-H.; Shaala, L.A.; Youssef, D.T.A.; Avery, M.A.; Chen, Z.-S.; El Sayed, K.A. Sipholane Triterpenoids: Chemistry, Reversal of ABCB1/P-Glycoprotein-Mediated Multidrug Resistance, and Pharmacophore Modeling. J. Nat. Prod. 2009, 72, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Al-Lihaibi, S.S.; Abdel-Lateff, A.; Alarif, W.M.; Nogata, Y.; Ayyad, S.-E.N.; Okino, T. Potent Antifouling Metabolites from Red Sea Organisms. Asian J. Chem. 2015, 27, 2252–2256. [Google Scholar] [CrossRef]
- Li, Y.-X.; Himaya, S.; Kim, S.-K. Triterpenoids of Marine Origin as Anti-Cancer Agents. Molecules 2013, 18, 7886–7909. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Laphookhieo, S.; Shi, Z.; Fu, L.-W.; Akiyama, S.-I.; Chen, Z.-S.; Youssef, D.T.A.; van Soest, R.W.M.; El Sayed, K.A. Reversal of P-Glycoprotein-Mediated Multidrug Resistance by Sipholane Triterpenoids. J. Nat. Prod. 2007, 70, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Jain, S.; Kim, I.-W.; Peng, X.-X.; Abraham, I.; Youssef, D.T.A.; Fu, L.-W.; El Sayed, K.; Ambudkar, S.V.; Chen, Z.-S. Sipholenol A, a marine-derived sipholane triterpene, potently reverses P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007, 98, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; Jain, S.; Wu, C.-P.; Khanfar, M.A.; Kuang, Y.; Dai, C.-L.; Shi, Z.; Chen, X.; Fu, L.; Ambudkar, S.V.; et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem. Pharmacol. 2010, 80, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Abraham, I.; El Sayed, K.; Chen, Z.-S.; Guo, H. Current Status on Marine Products with Reversal Effect on Cancer Multidrug Resistance. Mar. Drugs 2012, 10, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Lateff, A.; Al-Abd Ahmed, M.; Alahdal Abdulrahman, M.; Alarif Walied, M.; Ayyad Seif-Eldin, N.; Al-Lihaibi Sultan, S.; Hegazy Mohamed, E.; Al Mohammadi, A.; Abdelghany Tamer, M.; Abdel-Naim Ashraf, B.; et al. Antiproliferative effects of triterpenoidal derivatives, obtained from the marine sponge Siphonochalina sp. on human hepatic and colorectal cancer cells. Z. Naturforsch. C J. Biosci. 2016, 71, 29. [Google Scholar] [CrossRef] [PubMed]
- Carmely, S.; Kashman, Y. Neviotine-A, a new triterpene from the red sea sponge Siphonochalina siphonella. J. Org. Chem. 1986, 51, 784–788. [Google Scholar] [CrossRef]
- Angawi, R.; Saqer, E.; Abdel-Lateff, A.; Badria, F.; Ayyad, S.-E. Cytotoxic neviotane triterpene-type from the red sea sponge Siphonochalina siphonella. Pharmacogn. Mag. 2014, 10, 334–341. [Google Scholar] [CrossRef]
- Rudi, A.; Goldberg, I.; Stein, Z.; Benayahu, Y.; Schleyer, M.; Kashman, Y. Sodwanones A–C, three new triterpenoids from a marine sponge. Tetrahedron Lett. 1993, 34, 3943–3944. [Google Scholar] [CrossRef]
- Rudi, A.; Kashman, Y.; Benayahu, Y.; Schleyer, M. Sodwanones A–F, New Triterpenoids from the Marine Sponge Axinella weltneri. J. Nat. Prod. 1994, 57, 1416–1423. [Google Scholar] [CrossRef]
- Rudi, A.; Stein, Z.; Goldberg, I.; Yosief, T.; Kashman, Y.; Schleyer, M. Yardenone and abudinol two new triterpenes from the marine sponge Ptilocaulis spiculifer. Tetrahedron Lett. 1998, 39, 1445–1448. [Google Scholar] [CrossRef]
- Carletti, I.; Long, C.; Funel, C.; Amade, P. Yardenone A and B: New Cytotoxic Triterpenes from the Indian Ocean Sponge Axinella cf. bidderi. J. Nat. Prod. 2003, 66, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Aknin, M.; Gaydou, E.M.; Kashman, Y. Sodwanones K, L, and M; New Triterpenes from the Marine Sponge Axinella weltneri. J. Nat. Prod. 1997, 60, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Goldberg, I.; Stein, Z.; Kashman, Y.; Benayahu, Y.; Schleyer, M.; Gravalos, M.D.G. Sodwanones G, H, and I, New Cytotoxic Triterpenes from a Marine Sponge. J. Nat. Prod. 1995, 58, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Yosief, T.; Schleyer, M.; Kashman, Y. Several new isoprenoids from two marine sponges of the family Axinellidae. Tetrahedron 1999, 55, 5555–5566. [Google Scholar] [CrossRef]
- Funel-Le Bon, C.; Berrué, F.; Thomas, O.P.; Reyes, F.; Amade, P. Sodwanone S, a Triterpene from the Marine Sponge Axinella weltneri. J. Nat. Prod. 2005, 68, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Fishback, J.A.; Zhou, Y.-D.; Nagle, D.G. Sodwanone and Yardenone Triterpenes from a South African Species of the Marine Sponge Axinella Inhibit Hypoxia-Inducible Factor-1 (HIF-1) Activation in Both Breast and Prostate Tumor Cells. J. Nat. Prod. 2006, 69, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Whibley, C.E.; Keyzers, R.A.; Soper, A.G.; Davies-Coleman, M.T.; Samaai, T.; Hendricks, D.T. Antiesophageal Cancer Activity from Southern African Marine Organisms. Ann. N. Y. Acad. Sci. 2005, 1056, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.; Kashman, Y. Shaagrockol B and C; two hexaprenylhydroquinone disulfates from the red sea sponge toxiclona toxius. Tetrahedron Lett. 1992, 33, 2227–2230. [Google Scholar] [CrossRef]
- Loya, S.; Tal, R.; Hizi, A.; Issacs, S.; Kashman, Y.; Loya, Y. Hexaprenoid Hydroquinones, Novel Inhibitors of the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. J. Nat. Prod. 1993, 56, 2120–2125. [Google Scholar] [CrossRef] [PubMed]
- Kornprobst, J.-M.; Sallenave, C.; Barnathan, G. Sulfated Compounds from Marine Organisms. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 119, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Prinsep, M.R. Sulfur-Containing Natural Products from Marine Invertebrates. In Studies in Natural Products Chemistry; Rahman, A.-U., Ed.; Elsevier: Amsterdam, The Nederlands, 2003; Volume 28, pp. 617–751. [Google Scholar]
- Bokesch, H.R.; Stull, A.C.; Pannell, L.K.; McKee, T.C.; Boyd, M.R. A new pentacyclic sulfated hydroquinone from the marine sponge Haliclona sp. Tetrahedron Lett. 2002, 43, 3079–3081. [Google Scholar] [CrossRef]
- Menhour, B.; Aclinou, P.; Pete, J.-P. Synthesis of bicyclic oxepanes: An enantioselective approach to the western part of Shaagrockol C. Med. J. Chem. 2013, 2, 522–540. [Google Scholar] [CrossRef]
- Puliti, R.; Trivellone, E.; Crispino, A.; Cimino, G. Raspacionin, a New Tetracyclic Triterpenoid from the Sponge Raspaciona aculeata: A Structure Containing Disordered Solvent. Acta Crystallogr. C 1991, 47, 2609–2612. [Google Scholar] [CrossRef]
- Cimino, G.; Crispino, A.; Epifanio, R.d.A.; Madaio, A.; Mattia, C.A.; Mazzarella, L.; Puliti, R.; Enrico, T.; Maria, U. Raspacionin-A: A novel rearranged triterpenoid from the mediterranean sponge raspaciona aculeata. Tetrahedron 1992, 48, 9013–9022. [Google Scholar] [CrossRef]
- Cimino, G.; Crispino, A.; Madaio, A.; Trivellone, E.; Uriz, M. Raspacionin B, a Further Triterpenoid from the Mediterranean Sponge Raspaciona aculeata. J. Nat. Prod. 1993, 56, 534–538. [Google Scholar] [CrossRef]
- Cimino, G.; Epifanio, R.D.A.; Madaio, A.; Puliti, R.; Trivellone, E. Absolute Stereochemistry of Raspacionin, the Main Triterpenoid from the Marine Sponge Raspaciona Aculeata. J. Nat. Prod. 1993, 56, 1622–1626. [Google Scholar] [CrossRef]
- Cimino, G.; Madaio, A.; Trivellone, E.; Uriz, M. Minor Triterpenoids from the Mediterranean Sponge, Raspaciona aculeata. J. Nat. Prod. 1994, 57, 784–790. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Scognamiglio, G.; Trivellone, E.; Bisogno, T.; Cimino, G. New additional triterpenoids from the Mediterranean sponge Raspaciona aculeata. Tetrahedron 2002, 58, 4943–4948. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Van Rooyen, P.H.; Vleggaar, R.; Rabie, C.J. Structures of the austalides A–E, five noval toxic metabolites from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1981, 1265–1267. [Google Scholar] [CrossRef]
- Horak, R.M.; Steyn, P.S.; Vleggaar, R.; Rabie, C.J. Metabolites of Aspergillus ustus. Part 3. Structure elucidation of austalides G–L. J. Chem. Soc. Perkin Trans. 1 1985, 363–367. [Google Scholar] [CrossRef]
- Zhou, Y.; Mándi, A.; Debbab, A.; Wray, V.; Schulz, B.; Müller, W.E.G.; Lin, W.; Proksch, P.; Kurtán, T.; Aly, A.H. New Austalides from the Sponge-Associated Fungus Aspergillus sp. Eur. J. Org. Chem. 2011, 2011, 6009–6019. [Google Scholar] [CrossRef]
- Zhou, Y.; Debbab, A.; Wray, V.; Lin, W.; Schulz, B.; Trepos, R.; Pile, C.; Hellio, C.; Proksch, P.; Aly, A.H. Marine bacterial inhibitors from the sponge-derived fungus Aspergillus sp. Tetrahedron Lett. 2014, 55, 2789–2792. [Google Scholar] [CrossRef] [Green Version]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the Alga-Derived Fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.-G.; Wu, Z.-Y.; Pang, W.-W.; Ma, L.-F.; Ying, Y.-M.; Zhan, Z.-J. α-Glucosidase Inhibitors from the Fungus Aspergillus terreus 3.05358. Chem. Biodivers. 2015, 12, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, X.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Austalides S–U, New Meroterpenoids from the Sponge-Derived Fungus Aspergillus aureolatus HDN14-107. Mar. Drugs 2016, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, A.E.; Horak, R.M.; Steyn, P.S.; Vleggaar, R. Biosynthesis of austalide D, a meroterpenoid mycotoxin from Aspergillus ustus. J. Chem. Soc. Chem. Commun. 1983, 716–718. [Google Scholar] [CrossRef]
- De Jesus, A.E.; Horak, R.M.; Steyn, P.S.; Vleggaar, R. Metabolites of Aspergillus ustus. Part 4. Stable-isotope labelling studies on the biosynthesis of the austalides. J. Chem. Soc. Perkin Trans. 1 1987, 2253–2257. [Google Scholar] [CrossRef]
- Dillen, J.L.M.; Horak, R.M.; Maharaj, V.J.; Marais, S.F.; Vleggaar, R. Absolute configuration and biosynthesis of the austalides, meroterpenoid metabolites of Aspergillus ustus: Mode of cyclisation of the farnesyl moiety. J. Chem. Soc. Chem. Commun. 1989, 393–394. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Enantioselective synthesis of natural (−)-austalide B, an unusual ortho ester metabolite produced by toxigenic cultures of Aspergillus ustus. J. Am. Chem. Soc. 1994, 116, 2665–2666. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.-Z.; Sivik, M.R. Total Synthesis of (−)-Austalide B. A Generic Solution to Elaboration of the Pyran/p-Cresol/Butenolide Triad. J. Am. Chem. Soc. 1994, 116, 11323–11334. [Google Scholar] [CrossRef]
- Rotem, M.; Carmely, S.; Kashman, Y.; Loya, Y. Two new antibiotics from the red sea sponge Psammaplysilla purpurea. Tetrahedron 1983, 39, 667–676. [Google Scholar] [CrossRef]
- Roll, D.M.; Chang, C.W.J.; Scheuer, P.J.; Gray, G.A.; Shoolery, J.N.; Matsumoto, G.K.; Van Duyne, G.D.; Clardy, J. Structure of the psammaplysins. J. Am. Chem. Soc. 1985, 107, 2916–2920. [Google Scholar] [CrossRef]
- Mándi, A.; Mudianta, I.W.; Kurtán, T.; Garson, M.J. Absolute Configuration and Conformational Study of Psammaplysins A and B from the Balinese Marine Sponge Aplysinella strongylata. J. Nat. Prod. 2015, 78, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Copp, B.R.; Ireland, C.M.; Barrows, L.R. Psammaplysin C: A New Cytotoxic Dibromotyrosine-Derived Metabolite from the Marine Sponge Druinella (=Psammaplysilla) purpurea. J. Nat. Prod. 1992, 55, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Ichiba, T.; Scheuer, P.J.; Kelly-Borges, M. Three bromotyrosine derivatives, one terminating in an unprecedented diketocyclopentenylidene enamine. J. Org. Chem. 1993, 58, 4149–4150. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Kato, H.; Hirota, H.; Fusetani, N. Ceratinamides A and B: New antifouling dibromotyrosine derivatives from the marine sponge Pseudoceratina purpurea. Tetrahedron 1996, 52, 8181–8186. [Google Scholar] [CrossRef]
- Liu, S.; Fu, X.; Schmitz, F.J.; Kelly-Borges, M. Psammaplysin F, a New Bromotyrosine Derivative from a Sponge, Aplysinella sp. J. Nat. Prod. 1997, 60, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, G.M.; Eckman, L.L.; Ray, S.; Hughes, R.O.; Pfefferkorn, J.A.; Barluenga, S.; Nicolaou, K.C.; Bewley, C.A. Bromotyrosine-Derived Natural and Synthetic Products as Inhibitors of Mycothiol-S-Conjugate Amidase. Bioorg. Med. Chem. Lett. 2002, 12, 2487–2490. [Google Scholar] [CrossRef]
- Nicholas, G.M.; Eckman, L.L.; Newton, G.L.; Fahey, R.C.; Ray, S.; Bewley, C.A. Inhibition and kinetics of mycobacterium tuberculosis and mycobacterium smegmatis mycothiol-S-conjugate amidase by natural product inhibitors. Bioorg. Med. Chem. 2003, 11, 601–608. [Google Scholar] [CrossRef]
- Yang, X.; Davis, R.A.; Buchanan, M.S.; Duffy, S.; Avery, V.M.; Camp, D.; Quinn, R.J. Antimalarial Bromotyrosine Derivatives from the Australian Marine Sponge Hyattella sp. J. Nat. Prod. 2010, 73, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Andrews, K.T.; Birrell, G.W.; Tran, T.L.; Camp, D.; Davis, R.A.; Quinn, R.J. Psammaplysin H, a new antimalarial bromotyrosine alkaloid from a marine sponge of the genus Pseudoceratina. Bioorg. Med. Chem. Lett. 2011, 21, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.; Schupp, P.J.; Schrör, J.-P.; Engemann, A.; Rohde, S.; Kelman, D.; de Voogd, N.; Carroll, A.; Motti, C.A. Twilight Zone Sponges from Guam Yield Theonellin Isocyanate and Psammaplysins I and J. J. Nat. Prod. 2012, 75, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Mudianta, I.W.; Skinner-Adams, T.; Andrews, K.T.; Davis, R.A.; Hadi, T.A.; Hayes, P.Y.; Garson, M.J. Psammaplysin Derivatives from the Balinese Marine Sponge Aplysinella strongylata. J. Nat. Prod. 2012, 75, 2132–2143. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Han, S.; Lee, H.-S.; Kang, J.S.; Yun, J.; Sim, C.J.; Shin, H.J.; Lee, J.S. Cytotoxic Psammaplysin Analogues from a Suberea sp. Marine Sponge and the Role of the Spirooxepinisoxazoline in Their Activity. J. Nat. Prod. 2013, 76, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Kossuga, M.H. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2008, 25, 919–954. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, I.; Kusumi, T.; Kakisawa, H.; Kashman, Y.; Hirsh, S. Structure and chemical properties of ptilomycalin A. J. Am. Chem. Soc. 1992, 114, 8472–8479. [Google Scholar] [CrossRef]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Ingrum, A.L.; Carney, J.R.; Rinehart, K.L.; Sakai, R. Polycyclic guanidine-containing compounds from the Mediterranean sponge Crambe crambe: The structure of 13,14,15-isocrambescidin 800 and the absolute stereochemistry of the pentacyclic guanidine moieties of the crambescidins. J. Org. Chem. 1993, 58, 4805–4808. [Google Scholar] [CrossRef]
- Palagiano, E.; De Marino, S.; Minale, L.; Riccio, R.; Zollo, F.; Iorizzi, M.; Carré, J.B.; Debitus, C.; Lucarain, L.; Provost, J. Ptilomycalin A, crambescidin 800 and related new highly cytotoxic guanidine alkaloids from the starfishes Fromia monilis and Celerina heffernani. Tetrahedron 1995, 51, 3675–3682. [Google Scholar] [CrossRef]
- Venkateswarlu, Y.; Reddy, M.V.R.; Ramesh, P.; Rao, J.V. Neofolitispates, pentacyclic guanidine alkaloids from the sponge Neofolitispa dianchora. Indian J. Chem. 1999, 38B, 254–256. [Google Scholar] [CrossRef]
- Braekman, J.C.; Daloze, D.; Tavares, R.; Hajdu, E.; Van Soest, R.W.M. Novel Polycyclic Guanidine Alkaloids from Two Marine Sponges of the Genus Monanchora. J. Nat. Prod. 2000, 63, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Makarieva, T.N.; Tabakmaher, K.M.; Guzii, A.G.; Denisenko, V.A.; Dmitrenok, P.S.; Shubina, L.K.; Kuzmich, A.S.; Lee, H.-S.; Stonik, V.A. Monanchocidins B–E: Polycyclic Guanidine Alkaloids with Potent Antileukemic Activities from the Sponge Monanchora pulchra. J. Nat. Prod. 2011, 74, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Moazami, Y.; Pierce, J.G. Structure, synthesis and biological properties of the pentacyclic guanidinium alkaloids. Bioorg. Med. Chem. 2017, 25, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, J.E.H.; Nitcheu, J.; Mahmoudi, N.; Ibana, J.A.; Mangalindan, G.C.; Black, G.P.; Howard-Jones, A.G.; Moore, C.G.; Thomas, D.A.; Mazier, D.; et al. Antimalarial Activity of Crambescidin 800 and Synthetic Analogues against Liver and Blood Stage of Plasmodium sp. J. Antibiot. 2006, 59, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.; Ternon, E.; López-Alonso, H.; Thomas, O.; Vega, F.; Vieytes, M.; Botana, L. Crambescidin-816 Acts as a Fungicidal with More Potency than Crambescidin-800 and -830, Inducing Cell Cycle Arrest, Increased Cell Size and Apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kong, D.; Matsui, K.; Kobayashi, M. Erythroid Differentiation in K562 Chronic Myelogenous Cells Induced by Crambescidin 800, a Pentacyclic Guanidine Alkaloid. Anticancer Res. 2004, 24, 2325–2330. [Google Scholar] [PubMed]
- Aron, Z.D.; Pietraszkiewicz, H.; Overman, L.E.; Valeriote, F.; Cuevas, C. Synthesis and anticancer activity of side chain analogs of the crambescidin alkaloids. Bioorg. Med. Chem. Lett. 2004, 14, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Gustafson, K.R. Marine pharmacology in 2003–2004: Anti-tumour and cytotoxic compounds. Eur. J. Cancer 2006, 42, 2241–2270. [Google Scholar] [CrossRef] [PubMed]
- Rubiolo, J.A.; López-Alonso, H.; Roel, M.; Vieytes, M.R.; Thomas, O.; Ternon, E.; Vega, F.V.; Botana, L.M. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharmacol. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The Marine Guanidine Alkaloid Crambescidin 816 Induces Calcium Influx and Cytotoxicity in Primary Cultures of Cortical Neurons through Glutamate Receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Braekman, J.C.; Daloze, D.; Bruno, I.; Riccio, R.; Ferri, S.; Spampinato, S.; Speroni, E. Polycyclic Guanidine Alkaloids from the Marine Sponge Crambe crambe and Ca++ Channel Blocker Activity of Crambescidin 816. J. Nat. Prod. 1993, 56, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Ohizumi, Y.; Sasaki, S.; Kusumi, T.; Ohtani, I.I. Ptilomycalin A, a novel Na(+), K(+)- or Ca2(+)-ATPase inhibitor, competitively interacts with ATP at its binding site. Eur. J. Pharmacol. 1996, 310, 95–98. [Google Scholar] [CrossRef]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
- Donahue, M.G. Chapter 1—Recent Developments in the Synthesis of Cyclic Guanidine Alkaloids. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Nederlands, 2014; Volume 26, pp. 1–28. [Google Scholar]
- Ma, Y.; De, S.; Chen, C. Syntheses of cyclic guanidine-containing natural products. Tetrahedron 2015, 71, 1145–1173. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Williams, H.L. Synthesis of a pentacyclic model of ptilomycalin A. J. Chem. Soc. Chem. Commun. 1994, 819–820. [Google Scholar] [CrossRef]
- Murphy, P.J.; Williams, H.L.; Hursthouse, M.B.; Malik, K.M.A. Synthetic studies towards ptilomycalin a using A biomimetic approach. J. Chem. Soc. Chem. Commun. 1994, 119–120. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Walshe, N.D.A.; Hibbs, D.E.; Hursthouse, M.B.; Abdul Malik, K.M. A short synthetic route to the tricyclic guanidinium core of the batzelladine alkaloids. Tetrahedron Lett. 1996, 37, 6943–6946. [Google Scholar] [CrossRef]
- Murphy, P.J.; Harri Lloyd, W.; Hibbs, D.E.; Hursthouse, M.B.; Abdul Malik, K.M. Biomimetic model studies towards ptilomycalin A. Tetrahedron 1996, 52, 8315–8332. [Google Scholar] [CrossRef]
- Murphy, P.J.; Williams, H.L.; Hibbs, D.E.; Hursthouse, M.B.; Malik, K.M.A. Crystallographic evidence for the proposed host behaviour of ptilomycalin A. Chem. Commun. 1996, 445–447. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Walshe, N.D.A. A short synthetic route to the tricyclic guanidinium core of the batzelladine alkaloids. Tetrahedron 1998, 54, 9481–9488. [Google Scholar] [CrossRef]
- Black, G.P.; Murphy, P.J.; Thornhill, A.J.; Walshe, N.D.A.; Zanetti, C. Synthesis of the left hand unit of batzelladine F; Revision of the reported relative stereochemistry. Tetrahedron 1999, 55, 6547–6554. [Google Scholar] [CrossRef]
- Overman, L.E.; Rabinowitz, M.H.; Renhowe, P.A. Enantioselective Total Synthesis of (−)-Ptilomycalin A. J. Am. Chem. Soc. 1995, 117, 2657–2658. [Google Scholar] [CrossRef]
- Coffey, D.S.; McDonald, A.I.; Overman, L.E.; Rabinowitz, M.H.; Renhowe, P.A. A Practical Entry to the Crambescidin Family of Guanidine Alkaloids. Enantioselective Total Syntheses of Ptilomycalin A, Crambescidin 657 and Its Methyl Ester (Neofolitispates 2), and Crambescidin 800. J. Am. Chem. Soc. 2000, 122, 4893–4903. [Google Scholar] [CrossRef]
- Coffey, D.S.; Overman, L.E.; Stappenbeck, F. Enantioselective Total Syntheses of 13,14,15-Isocrambescidin 800 and 13,14,15-Isocrambescidin 657. J. Am. Chem. Soc. 2000, 122, 4904–4914. [Google Scholar] [CrossRef]
- Overman, L.E.; Rhee, Y.H. Total Synthesis of (−)-Crambidine and Definition of the Relative Configuration of Its Unique Tetracyclic Guanidinium Core. J. Am. Chem. Soc. 2005, 127, 15652–15658. [Google Scholar] [CrossRef] [PubMed]
- Perl, N.R.; Ide, N.D.; Prajapati, S.; Perfect, H.H.; Durón, S.G.; Gin, D.Y. Annulation of Thioimidates and Vinyl Carbodiimides to Prepare 2-Aminopyrimidines, Competent Nucleophiles for Intramolecular Alkyne Hydroamination. Synthesis of (−)-Crambidine. J. Am. Chem. Soc. 2010, 132, 1802–1803. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Georgieva, A.; Koshino, H.; Nakata, T.; Kita, T.; Hashimoto, Y. Total Synthesis of Crambescidin 359. Org. Lett. 2002, 4, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.G.; Murphy, P.J.; Williams, H.L.; McGown, A.T.; Smith, N.K. A synthesis of crambescidin 359. Tetrahedron Lett. 2003, 44, 251–254. [Google Scholar] [CrossRef]
- Aron, Z.D.; Overman, L.E. Total Synthesis and Properties of the Crambescidin Core Zwitterionic Acid and Crambescidin 359. J. Am. Chem. Soc. 2005, 127, 3380–3390. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.; Lefranc, F.; Kijjoa, A.; Kiss, R. Can Some Marine-Derived Fungal Metabolites Become Actual Anticancer Agents? Mar. Drugs 2015, 13, 3950–3991. [Google Scholar] [CrossRef] [PubMed]
- Cutler, H.G.; Springer, J.P.; Arrendale, R.F.; Arison, B.H.; Cole, P.D.; Roberts, R.G. Cinereain: A Novel Metabolite with Plant Growth Regulating Properties from Botrytis cinerea. Agric. Biol. Chem. 1988, 52, 1725–1733. [Google Scholar] [CrossRef]
- Rahbæk, L.; Breinholt, J.; Frisvad, J.C.; Christophersen, C. Circumdatin A, B, and C: Three New Benzodiazepine Alkaloids Isolated from a Culture of the Fungus Aspergillus ochraceus. J. Org. Chem. 1999, 64, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Ookura, R.; Kito, K.; Ooi, T.; Namikoshi, M.; Kusumi, T. Structure Revision of Circumdatins A and B, Benzodiazepine Alkaloids Produced by Marine Fungus Aspergillus ostianus, by X-ray Crystallography. J. Org. Chem. 2008, 73, 4245–4247. [Google Scholar] [CrossRef] [PubMed]
- González-Jartı́n, J.M.; Alfonso, A.; Sainz, M.J.; Vieytes, M.R.; Botana, L.M. UPLC-MS-IT-TOF Identification of Circumdatins Produced by Aspergillus ochraceus. J. Agric. Food. Chem. 2017, 65, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Belofsky, G.N.; Anguera, M.; Jensen, P.R.; Fenical, W.; Köck, M. Oxepinamides A–C and Fumiquinazolines H–I: Bioactive Metabolites from a Marine Isolate of a Fungus of the Genus Acremonium. Chem. Eur. J. 2000, 6, 1355–1360. [Google Scholar] [CrossRef]
- Sprogøe, K.; Manniche, S.; Larsen, T.O.; Christophersen, C. Janoxepin and brevicompanine B: Antiplasmodial metabolites from the fungus Aspergillus janus. Tetrahedron 2005, 61, 8718–8721. [Google Scholar] [CrossRef]
- Doveston, R.G.; Steendam, R.; Jones, S.; Taylor, R.J.K. Total Synthesis of an Oxepine Natural Product, (±)-Janoxepin. Org. Lett. 2012, 14, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Asami, Y.; Lee, D.; Jang, J.-H.; Ahn, J.S.; Oh, H. Protuboxepins A and B and Protubonines A and B from the Marine-Derived Fungus Aspergillus sp. SF-5044. J. Nat. Prod. 2011, 74, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Jang, J.-H.; Soung, N.-K.; He, L.; Moon, D.O.; Kim, J.W.; Oh, H.; Muroi, M.; Osada, H.; Kim, B.Y.; et al. Protuboxepin A, a marine fungal metabolite, inducing metaphase arrest and chromosomal misalignment in tumor cells. Bioorg. Med. Chem. 2012, 20, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Mándi, A.; Li, X.-M.; Du, F.-Y.; Wang, J.-N.; Li, X.; Kurtán, T.; Wang, B.-G. Varioxepine A, a 3H-Oxepine-Containing Alkaloid with a New Oxa-Cage from the Marine Algal-Derived Endophytic Fungus Paecilomyces variotii. Org. Lett. 2014, 16, 4834–4837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, X.-M.; Wang, J.-N.; Wang, B.-G. Oxepine-Containing Diketopiperazine Alkaloids from the Algal-Derived Endophytic Fungus Paecilomyces variotii EN-291. Helv. Chim. Acta 2015, 98, 800–804. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Huang, X.; Tian, X.; Liao, S.; Yang, B.; Wang, F.; Zhou, X.; Liu, Y. Antifungal New Oxepine-Containing Alkaloids and Xanthones from the Deep-Sea-Derived Fungus Aspergillus versicolor SCSIO 05879. J. Agric. Food. Chem. 2016, 64, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Shi, Y.; Chen, X.; Chen, C.-T.A.; Tao, X.; Wu, B. New compounds from a hydrothermal vent crab-associated fungus Aspergillus versicolor XZ-4. Org. Biomol. Chem. 2017, 15, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.M.; Waring, P.; Howlett, B.J. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis. Microbiology 2005, 151, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Kittakoop, P.; Kirtikara, K.; Hywel-Jones, N.L.; Thebtaranonth, Y. Bioactive Substances from Insect Pathogenic Fungi. Acc. Chem. Res. 2005, 38, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, A.D. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem. Rev. 2012, 112, 3641–3716. [Google Scholar] [CrossRef] [PubMed]
- Munday, R. Harmful and Beneficial Effects of Organic Monosulfides, Disulfides, and Polysulfides in Animals and Humans. Chem. Res. Toxicol. 2012, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bladt, T.; Frisvad, J.; Knudsen, P.; Larsen, T. Anticancer and Antifungal Compounds from Aspergillus, Penicillium and Other Filamentous Fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.-M.; Yi, X.-X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.-H. An Update on 2,5-Diketopiperazines from Marine Organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Yusoff, M.M.; Maniam, G.P.; Ichwan, S.J.A.; Soundharrajan, I.; Govindan, N. Nutraceuticals as potential therapeutic agents for colon cancer: A review. Acta Pharm. Sin. B 2014, 4, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Welch, T.R.; Williams, R.M. Epidithiodioxopiperazines. occurrence, synthesis and biogenesis. Nat. Prod. Rep. 2014, 31, 1376–1404. [Google Scholar] [CrossRef] [PubMed]
- Nadumane, V.K.; Venkatachalam, P.; Gajaraj, B. Chapter 19—Aspergillus Applications in Cancer Research A2. In New and Future Developments in Microbial Biotechnology and Bioengineering; Vijai, G., Ed.; Elsevier: Amsterdam, The Nederlands, 2016; pp. 243–255. [Google Scholar]
- Nagarajan, R.; Huckstep, L.L.; Lively, D.H.; DeLong, D.C.; Marsh, M.M.; Neuss, N. Aranotin and related metabolites from Arachniotus aureus. I. Determination of structure. J. Am. Chem. Soc. 1968, 90, 2980–2982. [Google Scholar] [CrossRef]
- Neuss, N.; Nagarajan, R.; Molloy, B.B.; Huckstep, L.L. Aranotin and related metabolites. II. Isolation, characterization, and structures of two new metabolites. Tetrahedron Lett. 1968, 9, 4467–4471. [Google Scholar] [CrossRef]
- Nagarajan, R.; Woody, R.W. Circular dichroism of gliotoxin and related epidithiapiperazinediones. J. Am. Chem. Soc. 1973, 95, 7212–7222. [Google Scholar] [CrossRef] [PubMed]
- Murdock, K.C. Antiviral agents. Chemical modifications of a disulfide antibiotic, acetylaranotin. J. Med. Chem. 1974, 17, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Sakai, H.; Hirota, A. Isolation of Acetylaranotin, Bisdethiodi(methylthio)-acetylaranotin and Terrein as Plant Growth Inhibitors from a Strain of Aspergillus terreus. Agric. Biol. Chem. 1983, 47, 2637–2638. [Google Scholar] [CrossRef]
- Guo, C.-J.; Yeh, H.-H.; Chiang, Y.-M.; Sanchez, J.F.; Chang, S.-L.; Bruno, K.S.; Wang, C.C.C. Biosynthetic Pathway for the Epipolythiodioxopiperazine Acetylaranotin in Aspergillus terreus Revealed by Genome-Based Deletion Analysis. J. Am. Chem. Soc. 2013, 135, 7205–7213. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Park, J.S.; Kim, Y.J.; Jung, J.H.; Lee, J.K.; Kwon, H.C.; Yang, H.O. Apoptosis-inducing effect of diketopiperazine disulfides produced by Aspergillus sp. KMD 901 isolated from marine sediment on HCT116 colon cancer cell lines. J. Appl. Microbiol. 2011, 110, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Seya, H.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Structure and absolute configuration of emestrin, a new macrocyclic epidithiodioxopiperazine from Emericell striata. J. Chem. Soc. Chem. Commun. 1985, 657–658. [Google Scholar] [CrossRef]
- Seya, H.; Nozawa, K.; Nakajima, S.; Kawai, K.-I.; Udagawa, S.-I. Studies on fungal products. Part 8. Isolation and structure of emestrin, a novel antifungal macrocyclic epidithiodioxopiperazine from Emericella striata. X-ray molecular structure of emestrin. J. Chem. Soc. Perkin Trans. 1 1986, 109–116. [Google Scholar] [CrossRef]
- Seya, H.; Nozawa, K.; Udagawa, S.-I.; Nakajima, S.; Kawai, K.-I. Studies on Fungal Products. IX. Dethiosecoemestrin, a New Metabolite Related to Emestrin, from Emericella striata. Chem. Pharm. Bull. 1986, 34, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Ooike, M.; Nozawa, K.; Kawai, K.-I. An epitetrathiodioxopiperazine related to emestrin from Emericella foveolata. Phytochemistry 1997, 46, 123–126. [Google Scholar] [CrossRef]
- Kawahara, N.; Nakajima, S.; Yamazaki, M.; Kawai, K. Structure of a Novel Epidithiodioxopiperazine, Emethallicin A, a Potent Inhibitor of Histamine Release, from Emericella heterothallica. Chem. Pharm. Bull. 1989, 37, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, N.; Nozawa, K.; Yamazaki, M.; Nakajima, S.; Kawai, K.-I. Structures of Novel Epipolythiodioxopiperazines, Emethallicins B, C, and D, Potent Inhibitors of Histamine Release, from Emericella heterothallica. Chem. Pharm. Bull. 1990, 38, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.M.; Kishi, Y. Experimental Support for the Primary Stereoelectronic Effect Governing Baeyer–Villiger Oxidation and Criegee Rearrangement. J. Am. Chem. Soc. 1998, 120, 9392–9393. [Google Scholar] [CrossRef]
- Peng, J.; Clive, D.L.J. Synthesis of Dihydrooxepin Models Related to the Antitumor Antibiotic MPC1001. Org. Lett. 2007, 9, 2939–2941. [Google Scholar] [CrossRef] [PubMed]
- Gross, U.; Nieger, M.; Bräse, S. A Unified Strategy Targeting the Thiodiketopiperazine Mycotoxins Exserohilone, Gliotoxin, the Epicoccins, the Epicorazines, Rostratin A and Aranotin. Chem. Eur. J. 2010, 16, 11624–11631. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Lu, M.; Totokotsopoulos, S.; Heretsch, P.; Giguère, D.; Sun, Y.-P.; Sarlah, D.; Nguyen, T.H.; Wolf, I.C.; Smee, D.F.; et al. Synthesis and Biological Evaluation of Epidithio-, Epitetrathio-, and bis-(Methylthio)diketopiperazines: Synthetic Methodology, Enantioselective Total Synthesis of Epicoccin G, 8,8′-epi-ent-Rostratin B, Gliotoxin, Gliotoxin G, Emethallicin E, and Haematocin and Discovery of New Antiviral and Antimalarial Agents. J. Am. Chem. Soc. 2012, 134, 17320–17332. [Google Scholar] [CrossRef] [PubMed]
- Schuber, P.T.; Williams, R.M. Synthetic studies toward MPC1001: Preparation of a β-hydroxyl-tyrosine derivative. Tetrahedron Lett. 2012, 53, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, H.F.; Carreira, E.M. An Efficient Synthesis Strategy to the Core Structure of 6-5-6-5-6-Membered Epipolythiodiketopiperazines. Org. Lett. 2014, 16, 2854–2857. [Google Scholar] [CrossRef] [PubMed]
- Belov, D.S.; Ratmanova, N.K.; Andreev, I.A.; Kurkin, A.V. Synthesis of Bicyclic Proline Derivatives by the Aza-Cope-Mannich Reaction: Formal Synthesis of (±)-Acetylaranotin. Chem. Eur. J. 2015, 21, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Liang, Y.-M. Copper-Catalyzed [2 + 2 + 3] Annulation of 1,6-Enynes with α-Bromo-1,3-Dicarbonyl Compounds: Synthesis of Dihydrooxepines. J. Org. Chem. 2017, 82, 7000–7007. [Google Scholar] [CrossRef] [PubMed]
- Codelli, J.A.; Puchlopek, A.L.A.; Reisman, S.E. Enantioselective Total Synthesis of (−)-Acetylaranotin, a Dihydrooxepine Epidithiodiketopiperazine. J. Am. Chem. Soc. 2012, 134, 1930–1933. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Hasegawa, A.; Tsumagari, N.; Nakai, R.; Ogawa, T.; Kanda, Y. MPC1001 and Its Analogues: New Antitumor Agents from the Fungus Cladorrhinum Species. Org. Lett. 2004, 6, 4101–4104. [Google Scholar] [CrossRef] [PubMed]
- Tsumaragi, N.; Nakai, R.; Onodera, H.; Hasegawa, A.; Rahayu, E.S.; Ando, K.; Yamashita, Y. MPC1001, a New Antitumor Antibiotic Produced by Cladorrhinum sp. J. Antibiot. 2004, 57, 532–534. [Google Scholar] [CrossRef]
- Wang, H.; Regan, C.J.; Codelli, J.A.; Romanato, P.; Puchlopek-Dermenci, A.L.A.; Reisman, S.E. Enantioselective Synthesis of (−)-Acetylapoaranotin. Org. Lett. 2017, 19, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of aplysistatin.

Figure 2.
Main structures in the palisadin family.
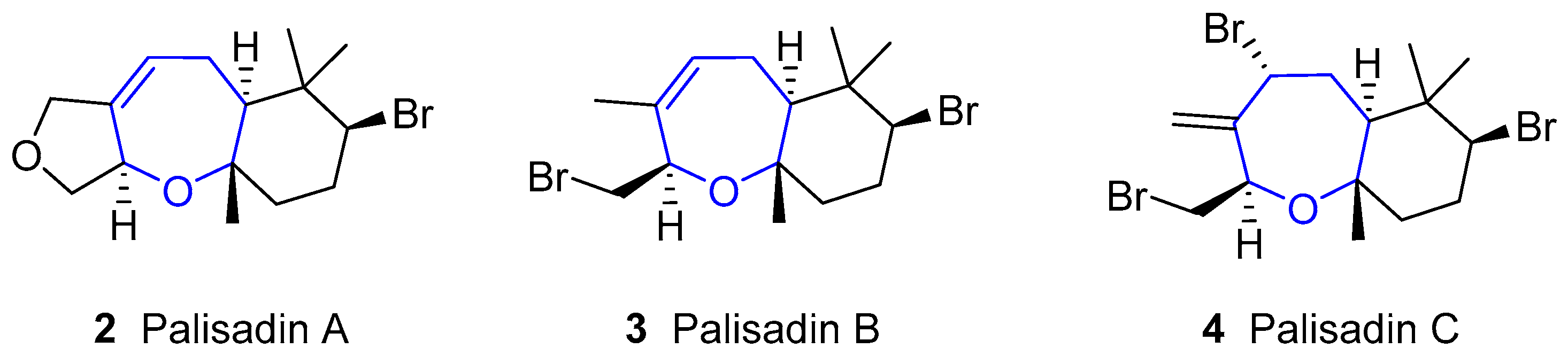
Figure 3.
Structure of 5β-hydroxypalisadin B.
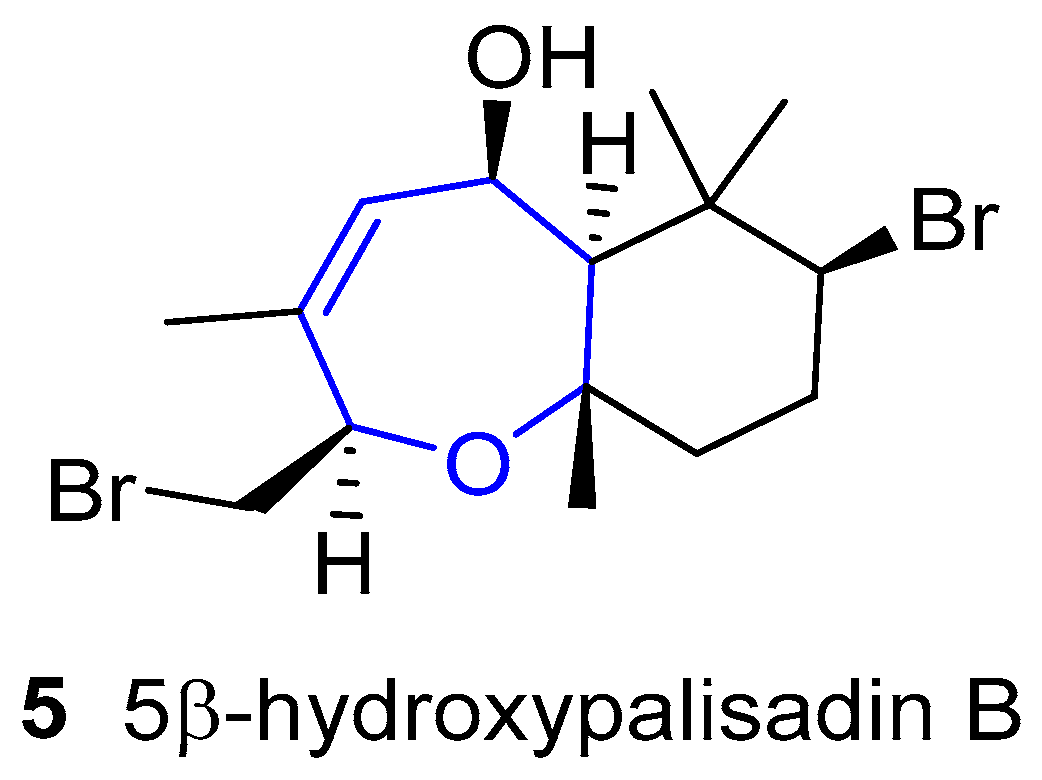
Scheme 1.
Overview of Hoye’s synthesis of (±)-aplysistatin.
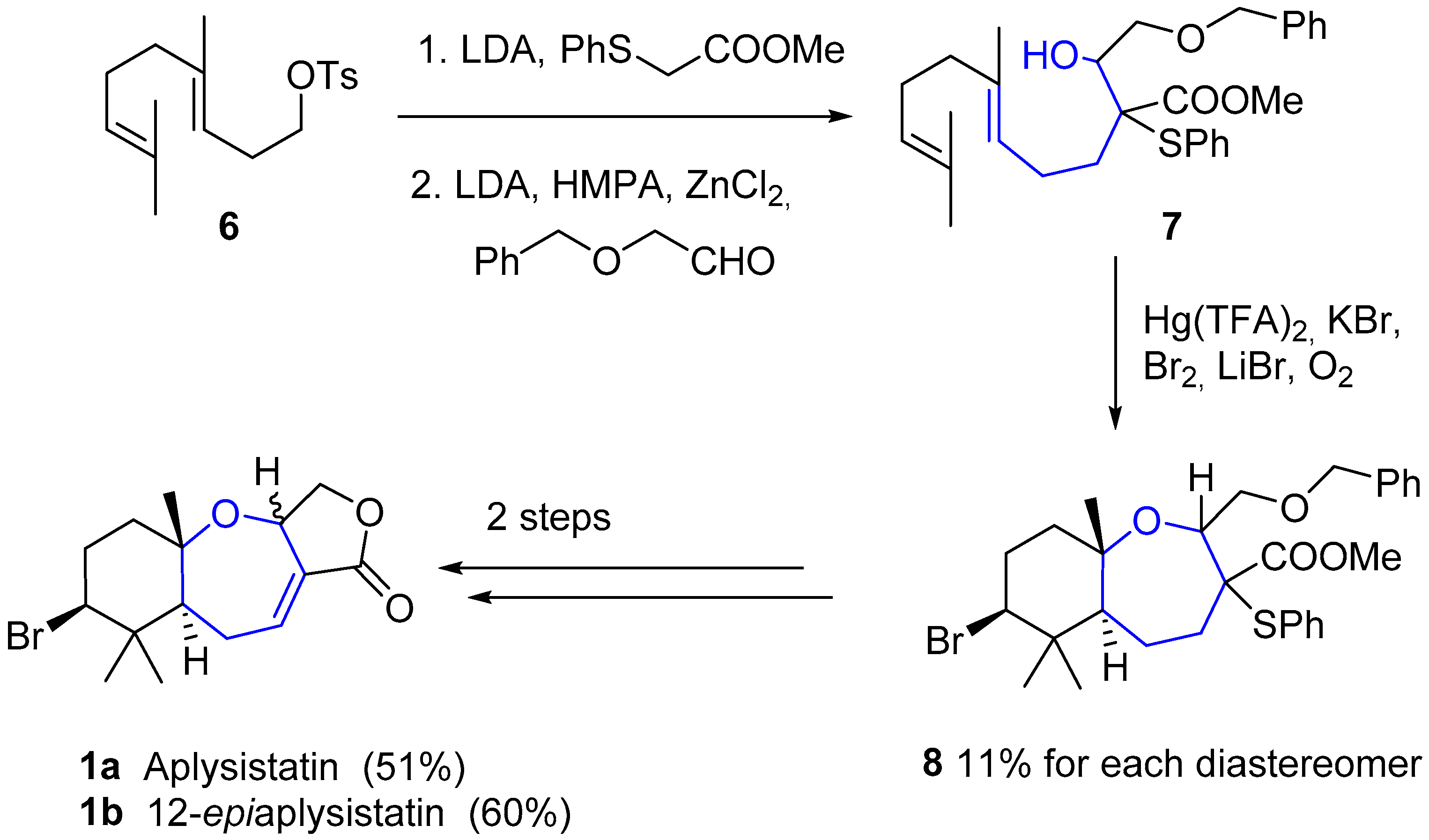
Scheme 2.
Couladouros’ retrosynthetic analysis of his key intermediate.
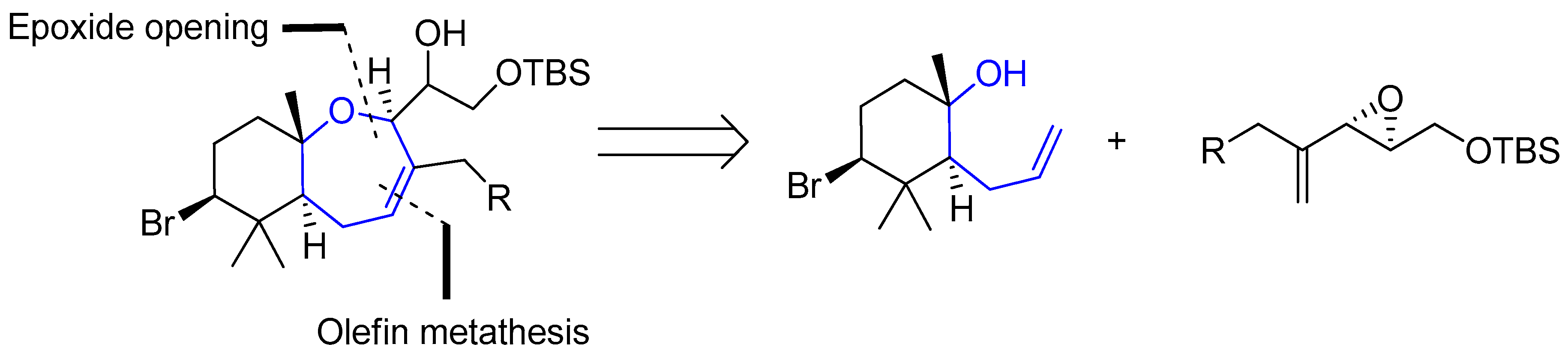
Scheme 3.
Couladouros’ synthesis of (−)-Aplysistatin, (+)-Palisadin A, (+)-Palisadin B, and (+)-12-hydroxy-Palisadin B. (a) NaIO4; then NaBH4; (b) TsCl; (c) LiBr, HMPA; (d) DDQ; (e) K2CO3; (f) MnO2; (g) PCC; (h) SeO2, NaBH4.
Scheme 3.
Couladouros’ synthesis of (−)-Aplysistatin, (+)-Palisadin A, (+)-Palisadin B, and (+)-12-hydroxy-Palisadin B. (a) NaIO4; then NaBH4; (b) TsCl; (c) LiBr, HMPA; (d) DDQ; (e) K2CO3; (f) MnO2; (g) PCC; (h) SeO2, NaBH4.
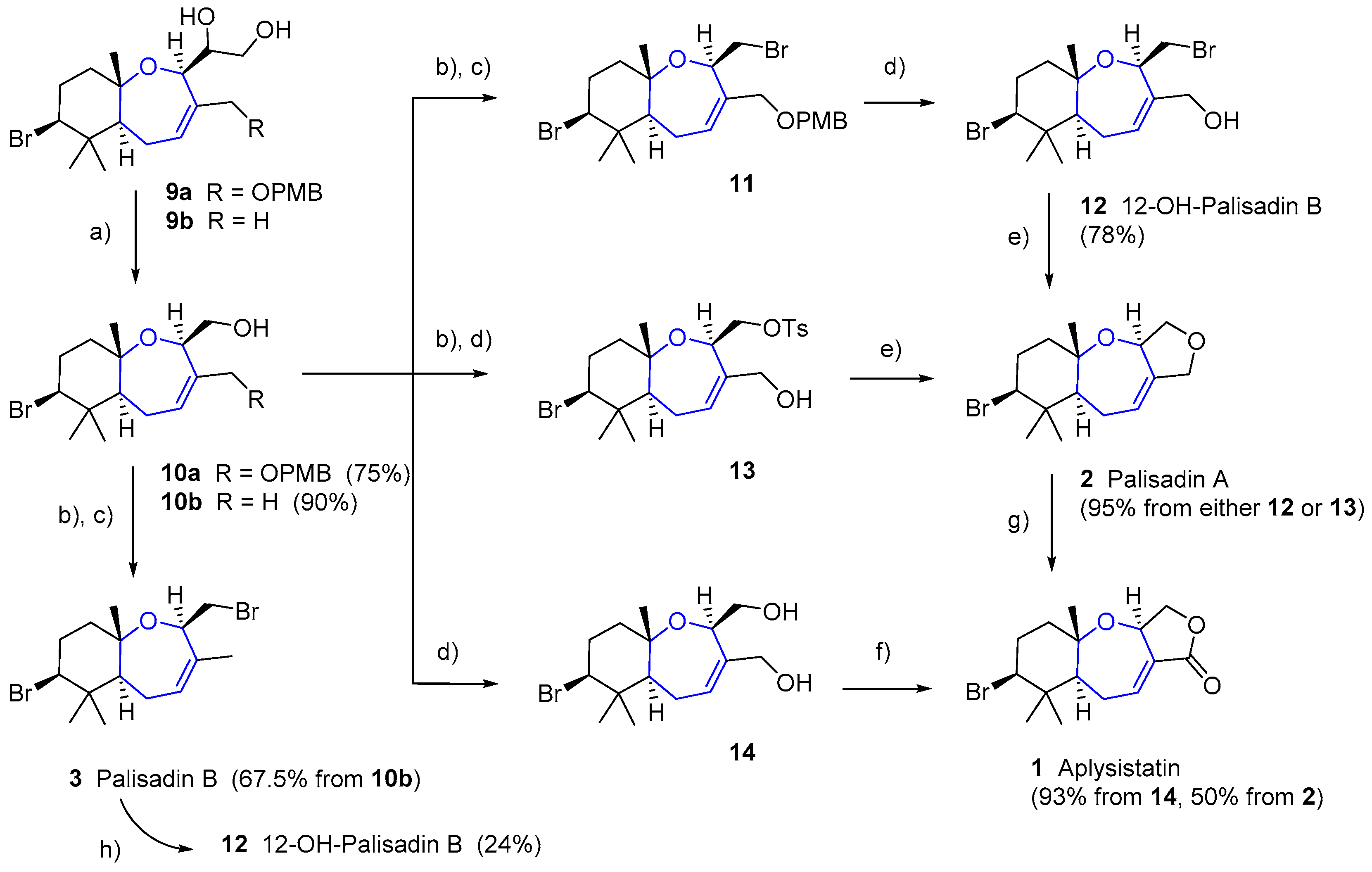
Figure 4.
Chemical structures of β-elemene (19), common scaffold for all lobanes; and of oxepin lobatrienol (20) with oxepine structure highlighted.
Figure 4.
Chemical structures of β-elemene (19), common scaffold for all lobanes; and of oxepin lobatrienol (20) with oxepine structure highlighted.
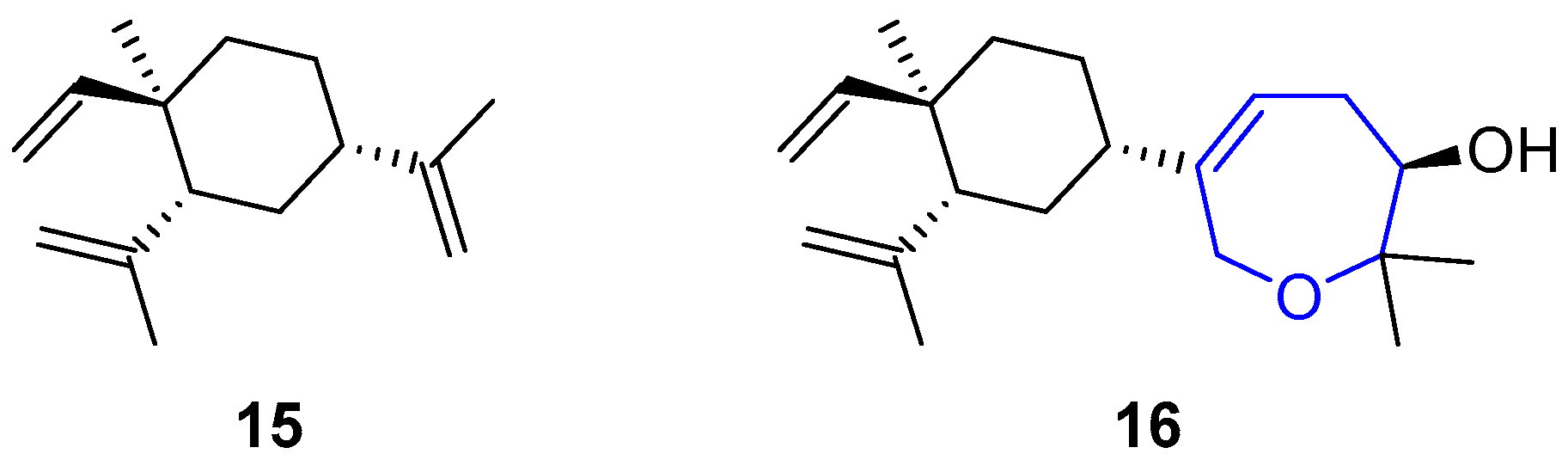
Figure 5.
Molecular structures of sipholenol A and the two main scaffolds in the sipholenol family.
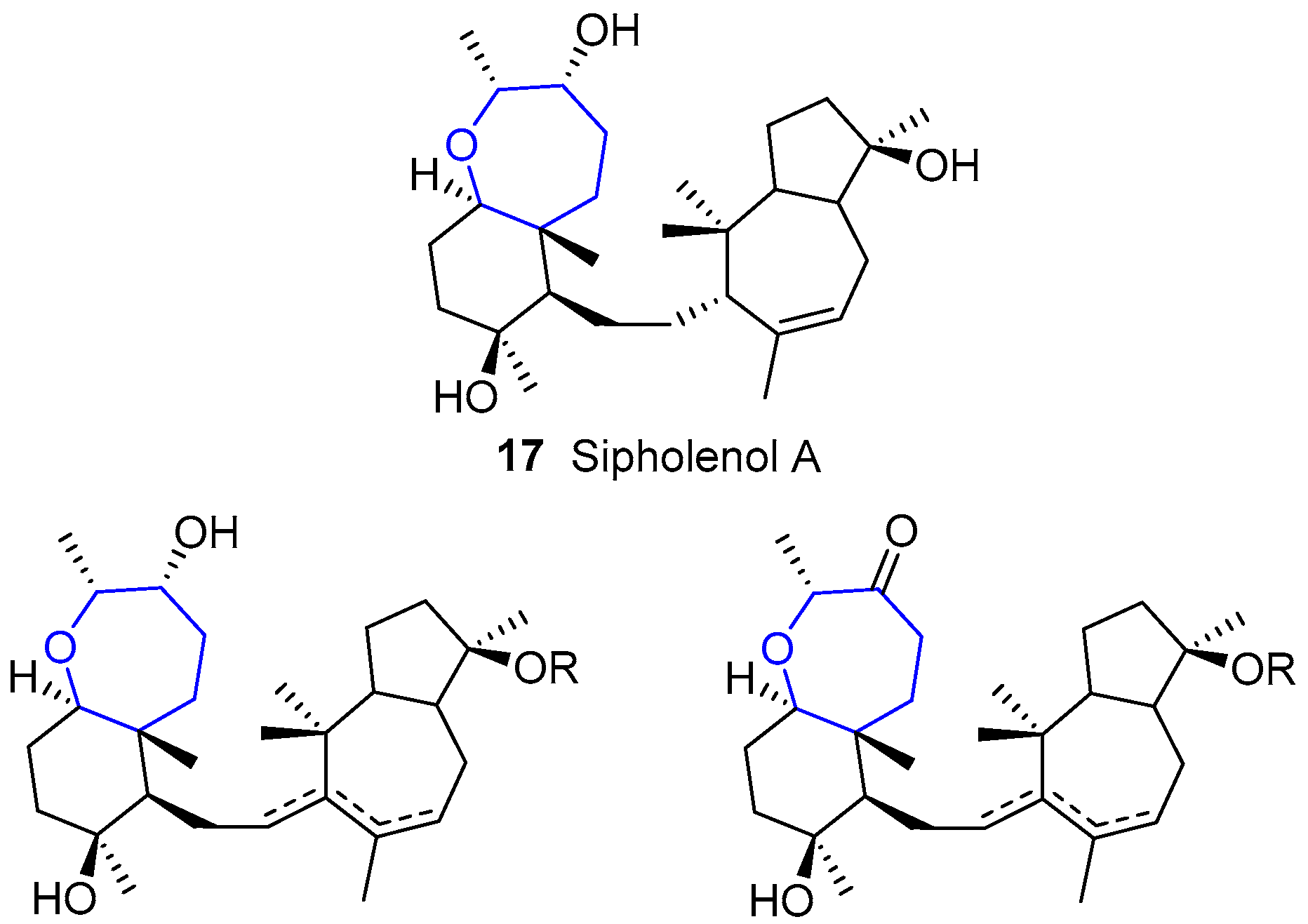
Figure 6.
Neviotanes and Dahabinone A discovered by Kashman during their studies with Siphonochalina siphonella.
Figure 6.
Neviotanes and Dahabinone A discovered by Kashman during their studies with Siphonochalina siphonella.
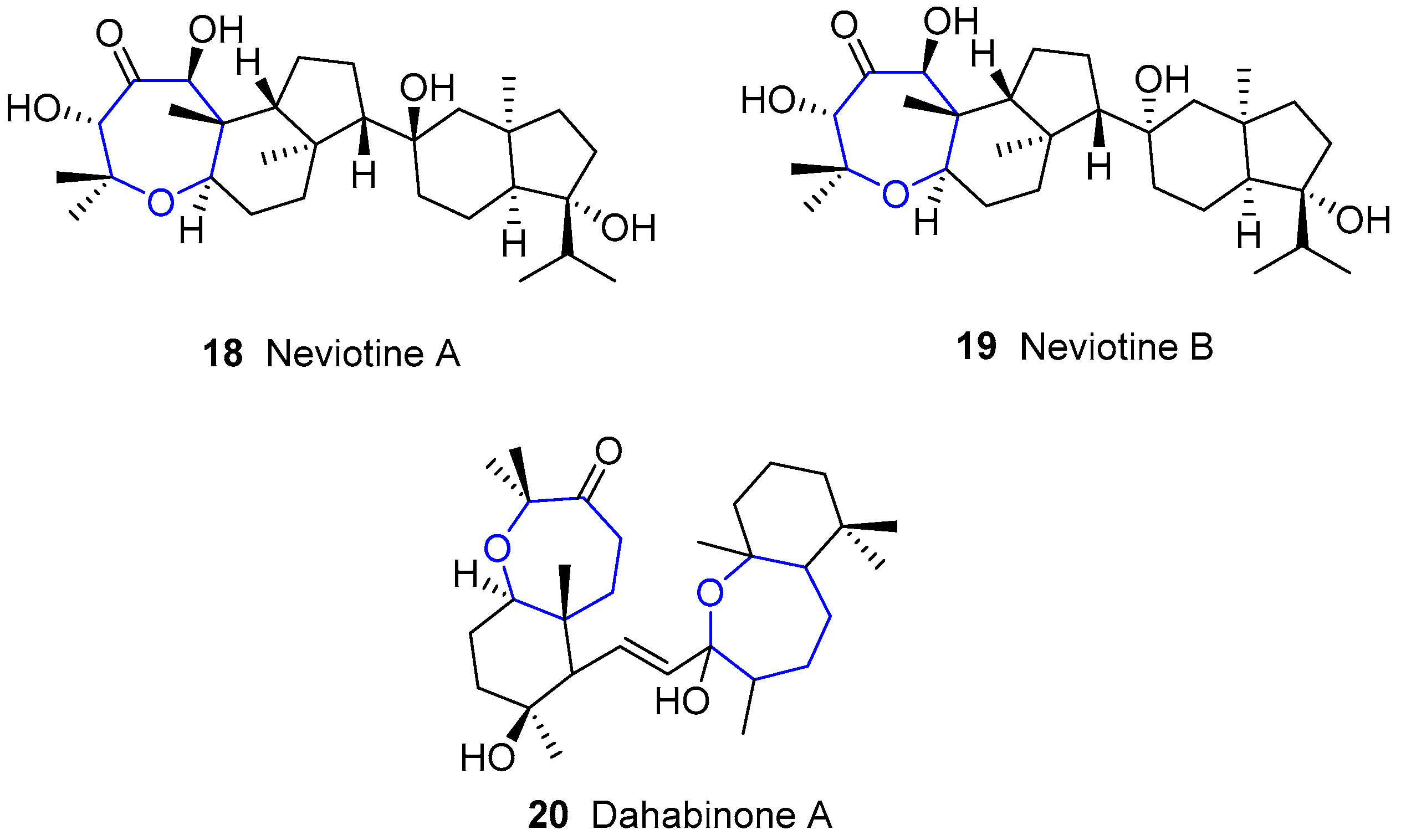
Figure 7.
Structure of Sodwanone A.
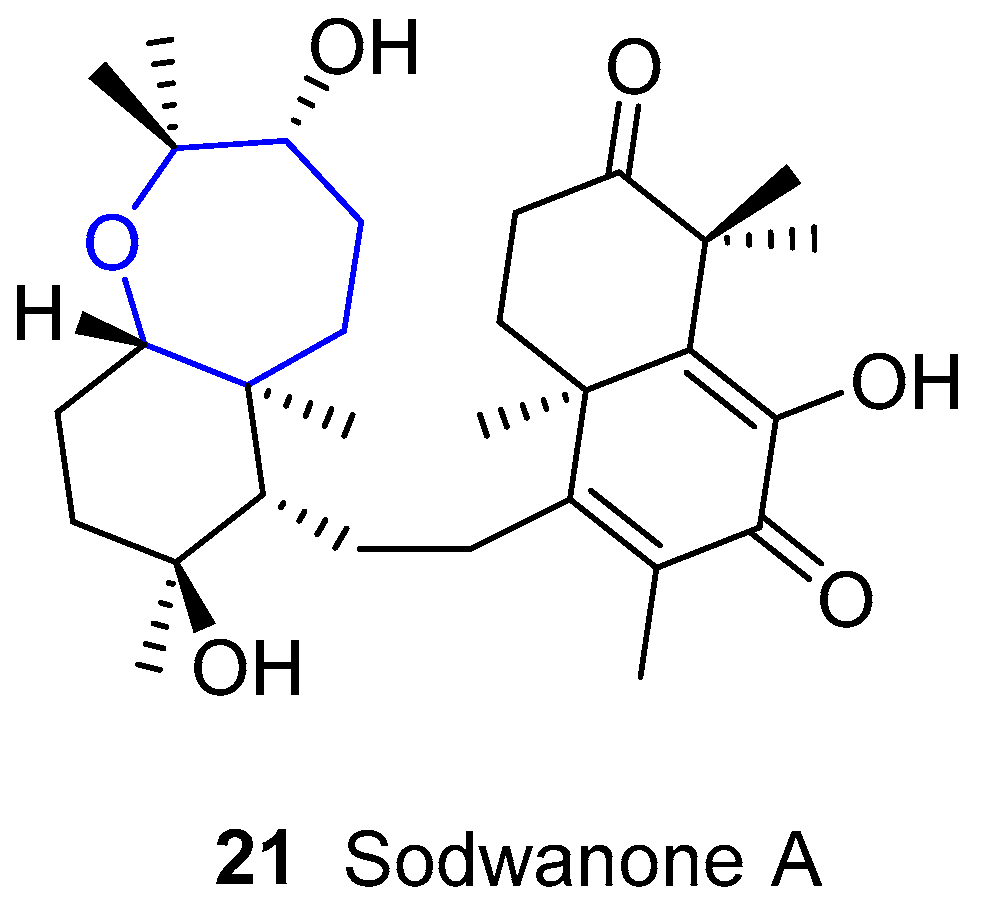
Figure 8.
Shaagrockols isolated by Kashman’s group.

Figure 9.
Structures of early raspacionins reported by Puliti and Madaio.
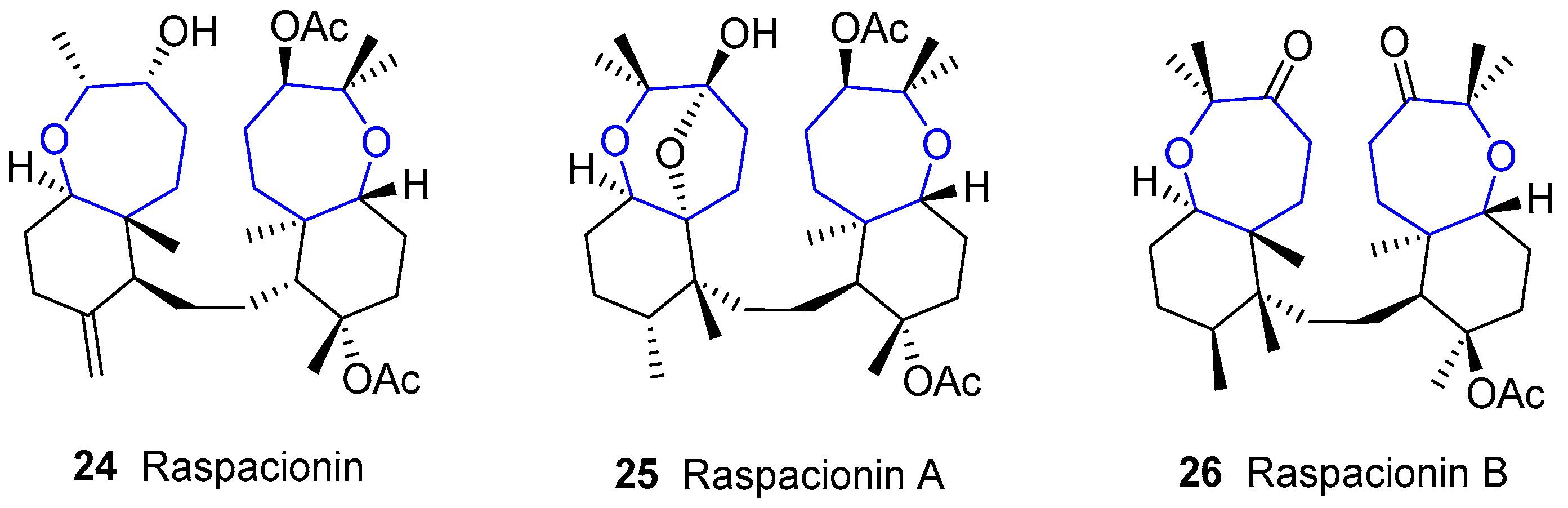
Figure 10.
The whole family of oxepane-containing austalides.

Scheme 4.
Retrosynthetic analysis towards (−)-Austalide B.
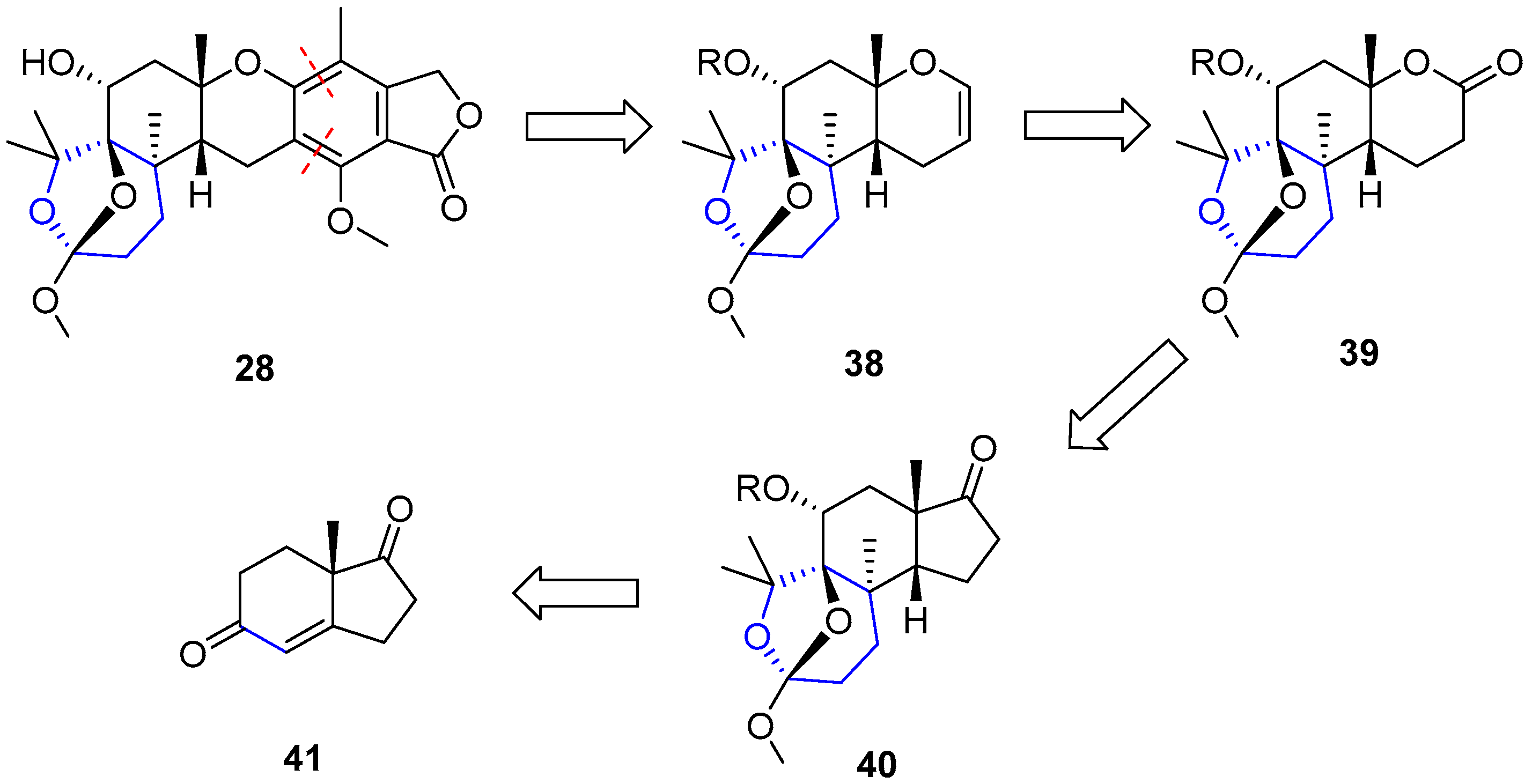
Scheme 5.
First steps towards the synthesis of the western section of (−)-Austalide B.
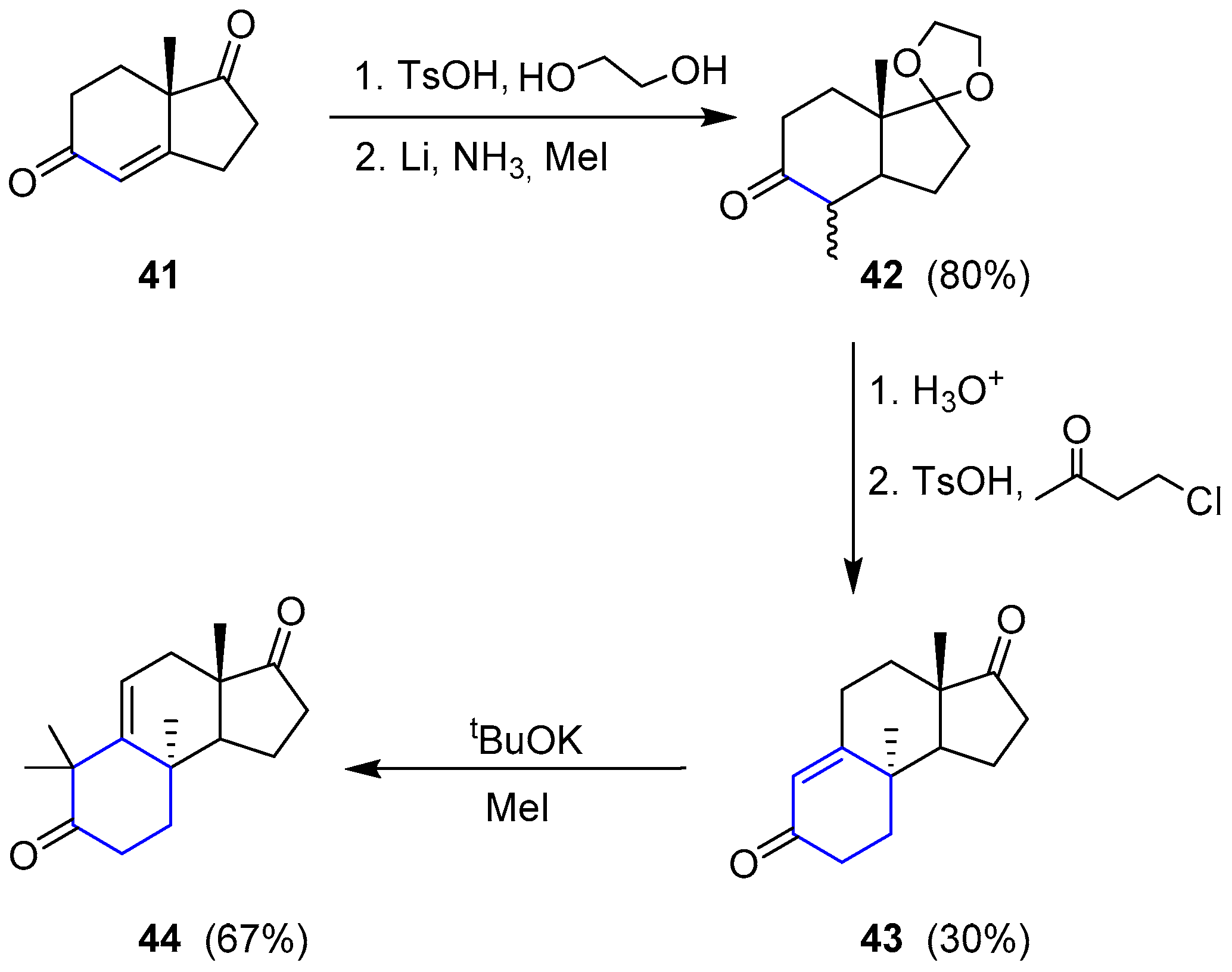
Scheme 6.
Second stage towards the synthesis of the western section of (−)-Austalide B.

Scheme 7.
Successful eastern section construction and final obtention of (−)-Austalide B.
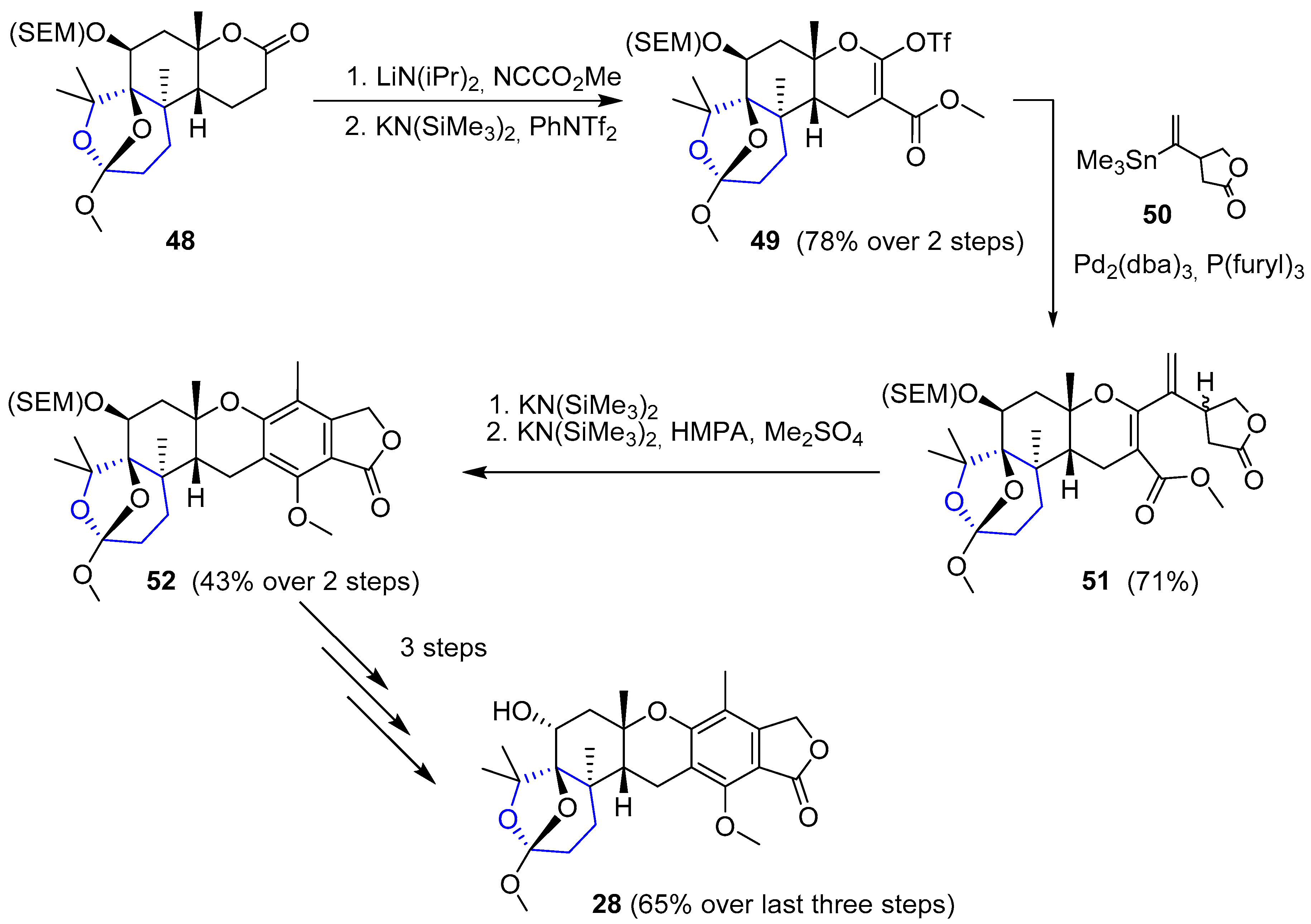
Figure 11.
Kashman’s assigned structure of psammaplysins A and B (wrong), and later correction by Scheuer, Clardy and coworkers.
Figure 11.
Kashman’s assigned structure of psammaplysins A and B (wrong), and later correction by Scheuer, Clardy and coworkers.
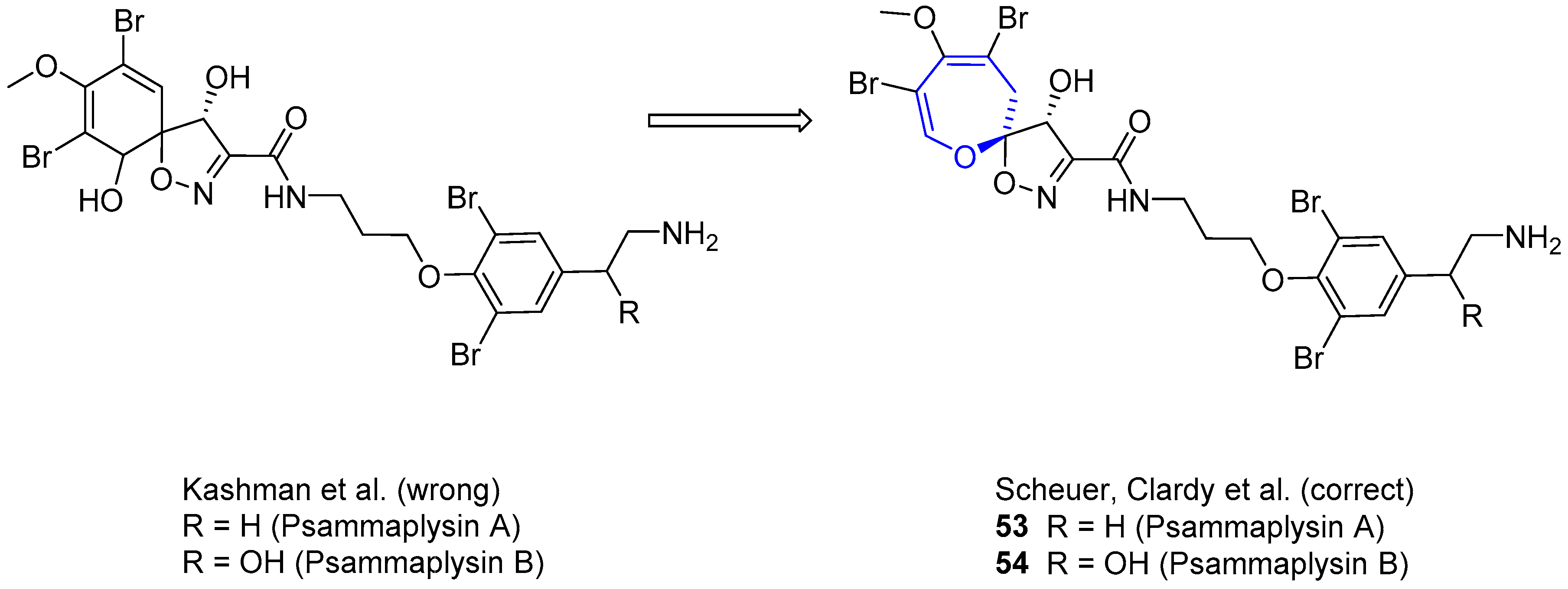
Figure 12.
Psammaplysins A–F and Ceratinamides A and B.
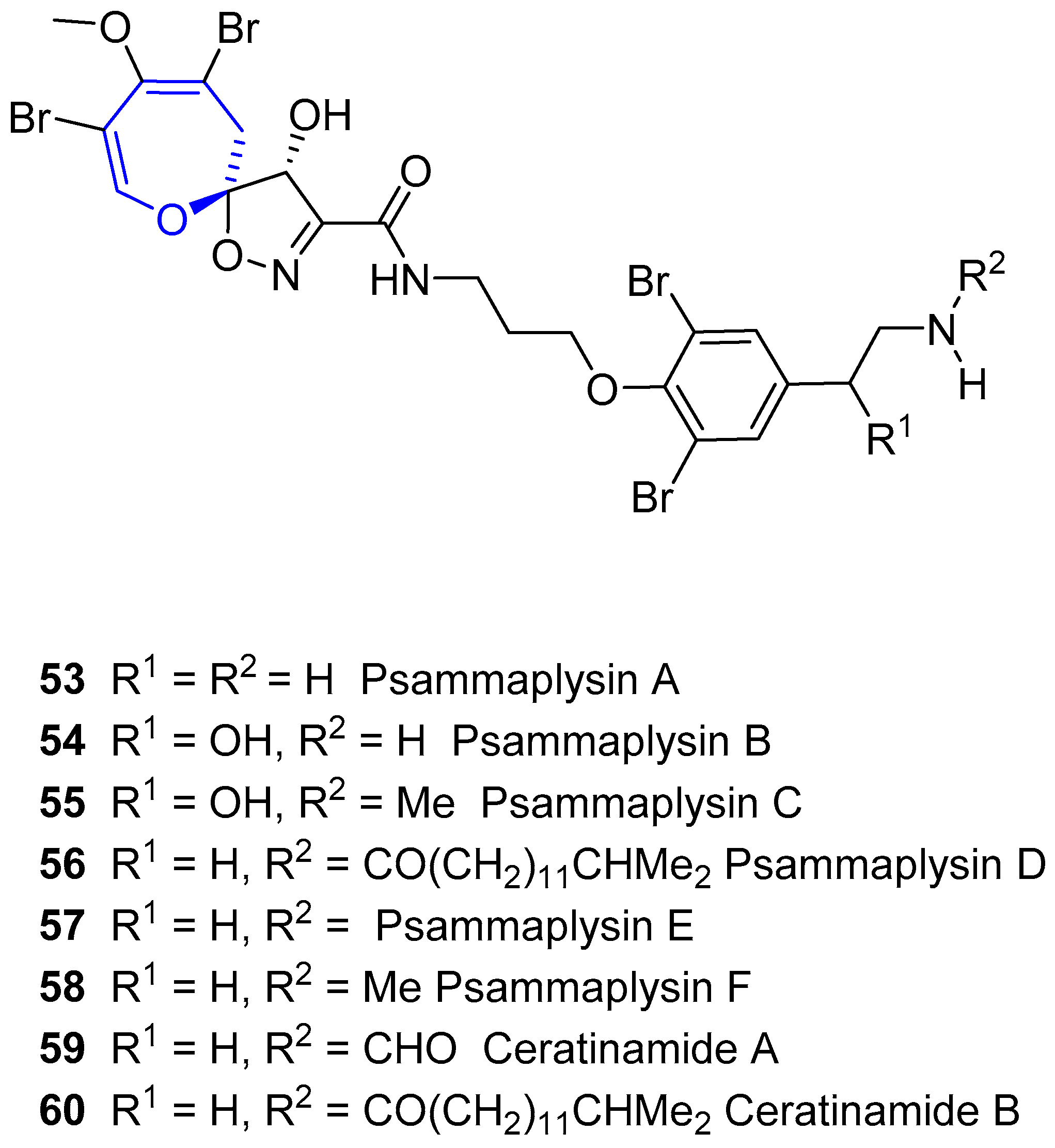
Figure 13.
Basic structures present in moloka’iamines, ceratinamines and psammaplysins. The spirooxepinisoxazoline moiety present in the latter (red square) could be the key for their anticancer properties.
Figure 13.
Basic structures present in moloka’iamines, ceratinamines and psammaplysins. The spirooxepinisoxazoline moiety present in the latter (red square) could be the key for their anticancer properties.

Figure 14.
Common scaffolds for oxepane-containing guanidinium alkaloids. Vessel units (61, 62 and 63); and typical anchor units (R2). Note that not all the structures are addressed here as R2 could contain other spermidine moieties or even being other functional groups (a simple alkyl chain, for instance). For a complete list of known compounds so far, see Ref. [121].
Figure 14.
Common scaffolds for oxepane-containing guanidinium alkaloids. Vessel units (61, 62 and 63); and typical anchor units (R2). Note that not all the structures are addressed here as R2 could contain other spermidine moieties or even being other functional groups (a simple alkyl chain, for instance). For a complete list of known compounds so far, see Ref. [121].

Scheme 8.
Retrosynthetic analysis towards Crambescidin 359 through a double 1,3-dipolar cycloaddition between a nitrone and two terminal olefins.
Scheme 8.
Retrosynthetic analysis towards Crambescidin 359 through a double 1,3-dipolar cycloaddition between a nitrone and two terminal olefins.
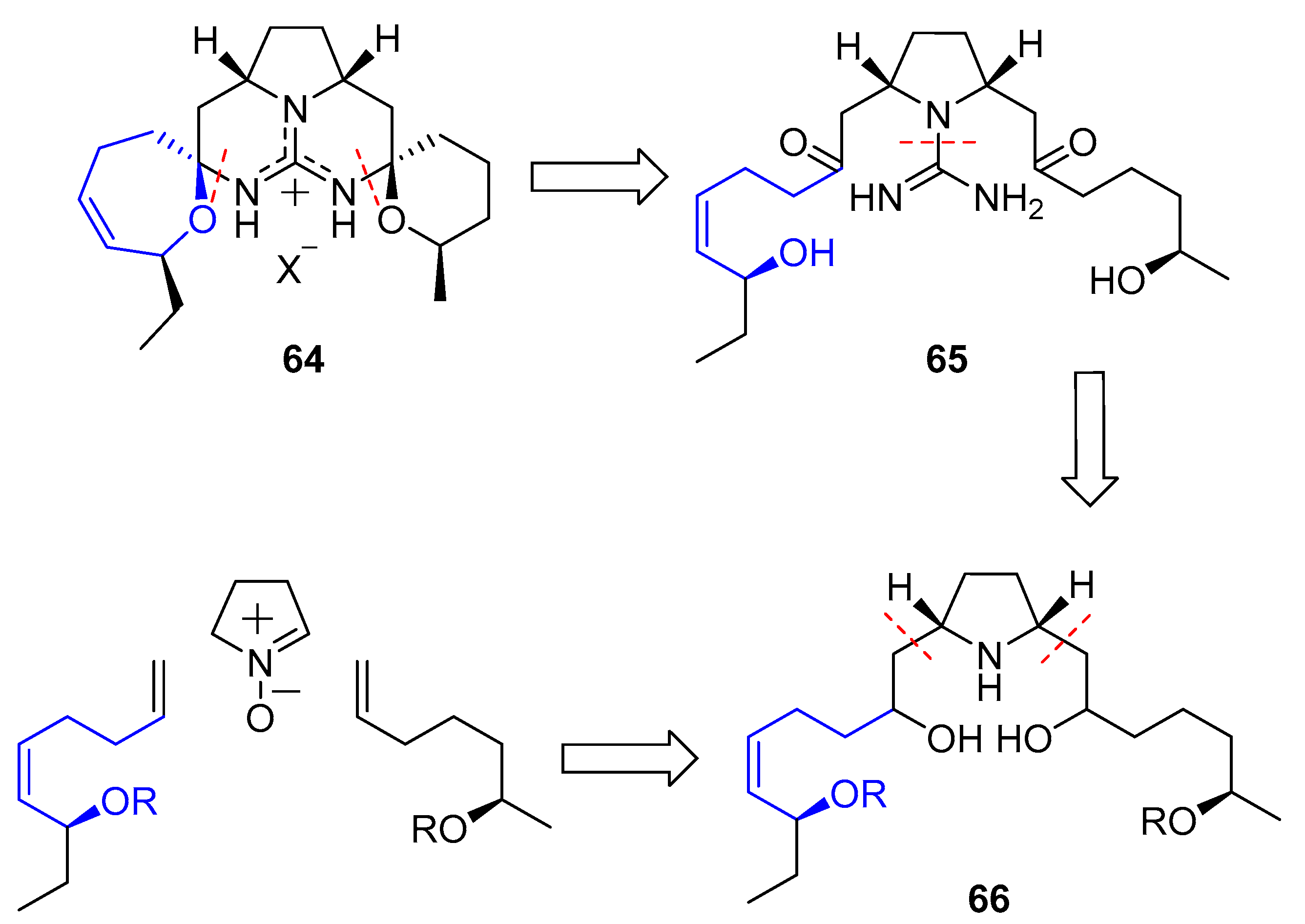
Scheme 9.
Summarized synthesis of Crambescidin 359 by Nagasawa’s group. a) Compound 70 was directly used for the next reaction and yield is not reported.
Scheme 9.
Summarized synthesis of Crambescidin 359 by Nagasawa’s group. a) Compound 70 was directly used for the next reaction and yield is not reported.
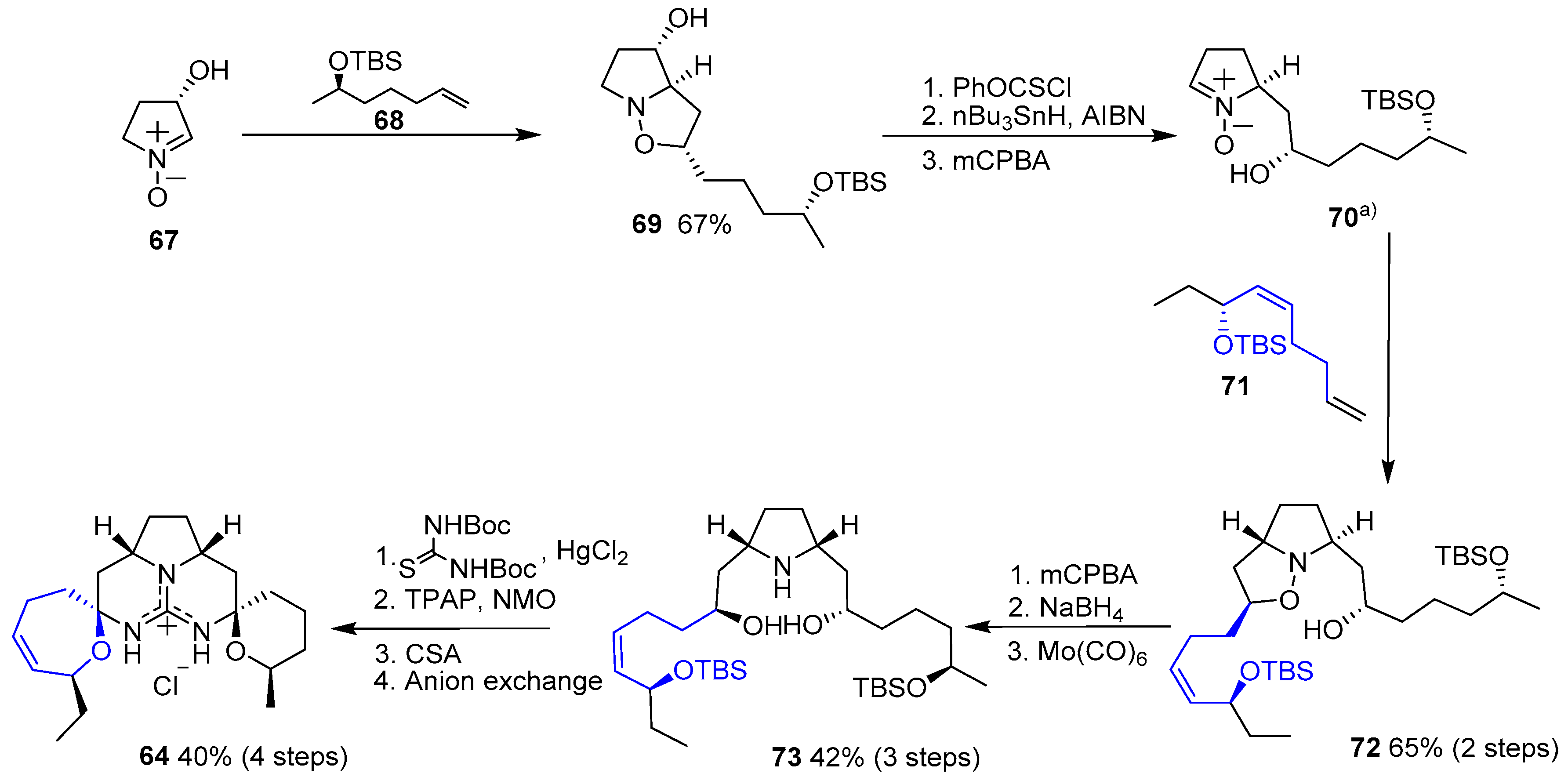
Scheme 10.
Alternative synthesis developed by Murphy towards Crambescidin 359.
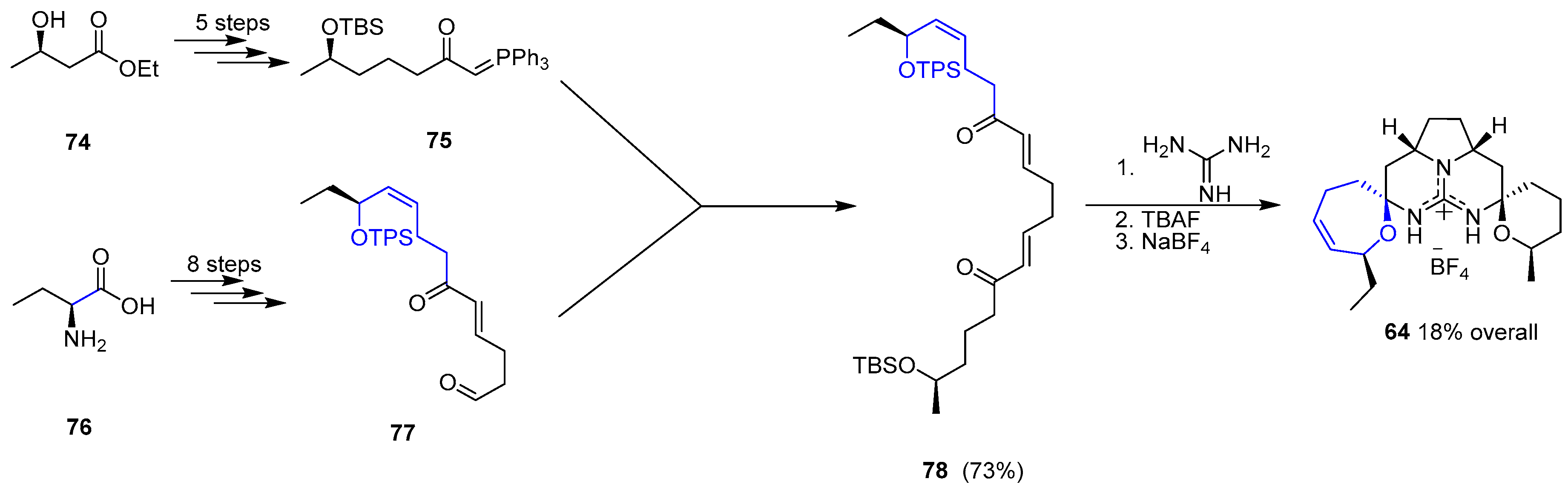
Scheme 11.
Overman’s synthesis of Crambescidin 359 upon decarboxylation of precursor 81.
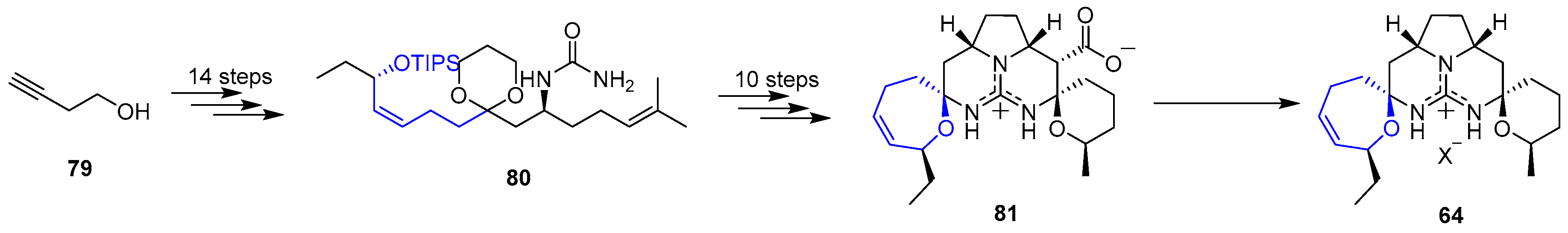
Figure 15.
Circumdatins A and B. Original zwitterionic structure proposals by Christophersen (Left), and revised definitive structures by Kusumi (Right).
Figure 15.
Circumdatins A and B. Original zwitterionic structure proposals by Christophersen (Left), and revised definitive structures by Kusumi (Right).

Figure 16.
Chemical structures of Oxepinamides A–C isolated by Belofsky and Köck.
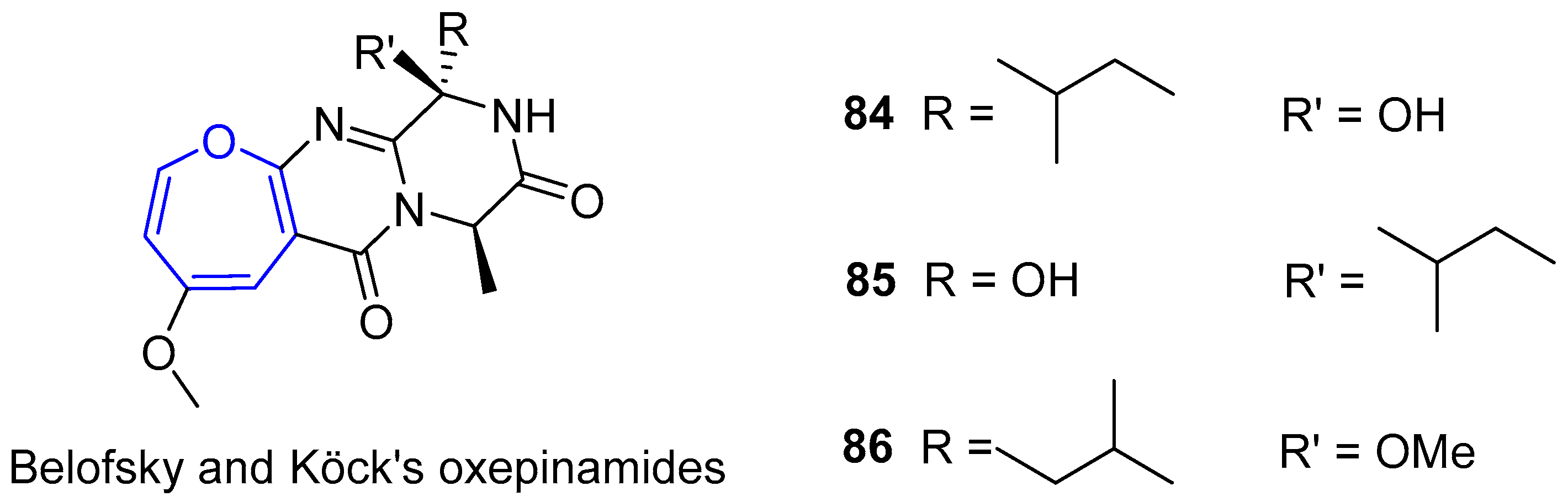
Figure 17.
Comparison of Cinereanin (Left) and Janoxepin (Right) structures.

Scheme 12.
Retrosynthetic analysis of Janoxepin.
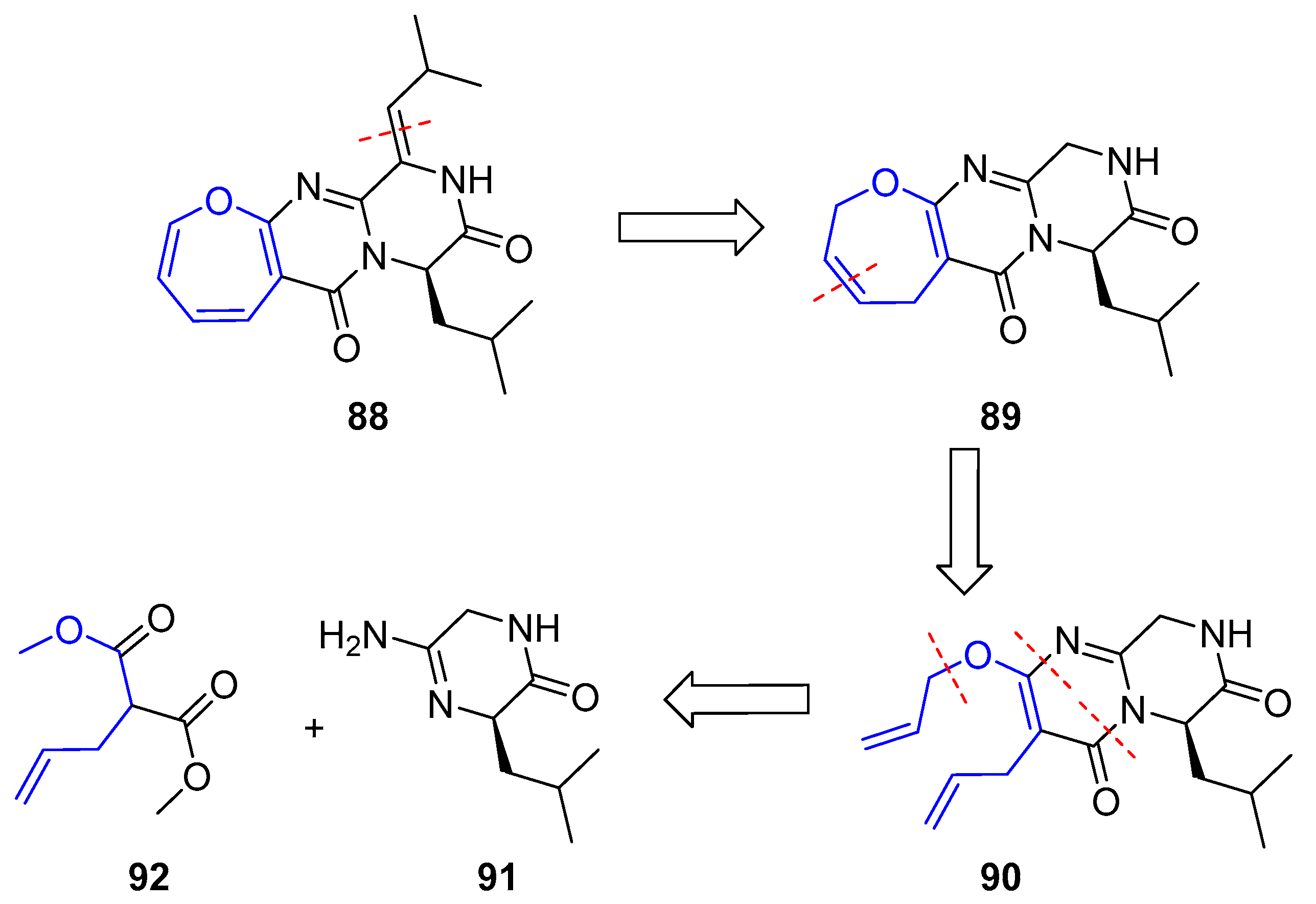
Scheme 13.
Main sequence towards diallylated pyrimidone 90 starting from N-Boc-d-leucine 93.
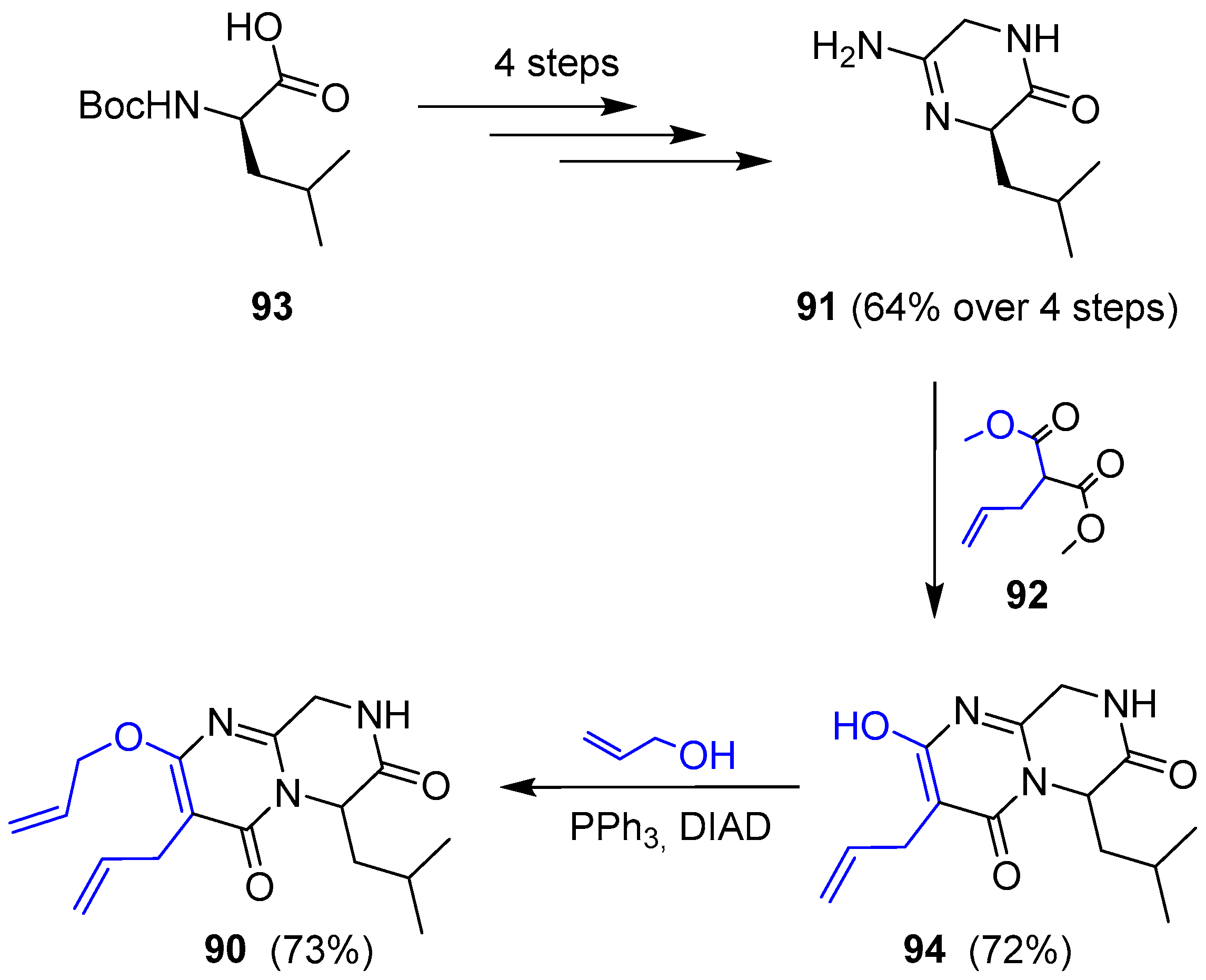
Scheme 14.
Synthesis sequence towards dihydro-oxepin and side chain incorporation.

Scheme 15.
Final steps towards (±)-Janoxepin (88).
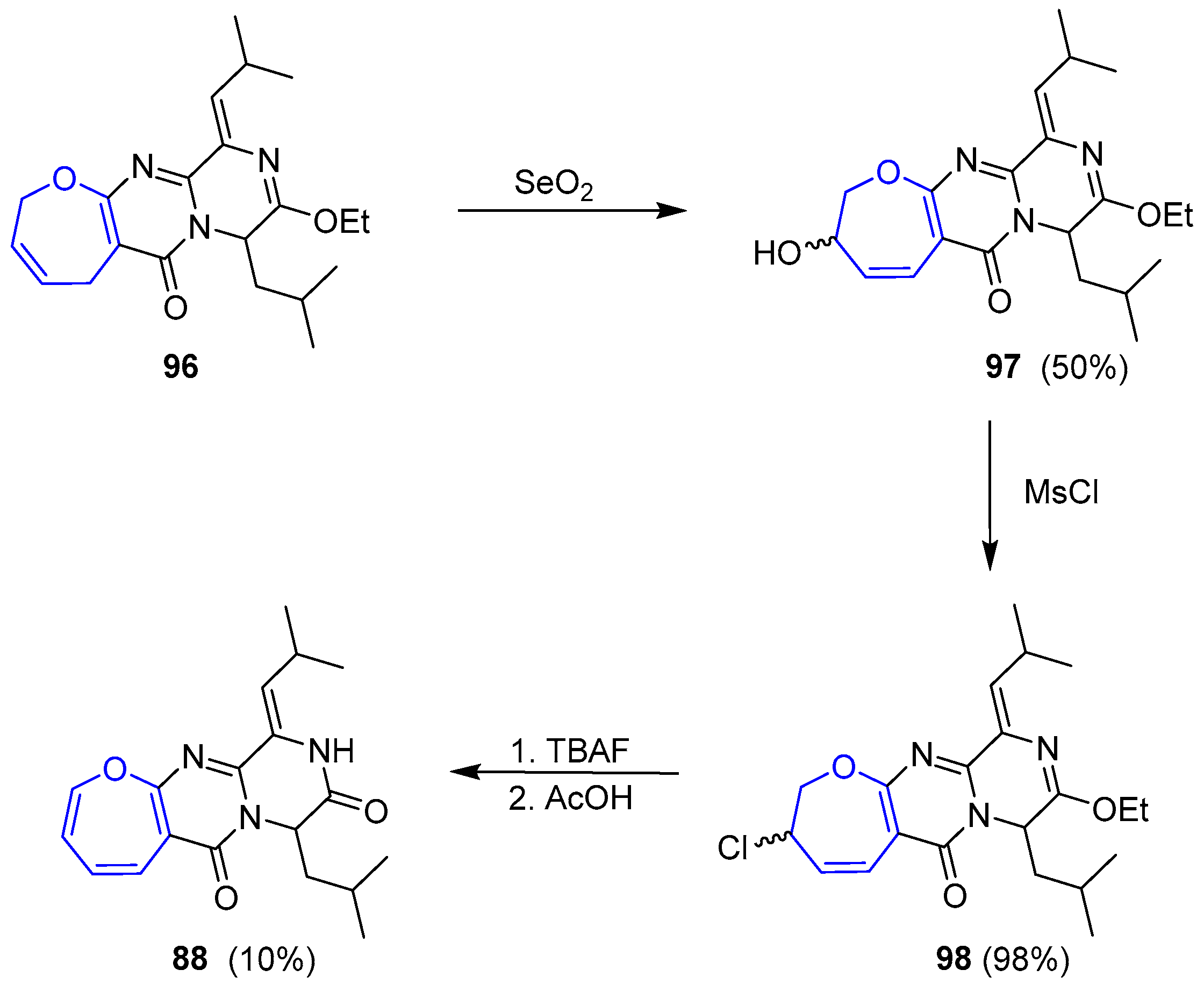
Figure 18.
Protuboxepins A and B from marine fungus Aspergillus sp. SF-5044.

Figure 19.
Latest oxepinamides. (101) Varioxepin A. (102) and (103) oxepinamides from Aecilomyces variotii EN-291. (104) and (105) oxepinamides from Aspergillus versicolor SCSIO 05879. (106) oxepinamides from vent-crab associated Aspergillus versicolor XZ-4.
Figure 19.
Latest oxepinamides. (101) Varioxepin A. (102) and (103) oxepinamides from Aecilomyces variotii EN-291. (104) and (105) oxepinamides from Aspergillus versicolor SCSIO 05879. (106) oxepinamides from vent-crab associated Aspergillus versicolor XZ-4.
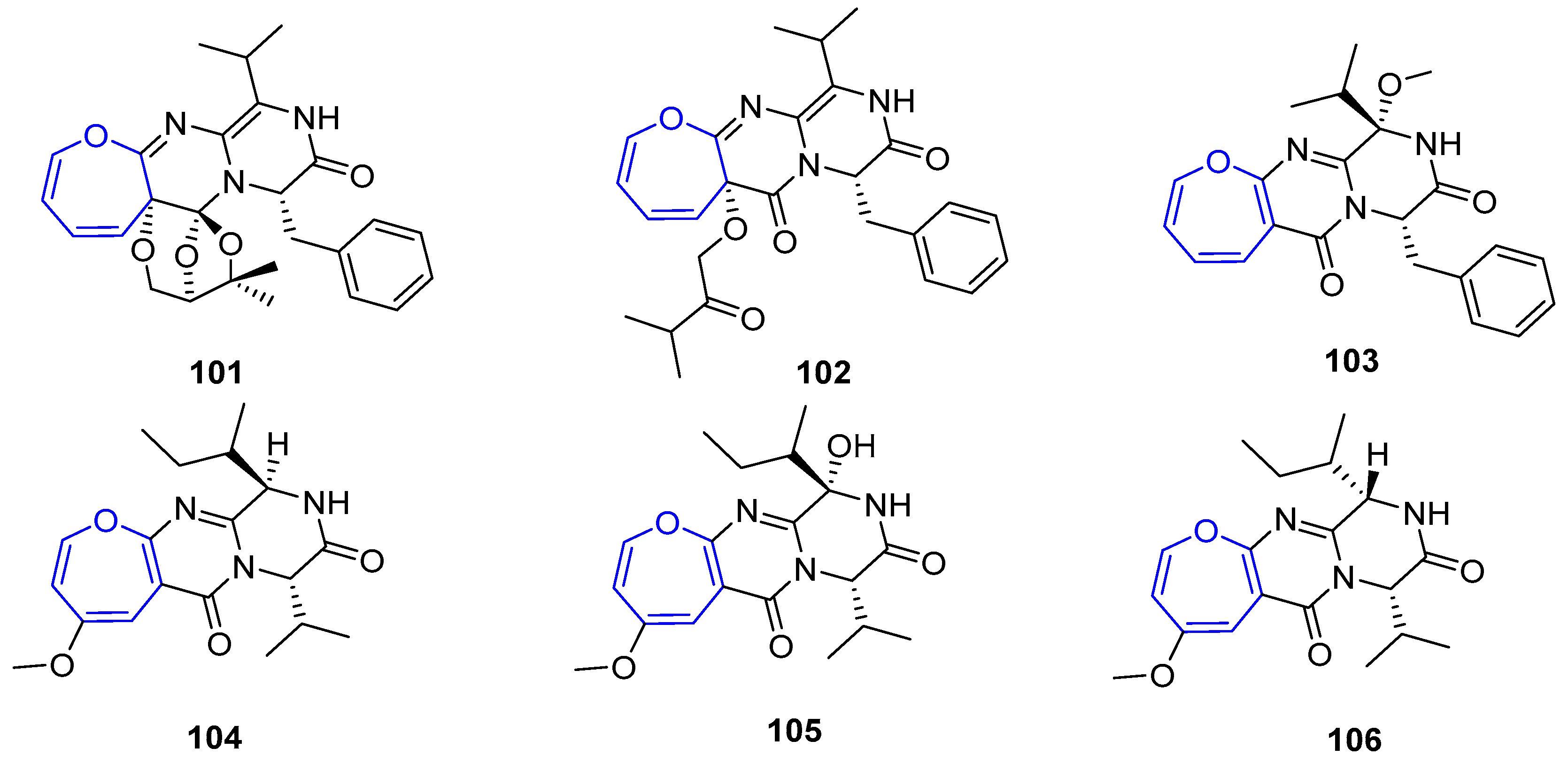
Figure 20.
First set of oxepin-containing diketopiperazine disulfides isolated from fungus Arachniotus aureus.
Figure 20.
First set of oxepin-containing diketopiperazine disulfides isolated from fungus Arachniotus aureus.

Figure 21.
Emestrin and its acetylated derivative isolated from Emericella striata.
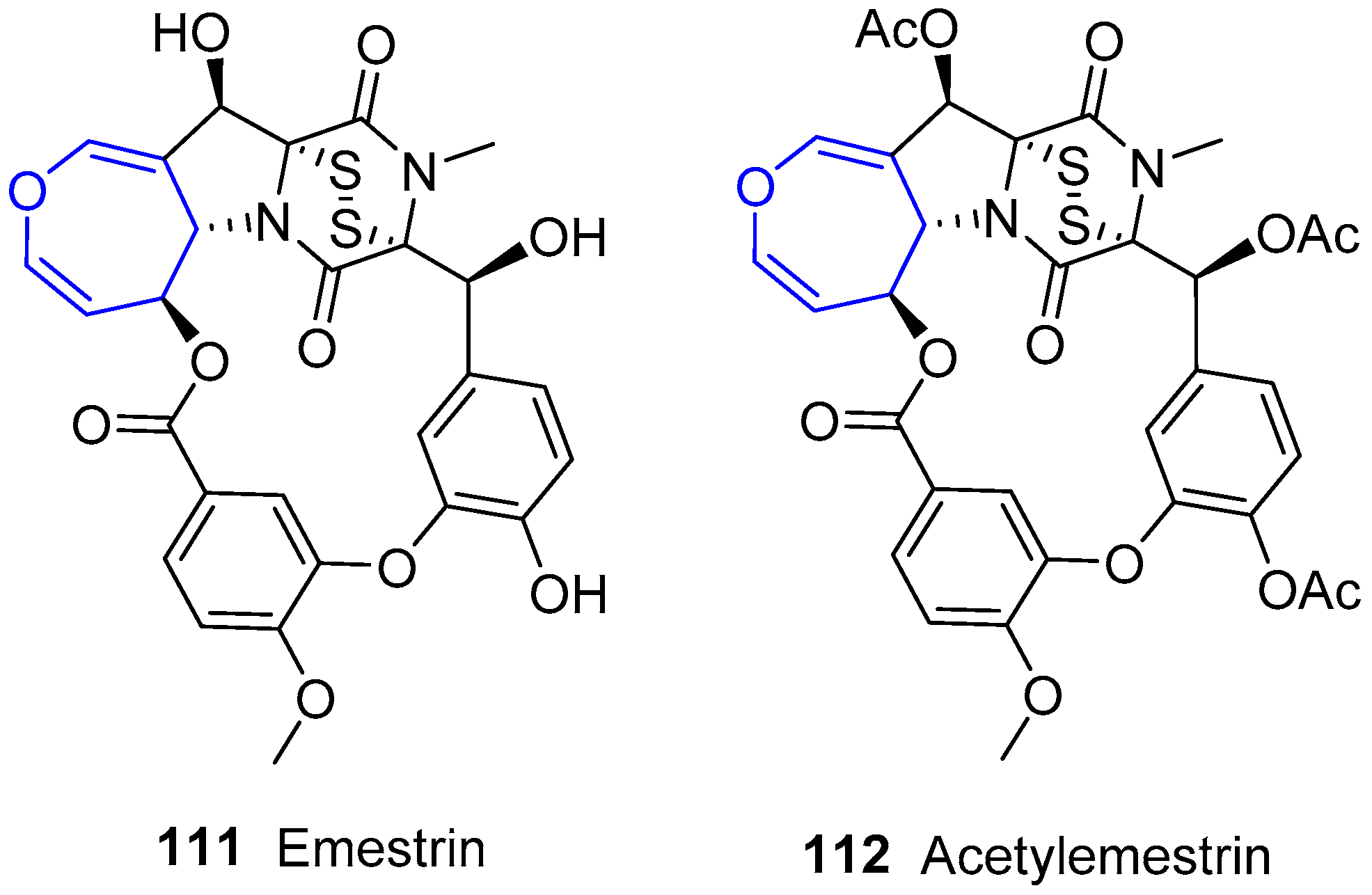
Figure 22.
Structures of oxepin-containing emethallicins, their acetylated forms being R = OAc.
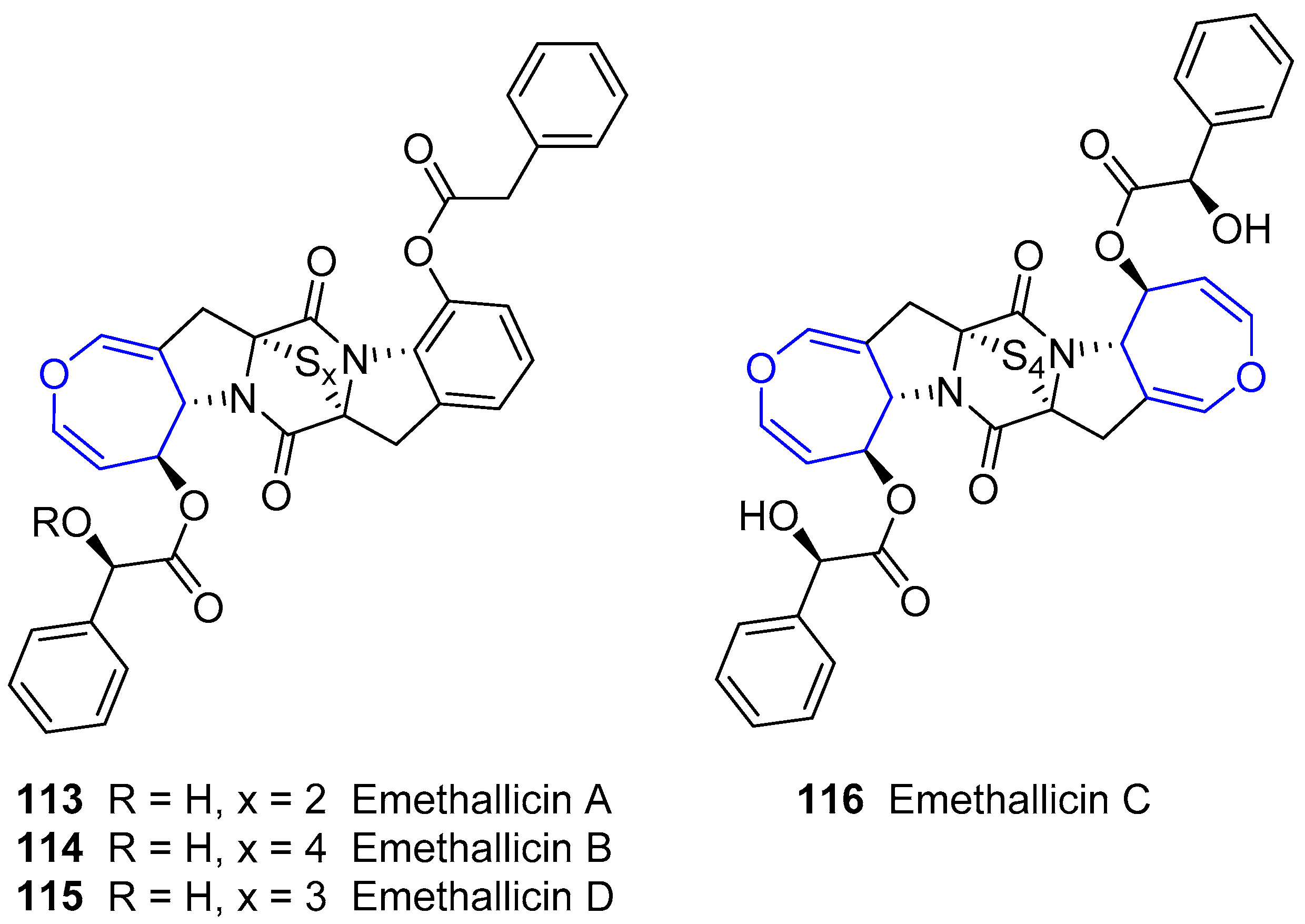
Scheme 16.
Retrosynthetic analysis of (−)-Acetylaranotin.
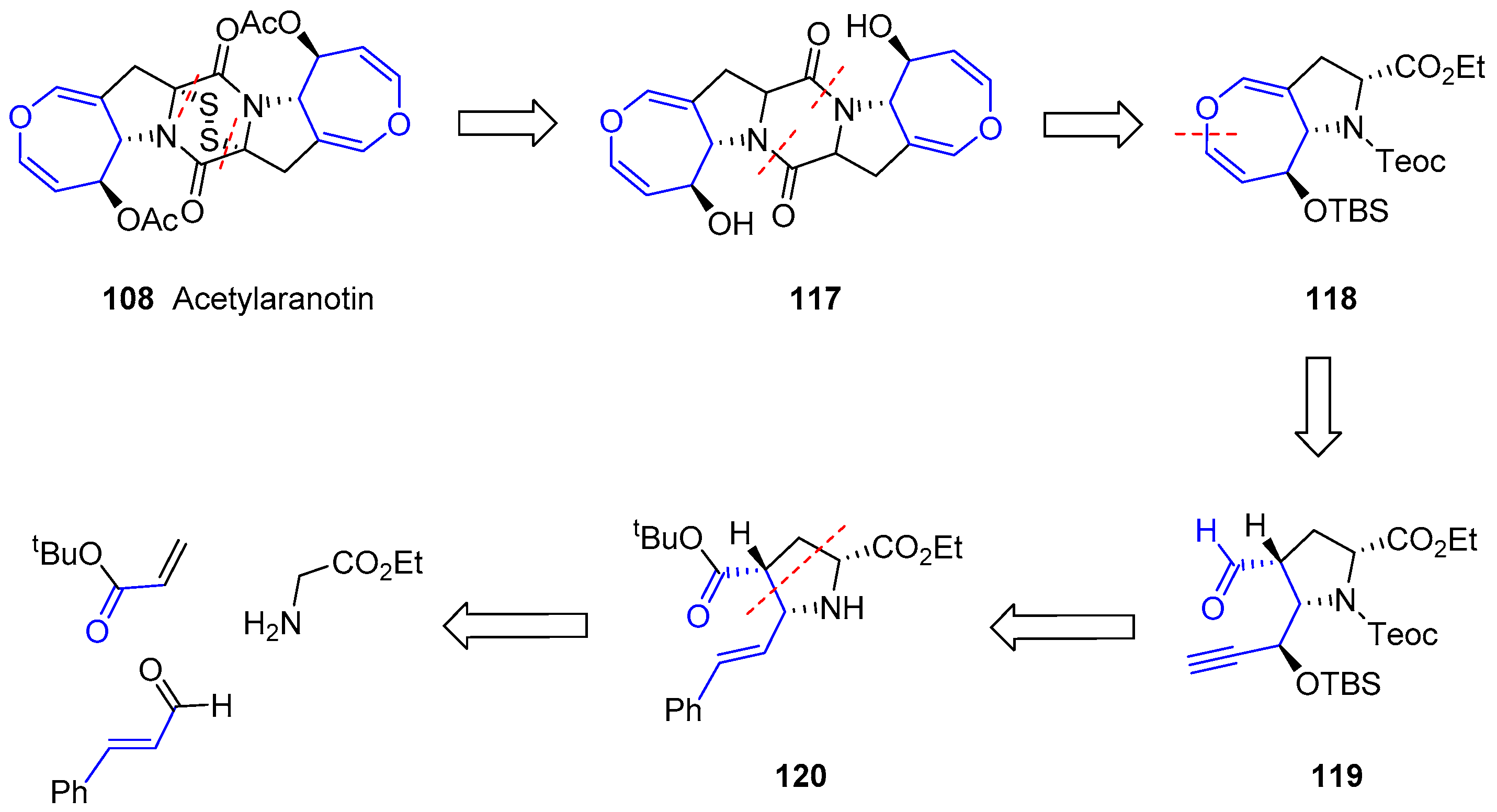
Scheme 17.
First stage of Acetylaranotin preparation. Synthetic sequence towards alkynol proline 123.
Scheme 17.
First stage of Acetylaranotin preparation. Synthetic sequence towards alkynol proline 123.
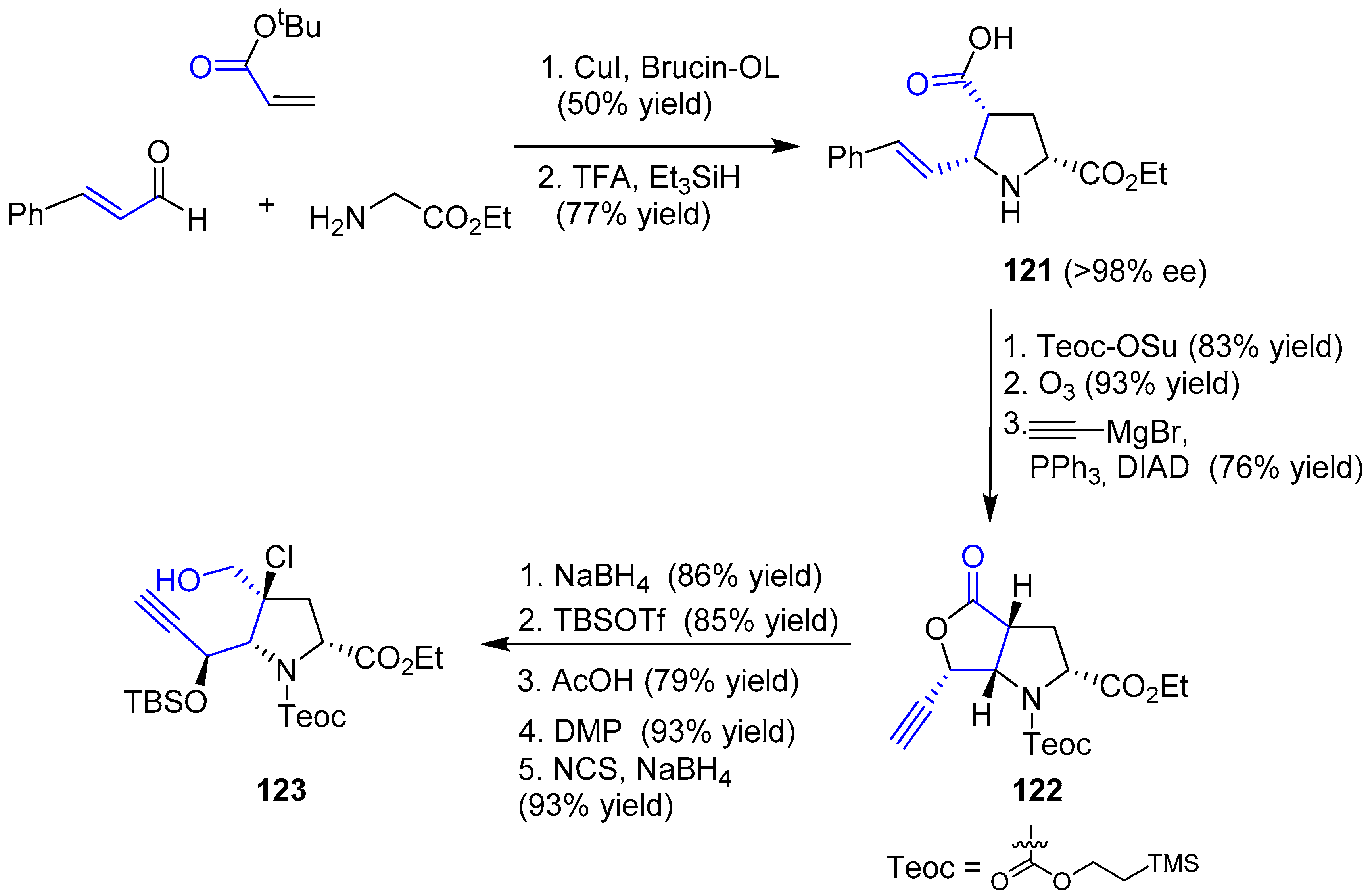
Scheme 18.
Second stage of Acetylaranotin preparation.
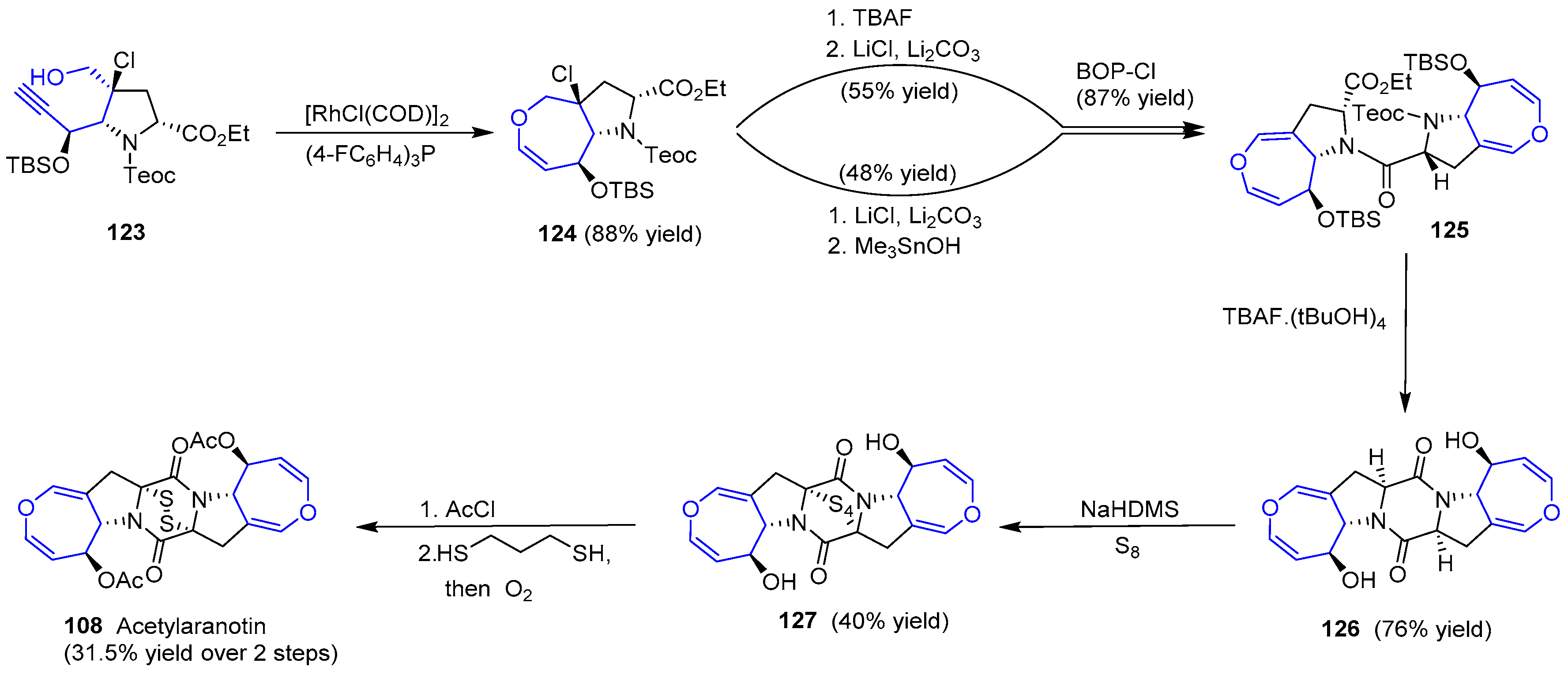
Scheme 19.
Retrosynthetic analyses for the formation of MPC-1001B. Note that compound 129 is considered a key intermediate present in both schemes. Anisic acid derivatives 130 and 131 were easily prepared.
Scheme 19.
Retrosynthetic analyses for the formation of MPC-1001B. Note that compound 129 is considered a key intermediate present in both schemes. Anisic acid derivatives 130 and 131 were easily prepared.
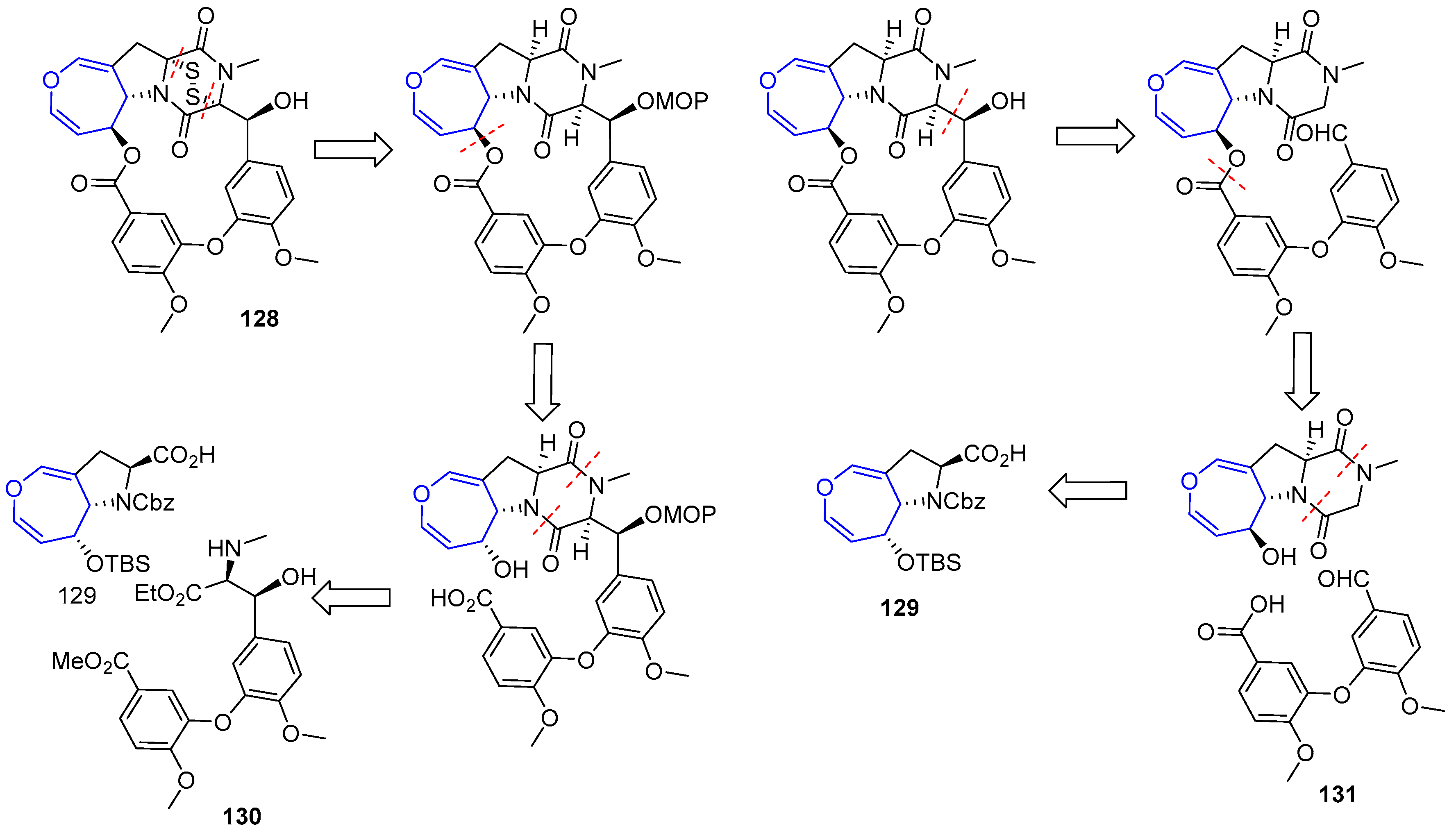
Scheme 20.
First unfruitful approach towards 15-membered macrolactone 133.
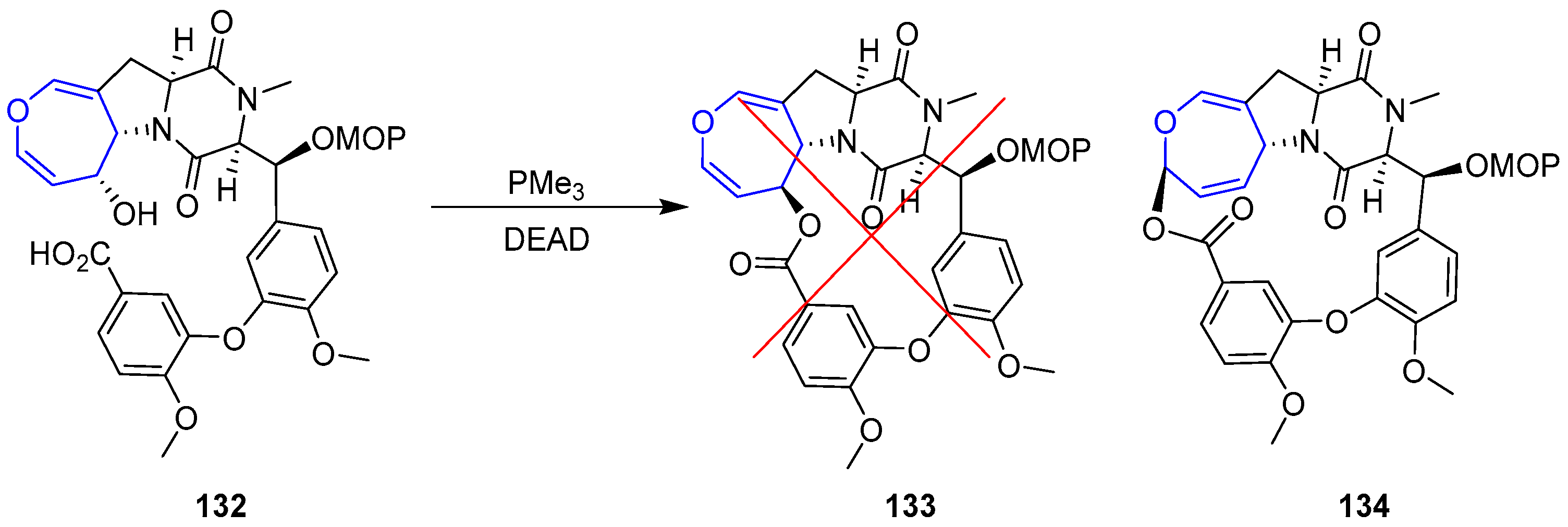
Scheme 21.
Total synthesis of (+)-MPC10001B (128).
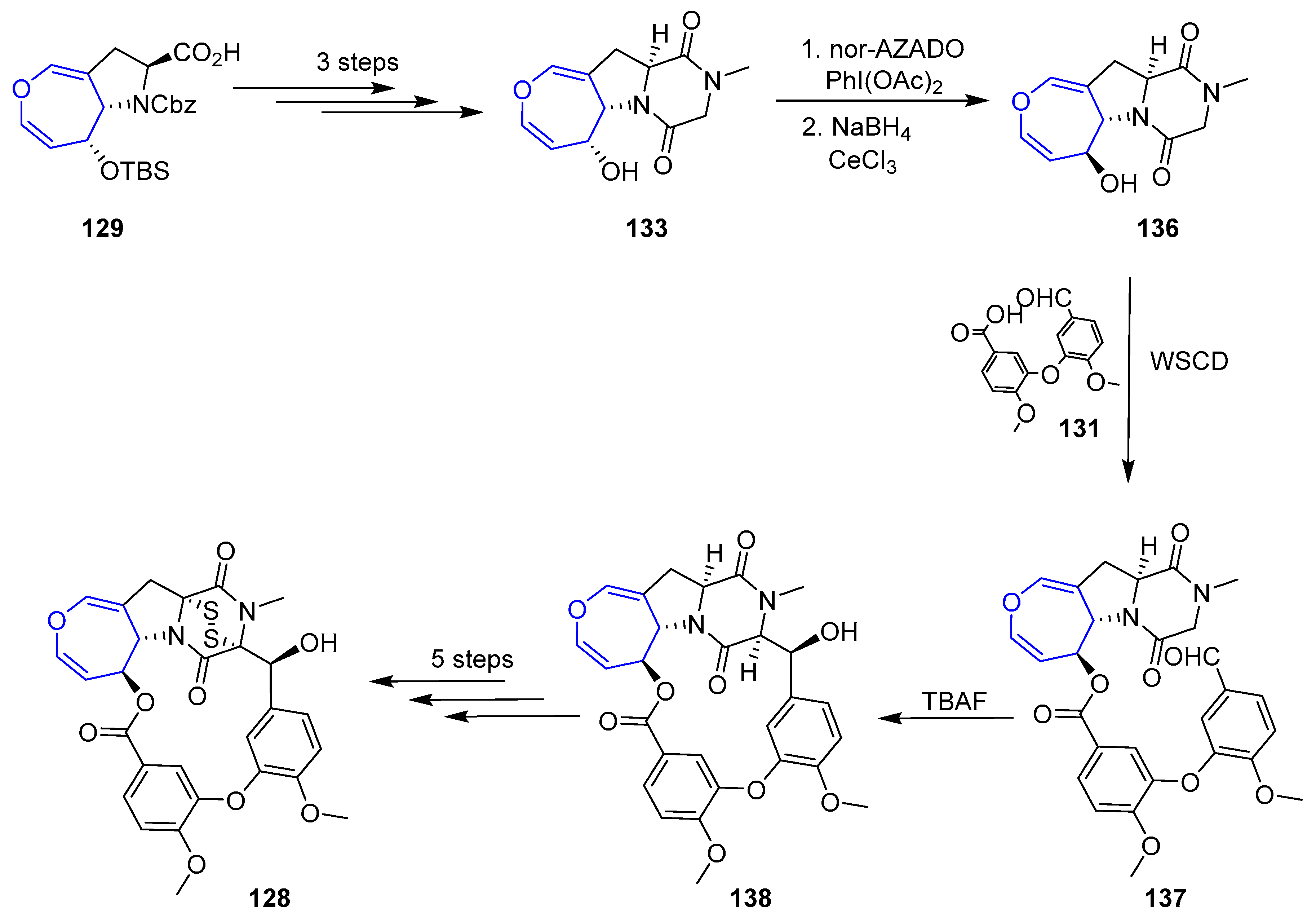
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barbero, H.; Díez-Poza, C.; Barbero, A. The Oxepane Motif in Marine Drugs. Mar. Drugs 2017, 15, 361. https://doi.org/10.3390/md15110361
AMA Style
Barbero H, Díez-Poza C, Barbero A. The Oxepane Motif in Marine Drugs. Marine Drugs. 2017; 15(11):361. https://doi.org/10.3390/md15110361
Chicago/Turabian StyleBarbero, Héctor, Carlos Díez-Poza, and Asunción Barbero. 2017. "The Oxepane Motif in Marine Drugs" Marine Drugs 15, no. 11: 361. https://doi.org/10.3390/md15110361
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.