
QM-HiFSA-Aided Structure Determination of Succinilenes A–D, New Triene Polyols from a Marine-Derived Streptomyces sp.

 , and
, and
Abstract
:1. Introduction
2. Results
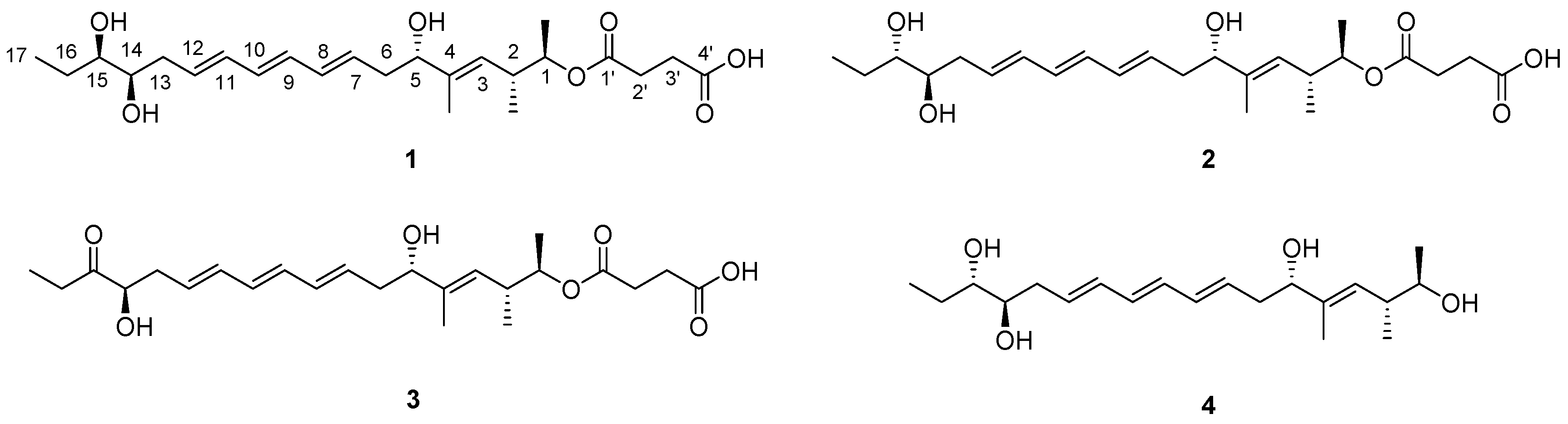
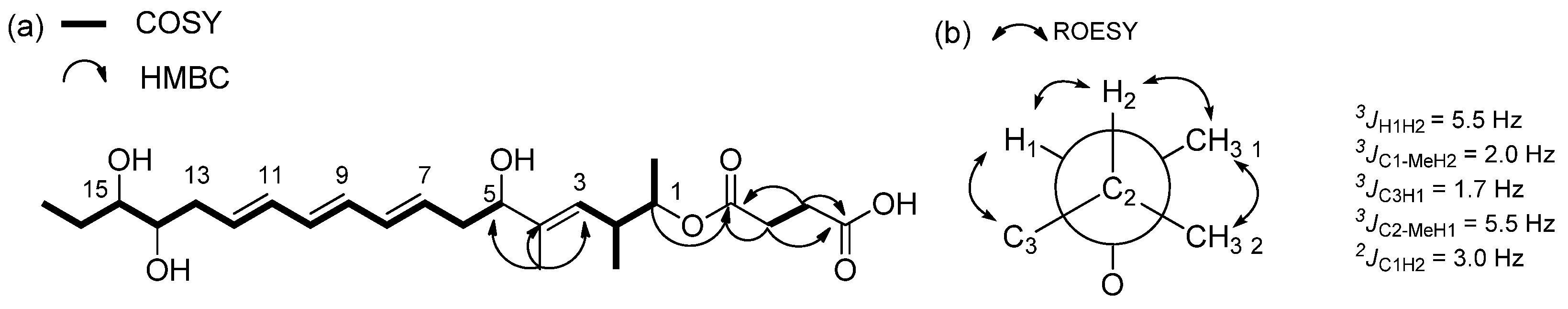
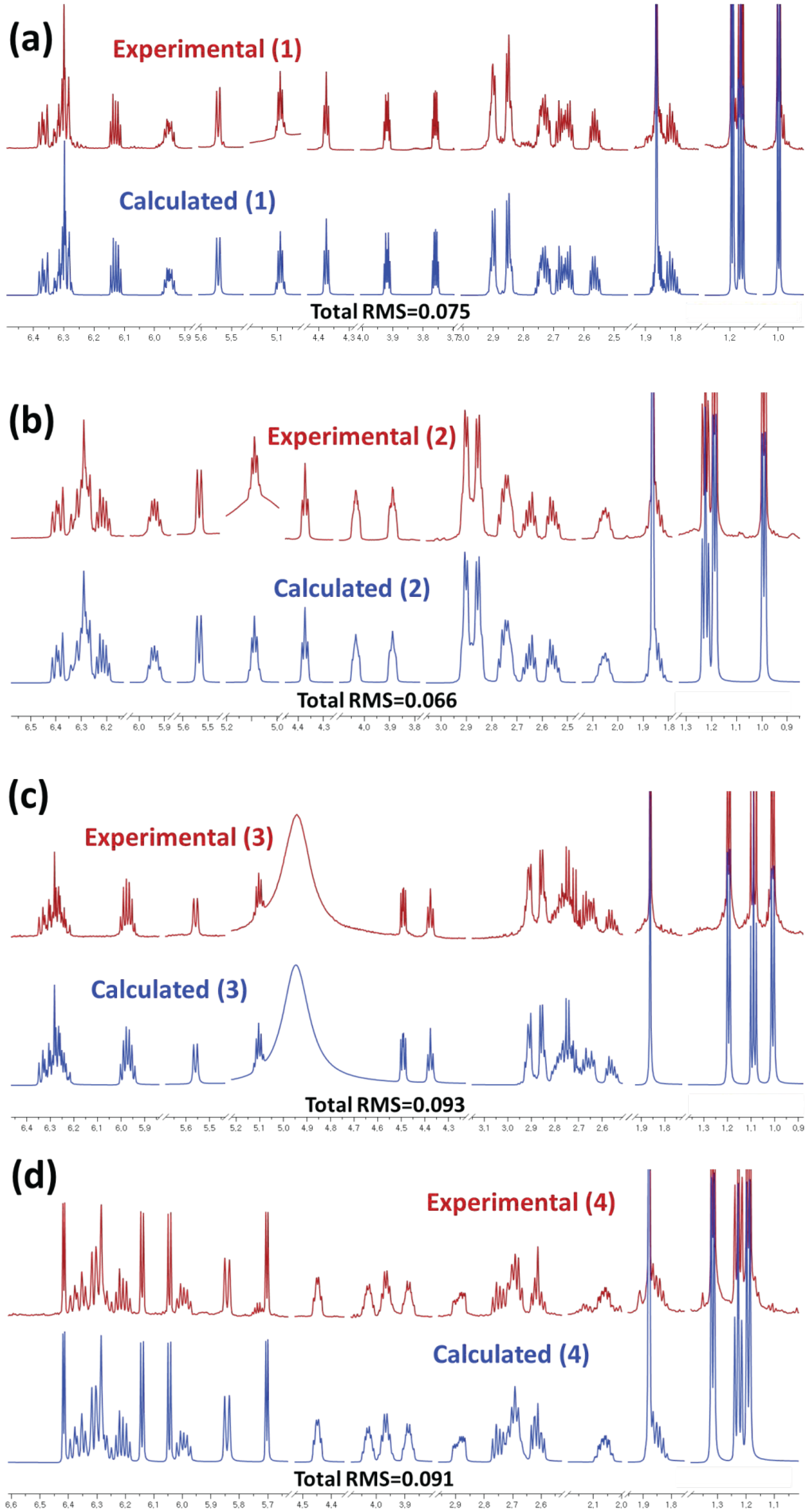
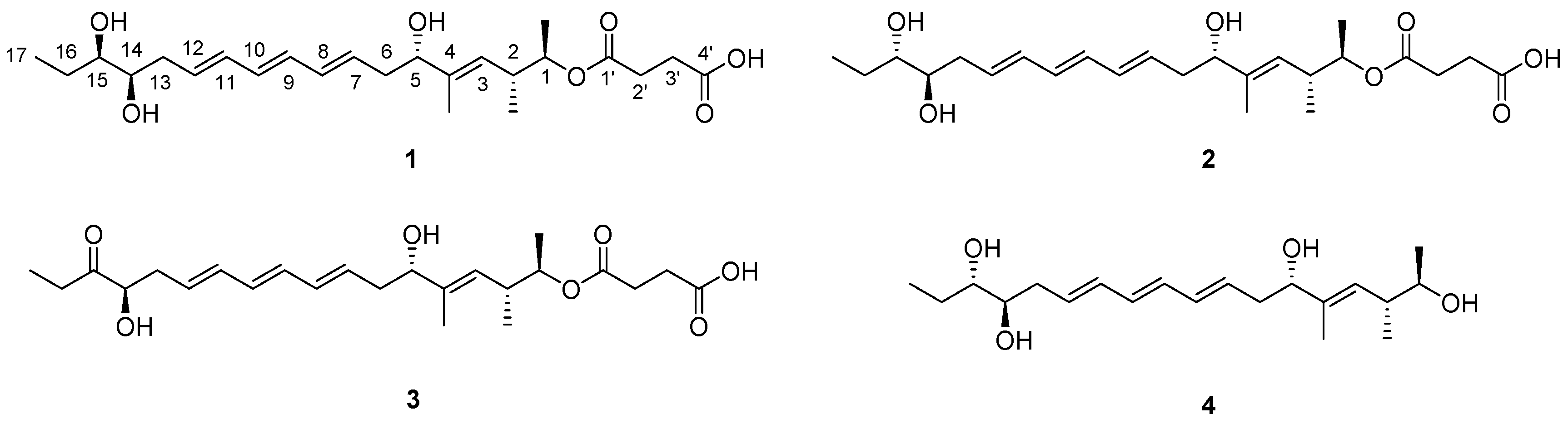
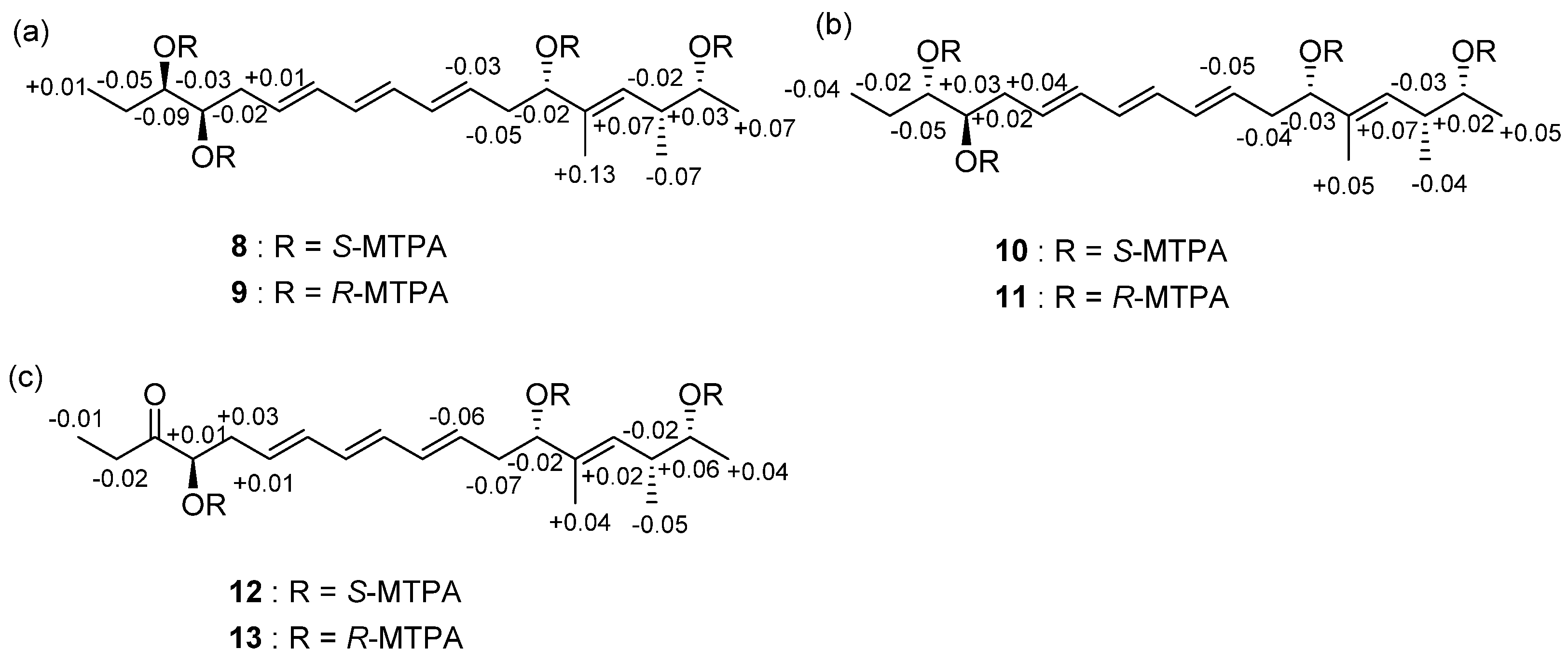
2.1. Structural Elucidation
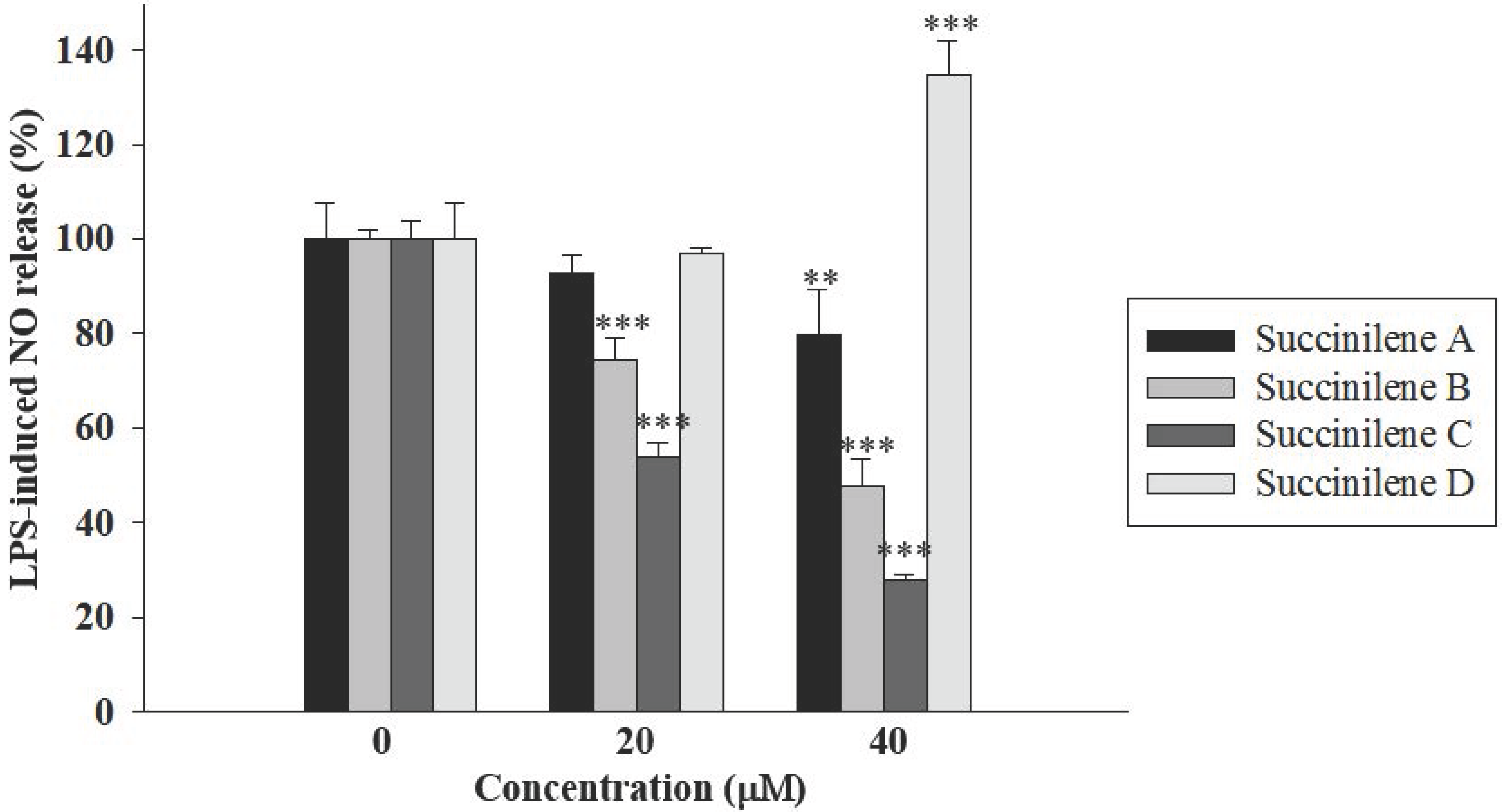
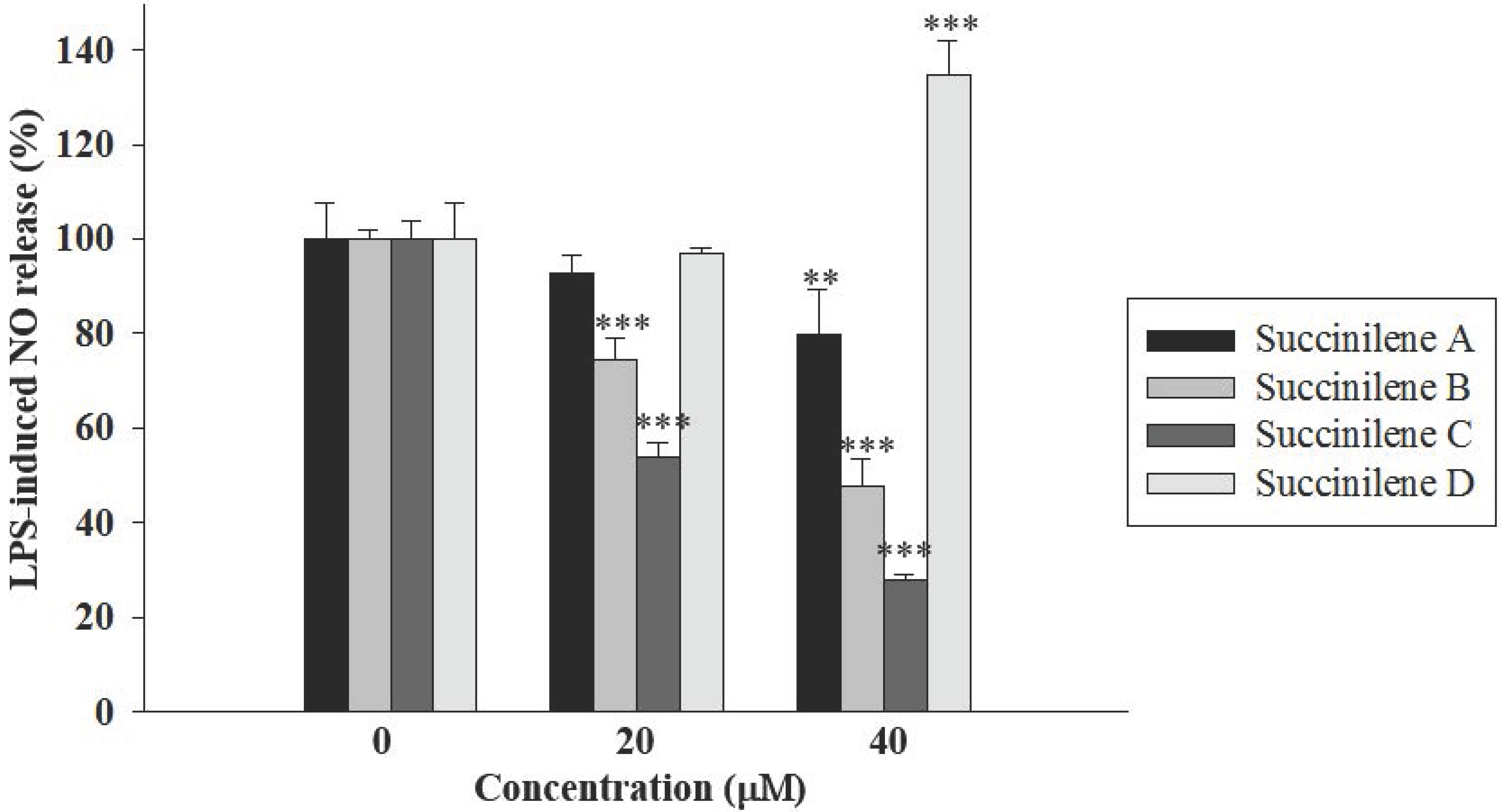
2.2. Bioactivities of Succinilenes A–D
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation of Bacteria, Cultivation, and Extraction
3.3. Isolation of Succinilenes A–D
3.3.1. Succinilene A (1)
3.3.2. Succinilene B (2)
3.3.3. Succinilene C (3)
3.3.4. Succinilene D (4)
3.4. Methanolysis of Succinilenes A–C
3.5. MTPA Esterification of Succinilenes A–C
3.5.1. Tetra S-MTPA Ester (8) of Methanolysis Product (5) of Succinilene A (1)
3.5.2. Tetra-R-MTPA Ester (9) of Methanolysis Product (5) of Succinilene A (1)
3.5.3. Tetra-S-MTPA Ester (10) of Methanolysis Product (6) of Succinilene B (2)
3.5.4. Tetra-R-MTPA Ester (11) of Methanolysis Product (6) of Succinilene B (2)
3.5.5. Tetra-S-MTPA Ester (12) of Methanolysis Product (7) of Succinilene C (3)
3.5.6. Tetra-R-MTPA Ester (13) of Methanolysis Product (7) of Succinilene C (3)
3.6. Evaluation of Antiproliferative Activity
3.7. Evaluation of i-NOS Assay
3.7.1. Materials
3.7.2. Cell Culture
3.7.3. Nitrite Assay
3.7.4. Statistical Analysis
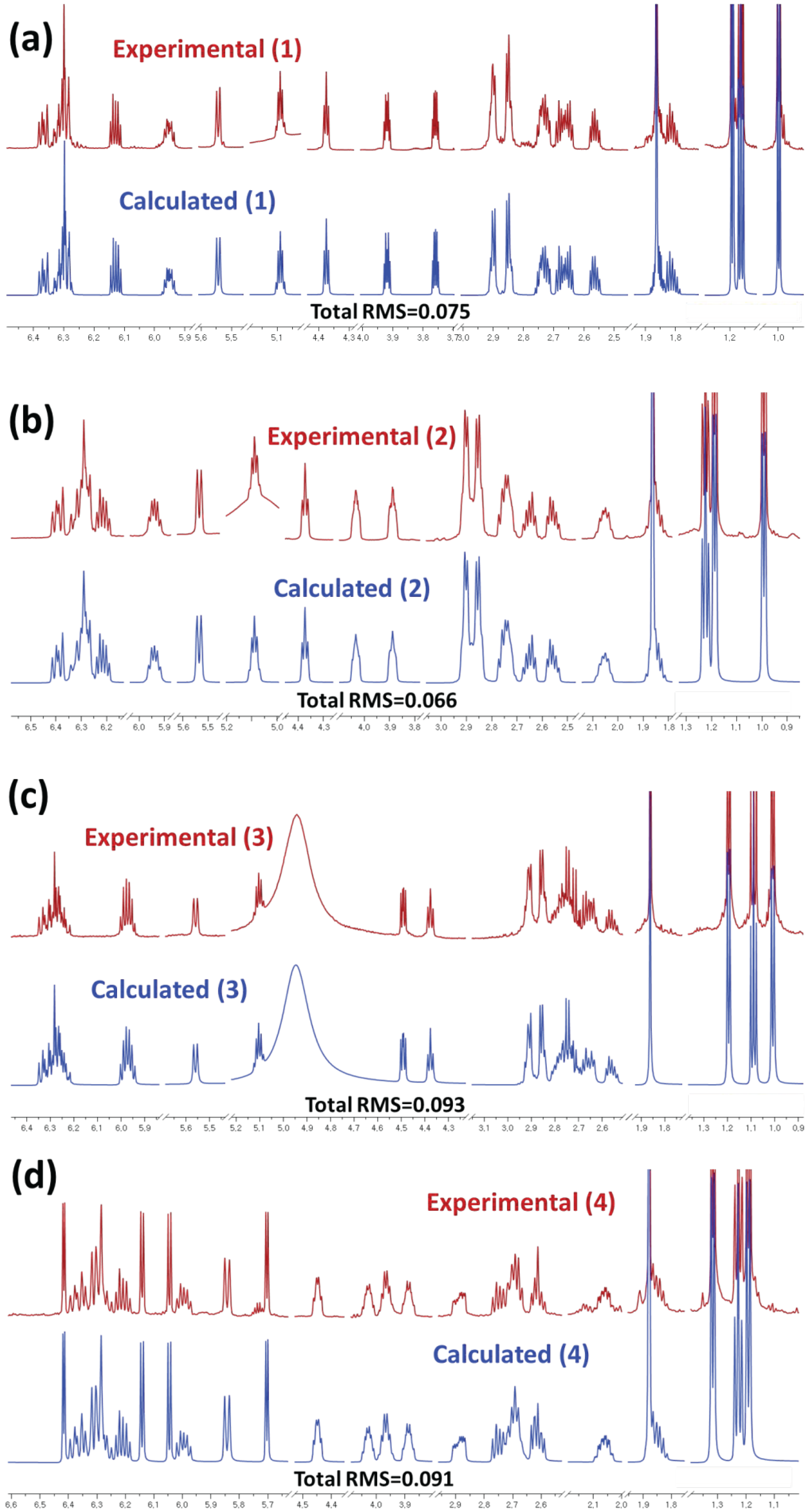
3.8. Computer-Aided NMR Spectral Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Napolitano, J.G.; Gödecke, T.; Rodriguez-Brasco, M.F.; Jaki, B.U.; Chen, S.-N.; Lankin, D.C.; Pauli, G.F. The tandem of full spin analysis and qHNMR for the quality control of botanicals exemplified with Ginkgo biloba. J. Nat. Prod. 2012, 75, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, J.G.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Complete 1H NMR spectral analysis of ten chemical markers of Ginkgo biloba. Magn. Reson. Chem. 2012, 50, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Niemitz, M.; Laatikainen, R.; Chen, S.-N.; Kleps, R.; Kozikowski, A.P.; Pauli, G.F. Complete 1H NMR spectral fingerprint of huperzine A. Magn. Reson. Chem. 2007, 45, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Shin, Y.; Lee, S.-H.; Oh, K.-B.; Lee, S.K.; Shin, J.; Oh, D.-C. Salternamides A–D from a halophilic Streptomyces sp. actinobacterium. J. Nat. Prod. 2015, 78, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Shin, Y.; Lee, S.K.; Shin, J.; Oh, D.-C. Salternamide E from a saltern-derived marine actinomycete Streptomyces sp. Nat. Prod. Sci. 2015, 21, 273–277. [Google Scholar] [CrossRef]
- Kim, S.-H.; Ha, T.-K.-Q.; Oh, W.K.; Shin, J.; Oh, D.-C. Antiviral indolosesquiterpenoid xiamycins C–E from a halophilic Streptomyces sp. actinomycete. J. Nat. Prod. 2016, 79, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Kim, H.; Moon, K.; Nam, S.-J.; Shin, J.; Oh, K.-B.; Oh, D.-C. Mohangamides A and B, new dilactone-tethered pseudo-dimeric peptides inhibiting Candida albicans isocitrate lyase. Org. Lett. 2015, 17, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Chung, B.; Oh, K.-B.; Shin, J.; Oh, D.-C. Hormaomycins B and C: New antibiotic cyclic depsipeptides from a marine mudflat-derived Streptomyces sp. Mar. Drugs 2015, 13, 5187–5200. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Ahn, S.; Noh, M.; Shin, J.; Oh, D.-C. Suncheonosides A–D, benzothioate glycosides from a marine-derived Streptomyces sp. J. Nat. Prod. 2015, 78, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Um, S.; Choi, T.J.; Lee, S.-H.; Kim, B.Y.; Kim, S.-H.; Lee, S.K.; Oh, K.-B.; Shin, J.; Oh, D.-C. Ohmyungsamycins A and B: cytotoxic and antimicrobial cyclic peptides produced by Streptomyces sp. from a volcanic island. J. Org. Chem. 2013, 78, 12321–12329. [Google Scholar] [CrossRef] [PubMed]
- Um, S.; Kim, Y.-J.; Kwon, H.; Wen, H.; Kim, S.-H.; Kwon, H.C.; Park, S.; Shin, J.; Oh, D.-C. Sungsanpin, a lasso peptide from a deep-sea streptomycete. J. Nat. Prod. 2013, 76, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Moon, K.; Bang, H.-S.; Kim, S.-H.; Kim, D.-G.; Oh, K.-B.; Shin, J.; Oh, D.-C. Tripartilactam, a cyclobutane-bearing tricyclic lactam from a Streptomyces sp. in a dung beetle’s brood ball. Org. Lett. 2012, 14, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-C.; Poulsen, M.; Currie, C.R.; Clardy, J. Sceliphrolactam, a polyene macrocyclic lactam from a wasp-associated Streptomyces sp. Org. Lett. 2011, 13, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Kurz, M.; Schmieder, P.; Kessler, H. HETLOC, an efficient method for the determination of heteronuclear long-range coupling of heteronuclei in natural abundance. Angew. Chem. Int. Ed. 1991, 30, 1329–1331. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Félix, F.; Seco, J.M.; Quiñoá, E.; Riguera, R. Determining the absolute stereochemistry of secondary/secondary diols by 1H NMR: Basis and applications. J. Org. Chem. 2005, 70, 3778–3790. [Google Scholar]
- Pauli, G.F.; Chen, S.-N.; Lankin, D.C.; Bisson, J.; Case, R.J.; Chadwick, L.R.; Gödecke, T.; Inui, T.; Krunic, A.; Jaki, B.U.; et al. Essential parameters for structural analysis and dereplication by 1H NMR Spectroscopy. J. Nat. Prod. 2014, 77, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Friebolin, H. Basic One- and Two- Dimensional NMR Spectroscopy, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Fujihara, M.; Muroi, M.; Tanamoto, K.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: roles of the receptor complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Rietschel, E.T.; Brade, H. Bacterial endotoxins. Sci. Am. 1992, 267, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.J.; Hume, D.A. Endotoxin signal transduction in macrophages. J. Leukoc. Biol. 1996, 60, 8–26. [Google Scholar] [PubMed]
- Napolitano, J.G.; Lankin, D.C.; Graf, T.N.; Friesen, J.B.; Chen, S.-N.; McAlpine, J.B.; Oberlies, N.H.; Pauli, G.F. HiFSA fingerprinting applied to isomers with near-identical NMR spectra: The silybin/isosilybin case. J. Org. Chem. 2013, 78, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, J.G.; Lankin, D.C.; McAlpine, J.B.; Niemitz, M.; Korhonen, S.-P.; Chen, S.-N.; Pauli, G.F. Proton fingerprints portray molecular structures: enhanced description of the 1D 1H NMR spectra of small molecules. J. Org. Chem. 2013, 78, 9963–9968. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-W.; Phansalkar, R.S.; Lankin, D.C.; Bisson, J.; McAlpine, J.B.; Leme, A.A.; Vidal, C.M.P.; Ramirez, B.; Niemitz, M.; Bedran-Russo, A.; et al. Subtle chemical shifts explain the NMR fingerprints of oligomeric proanthocyanidins with high dentin biomodification potency. J. Org. Chem. 2015, 80, 7495–7507. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Tian, L.; Xu, M.; Deng, Z.; Lin, W. A new 24-membered lactone and a new polyene δ-lactone from the marine bacterium Bacillus marinus. J. Antibiot. 2008, 61, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Koshino, H.; Isono, K.; Takahashi, H.; Kawanishi, G. Reveromycin A, a new antibiotic which inhibits the mitogenic activity of epidermal growth factor. J. Antibiot. 1991, 44, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Michel, K.H.; Demarco, P.V.; Nagarajan, R. The isolation and structure elucidation of macrocyclic lactone antibiotic, A26771B. J. Antibiot. 1977, 30, 571–575. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| δH a | Mult (J in Hz) | δC b | δH c | Mult (J in Hz) | δC d | |
| 1 | 5.0898 | dq (6.35, 5.50) | 74.1, d | 5.0882 | dq (6.35, 5.75) | 74.1, d |
| 1-Me | 1.1907 | d (6.35) | 17.2, q | 1.1862 | d (6.35) | 17.2, q |
| 2 | 2.7397 | ddq (9.74, 6.82, 5.50) | 37.1, d | 2.7356 | ddq (9.60, 6.76, 5.75) | 37.2, d |
| 2-Me | 0.9973 | d (6.82) | 16.6, q | 0.9916 | d (6.76) | 16.7, q |
| 3 | 5.5426 | d (9.74) | 127.2, d | 5.5359 | d (9.60) | 127.2, d |
| 4 | – | – | 140.1, s | – | – | 140.1, s |
| 4-Me | 1.8616 | s | 12.2, q | 1.8618 | s | 12.1, q |
| 5 | 4.3745 | dd (7.04, 6.04) | 76.8, d | 4.3721 | dd (6.69, 6.34) | 76.9, d |
| 6 | 2.6519 | ddd (−14.00, 7.30, 7.04) | 39.8, t | 2.6498 | ddd (−14.11, 7.47, 6.69) | 39.8, t |
| 2.5631 | ddd (−14.00, 7.50, 6.04) | 2.5587 | ddd (−14.11, 7.10, 6.34) | |||
| 7 | 5.9506 | ddd (14.65, 7.50, 7.30) | 132.0, d | 5.9395 | ddd (14.81, 7.47, 7.10) | 131.9, d |
| 8 | 6.2885 | dd (14.65, 10.77) | 132.5, d | 6.2814 | dd (14.81, 10.73) | 132.6, d |
| 9 | 6.2929 | dd (14.03, 10.77) | 131.6, d | 6.2773 | dd (14.79, 10.73) | 131.6, d |
| 10 | 6.3158 | dd (14.03, 10.49) | 131.7, d | 6.3179 | dd (14.79, 10.53) | 131.7, d |
| 11 | 6.3664 | dd (15.19, 10.49) | 132.6, d | 6.3913 | dd (15.19, 10.53) | 132.7, d |
| 12 | 6.1280 | ddd (15.19, 7.50, 7.12) | 132.8, d | 6.2145 | ddd (15.19, 7.45, 7.24) | 133.0, d |
| 13 | 2.7253 | ddd (−14.36, 7.50, 4.66) | 38.0, t | 2.8829 | ddd (−14.13, 7.24, 3.36) | 37.5, t |
| 2.6745 | ddd (−14.36, 8.02, 7.12) | 2.7481 | ddd (−14.13, 8.12, 7.45) | |||
| 14 | 3.9176 | ddd (8.02, 4.66, 4.33) | 74.2, d | 4.0324 | ddd (8.12, 5.84, 3.36) | 74.9, d |
| 15 | 3.7579 | ddd (8.68, 4.33, 3.88) | 75.5, d | 3.8871 | ddd (8.72, 5.84, 2.69) | 76.1, d |
| 16 | 1.8578 | ddq (−13.87, 7.38, 3.88) | 27.5, t | 2.0541 | ddq (−13.61, 7.36, 2.69) | 26.5, t |
| 1.8105 | ddq (−13.87, 8.68, 7.49) | 1.8504 | ddq (−13.61, 8.72, 7.42) | |||
| 17 | 1.1619 | dd (7.49, 7.38) | 10.9, q | 1.2234 | dd (7.42, 7.36) | 10.9, q |
| 1′ | – | – | 172.7, s | – | – | 173.0, s |
| 2′ | 2.8516 | ddd (−11.48, 7.29, 6.04) | 30.3, t | 2.8496 | ddd (−18.10, 7.13, 6.33) | 30.6, t |
| 2.8436 | ddd (−11.48, 7.54, 5.82) | 2.8496 | ddd (−18.10, 8.56, 4.83) | |||
| 3′ | 2.9046 | ddd (−16.39, 7.29, 5.82) | 30.4, t | 2.9078 | ddd (−15.38, 8.56, 7.13) | 30.8, t |
| 2.8954 | ddd (−16.39, 7.54, 6.04) | 2.9022 | ddd (−15.38, 6.33, 4.83) | |||
| 4′ | – | – | 175.7, s | – | – | 176.2, s |
| Position | 3 | 4 | ||||
|---|---|---|---|---|---|---|
| δH a | Mult (J in Hz) | δC b | δH a | Mult (J in Hz) | δC b | |
| 1 | 5.1041 | dq (6.33, 5.27) | 74.2, d | 3.9666 | ddq (6.23, 5.00, 4.60) | 70.8, d |
| 1-Me | 1.1944 | d (6.33) | 17.3, q | 1.3158 | d (6.23) | 21.0, q |
| 2 | 2.7403 | ddq (9.97, 6.86, 5.27) | 37.2, d | 2.6988 | ddq (9.62, 6.85, 5.00) | 39.6, d |
| 2-Me | 1.0069 | d (6.86) | 16.6, q | 1.1905 | d (6.85) | 17.0, q |
| 3 | 5.5580 | d (9.97) | 127.4, d | 5.8422 | d (9.62) | 128.6, d |
| 4 | – | – | 140.3, s | – | – | 138.8, s |
| 4-Me | 1.8616 | s | 12.4, q | 1.8782 | s | 12.1, q |
| 5 | 4.3767 | dd (7.10, 6.06) | 76.9, d | 4.4482 | ddd (7.07, 5.51, 3.83) | 77.2, d |
| 6 | 2.6443 | ddd (−13.79, 6.93, 6.06) | 39.8, t | 2.6890 | ddd (−13.68, 7.07, 7.00) | 39.9, t |
| 2.5606 | ddd (−13.79, 7.12, 7.10) | 2.6130 | ddd (−13.68, 7.99, 5.51) | |||
| 7 | 5.9652 | ddd (14.66, 7.12, 6.93) | 132.6, d | 5.9948 | ddd (14.80, 7.99, 7.00) | 132.1, d |
| 8 | 6.2708 | dd (14.66, 10.63) | 132.5, d | 6.2974 | dd (14.80, 10.62) | 132.7, d |
| 9 | 6.2902 | dd (15.05, 10.63) | 132.4, d | 6.2738 | dd (14.86, 10.62) | 131.8, d |
| 10 | 6.2414 | dd (15.05, 10.72) | 131.2, d | 6.3186 | dd (14.86, 10.55) | 132.1, d |
| 11 | 6.3255 | dd (15.14, 10.72) | 133.7, d | 6.3706 | dd (15.21, 10.55) | 133.0, d |
| 12 | 5.9778 | ddd (15.14, 7.41, 7.00) | 129.8, d | 6.2091 | ddd (15.21, 7.38, 7.16) | 133.2, d |
| 13 | 2.7803 | ddd (−13.49, 7.27, 7.00) | 38.2, t | 2.8870 | ddd (−14.32, 7.38, 3.45) | 37.6, t |
| 2.6591 | ddd (−13.49, 7.41, 4.88) | 2.7460 | ddd (−14.32, 8.46, 7.16) | |||
| 14 | 4.4922 | dd (7.27, 4.88) | 77.2, d | 4.0282 | dddd (8.46, 5.85, 5.74, 3.45) | 75.0, d |
| 15 | – | – | 214.0, s | 3.8872 | dddd (8.87, 5.85, 5.90, 3.10) | 76.2, d |
| 16 | 2.7549 | dq (−14.89, 7.05) | 31.7, t | 2.0620 | ddq (−13.56, 7.35, 3.10) | 26.5, t |
| 2.7206 | dq (−14.89, 7.45) | 1.8533 | ddq (−13.56, 8.87, 7.37) | |||
| 17 | 1.0890 | dd (7.45, 7.05) | 7.8, q | 1.2266 | dd (7.37, 7.35) | 10.8, q |
| 1′ | – | – | 172.7, s | |||
| 2′ | 2.8525 | ddd (−16.88, 7.01, 6.26) | 30.3, t | |||
| 2.8525 | ddd (−16.88, 8.79, 4.47) | |||||
| 3′ | 2.9172 | ddd (−16.89, 8.79, 7.01) | 30.0, t | |||
| 2.9060 | ddd (−16.89, 6.26, 4.47) | |||||
| 4′ | – | – | 175.3, s | |||
| OH-1 | 5.7021 | d (4.60) | ||||
| OH-5 | 6.4159 | d (3.83) | ||||
| OH-14 | 6.1408 | d (5.74) | ||||
| OH-15 | 6.0448 | d (5.90) | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, M.; Park, S.H.; Kwon, Y.; Lee, S.K.; Shin, J.; Nam, J.-W.; Oh, D.-C. QM-HiFSA-Aided Structure Determination of Succinilenes A–D, New Triene Polyols from a Marine-Derived Streptomyces sp. Mar. Drugs 2017, 15, 38. https://doi.org/10.3390/md15020038
Bae M, Park SH, Kwon Y, Lee SK, Shin J, Nam J-W, Oh D-C. QM-HiFSA-Aided Structure Determination of Succinilenes A–D, New Triene Polyols from a Marine-Derived Streptomyces sp. Marine Drugs. 2017; 15(2):38. https://doi.org/10.3390/md15020038
Chicago/Turabian StyleBae, Munhyung, So Hyun Park, Yun Kwon, Sang Kook Lee, Jongheon Shin, Joo-Won Nam, and Dong-Chan Oh. 2017. "QM-HiFSA-Aided Structure Determination of Succinilenes A–D, New Triene Polyols from a Marine-Derived Streptomyces sp." Marine Drugs 15, no. 2: 38. https://doi.org/10.3390/md15020038