Three New Malyngamides from the Marine Cyanobacterium Moorea producens
by
 , and
, and
Kosuke Sueyoshi
1, 
Aki Yamano
1,
Kaori Ozaki
1,
Shimpei Sumimoto
2,
Arihiro Iwasaki
2,
Kiyotake Suenaga
2 and
Toshiaki Teruya
1,* 1
Faculty of Education, University of the Ryukyus, 1 Senbaru, Nishihara, Okinawa 903-0213, Japan
2
Department of Chemistry, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, Kanagawa 223-8522, Japan
*
Author to whom correspondence should be addressed.
Mar. Drugs 2017, 15(12), 367; https://doi.org/10.3390/md15120367
Submission received: 28 September 2017
/
Revised: 2 November 2017
/
Accepted: 16 November 2017
/
Published: 29 November 2017
(This article belongs to the Special Issue Marine Bioactive Natural Product Studies in Asia)
Abstract
:Three new compounds of the malyngamide series, 6,8-di-O-acetylmalyngamide 2 (1), 6-O-acetylmalyngamide 2 (2), and N-demethyl-isomalyngamide I (3), were isolated from the marine cyanobacterium Moorea producens. Their structures were determined by spectroscopic analysis and chemical derivatization and degradation. These compounds stimulated glucose uptake in cultured L6 myotubes. In particular, 6,8-di-O-acetylmalyngamide 2 (1) showed potent activity and activated adenosine monophosphate-activated protein kinase (AMPK).
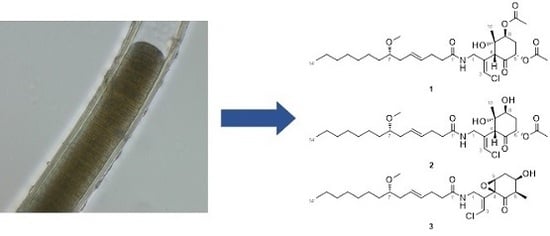
1. Introduction
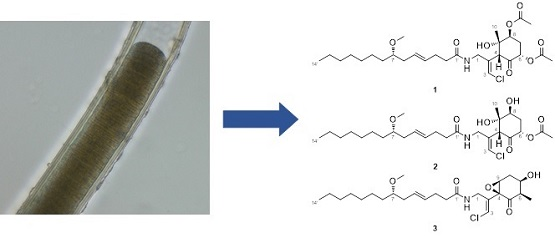
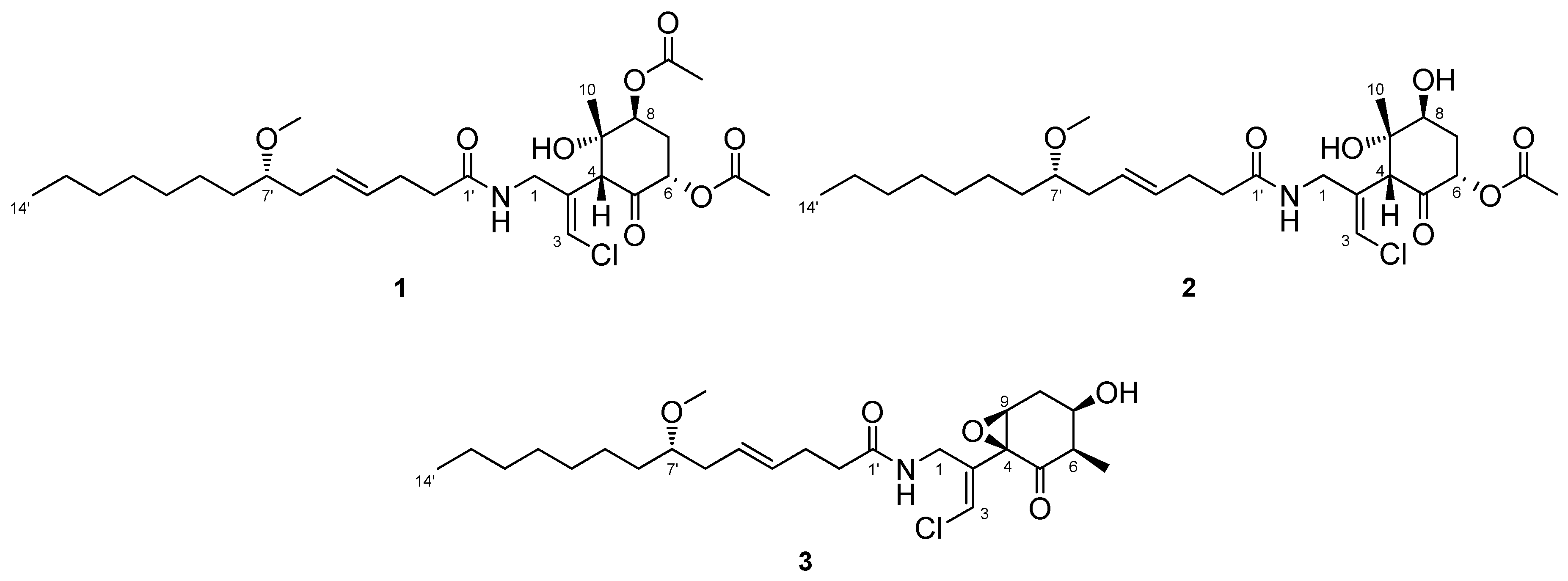
The ocean covers more than 70% of the Earth’s surface and hosts huge biological and chemical diversity. Because marine environmental conditions are quite different from terrestrial ones, natural products from marine organisms have unique structures and biological activities. Marine cyanobacteria, in particular, are known to produce various secondary metabolites and have been recognized as a source of pharmaceutical lead compounds [1,2,3]. For example, bisebromoamide, isolated from Lyngbya sp., showed potent cytotoxicity against HeLa S3 cells [4]. Bisebromoamide inhibited the phosphorylation of extracellular signal-regulated protein kinase (ERK) and was identified as an actin filament stabilizer [5]. Meanwhile, hoshinolactam was found to possess both a cyclopropane ring and γ-lactam ring, and exhibited potent antitrypanosomal activity without cytotoxicity against human fetal lung fibroblast MRC-5 cells [6]. The malyngamide series of natural products have been isolated from various marine filamentous cyanobacteria. Malynagmide A, the first compound of this group, was isolated from Lyngbya majuscule in 1979 [7]; since then, over 30 malyngamide analogs have been isolated [8]. As part of our ongoing effort to identify novel bioactive natural products, we have focused on the constituents of marine cyanobacteria and isolated odoamide [9,10] and odobromoamide [11]. We recently discovered three new malyngamides, 6,8-di-O-acetylmalyngamide 2 (1), 6-O-acetylmalyngamide 2 (2), and N-demethyl-isomalyngamide I (3), from the Okinawan cyanobacterium belonging to the genus Moorea producens. (Figure 1). Herein, we report the isolation, structure determination, and biological evaluation of these compounds.
2. Results
2.1. 6,8-Di-O-Acetylmalyngamide 2 (1) and 6-O-Acetylmalyngamide 2 (2)
The marine cyanobacterium M. producens (30 g, wet weight) was collected at Bise, Okinawa Prefecture, Japan, and extracted with methanol. The extract was filtered and concentrated, and then the residue was partitioned between EtOAc and H2O. The organic layer was further partitioned between 90% aqueous MeOH and n-hexane. The material obtained from the 90% aqueous portion was fractionated by octadecylsilyl (ODS) column chromatography and subjected to reversed-phase high-performance liquid chromatography (HPLC) to give compound 1 (12.0 mg), 2 (26.8 mg), and 3 (12.6 mg).
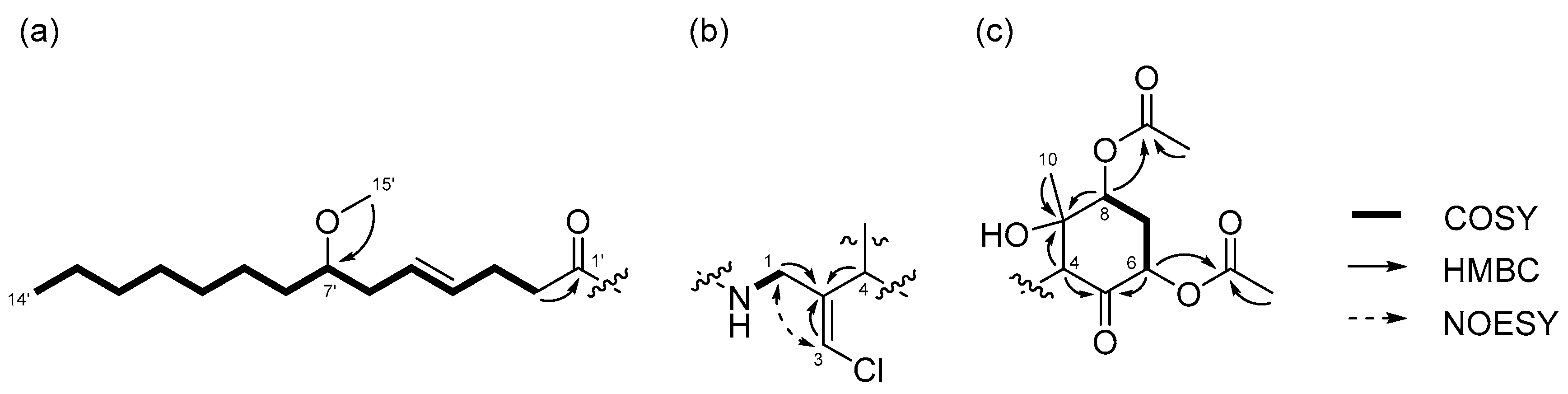
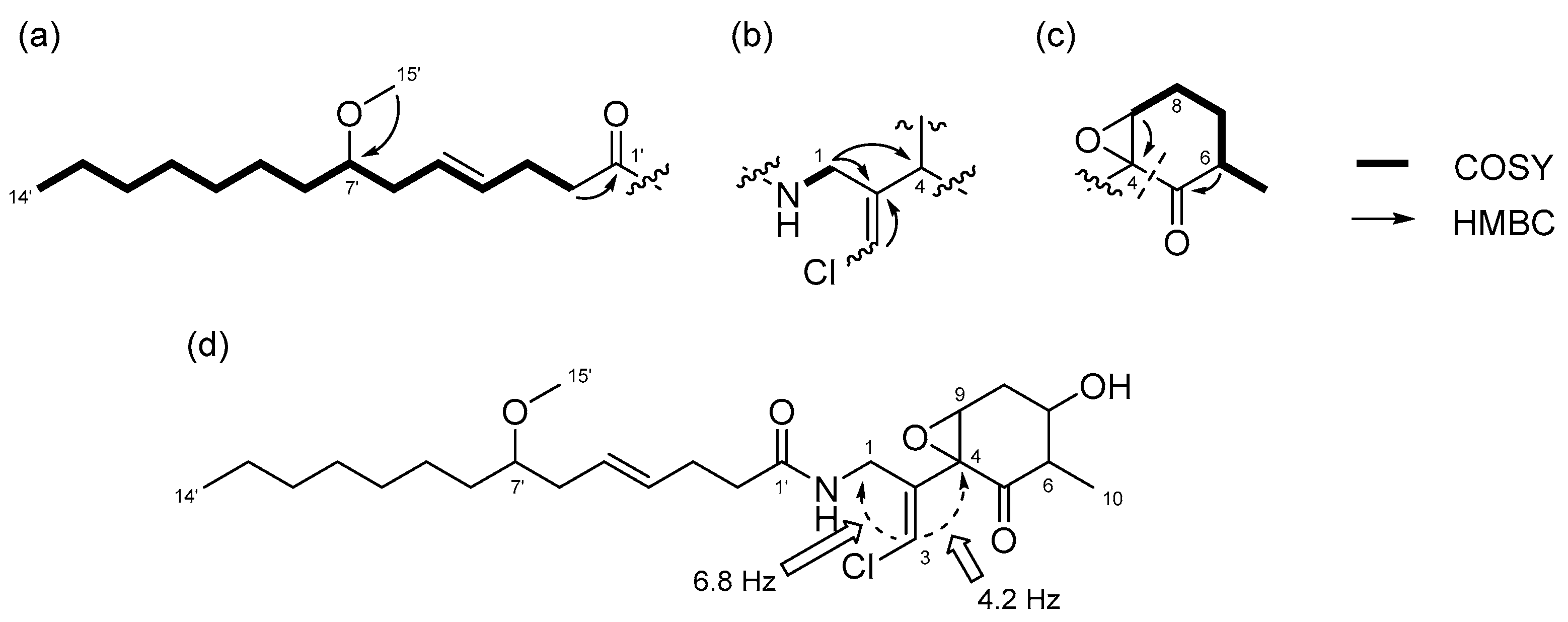
Compound 1 was obtained as a colorless oil. The molecular formula of 1 was determined to be C29H46ClNO8 on the basis of the 13C NMR spectrum (29 carbon signals, Figure S2) and high-resolution electrospray ionization mass spectrometry (HRESIMS) (m/z 572.2989 [M + H]+, calcd. 572.2985). The nuclear magnetic resonance (NMR) data for 1 are summarized in Table 1. These data indicated the presence of a secondary amide (δH 6.68, δC 173.8), disubstituted olefin (δH 5.45, 5.47, δC 128.3, 130.7), vinyl chloride (δH 6.29, δC 121.6) [12], and O-methyl group (δH 3.31, δC 56.5), which are characteristic resonances of the malyngamide series [7,13,14,15]. A detailed 2D NMR analysis using COSY, HSQC, and HMBC data (Figures S3–S5) revealed the presence of 7-methoxytetradec-4(E)-enoic acid (lyngbic acid, Figure 2a). The E-geometry of the C-4’/C-5’ olefin was assigned based on the comparison of the 13C NMR chemical shifts of C-3’ (δC 28.7) and C-6’ (δC 36.3) with those of other known malyngamides [7,13,14,15].
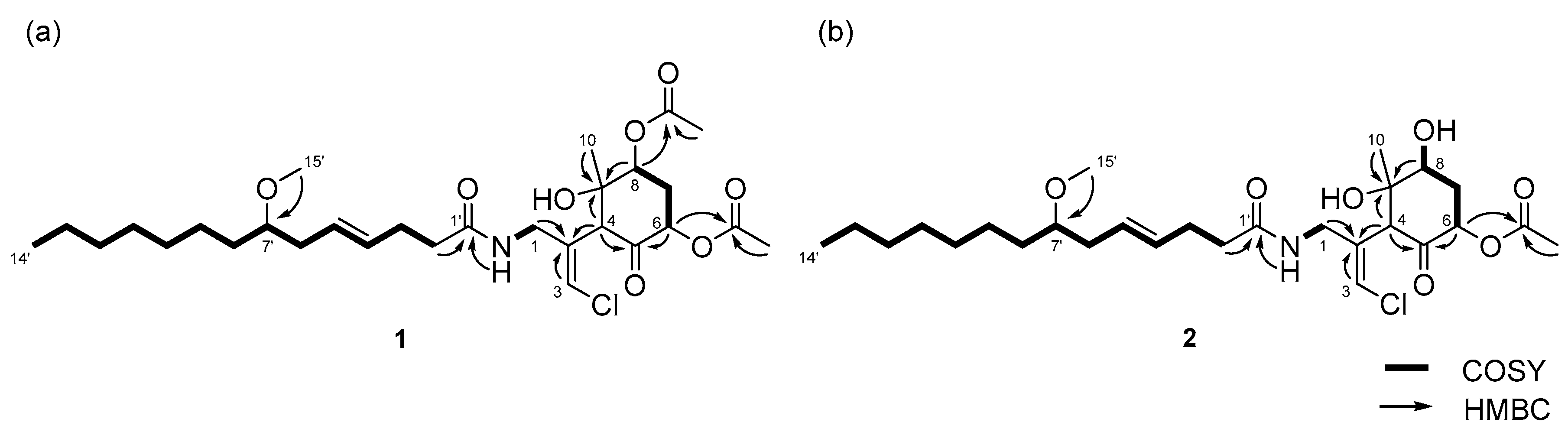
The structure of the remaining C14H19ClNO6 unit was determined as follows. COSY correlation between H-1/NH and HMBC correlations between H-1/C-2, H-3/C-2, and H-4/C-2 led to the partial structure C-1 to C-4 containing the chloromethylene moiety (Figure 2b). The geometry of the vinyl chloride was determined to be E by NOESY correlation between H-3/H-1. Additionally, COSY correlations between H-6/H-7 and H-7/H-8, and HMBC correlations between H-4/C-5, H-4/C-9, H-6/C-5, and H-8/C-9, allowed the assignment of the cyclohexanone ring. The chemical shifts of H-6 (δH 5.41), C-6 (δC 72.4), 7 and HMBC correlation from H-6 to the quaternary carbon (δC 170.3) connected the acetyl group (δH 2.16, δC 20.8) to C-6 via the oxygen atom. Similarly, another acetyl group (δH 2.19, δC 21.3) was connected to C-8 via the oxygen atom. The remaining methyl group (δH 1.26, δC 23.5) and OH group (δH 5.69) were connected to C-9 by HMBC correlations between H-10/C-9 and OH/C-9 (Figure 2c). The connections of these partial structures (Figure 2a–c) were determined on the basis of HMBC data. The NH proton showed a cross peak to the C-1’ carbonyl carbon, and H-4 correlated with the C-2 olefinic carbon. Thus, the gross structure of 1 was determined to be that shown in Figure 3a.
Compound 2 was obtained as a colorless oil. The molecular formula of 2 was determined to be C27H44ClNO7 on the basis of the 13C NMR spectrum (27 carbon signals, Figure S8) and HRESIMS (m/z 552.2692 [M + Na]+, calcd. 552.2699). The 1H NMR features of 2 (Figure S7) were very similar to those of 1, but there was only one signal from an acetyl proton. In the 13C NMR spectrum of 2, there was one less carbonyl carbon compared with that of 1, and thus, compound 2 was thought to be a deacetylated version of compound 1. COSY correlations between H-6/H-7, H-7/H-8, and H-8/OH, and HMBC correlations between H-6/C-5 and H-8/C-9 revealed the position of the acetyl group at C-6. Therefore, the gross structure of 2 was established to be that depicted in Figure 3b.
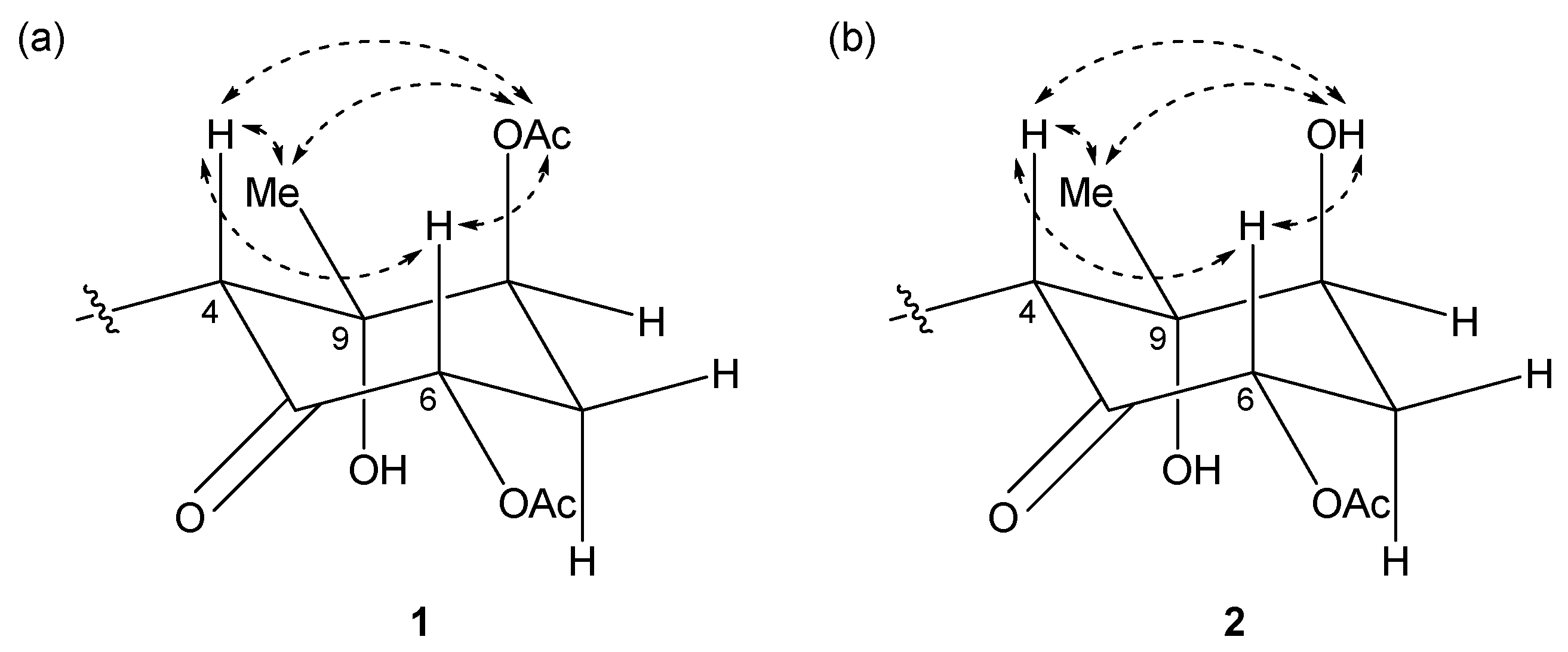
The relative structures of the cyclohexanone rings in 1 and 2 were determined by NOESY experiments. The NOESY spectrum of 1 indicated that H-4, H-6, and 8-OAc were in axial positions of the ring (Figure 4a). NOESY correlations between H-4/H3-10 and 8-OAc/H3-10 indicated the methyl group at C-9 was in an equatorial position, and thus, the relative configuration of the ring moiety in 1 was 4R*, 6S*, 8S*, and 9S*. The relative configuration of the cyclohexanone ring in 2 was revealed to be 4R*, 6S*, 8S*, and 9S* by NOESY correlations (Figure 4b), and was the same as that 1.
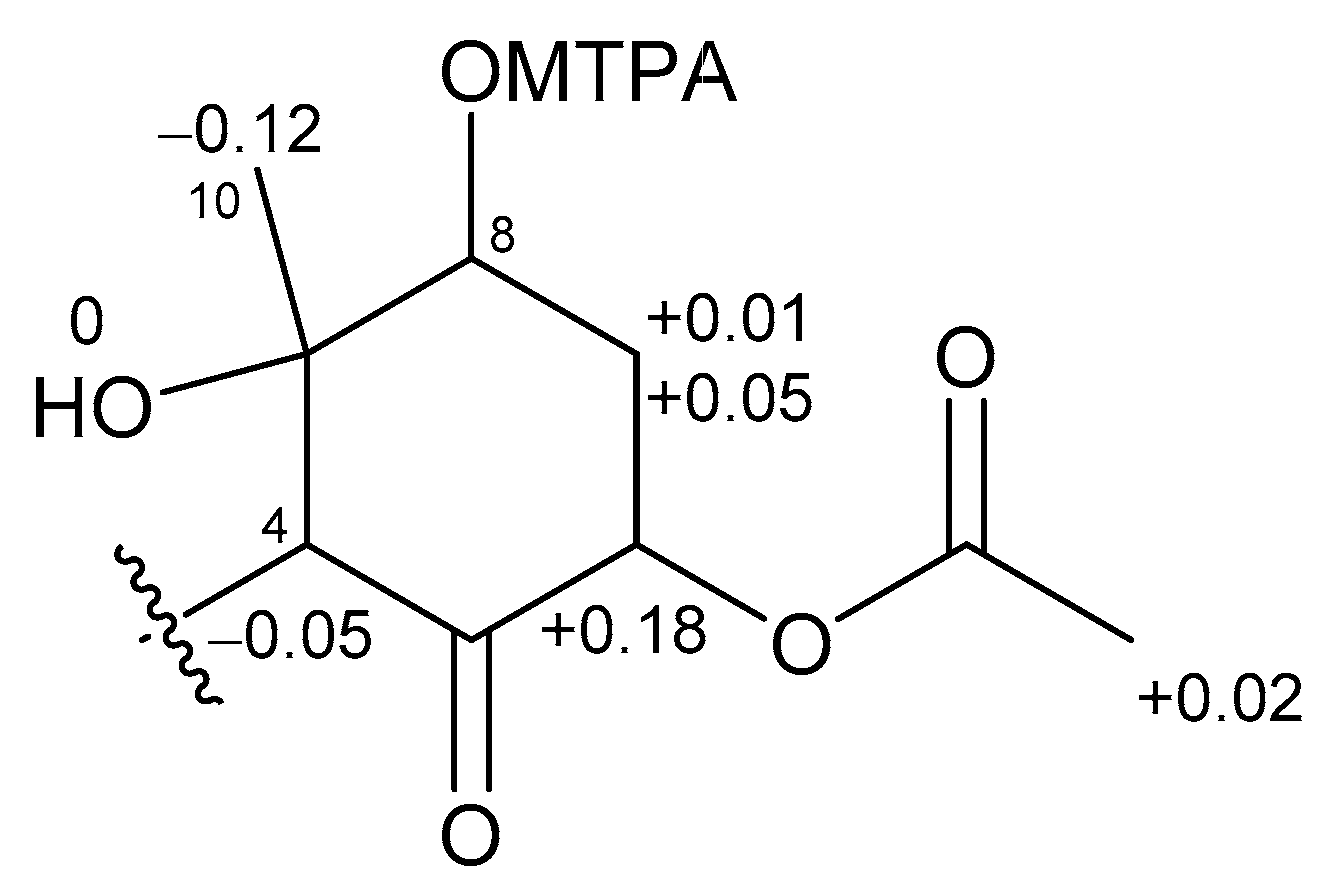
The absolute stereochemistries of the cyclohexanone rings in 1 and 2 were determined as follows. Compound 2 was treated with (R)- and (S)-MTPACl to give (S)- and (R)-MTPA esters, respectively. The 1H NMR chemical shifts of these esters (Figures S25 and S26) were assigned on the basis of the COSY spectrum. Calculation of the Δδ(S−R) values (Figure 5) revealed that C-8 existed in the S configuration [16], and the absolute configuration of the ring moiety in 2 was therefore determined to be 4R, 6S, 8S, and 9S. Compound 2 was derivatized with Ac2O to give an acetylated derivative of compound 2. The optical rotation value of this compound ( +5.5) was identical to that of 1 ( +4.8). Thus, the absolute configuration of the cyclohexanone ring in 1 was determined to be 4R, 6S, 8S, and 9S.
To confirm the absolute configuration of C-7’ in 2, compound 2 was hydrolyzed under basic conditions to yield lyngbic acid. The optical rotation of the product ( −10.8) was comparable to the reported value for 7(S)-methoxytetradec-4(E)-enoic acid ( −11.1 [17]), thus establishing the S configuration at the C-7’ position in 2. Because the 1H NMR spectrum (Figure S24) and optical rotation of the acetylated compound of 2 were identical to those of 1 described above, the absolute configuration of C-7’ in 1 was determined to be S. Therefore, the complete stereostructures of compound 1 and 2 were established to be those shown in Figure 1.
2.2. N-Demethyl-isomalyngamide I (3)
Compound 3 was obtained as a colorless oil. The molecular formula of 3 was determined to be C25H40ClNO5 on the basis of the 13C NMR spectrum (25 carbon signals, Figure S14) and HRESIMS (m/z 460.2692 [M + H]+, calcd. 470.2668). The NMR data for 3 are summarized in Table 2. The 1D and 2D NMR spectra of 3 revealed that it had a lyngbic acid moiety (Figure 6a), like compound 1 and 2. The structure of the remaining C10H13ClNO3 unit was determined as follows. COSY correlation between H-1/NH and HMBC correlations between H-1/C-2, H-1/C-4, and H-3/C-2 led to the partial structure C-1 to C-4 containing a chloromethylene moiety (Figure 6b). Because the geometry of the vinyl chloride could not be determined by NOESY experiments, we conducted HSQMBC NMR experiments [18]. We observed a 6.8 Hz 3J coupling from H-3 to C-1 and 4.2 Hz 3J coupling from H-3 to C-4. These coupling constants and comparison with the reported results for malyngamide R [14] revealed the E geometry of this double bond. Additionally, COSY correlations between H-10/H-6, H-6/H-7, H-7/H-8, and H-8/H-9, and HMBC correlations between H-6/C-5 and H-9/C-4 led to the partial structure from C-5 to C-4 (Figure 6c). The chemical shifts of H-7 (δH 3.81) and C-7 (δC 68.7) were consistent with the presence of a hydroxy group at C-7. The remaining component and the high-field chemical shifts of C-4 and C-9 (δC 61.4 and 62.1, respectively) indicated the presence of an epoxy group at C-4 and C-9 [19,20]. Although HMBC correlations between H-6/C-5 and H-9/C-5 were not observed, C-4 and C-5 should be connected considering the degree of unsaturation of 3. Thus, it became apparent that compound 3 had a cyclohexanone ring, and the gross structure of 3 was determined to be that displayed in Figure 6d.
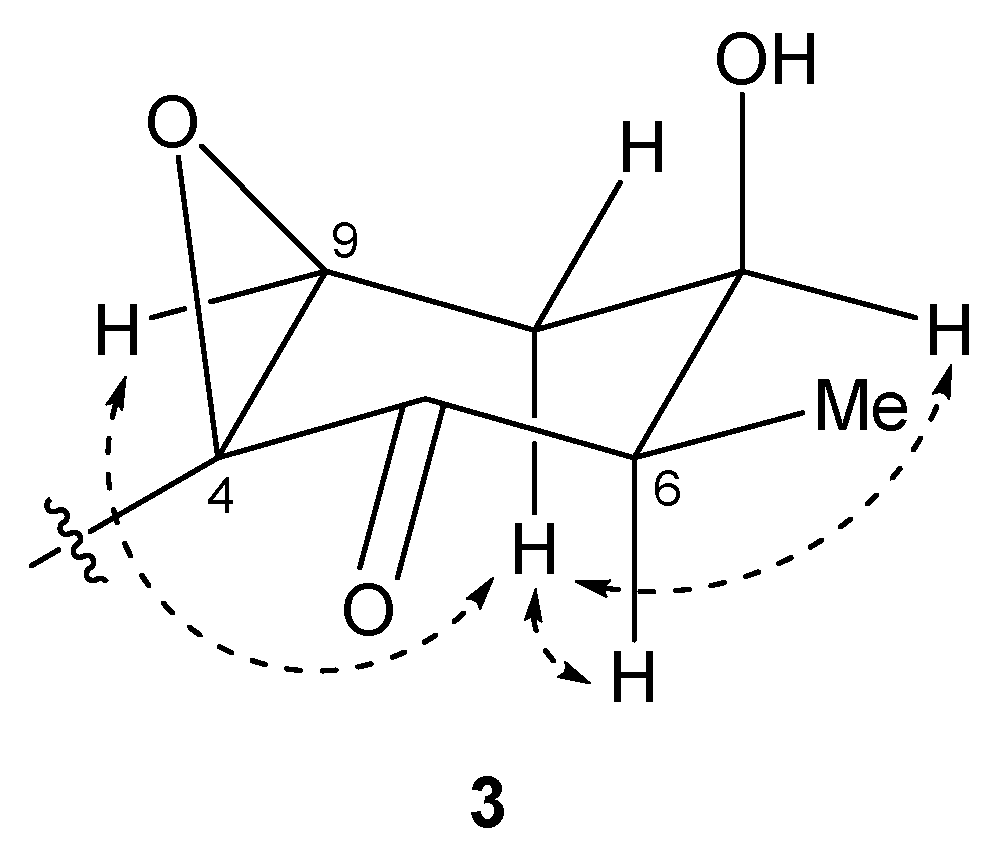
The relative structure of the cyclohexanone ring in 3 was determined by a NOESY experiment. The NOESY spectrum of 3 (Figure S18) indicated that H-6 and H-8b were in axial positions of the ring. The coupling constant of H-9 (2.6 Hz) and NOESY correlations between H-8b/H-7 and H-8b/H-9 indicated that H-7 and H-9 were in equatorial positions of the ring. Therefore, the relative configuration of the ring moiety in 3 was deduced to be 4S*, 6R*, 7R*, and 9S* (Figure 7).
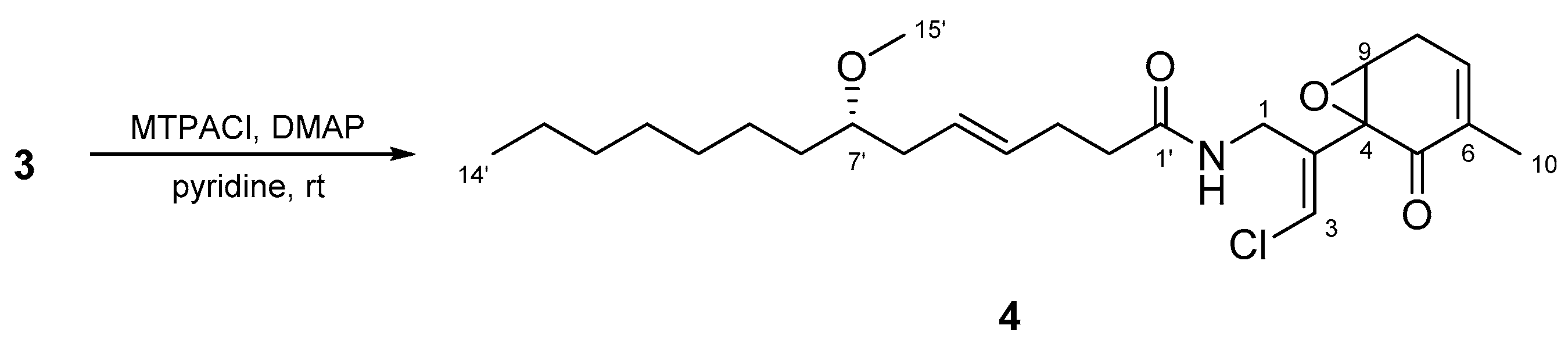
To determine the absolute configuration of C-7, 3 was treated with MTPACl. However, the MTPA ester of 3 was not obtained; compound 4 was obtained instead (Figure 8). Accordingly, the absolute stereochemistry of the cyclohexanone ring in 3 was not determined. The absolute configuration of C-7’ was established to be S using the same method as described above for 2.
Structurally similar compounds, malyngamide I [21] and 8-epi-malyngamide C [20], have been reported. The formation of the carbon framework of compound 3 is predicted to proceed in a similar fashion to those of 8-epi-malyngamide C and jamaicamides, and the branching methyl group C-6 should originate from methionine [12].
2.3. Biological Activities
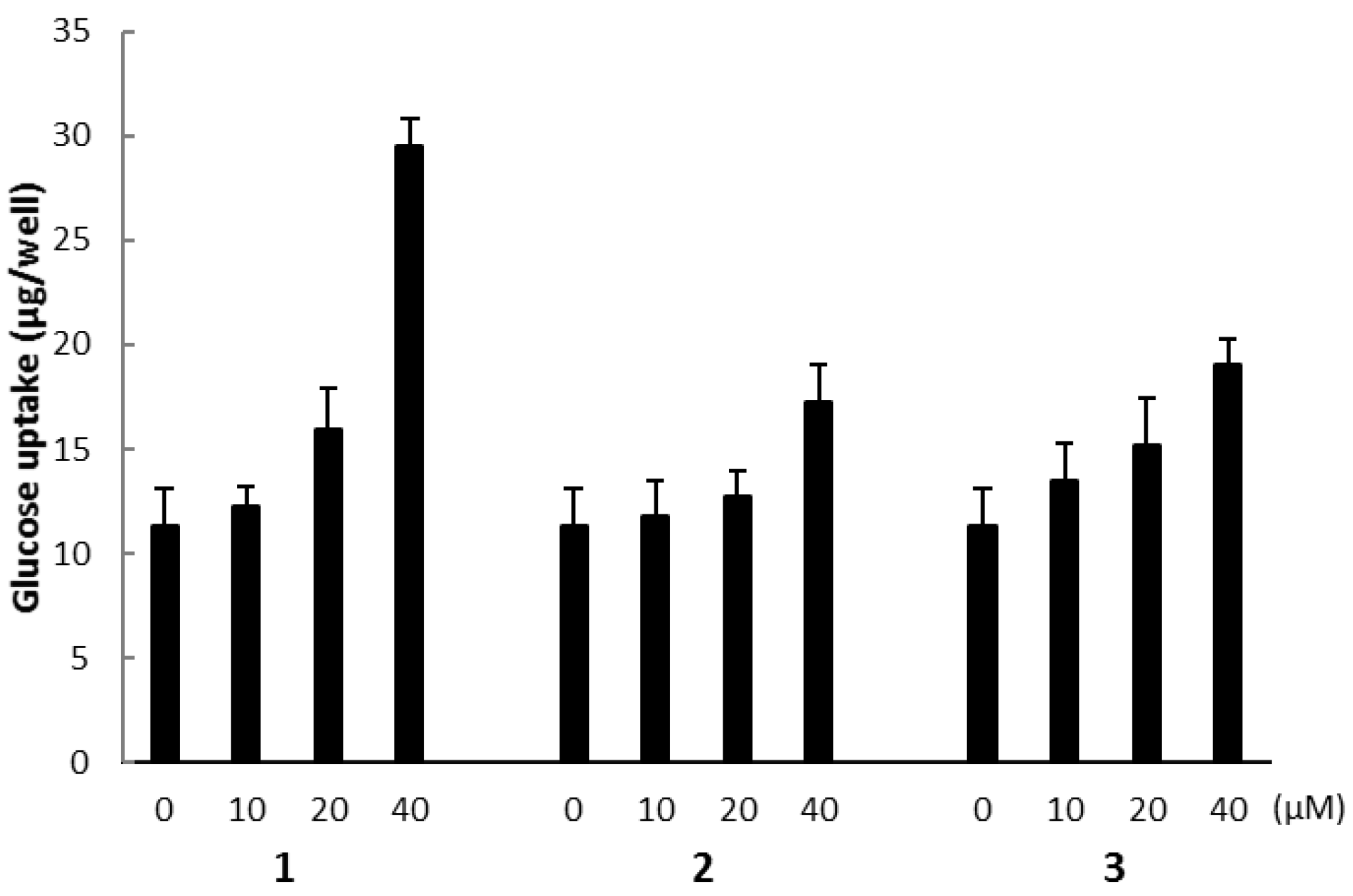
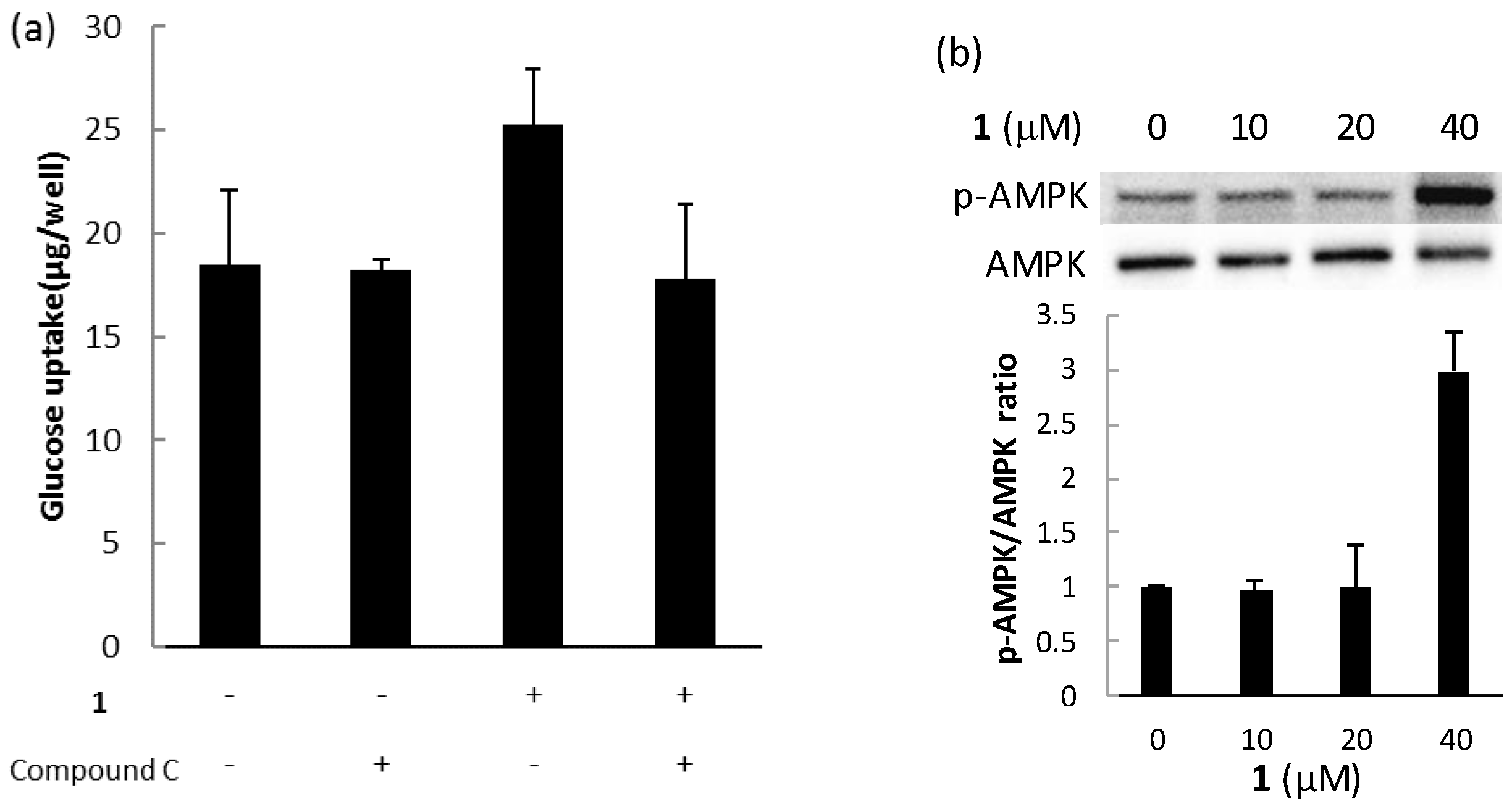
The biological activities of compounds 1, 2, and 3 were evaluated using a glucose uptake assay in cultured L6 myotubes. Compound 1 stimulated glucose uptake in a dose-dependent and insulin-independent manner, and compounds 2 and 3 showed weak activity for glucose uptake (Figure 9). To confirm the involvement of AMP-activated protein kinase (AMPK), which increases insulin-independent glucose uptake in skeletal muscle [22,23], we examined the effect of compound C, a selective AMPK inhibitor and performed western blotting with anti-AMPK and anti-phosphorylated AMPK (p-AMPK) antibodies. Compound C markedly lowered glucose uptake stimulated by compound 1 in cultured L6 myotubes (Figure 10a). The expression of p-AMPK (the activated form of AMPK) increased in the cells treated with 40 μM of 1 (Figure 10b). These results indicate that compound 1 stimulated glucose uptake in cultured L6 myotubes via the AMPK pathway, regulating cellular metabolism.
3. Experimental Section
3.1. General Experimental Procedures
Chemicals and solvents were the best grade available and used as received from commercial sources. Rat L6 myoblasts were purchased from JCRB Cell Bank (Osaka, Japan). Optical rotations were measured on a JASCO P-1010 polarimeter (JASCO Corporation, Tokyo, Japan). IR spectra were measured on a JASCO FT/IR-6100 spectrometer (JASCO Corporation, Tokyo, Japan). All NMR spectra were recorded on a Bruker AVANCE III 500 NMR spectrometer (500 and 125 MHz for 1H and 13C NMR, respectively, Bruker BioSpin Corporation, Billerica, MA, USA). Chemical shifts were reported as δ values in parts per million (ppm) relative to the residual solvent signals (CD3Cl: δH 7.26, δC 77.0), and coupling constants were in hertz (Hz). ESIMS data were obtained using a Waters Quattro micro API mass spectrometer (Waters Corporation, Milford, MA, USA), HRESIMS data were obtained using a Waters Micromass Q-TOF spectrometer (Waters Corporation, Milford, MA, USA). HPLC was carried out on a JASCO PU-2080 Plus Intelligent HPLC pump (JASCO Corporation, Tokyo, Japan) and a JASCO UV-2075 Plus Intelligent UV/VIS detector (JASCO Corporation, Tokyo, Japan). Absorbance of assay mixture was completed using a BioTek ELx800 absorbance microplate reader (BioTek Instruments Inc., Winooski, VT, USA).
3.2. Collection, Extraction, and Isolation
Samples of the marine cyanobacterium, Moorea producens were collected by hand from the coast of Bise, Okinawa Prefecture, Japan, in April 2016. The cyanobacterium was identified by 16S rRNA sequence analysis. Approximately 30 g (wet weight) of the samples were extracted with MeOH (1.0 L). The extract was filtered, and the filtrate was concentrated. The residue was partitioned between H2O (0.2 L) and EtOAc (0.2 L × 3). The material obtained from the organic layer was further partitioned between 90% aqueous MeOH (0.1 L) and n-hexane (0.1 L × 3). The aqueous MeOH fraction (0.23 g) was separated by column chromatography on ODS (2.0 g) using 60% aqueous MeOH, 80% aqueous MeOH, and MeOH. The fraction (131.4 mg) eluted with 80% aqueous MeOH was subjected to reversed-phase HPLC [Cosmosil 5C18-AR-II (20 mm × 250 mm), 85% MeOH at 5.0 mL/min, and UV detection at 215 nm] to give five fractions (Fractions 1–5). Fraction 1 was subjected to further HPLC [Cosmosil 5C18-AR-II (20 mm × 250 mm), 80% MeOH at 5.0 mL/min, and UV detection at 215 nm] to yield compound 2 (26.8 mg, tR = 34.2 min). Fraction 2 was subjected to further HPLC [Cosmosil 5C18-AR-II (20 mm × 250 mm), 70% MeCN at 5.0 mL/min, and UV detection at 215 nm] to yield compound 3 (12.6 mg, tR = 38.4 min) and two fractions (Fractions 2-1 and 2-2). Fraction 2-2 was further subjected to HPLC [Cosmosil π NAP (20 mm × 250 mm), 65% MeCN at 5.0 mL/min, and UV detection at 215 nm] to yield compound 1 (12.0 mg, tR = 35.1 min).
6,8-Di-O-Acetylmalyngamide 2 (1): Colorless oil; +4.8 (c 1.20, MeOH); IR (neat) 3302, 2929, 2856, 1731, 1651, 1539, 1445, 1373, 1232, 1132, 1096 cm−1; 1H NMR, 13C NMR and HMBC data, see Table 1; HRESIMS m/z 572.2989 [M + H]+ (calcd. for C29H47ClNO8 572.2985).
6-O-Acetylmalyngamide 2 (2): Colorless oil; +22.9 (c 2.68, MeOH); IR (neat) 3375, 2929, 2856, 1730, 1650, 1540, 1446, 1375, 1239, 1066 cm−1; 1H NMR, 13C NMR and HMBC data, see Table 1; HRESIMS m/z 552.2692 [M + Na]+ (calcd. for C27H44ClNO7Na 552.2699).
N-Demethyl-isomalyngamide I (3): Colorless oil; +108.2 (c 1.12, MeOH); IR (neat) 3313, 2928, 2856, 1714, 1647, 1541, 1456, 1433, 1094 cm−1; 1H NMR, 13C NMR and HMBC data, see Table 2; HRESIMS m/z 470.2692 [M + H]+ (calcd. for C25H41ClNO5 470.2668).
3.3. Identification of the Marine Cyanobacterium
Morphological observation was performed using a phase contrast microscopy ECLIPSE Ti-S (Nicon, Tokyo, Japan). The mean cell size and standard deviation of 50 cells were measured. The cell width was observed 36.3 ± 2.0 μm and length was observed 4.0 ± 0.9 μm. The cells were surrounded by thick (2.5–14.5 µm) firm and laminated sheaths. These morphological characters were consistent with description of Moorea producens [24]. Therefore, the marine cyanobacterium was identified as M. producens.
3.4. Preparation of MTPA Esters of 2
Compound 2 (1.1 mg) was reacted with R-MTPACl (10 μL) and DMAP (1.1 mg) in pyridine (50 μL), and the mixture was stirred for 5 h at room temperature. The reaction mixture was concentrated, and the residue was partitioned between EtOAc and 0.1 M NaHCO3 (1:1). The extract obtained from the organic layer was subjected to reversed-phase HPLC [Cosmosil 5C18-AR-II (20 mm × 250 mm), 85% MeCN at 5.0 mL/min, and UV detection at 215 nm] to yield S-MTPA ester (0.8 mg). Using the same procedure as described above, R-MTPA (0.5 mg) ester was obtained from 2 (1.0 mg).
S-MTPA ester: 1H NMR (500 MHz, CDCl3) δ 4.03 (H-4), 5.29 (H-6), 2.41 (H-7a), 2.66 (H-7b), 5.28 (H-8), 1.12 (H-10), 2.15 (6-OAc); ESIMS m/z [M + Na]+ 768.3.
R-MTPA ester: 1H NMR (500 MHz, CDCl3) δ 4.08 (H-4), 5.11 (H-6), 2.40 (H-7a), 2.61 (H-7b), 5.21 (H-8), 1.24 (H-10), 2.13 (6-OAc); ESIMS m/z [M + Na]+ 768.3.
3.5. Preparation of Acetylated Compound 2
Compound 2 (5.0 mg) was reacted with Ac2O (50 μL) in pyridine (50 μL), and the mixture was stirred for 2 h at room temperature. The reaction mixture was subjected to reversed-phase HPLC [Cosmosil 5C18-AR-II (10 mm × 250 mm), 85% MeOH at 4.0 mL/min, and UV detection at 215 nm] to yield acetylated compound of 2 (4.7 mg).
Acetylated compound of 2: +5.5 (c 1.20, MeOH); 1H NMR (500 MHz, CDCl3) δ 6.63 (1H, t, J = 6.0 Hz), 6.29 (1H, d, J = 1.6 Hz), 5.69 (1H, s), 5.47 (1H, m), 5.45 (1H, m), 5.41 (1H, dd, J = 12.9, 6.7 Hz), 5.11 (1H, t, J = 2.8 Hz), 4.27 (1H, s), 4.23 (1H, dd, J = 16.5, 6.7 Hz), 3.95 (1H, ddd, J = 16.5, 5.8, 2.0 Hz), 3.31 (3H, s), 3.17 (1H, m), 2.60 (1H, dt, J = 13.3, 2.6 Hz), 2.32 (1H, m), 2.28 (2H, m), 2.27 (2H, m), 2.19 (3H, s), 2.18 (2H, m), 2.16 (3H, s), 1.42 (2H, m), 1.27 (10H, m), 1.26 (3H, s), 0.87 (3H, t, J = 6.7 Hz); ESIMS m/z [M + H]+ 572.3.
3.6. Base Hydrolysis of Compounds 2 and 3
Compound 2 (7.3 mg) was dissolved in a 5.0 mL solution of 10% KOH in 80% aqueous EtOH and refluxed for 14 h. The reaction mixture was concentrated, and the residue was partitioned between EtOAc and H2O. The organic layer was subjected to reversed-phase HPLC [Cosmosil 5C18-AR-II (10 mm × 250 mm), 80% MeOH with 0.1% TFA at 5.0 mL/min, and UV detection at 215 nm] to yield lyngbic acid (1.6 mg). Using the same procedure as described above, lyngbic acid (4.7 mg) ester was obtained from 3 (13.5 mg).
Lyngbic acid from 2: −10.8 (c 0.16, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.49 (2H, m), 3.32 (3H, s), 3.15 (1H, m), 2.42 (2H, m), 2.35 (2H, m), 2.19 (2H, m), 1.43 (2H, m), 1.27 (10H, m), 0.88 (3H, t, J = 6.8 Hz).
Lyngbic acid from 3: −19.6 (c 0.47, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.49 (2H, m), 3.32 (3H, s), 3.15 (1H, m), 2.42 (2H, m), 2.35 (2H, m), 2.19 (2H, m), 1.43 (2H, m), 1.27 (10H, m), 0.88 (3H, t, J = 6.8 Hz).
3.7. Preparation of MTPA Ester of Compound 3
Compound 3 (7.3 mg) was reacted with R-MTPACl (10 μL) and DMAP (0.5 mg) in pyridine (50 μL), and the mixture was stirred for 3 h at room temperature. The reaction mixture was concentrated, and the residue was partitioned between EtOAc and 0.1M NaHCO3 (1:1). The extract obtained from the organic layer was subjected to reversed-phase HPLC [Cosmosil 5C18-AR-II (20 mm × 250 mm), 85% MeOH at 5.0 mL/min, and UV detection at 215 nm] to yield compound 4 (2.3 mg).
Compound 4: +40.0 (c 0.23, MeOH); 1H NMR, 13C NMR and HMBC data, see Table S1; HRESIMS m/z 474.2381 [M + Na]+ (calcd. for C25H38ClNO4Na 474.2382).
3.8. Culture of L6 Myoblasts
L6 myoblasts (5 × 103 cells/well in 96-well plates or 2 × 105 cells/well in 60-mm culture dishes) were maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), streptomycin (100 µg/mL), and penicillin G (100 units/mL) at 37 °C with 5% CO2. After reaching 80% confluence, the cells were cultured in low glucose DMEM containing 2% FBS for 1 week to differentiate into myotubes. The medium was renewed every 2 days.
3.9. Determination of Glucose Uptake
L6 myotubes were incubated in filter-sterilized Krebs-Henseleit buffer (1.2 mM MgSO4, 1.2 mM KH2PO4, 4.7 mK KCl, 119 mM NaCl, 2.5 mM CaCl2·2H2O, and 25 mM NaHCO3, pH 7.4) containing 0.1% bovine serum albumin (BSA), 10 mM HEPES, and 2 mM sodium pyruvate (KHH buffer) for 2 h. The myotubes were then cultured in KHH buffer containing 5 mM glucose with or without compounds 1, 2, and 3 (10–40 µM) for 16 h and without or with compound C (30 µM), an AMPK inhibitor for 6 h. Nepodin [25] was used as a positive control, and DMSO alone was used as a negative control. The concentrations of glucose remaining in KHH buffer were determined by a commercial assay kit (Glucose CII-Test Wako) and a microplate reader at 490 nm. The amounts of glucose uptake by myotubes were calculated from the differences in glucose concentrations between before and after culture.
3.10. Western Blotting
L6 myotubes were lysed in Blue Loading Buffer for 1 min after washing with ice-cold PBS. The lysates were sonicated for 10 s, boiled at 100 °C for 10 min and centrifuged at 15,000 rpm for 5 min. The protein concentrations of the supernatants were determined by a commercial assay kit (RC DC Protein Assay, Bio-Rad laboratories Inc., Hercules, CA, USA). Equal amounts of protein samples (50 µg/lane) were electrophoresed on 10% Mini-PROTEAN TGX precast gels (Bio-Rad laboratories Inc., Hercules, CA, USA) and transferred to nitrocellulose membranes (Bio-Rad laboratories Inc., Hercules, CA, USA). The membranes were washed with Tris buffered saline (TBS) for 15 min and blocked with 3% nonfat dry milk, or 80% BSA in TBS containing 0.05% Tween 20 (TBST), at room temperature for 1 h. The membranes were then washed with TBST and incubated with anti-AMPK or anti-phosphorylated AMPK primary antibodies (1:1000 or 1:2000 in blocking buffer, respectively) at 4 °C overnight. The membranes were then washed with TBST and incubated with HRP-linked anti-rabbit IgG secondary antibodies (1:2000 in blocking buffer) at room temperature for 1 h. After washing with TBST, immunoreactive bands were detected by a chemiluminescent reagent (Clarity Western ECL Substrate, Bio-Rad laboratories Inc., Hercules, CA, USA) and quantified by densitometry analysis using a ChemiDoc XRS Plus system (Bio-Rad laboratories Inc., Hercules, CA, USA) and Image Lab software (version 5.2, Bio-Rad laboratories Inc., Hercules, CA, USA).
4. Conclusions
Three new malyngamide analogs, 6,8-di-O-acetylmalyngamide 2 (1), 6-O-acetylmalyngamide 2 (2), and N-demethyl-isomalyngamide I (3) were isolated from the marine cyanobacterum Moorea producens collected at Okinawa Prefecture, Japan. The gross structures of these compounds were determined on the basis of spectroscopic analysis. The absolute stereostructures of 1 and 2 were established by NOESY experiments, and chemical derivatization and degradation. The relative configuration of the cyclohexanone ring in 3 was determined by a NOESY experiment. These compounds stimulated glucose uptake in cultured L6 myotubes. Compound 1 was found to activate an insulin-independent AMPK pathway, so 1 may have antidiabetic properties.
Supplementary Materials
The following are available online at www.mdpi.com/1660-3397/15/12/367/s1, Figures S1–S6: 1H NMR, 13C NMR, HSQC, HMBC, COSY, and NOESY spectrum of 6,8-di-O-acetylmalyngamide 2 (1) in CDCl3; Figures S7–S12: 1H NMR, 13C NMR, HSQC, HMBC, COSY, and NOESY spectrum of 6-O-acetylmalyngamide 2 (2) in CDCl3; Figures S13–S18: 1H NMR, 13C NMR, HSQC, HMBC, COSY, and NOESY spectrum of N-demethyl-isomalyngamide I (3) in CDCl3; Figures S19–S23: 1H NMR, 13C NMR, HSQC, HMBC, and COSY spectrum of compound 4 in CDCl3; Figure S24: 1H NMR spectrum of acetylated compound of 2 in CDCl3; Figure S25: 1H NMR spectrum of S-MTPA ester of 2 in CDCl3; Figure S26: 1H NMR spectrum of R-MTPA ester of 2 in CDCl3; Figure S27: 1H NMR spectrum of lyngbic acid from 2 in CDCl3; Figure S28: 1H NMR spectrum of lyngbic acid from 3 in CDCl3, Table S1: NMR spectral data for compound 4 in CDCl3.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Young Scientists (B) (21710237) from the Ministry of Education, Culture, Sports, Science and Technology, Japan; University of the Ryukyus Strategic Research Grant.
Author Contributions
Kosuke Sueyoshi and Toshiaki Teruya iments; Kosuke Sueyoshi, Aki Yamano, Kaori Ozaki and Toshiaki Teruya analyzed the data; Shimpei Sumimoto, Arihiro Iwasaki and Kosuke Sueyoshi contributed identification of cyanobacteria; Kosuke Sueyoshi, Shimpei Sumimoto and Toshiaki Teruya wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Teruya, T.; Sasaki, H.; Fukazawa, H.; Suenaga, K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp.: Isolation, stereostructure, and biological activity. Org. Lett. 2009, 11, 5062–5065. [Google Scholar] [CrossRef] [PubMed]
- Sumiya, E.; Shimogawa, H.; Sasaki, H.; Tsutsumi, M.; Yoshita, K.; Ojika, M.; Suenaga, K.; Uesugi, M. Cell-morphology profiling of a natural product library identifies bisebromoamide and miuraenamide A as actin filament stabilizers. ACS Chem. Biol. 2011, 6, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Iwasaki, A.; Sumimoto, S.; Iwatsuki, M.; Ishiyama, A.; Hokari, R.; Otoguro, K.; Ōmura, S.; Suenaga, K. Isolation and total synthesis of hoshinolactam, an antitrypanosomal lactam from a marine cyanobacterium. Org. Lett. 2017, 19, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Cardellina, J.H., II; Marner, F.J.; Moore, R.E. Malyngamide A, a novel chlorinated metabolite of the marine cyanophyte Lyngbya majuscule. J. Am. Chem. Soc. 1979, 101, 240–242. [Google Scholar] [CrossRef]
- Gerwick, L.; Boudreau, P.; Choi, H.; Mascuch, S.; Villa, F.A.; Balunas, M.J.; Malloy, K.L.; Teasdale, M.E.; Rowley, D.C.; Gerwick, W.H. Interkingdom signaling by structurally related cyanobacterial and algal secondary metabolites. Phytochem. Rev. 2013, 12, 459–465. [Google Scholar] [CrossRef]
- Sueyoshi, K.; Kaneda, M.; Sumimoto, S.; Oishi, S.; Fujii, N.; Suenaga, K.; Teruya, T. Odoamide, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Okeania sp. Tetrahedron 2016, 72, 5472–5478. [Google Scholar] [CrossRef]
- Kaneda, M.; Sueyoshi, K.; Teruya, T.; Ohno, H.; Fujii, N.; Oishi, S. Total synthesis of odoamide, a novel cyclic depsipeptide, from an Okinawan marine cyanobacterium. Org. Biomol. Chem. 2016, 14, 9093–9104. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, K.; Kudo, T.; Yamano, A.; Sumimoto, S.; Iwasaki, A.; Suenaga, K.; Teruya, T. Odobromoamide, a terminal alkynyl bromide-containing cyclodepsipeptide from the marine cyanobacterium Okeania sp. Bull. Chem. Soc. Jpn. 2017, 90, 436–440. [Google Scholar] [CrossRef]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Ainslie, R.D.; Barchi, J.J.; Kuniyoshi, M.; Moore, R.E.; Mynderse, J.S. Structure of mlyngamide C. J. Org. Chem. 1985, 50, 2859–2862. [Google Scholar] [CrossRef]
- Milligan, K.E.; Márquez, B.L.; Williamson, R.T.; Davies-Coleman, M.; Gerwick, W.H. Two new malyngamides from a Madagascan Lyngbya majuscula. J. Nat. Prod. 2000, 63, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Shaala, L.A.; Youssef, D.T.A.; McPhail, K.L.; Elbandy, M. Malyngamide 4, a new lipopeptide from the Red Sea marine cyanobacterium Moorea producens (formerly Lyngbya majuscule). Phytochem. Lett. 2013, 6, 183–185. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Cardellina, J.H., II; Dalietos, D.; Marner, F.J.; Mynderse, J.S.; Moore, R.E. (−)-trans-7(S)-Methoxytetradec-4-enoic acid and related amides from the marine cyanophyte Lyngbya majuscule. Phytochemistry 1978, 17, 2091–2095. [Google Scholar] [CrossRef]
- Williamson, R.T.; Márquez, B.L.; Gerwick, W.H.; Kover, K.E. One- and two-dimensional gradient-selected HSQMBC NMR experiments for the efficient analysis of long-range heteronuclear coupling constants. Magn. Reson. Chem. 2000, 38, 265–273. [Google Scholar] [CrossRef]
- Kan, Y.; Fujita, T.; Nagai, H.; Sakamoto, B.; Hokama, Y. Malyngamides M and N from the Hawaiian red alga Gracilaria coronopifolia. J. Nat. Prod. 1998, 61, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.; McPhail, K.L.; Goeger, D.E.; Valeriote, F.A.; Gerwick, W.H. Two cytotoxic stereoisomers of malyngamide C, 8-epi-malyngamide C and 8-O-acetyl-8-epi-malyngamide C, from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2010, 71, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.S.; Gerwick, W.H. Malyngamide I from the tropical marine cyanobacterium Lyngbya majuscula and the probable structure revision of stylocheilamide. Tetrahedron Lett. 1995, 36, 7837–7840. [Google Scholar] [CrossRef]
- Hayashi, T.; Hirshman, M.F.; Kurth, E.J.; Winder, W.W.; Goodyear, L.J. Evidence for 5’AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 1998, 47, 1369–1373. [Google Scholar] [PubMed]
- Hayashi, T.; Hirshman, M.F.; Fujii, N.; Habinowski, S.N.; Witters, L.A.; Goodyear, L.J. Metabolic stress and altered glucose transport. Diabetes 2000, 49, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Rottacker, E.C.; Kaštovský, J.; Byrum, T.; Choi, H.; Ellisman, M.H.; Komárek, J.; Gerwick, W.H. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int. J. Syst. Evol. Microbiol. 2012, 62, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Ha, B.G.; Yonezawa, T.; Son, M.J.; Woo, J.T.; Ohba, S.; Chung, U.I.; Yagasaki, K. Antidiabetic effect of nepodin, a component of Rumex roots, and its modes of action in vitro and in vivo. Biofactors 2014, 40, 436–447. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of new malyngamides 1–3.
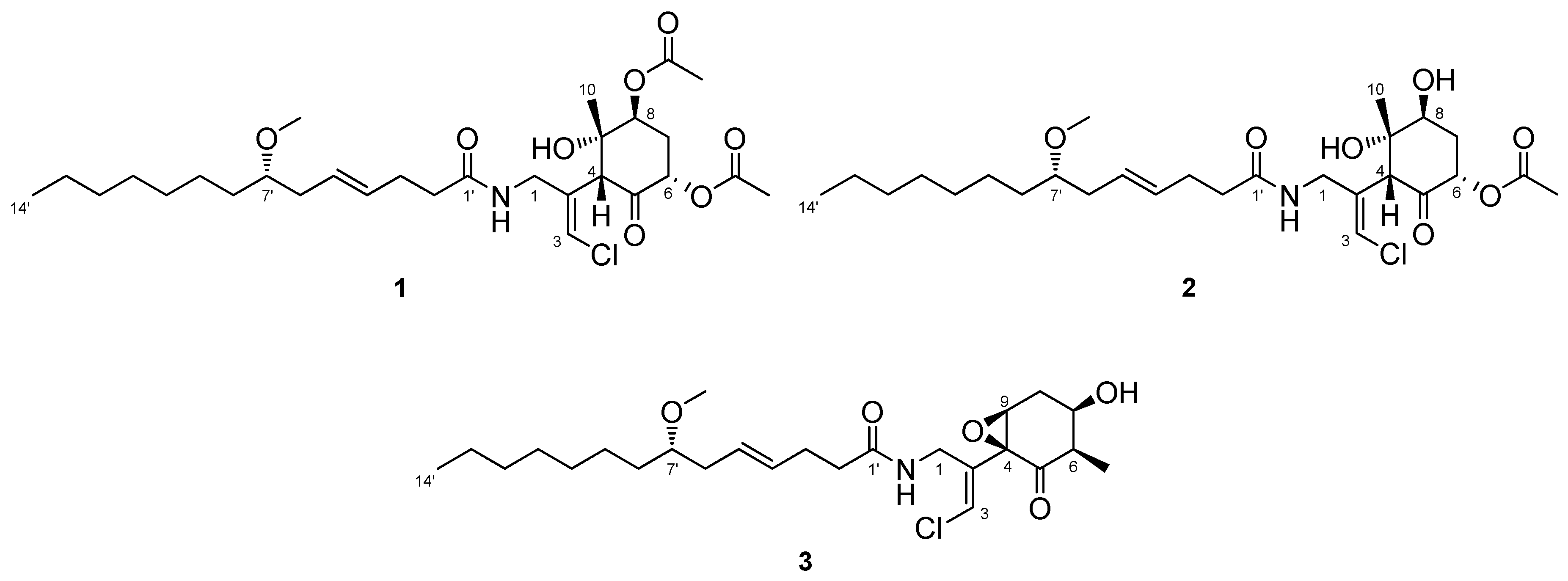
Figure 2.
Partial structures of compound 1: (a) lyngbic acid moiety; (b) chloromethylene moity; (c) cyclohexanone ring moiety of 1. Wavy lines were used to indicate partial structures of 1.
Figure 2.
Partial structures of compound 1: (a) lyngbic acid moiety; (b) chloromethylene moity; (c) cyclohexanone ring moiety of 1. Wavy lines were used to indicate partial structures of 1.
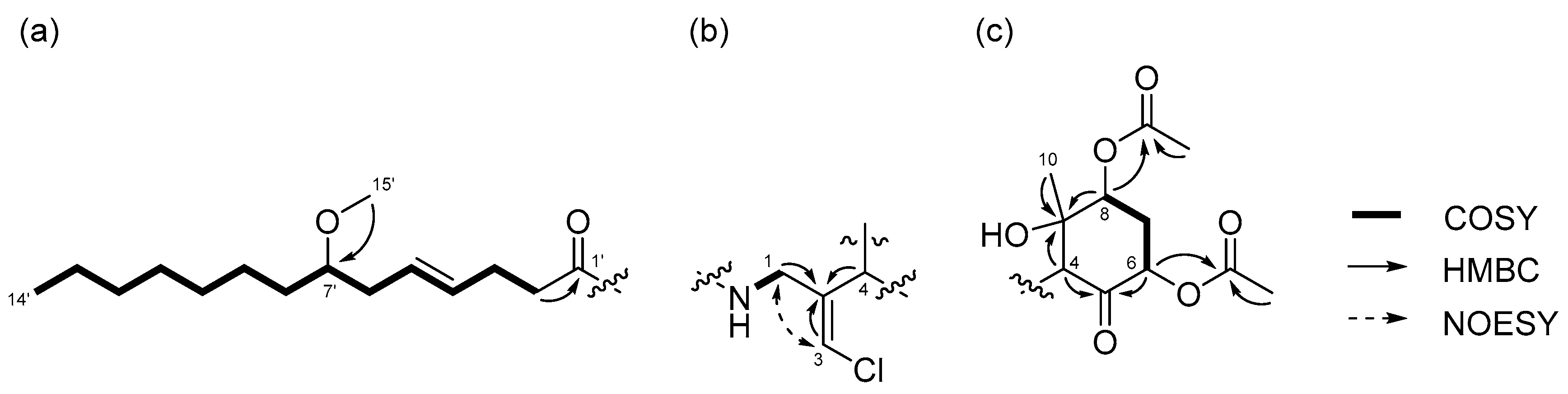
Figure 3.
Gross structures of (a) compound 1 and (b) 2.
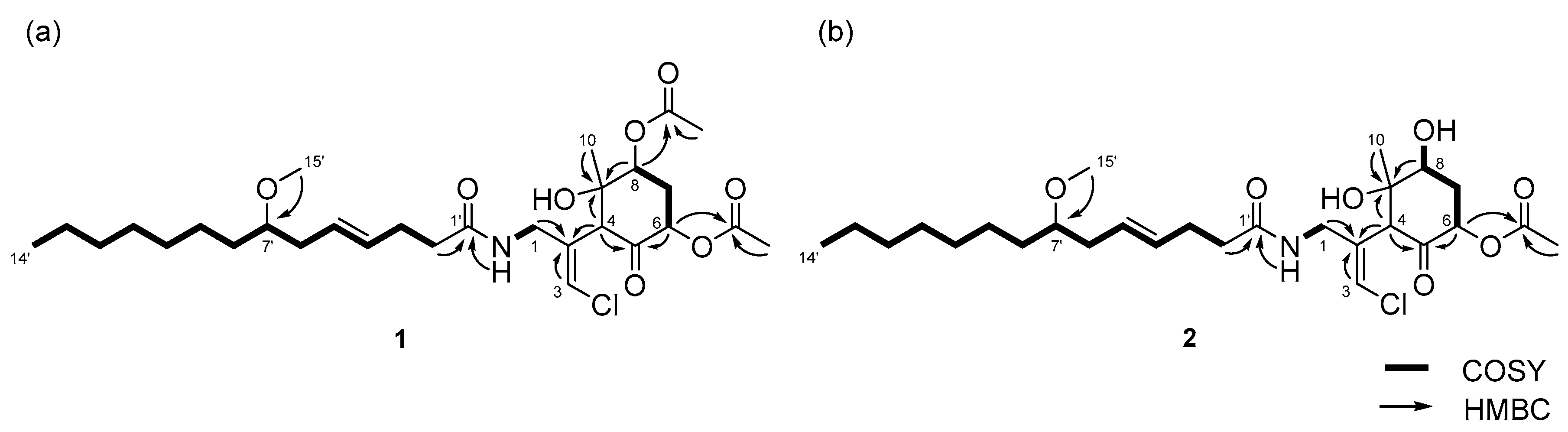
Figure 4.
Key NOESY correlations for (a) compound 1 and (b) 2.
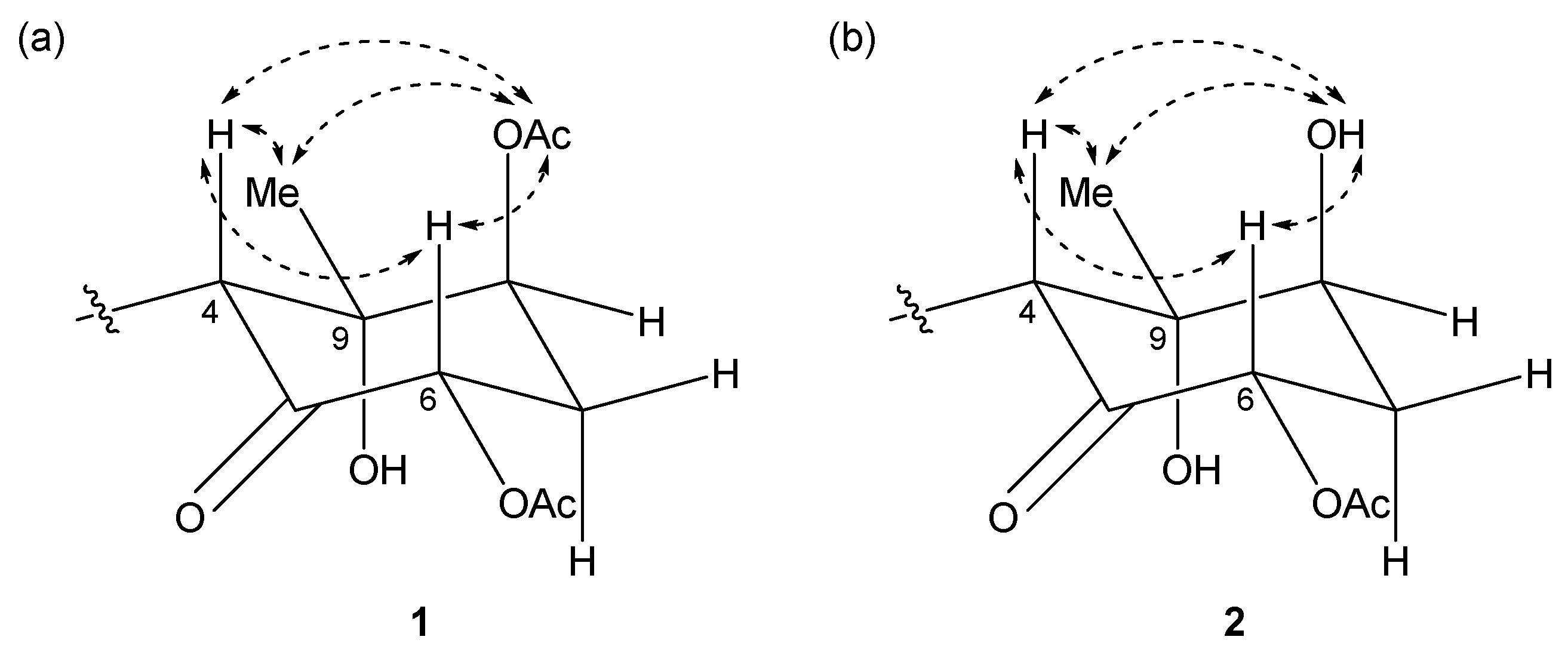
Figure 5.
Δδ values (δS − δR) in ppm for the MTPA ester of 2.
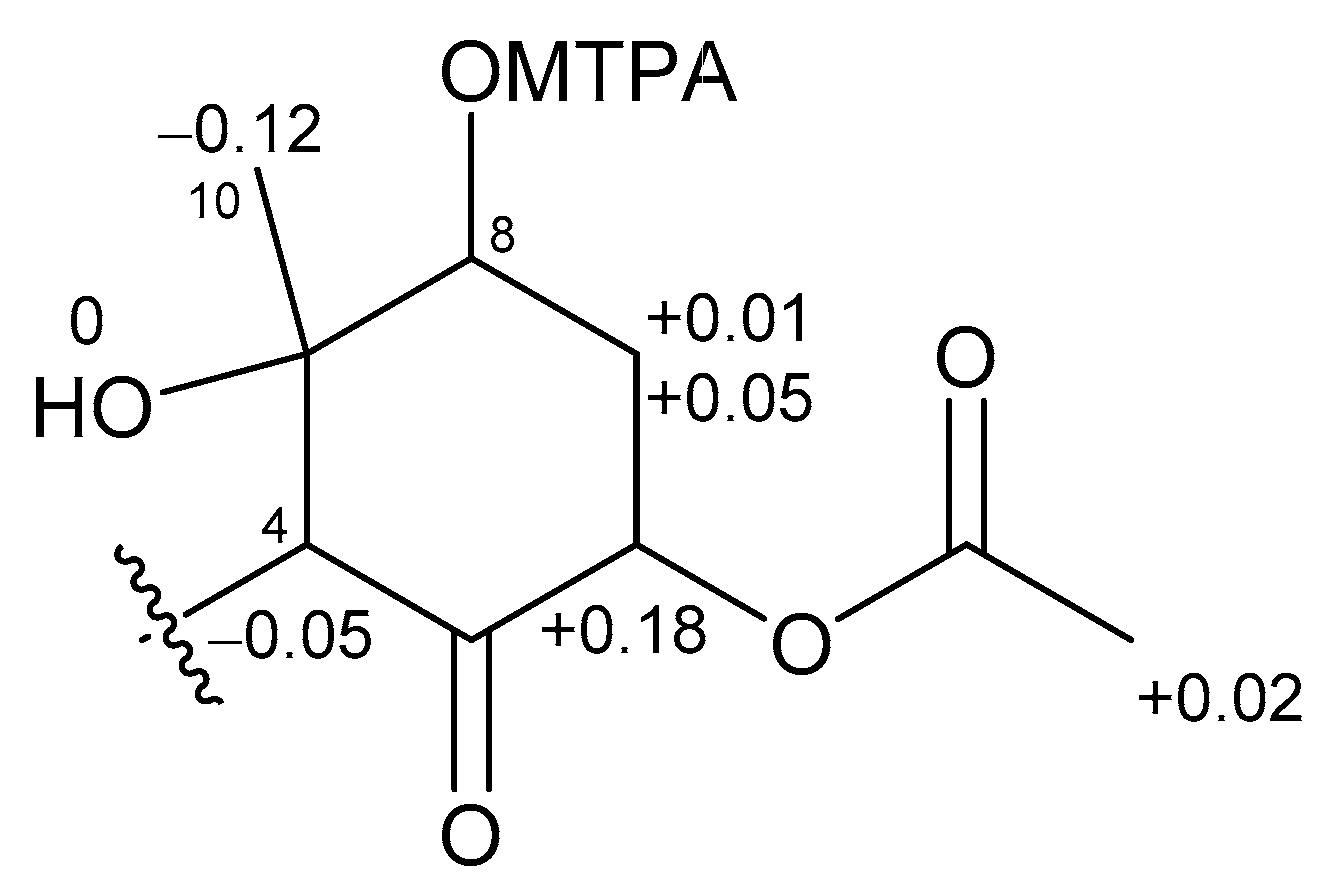
Figure 6.
Partial structures and gross structure of compound 3: (a) lyngbic acid moiety; (b) chloromethylene moiety; (c) cyclohexanone ring moiety; (d) gross structure of 3.
Figure 6.
Partial structures and gross structure of compound 3: (a) lyngbic acid moiety; (b) chloromethylene moiety; (c) cyclohexanone ring moiety; (d) gross structure of 3.
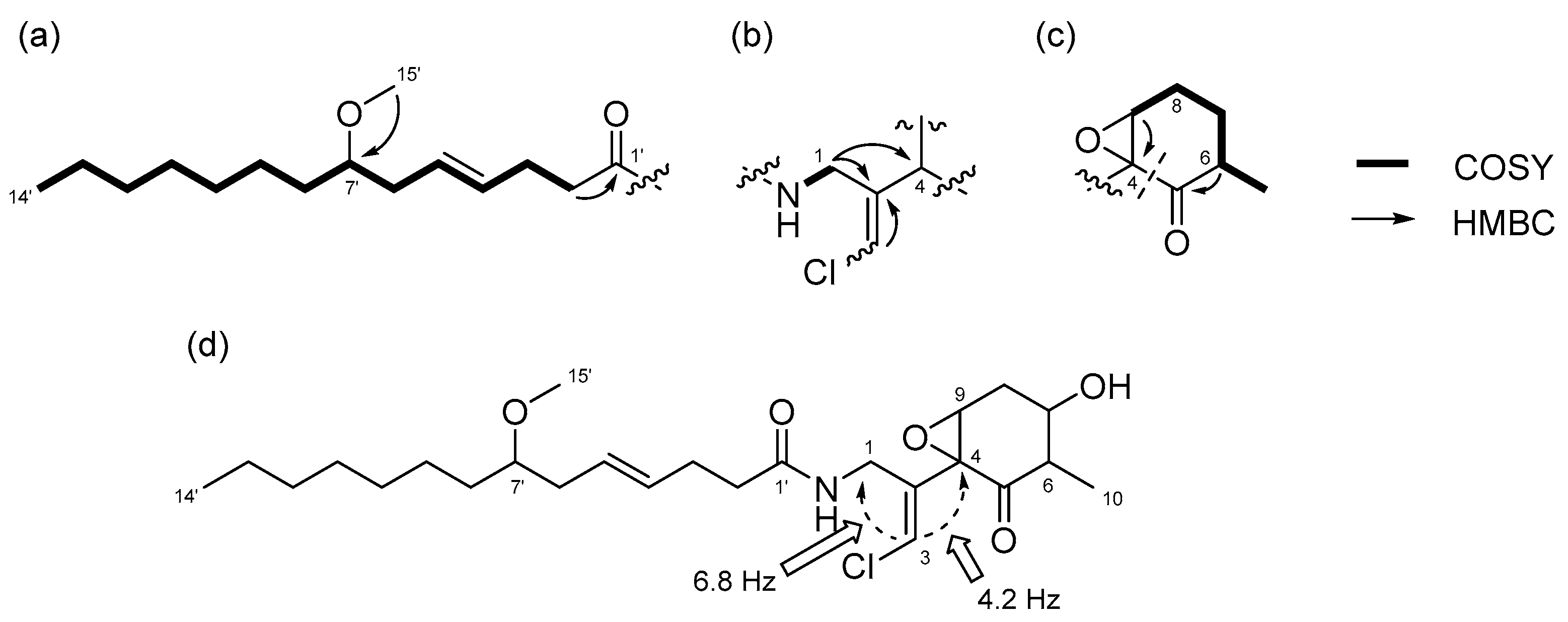
Figure 7.
Key NOESY correlations for compound 3.
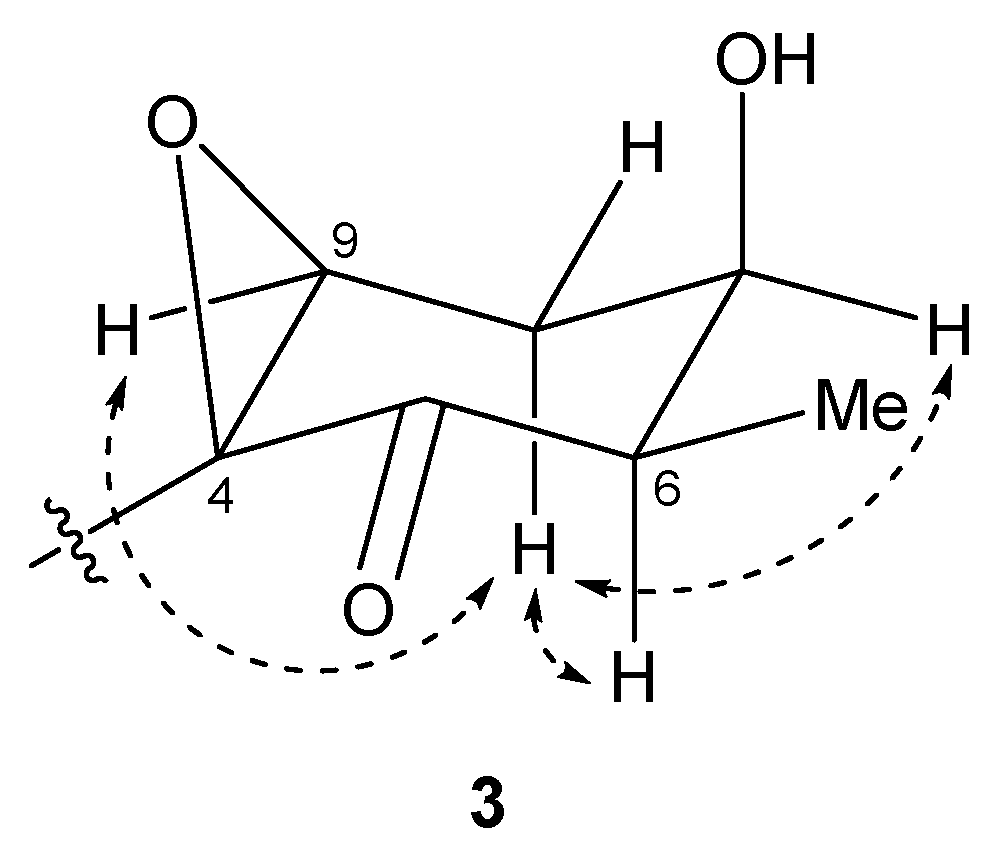
Figure 8.
Structure of compound 4.
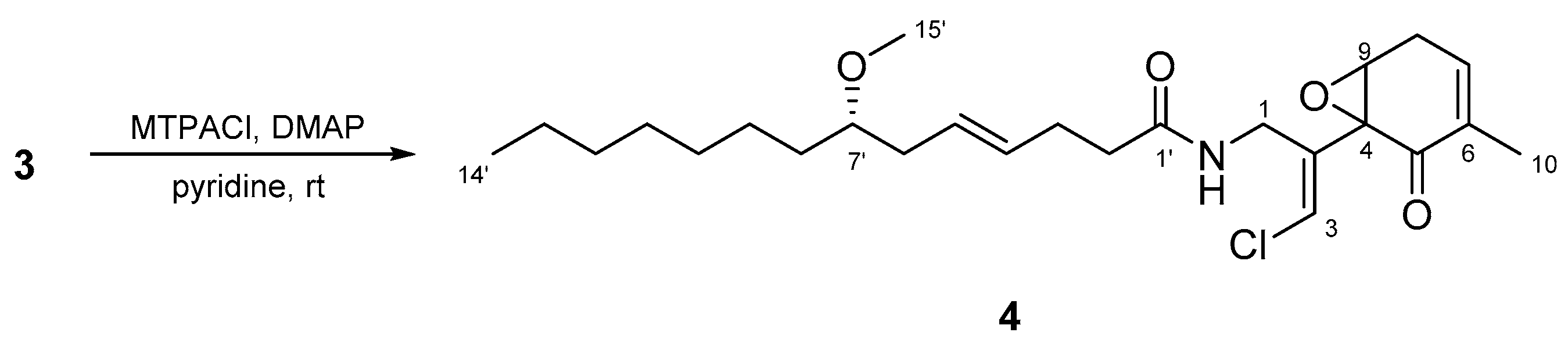
Figure 9.
The effect of compounds 1, 2, and 3 on glucose uptake in cultured L6 myotubes. Cells were preincubated in Krebs-Henseleit-HEPES buffer (KHH buffer) without glucose for 2 h. They were then incubated in KHH buffer containing 5 mM glucose with the indicated concentrations of compounds for 16 h. Glucose uptake was measured using a Glucose C-II Test kit. Values are the mean ± SD of quadruplicate determinations.
Figure 9.
The effect of compounds 1, 2, and 3 on glucose uptake in cultured L6 myotubes. Cells were preincubated in Krebs-Henseleit-HEPES buffer (KHH buffer) without glucose for 2 h. They were then incubated in KHH buffer containing 5 mM glucose with the indicated concentrations of compounds for 16 h. Glucose uptake was measured using a Glucose C-II Test kit. Values are the mean ± SD of quadruplicate determinations.
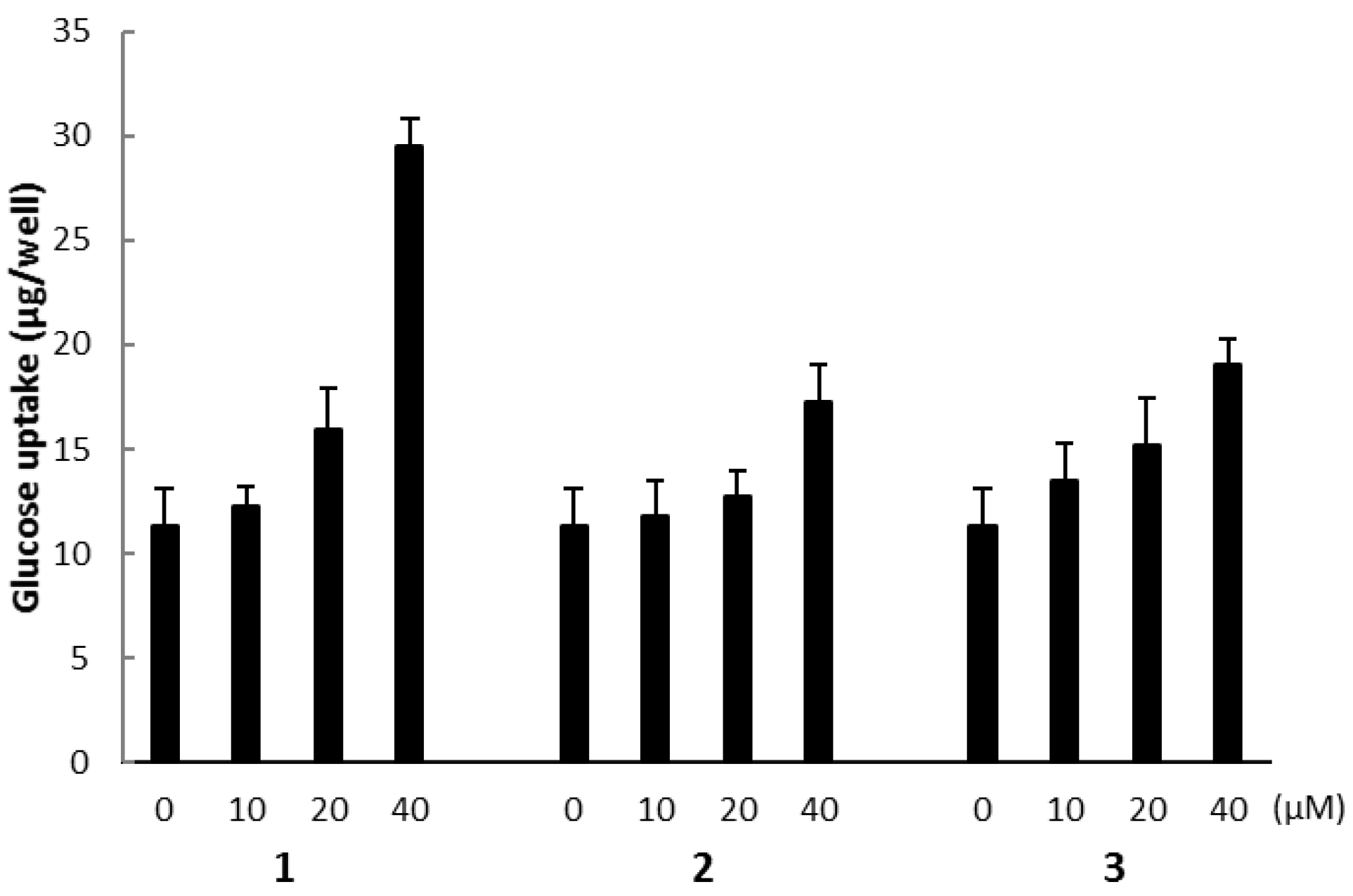
Figure 10.
The effect of compound 1 on the AMPK pathway in cultured L6 myotubes. (a) Cells were preincubated in KHH buffer without glucose for 2 h. They were then incubated in KHH buffer containing 5 mM glucose with or without 40 μM of compound 1 and with or without 30 μM of compound C for 8 h. Glucose uptake was measured using a Glucose C-II Test kit. Values are the mean ± SD of quadruplicate determinations. (b) Cells were treated with the indicated concentrations of compound 1. The lysates were analyzed by western blotting with anti-phosphorylated AMPK (p-AMPK) and anti-AMPK antibodies. Immunoreactive bands were quantified using Image Lab software and ratios of p-AMPK/AMPK are shown. The ratio of the control (0 μM of compound 1) was regarded as 1. Values are the mean ± SD of quadruplicate determinations.
Figure 10.
The effect of compound 1 on the AMPK pathway in cultured L6 myotubes. (a) Cells were preincubated in KHH buffer without glucose for 2 h. They were then incubated in KHH buffer containing 5 mM glucose with or without 40 μM of compound 1 and with or without 30 μM of compound C for 8 h. Glucose uptake was measured using a Glucose C-II Test kit. Values are the mean ± SD of quadruplicate determinations. (b) Cells were treated with the indicated concentrations of compound 1. The lysates were analyzed by western blotting with anti-phosphorylated AMPK (p-AMPK) and anti-AMPK antibodies. Immunoreactive bands were quantified using Image Lab software and ratios of p-AMPK/AMPK are shown. The ratio of the control (0 μM of compound 1) was regarded as 1. Values are the mean ± SD of quadruplicate determinations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Nuclear magnetic resonance (NMR) spectral data for compound 1 and 2 in CDCl3.
| Position | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δC a | δH, Mult (J in Hz) b | HMBC | δC a | δH, Mult (J in Hz) b | HMBC | |
| 1a | 41.6 | 3.95, ddd (16.5, 5.8, 1.8) | 2, 3, 4, 1’ | 41.7 | 3.95, ddd (16.3, 5.6, 1.6) | 2, 3, 4, 1’ |
| 1b | 4.23, dd (16.5, 6.7) | 2, 3, 4, 1’ | 4.24, dd (16.3, 6.8) | 2, 3, 4, 1’ | ||
| 2 | 136.3 | - | - | 136.5 | - | - |
| 3 | 121.6 | 6.29, d (1.8) | 1, 2, 4 | 121.4 | 6.27, d (1.6) | 1, 2, 4 |
| 4 | 56.1 | 4.27, s | 1, 2, 3, 5, 9, 10 | 55.1 | 4.36, s | 1, 2, 3, 5, 9, 10 |
| 5 | 201.9 | - | - | 203.3 | - | - |
| 6 | 72.4 | 5.41, dd (12.9, 6.7) | 5, 7, 6-OAc | 72.7 | 5.60, dd (12.9, 6.8) | 5, 7, 6-OAc |
| 7a | 31.3 | 2.32, m | 5, 6, 8, 9 | 34.1 | 2.26, m | 5, 6, 9 |
| 7b | 2.60, ddd (13.3, 12.9, 2.6) | 6 | 2.54, dt (12.9, 2.3) | 5, 6 | ||
| 8 | 74.8 | 5.11, dd (2.6, 2.6) | 4, 6, 9, 10, 8-OAc | 73.8 | 3.90, dd (6.1, 3.0) | 4, 6, 9, 10 |
| 9 | 78.9 | - | - | 80.4 | - | - |
| 10 | 23.5 | 1.26, s | 4, 5, 8, 9 | 23.9 | 1.34, s | 4, 8, 9 |
| NH | - | 6.68, dd (6.7, 5.8) | 1, 1’ | - | 6.84, dd (6.8, 5.6) | 1, 1’ |
| 1’ | 173.8 | - | - | 173.8 | - | - |
| 2’ | 36.2 | 2.27, m | 1’, 3’ | 36.2 | 2.27, m | 1’, 3’, 4’ |
| 3’ | 28.7 | 2.28, m | 4’ | 28.7 | 2.32, m | 1’, 2’, 4’, 5’ |
| 4’ | 130.7 | 5.45, m | 3’, 6’ | 130.7 | 5.45, m | 2’, 3’, 5’ |
| 5’ | 128.3 | 5.47, m | 4’, 7’ | 128.1 | 5.47, m | 3’, 4’, 7’ |
| 6’ | 36.3 | 2.18, m | 4’, 5’, 7’, 8’ | 36.3 | 2.19, m | 4’, 5’, 7’, 8’ |
| 7’ | 80.7 | 3.17, m | 5’, 8’, 9’, 15’ | 80.8 | 3.17, m | 5’, 8’, 9’, 15’ |
| 8’ | 33.3 | 1.42, m | 6’, 7’, 9’, 10’ | 33.4 | 1.43, m | 7’, 9’, 10’ |
| 9’ | 25.5 | 1.27, m e | - | 25.5 | 1.26, m f | - |
| 10’ | 29.9 | 1.27, m e | - | 29.9 | 1.26, m f | - |
| 11’ | 29.4 | 1.27, m e | - | 29.4 | 1.26, m f | - |
| 12’ | 32.0 c | 1.27, m e | - | 31.9 d | 1.26, m f | - |
| 13’ | 22.8 c | 1.27, m e | - | 22.8 d | 1.26, m f | - |
| 14’ | 14.2 | 0.87, t (6.7) | 12’, 13’ | 14.2 | 0.87, t (6.8) | 12’, 13’ |
| 15’ | 56.5 | 3.31, s | 7’ | 56.5 | 3.31, s | 7’ |
| 6-OAc | 170.3 | - | - | 170.4 | - | - |
| 20.8 | 2.16, s | 6-OAc | 20.9 | 2.15, s | 6-OAc | |
| 8-OAc/OH | 169.9 | - | - | - | 3.00, d (3.9) | 7, 8, 9 |
| 21.3 | 2.19, s | 8-OAc | - | - | - | |
| 9-OH | - | 5.69, s | 8, 9, 10 | - | 5.33, s | 8, 9, 10 |
a Recorded at 125 MHz. b Recorded at 500 MHz. c,d Assignments may be interchanged. e,f Overlapped signals.
Table 2.
NMR spectral data for compound 3 in CDCl3.
| Position | δC a | δH, mult (J in Hz) b | HMBC |
|---|---|---|---|
| 1 | 37.3 | 4.08, d (6.1) | 2, 3, 4, 1’ |
| 2 | 135.1 | - | - |
| 3 | 120.9 | 6.29, s | 1, 2, 4 |
| 4 | 61.4 | - | - |
| 5 | 204.2 | - | - |
| 6 | 51.0 | 2.41, m | 5, 7, 8, 10 |
| 7 | 68.7 | 3.81, m | 6, 8, 9, 10 |
| 8a | 31.7 | 2.08, ddd (14.9, 8.5, 2.0) | 6, 7, 9 |
| 8b | 2.61, ddd (14.9, 4.7, 2.6) | 4, 6, 7 | |
| 9 | 62.1 | 3.26, dd (2.6, 2.0) | 4, 7, 8 |
| 10 | 12.4 | 1.24, d (7.0) | 5, 6, 7 |
| NH | - | 6.03, t (6.1) | 1, 1’ |
| 1’ | 173.0 | - | - |
| 2’ | 36.4 | 2.21, m | 1’, 3’, 4’ |
| 3’ | 28.6 | 2.30, m | 2’, 4’, 5’ |
| 4’ | 130.7 | 5.45, m | 3’, 5’ 6’ |
| 5’ | 128.0 | 5.47, m | 3’, 4’, 6’, 7’ |
| 6’ | 36.4 | 2.18, m | 7’, 8’ |
| 7’ | 80.8 | 3.16, m | 5’, 8’, 9’, 15’ |
| 8’ | 33.5 | 1.42, m | 7’, 9’, 10’ |
| 9’ | 25.5 | 1.27, m d | - |
| 10’ | 29.9 | 1.27, m d | - |
| 11’ | 29.4 | 1.27, m d | - |
| 12’ | 32.0 c | 1.27, m d | - |
| 13’ | 22.8 c | 1.27, m d | - |
| 14’ | 14.2 | 0.88, t (6.8) | 12’, 13’ |
| 15’ | 56.6 | 3.31, s | 7’ |
a Recorded at 125 MHz. b Recorded at 500 MHz. c Assignments may be interchanged. d Overlapped signals.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sueyoshi, K.; Yamano, A.; Ozaki, K.; Sumimoto, S.; Iwasaki, A.; Suenaga, K.; Teruya, T. Three New Malyngamides from the Marine Cyanobacterium Moorea producens. Mar. Drugs 2017, 15, 367. https://doi.org/10.3390/md15120367
AMA Style
Sueyoshi K, Yamano A, Ozaki K, Sumimoto S, Iwasaki A, Suenaga K, Teruya T. Three New Malyngamides from the Marine Cyanobacterium Moorea producens. Marine Drugs. 2017; 15(12):367. https://doi.org/10.3390/md15120367
Chicago/Turabian StyleSueyoshi, Kosuke, Aki Yamano, Kaori Ozaki, Shimpei Sumimoto, Arihiro Iwasaki, Kiyotake Suenaga, and Toshiaki Teruya. 2017. "Three New Malyngamides from the Marine Cyanobacterium Moorea producens" Marine Drugs 15, no. 12: 367. https://doi.org/10.3390/md15120367
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.