
Inhibitors of BRD4 Protein from a Marine-Derived Fungus Alternaria sp. NH-F6

Abstract
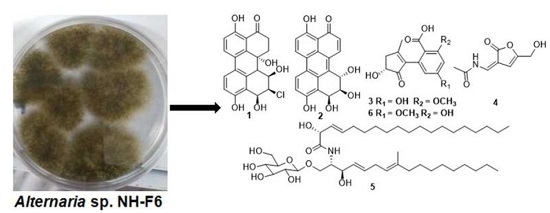
:
1. Introduction
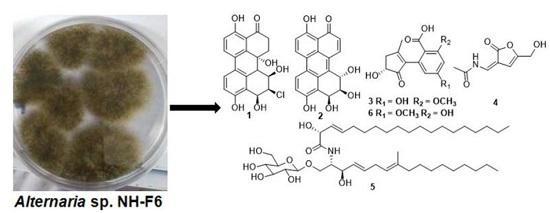
2. Results
2.1. Identification and Structure Elucidation
2.2. Inhibitory Activity against BRD4 Protein
3. Discussion
4. Experimental Section
4.1. General
4.2. Fungus Material
4.3. Fermentation and Isolation
4.4. Methanolysis of Compound 5
4.5. BRD4 Inhibitor Screening Assay
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2012, 29, 144–222. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2010, 27, 165–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Li, X.M.; Li, C.S.; Ji, N.Y.; Wang, B.G. Secondary metabolites from Penicillium pinophilum SD-272, a marine sediment-derived fungus. Mar. Drugs 2013, 11, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Quan, C.; Hou, X.; Fan, S. Potential pharmacological resources: Natural bioactive compounds from marine-derived fungi. Mar. Drugs 2016, 14, 76. [Google Scholar] [CrossRef]
- Vidler, L.R.; Brown, N.; Knapp, S.; Hoelder, S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Bamborough, P.; Diallo, H.; Goodacre, J.D.; Gordon, L.; Lewis, A.; Seal, J.T.; Wilson, D.M.; Woodrow, M.D.; Chung, C. Fragment-based discovery of bromodomain inhibitors part 2: Optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Conway, S.J. Bromodomains: Are readers right for epigenetic therapy? ACS Med. Chem. Lett. 2012, 3, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sanchez, R.; Zhou, M.M. Scaling the druggability landscape of human bromodomains, a new class of drug targets. J. Med. Chem. 2012, 55, 7342–7345. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Natsume, I.; Sawai, K.; Sawamura, K.; Furusaki, A.; Matsumoto, T. Structure of antifungal and phytotoxic pigments produced by Alternaria sps. Tetrahedron Lett. 1983, 24, 5653–5656. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Z.; Bai, J.; Wang, Y.; Zhang, L.; Wu, X.; Hua, H. A new perylenequinone from a halotolerant fungus, Alternaria sp. M6. Chin. J. Nat. Med. 2012, 10, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.H.; Edrada-Ebel, R.; Indriani, I.D.; Wray, V.; Müller, W.E.; Totzke, F.; Zirrgiebel, U.; Schächtele, C.; Kubbutat, M.H.; Lin, W.H.; et al. Cytotoxic metabolites from the fungal endophyte Alternaria sp. and their subsequent detectionin its host plant Polygonum senegalense. J. Nat. Prod. 2008, 71, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, Y.; Sun, K.; Liu, P.; Yin, X.; Zhu, W. Cerebrosides and 2-pyridone alkaloids from the halotolerant fungus Penicillium chrysogenum grown in a hypersaline medium. J. Nat. Prod. 2011, 74, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Wang, Y.; Tao, H.W.; Peng, X.P.; Liu, P.P.; Zhu, W.M. Cerebrosides of the halotolerant fungus Alternaria raphani isolated from a sea salt field. J. Nat. Prod. 2009, 72, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Keusgen, M.; Yu, C.M.; Curtis, J.M.; Brewer, D.; Ayer, S.W. A cerebroside from the marine fungus Microsphaeropsis olivacea (Bonord.) Höhn. Biochem. Syst. Ecol. 1996, 24, 465–468. [Google Scholar] [CrossRef]
- Naganuma, M.; Nishida, M.; Kuramochi, K.; Sugawara, F.; Yoshida, H.; Mizushina, Y. 1-Deoxyrubralactone, a novel specific inhibitor of families X and Y of eukaryotic DNA polymerases from a fungal strain derived from sea algae. Bioorg. Med. Chem. 2008, 16, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, Q.; Gao, Y.; Tang, J.; Zhang, A.; Gao, J. Secondary metabolites from the endophytic Botryosphaeria dothidea of Melia azedarach and their antifungal, antibacterial, antioxidant, and cytotoxic activities. J. Agric. Food Chem. 2014, 62, 3584–3590. [Google Scholar] [CrossRef] [PubMed]
- He, J.W.; Chen, G.D.; Gao, H.; Yang, F.; Li, X.X.; Peng, T.; Guo, L.D.; Yao, X.S. Heptaketides with antiviral activity from three endolichenic fungal strains Nigrospora sp., Alternaria sp. and Phialophora sp. Fitoterapia 2012, 83, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Kiet, T.T.; Schlegel, B.; Gräfe, U. New homoserine lipids from Xerocomus langbianensis. J. Basic Microbiol. 2002, 42, 133–136. [Google Scholar] [CrossRef]
- Ioannou, E.; Abdel-Razik, A.F.; Zervou, M.; Christofidis, D.; Alexi, X.; Vagias, C.; Alexis, M.N.; Roussis, V. 5α,8α-Epidioxysterols from the gorgonian Eunicella cavolini and the ascidian Trididemnum inarmatum: Isolation and evaluation of their antiproliferative activity. Steroids 2009, 74, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Piccialli, V.; Sica, D. Four new trihydroxylated sterols from the sponge Spongionella gracilis. J. Nat. Prod. 1987, 50, 915–920. [Google Scholar] [CrossRef]
- Singh, S.B.; Jayasuriya, H.; Dewey, R.; Polishook, J.D.; Dombrowski, A.W.; Zink, D.L.; Guan, Z.; Collado, J.; Platas, G.; Pelaez, F.; et al. Isolation, structure, and HIV-1-integrase inhibitory activity of structurally diverse fungal metabolites. J. Ind. Microbiol. Biotechnol. 2003, 30, 721–731. [Google Scholar] [PubMed]
- Kim, E.L.; Li, J.L.; Xiao, B.; Hong, J.; Yoo, E.S.; Yoon, W.D.; Jung, J.H. A new cyclic tetrapeptide from the jellyfish-derived fungus Phoma sp. Chem. Pharm. Bull. 2012, 60, 1590–1593. [Google Scholar] [PubMed]
- Yuan, L.; Zhao, P.; Ma, J.; Li, G.; Shen, Y. Tricycloalternarenes A–E: Five new mixed terpenoids from the endophytic fungal strain Alternaria alternata Ly83. Helv. Chim. Acta 2008, 91, 1588–1594. [Google Scholar] [CrossRef]
- Phaopongthai, J.; Wiyakrutta, S.; Meksuriyen, D.; Sriubolmas, N.; Suwanborirux, K. Azole-synergistic anti-candidal activity of altenusin, a biphenyl metabolite of the endophytic fungus Alternaria alternata isolated from Terminalia chebula Retz. J. Microbiol. 2013, 51, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Shiojima, K.; Nakajima, H.; Hamasaki, T. Structure and biological activity of plant growth regulators produced by Penicillium sp. No. 31f. Biosci. Biotechnol. Biochem. 1992, 56, 1138–1139. [Google Scholar] [CrossRef] [PubMed]
- Domondon, D.L.; He, W.; Kimpe, N.D.; Hofte, M.; Pöppe, J. β-adenosine, a bioactive compound in grass chaff stimulating mushroom production. Phytochemistry 2004, 65, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Han, L.; Kang, K.; Shan, C.; Wei, Y.; Zheng, C.; Guan, H. Secondary metabolites from green algae Ulva pertusa. Chem. Nat. Compd. 2010, 46, 828–830. [Google Scholar] [CrossRef]
- Teng, X.; Zhuang, Y.; Wang, Y.; Liu, P.; Xu, Z.; Zhu, W. Secondary metabolites from Penicillium sp. gxwz406 symbiotic with the gorgonian Echinogorgia flora. Chin. J. Mar. Drugs 2010, 29, 11–15. [Google Scholar]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain proteins in HIV infection. Viruses 2013, 5, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Philpott, M.; Muller, S.; Schulze, J.; Badock, V.; Eberspacher, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernandez-Montalvan, A.; et al. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Fu, L.; Peng, Y.; Zhou, L. Metabolites from Alternaria fungi and their bioactivities. Molecules 2013, 18, 5891–5935. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug discovery targeting bromodomain-containing protein 4 (BRD4). J. Med. Chem. 2017, 14. [Google Scholar] [CrossRef]
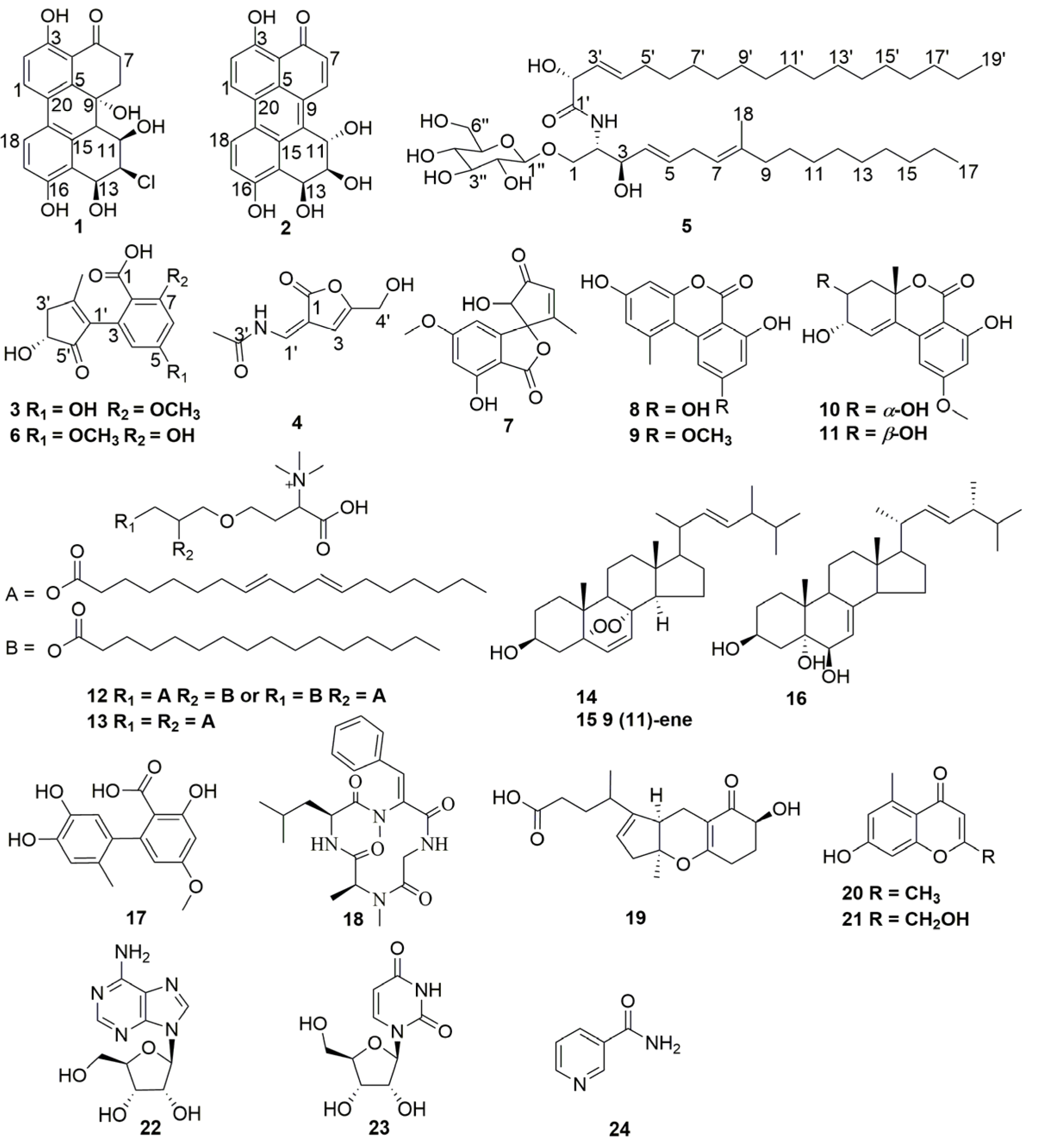
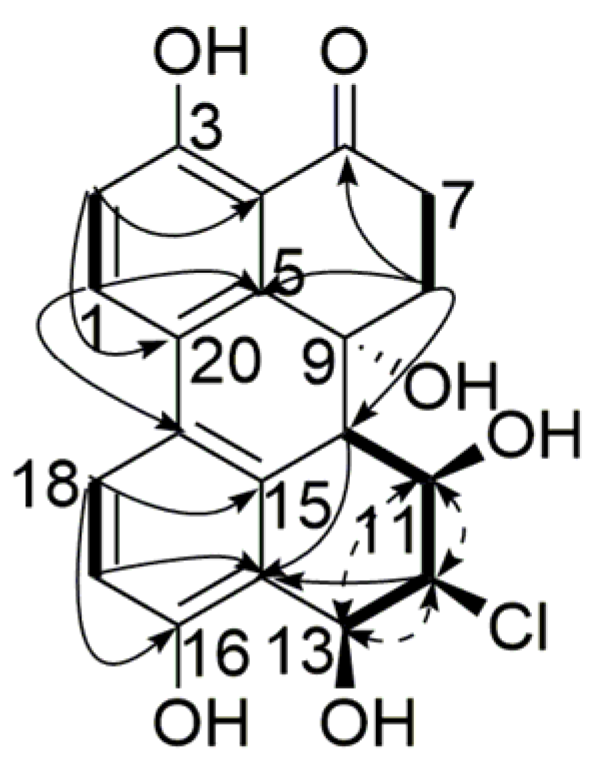
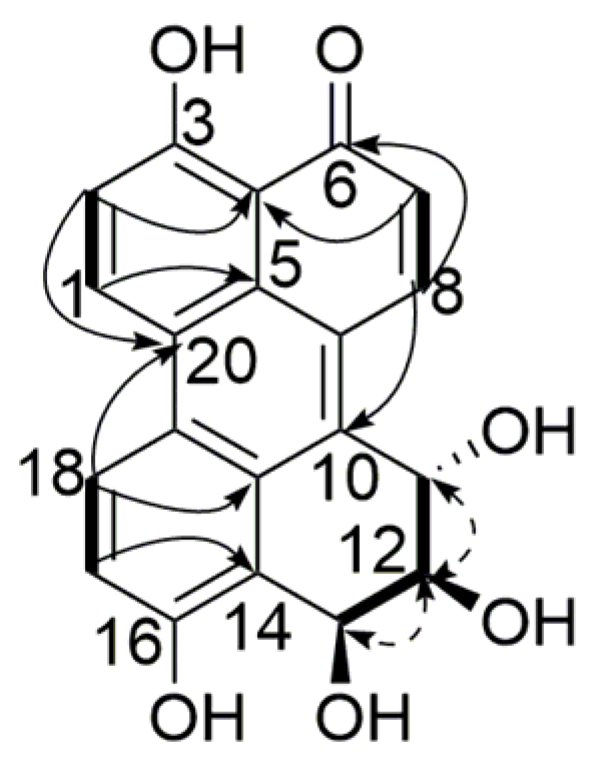
 ), 1H-1H COSY (
), 1H-1H COSY (  ) and NOESY (
) and NOESY (  ) correlations of compound 1.
) correlations of compound 1.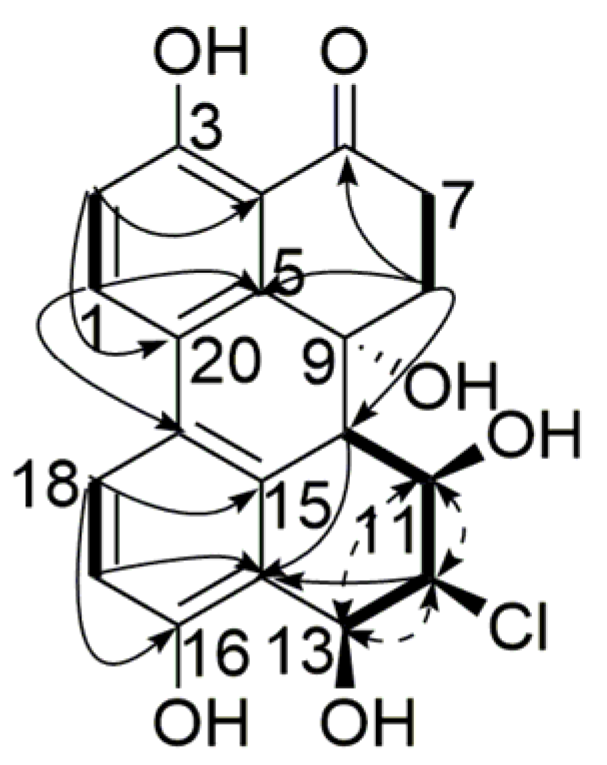 ), 1H-1H COSY ( ) and NOESY ( ) correlations of compound 2.
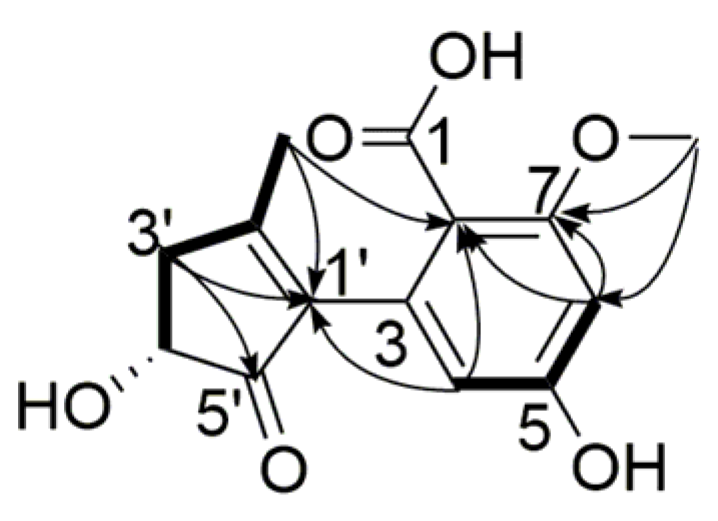
), 1H-1H COSY ( ) and NOESY ( ) correlations of compound 2. ) and 1H-1H COSY ( ) correlations of compound 3.
) and 1H-1H COSY ( ) correlations of compound 3.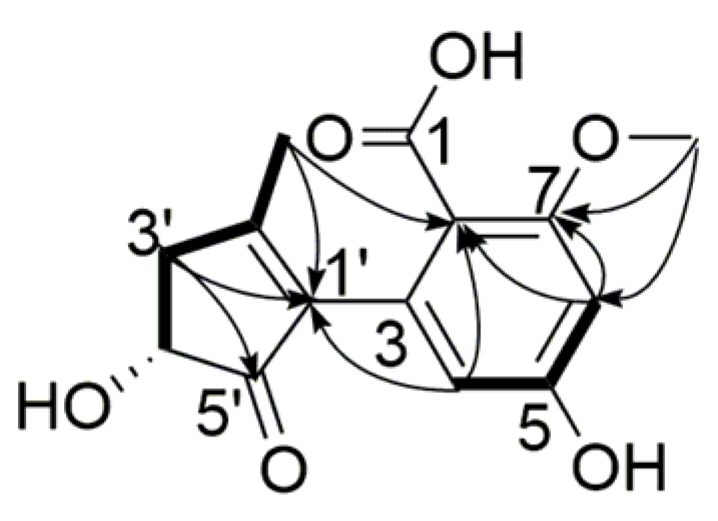 ) and 1H-1H COSY ( ) correlations of compound 4.
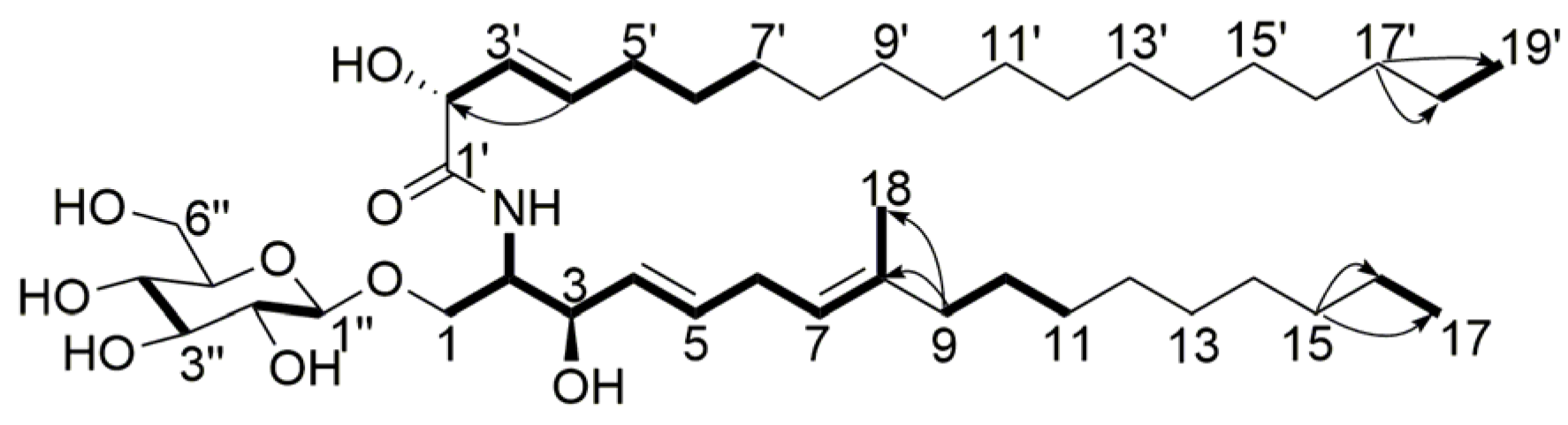
) and 1H-1H COSY ( ) correlations of compound 4.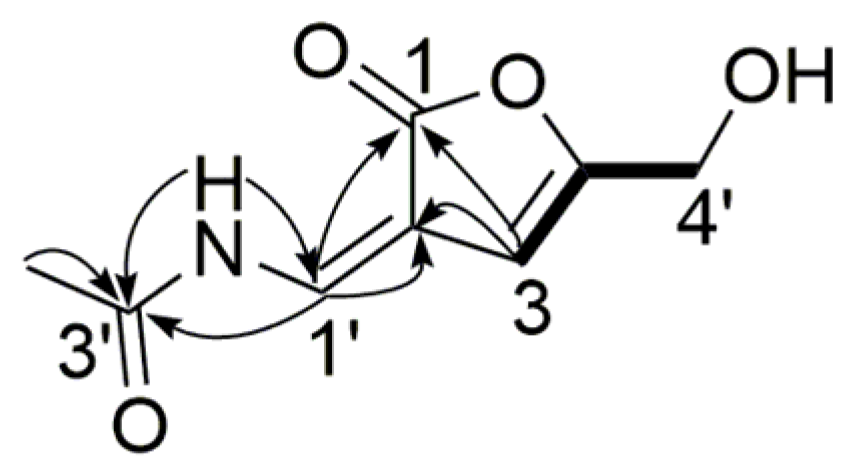 ) and 1H-1H COSY ( ) correlations of compound 5.
) and 1H-1H COSY ( ) correlations of compound 5.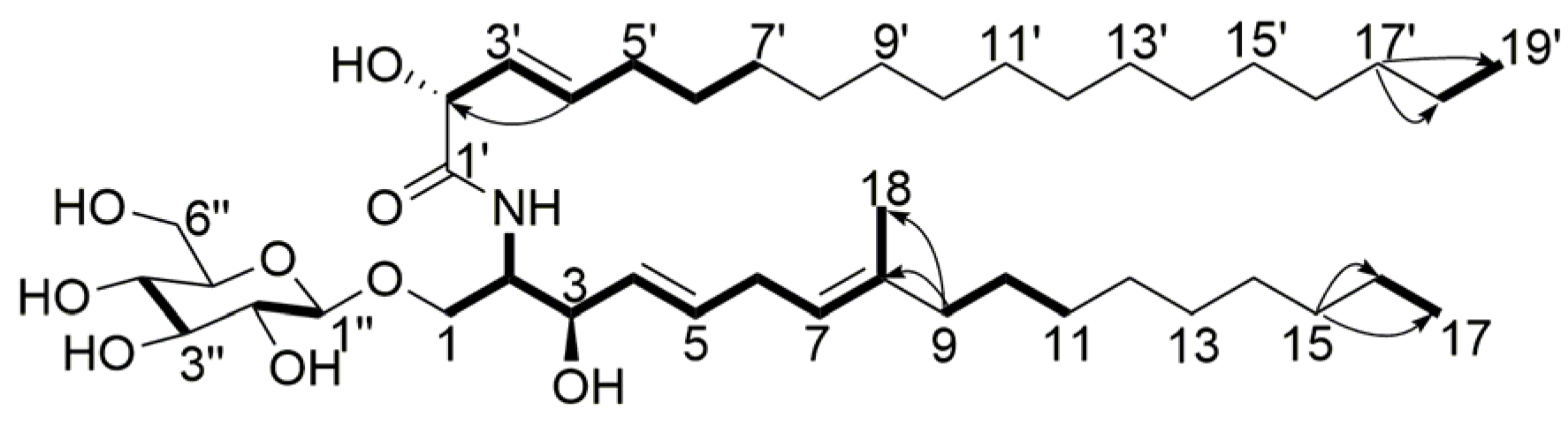

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Compound 1 a | Compound 2 a | ||||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC b | δC, Type | δH (J in Hz) | HMBC b | |
| 1 | 132.8, CH | 7.97, 1H, d (8.8) | 3, 5, 19 | 133.7, CH | 9.21, 1H, d (9.2) | 3, 5, 19 |
| 2 | 117.5, CH | 6.98, 1H, d (8.8) | 3, 4, 20 | 118.9, CH | 7.47, 1H, d (9.2) | 4, 20 |
| 3 | 160.4, C | 166.1, C | ||||
| 4 | 113.6, C | 111.1, C | ||||
| 5 | 140.8, C | 124.3, C | ||||
| 6 | 206.4, C | 187.9, C | ||||
| 7a | 33.6, CH2 | 3.06, 1H, td (17.5, 4.2) | 6 | 126.4, CH | 7.04, 1H, (10.0) | 4, 9 |
| 7b | 2.54, 1H, dt (17.5, 3.0) | 6, 9 | ||||
| 8a | 34.9, CH2 | 2.83, 1H, dt (14.4, 3.0) | 5, 6, 7, 9, 10 | 140.4, CH | 8.79, 1H, (10.0) | 5, 6, 10 |
| 8b | 2.27, 1H, td (14.4, 4.2) | 7 | ||||
| 9 | 67.8, C | 122.1, C | ||||
| 10 | 47.7, CH | 2.69, 1H, d (9.3) | 5, 9, 11, 14, 15 | 126.3, C | ||
| 11 | 62.6, CH | 4.76, 1H, ddd (9.3, 5.0, 2.9) | 10, 12 | 68.6, CH | 5.33, 1H, d (8.5) | 12 |
| 12 | 68.4, CH | 4.41, 1H, t (3.0) | 10, 11, 13, 14 | 73.0, CH | 3.92, 1H, ddd (8.5, 5.4, 3.3) | 11 |
| 13 | 65.9, CH | 5.03, 1H, d (3.0) | 11, 12, 14, 15, 16 | 66.3, CH | 5.65, 1H, dd, (5.3, 3.3) | 12 |
| 14 | 121.8, C | 118.8, C | ||||
| 15 | 132.4, C | 141.8, C | ||||
| 16 | 156.1, C | 154.8, C | ||||
| 17 | 113.4, CH | 6.80, 1H, d (8.5) | 14, 16, 19 | 121.8, CH | 7.43, 1H, d (9.2) | 14, 19 |
| 18 | 124.1, CH | 7.49, 1H, d (8.5) | 15, 16, 20 | 123.7, CH | 8.81, 1H, d (9.2) | 15, 16, 20 |
| 19 | 123.5, C | 125.7, C | ||||
| 20 | 125.0, C | 121.4, C | ||||
| 3-OH | 12.68, 1H, s | 2, 3, 4 | 15.23, 1H, s | 2, 3, 4 | ||
| 9-OH | 5.01, 1H, s | |||||
| 12-OH | 5.51, 1H, d (5.4) | |||||
| 11-OH | 5.12, 1H, d (5.0) | |||||
| 13-OH | 5.72, 1H, s | 5.72, 1H, d (5.3) | ||||
| 16-OH | 9.86, 1H, s | 10.54, 1H, s | ||||
| Position | Compound 3 a | ||
|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC b | |
| 1 | 172.3, C | ||
| 2 | 109.1, C | ||
| 3 | 136.3, C | ||
| 4 | 107.8, CH | 5.95, 1H, s | 2, 6, 7, 1′ |
| 5 | 160.5, C | ||
| 6 | 100.2, CH | 6.35, 1H, s | 2, 4, 7 |
| 7 | 162.0, C | ||
| 1′ | 141.0, C | ||
| 2′ | 164.9, C | ||
| 3′a | 40.5, CH2 | 2.90, 1H, dd (17.1, 6.5) | 1′, 4′, 5′, 2′-CH3 |
| 3′b | 2.41, 1H, d (17.1) | 1′, 4′ | |
| 4′ | 71.4, CH | 4.21, 1H, br s | 2′, 5′ |
| 5′ | 205.9, C | ||
| 7-OCH3 | 55.2, CH3 | 3.74, 3H, s | 1, 6, 7 |
| 2′-CH3 | 17.4, CH3 | 1.90, 3H, s | 2, 4, 1′, 2′, 3′, 5′ |
| Position | Compound 4 a | ||
|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC b | |
| 1 | 172.1, C | ||
| 2 | 128.0, C | ||
| 3 | 109.5, CH | 6.40, 1H, s | 4, 5, 6, 8 |
| 4 | 168.7, C | ||
| 1′ | 144.3, CH | 9.07, 1H, s | 1, 4, 5 |
| 2′-NH | 9.25, 1H, s | 1, 3 | |
| 3′ | 169.4, C | ||
| 4′ | 59.4, CH2 | 4.32, 2H, s | 6, 7 |
| 3′-CH3 | 23.3, CH3 | 2.10, 3H, s | 1 |
| 4′-OH | 5.73, 1H, s | ||
| Position | Compound 5 a | ||
|---|---|---|---|
| δC, Type | δH (J in Hz) | HMBC b | |
| 1a | 69.6, CH2 | 4.04, 1H, m | |
| 1b | 3.85, 1H, m | ||
| 2 | 53.5, CH | 4.04, 1H, m | |
| 3 | 72.4, CH | 4.10, 1H, m | |
| 4 | 128.6, CH | 5.45, 1H, dd (15.8, 6.0) | |
| 5 | 134.6, CH | 5.72, 1H, d (15.8) | |
| 6 | 27.9, CH2 | 2.02, 2H, m | |
| 7 | 123.3, CH | 5.08, 1H, t (6.0) | 6 |
| 8 | 136.1, C | ||
| 9 | 40.0, CH2 | 1.95, 2H, t (7.5) | 6, 8, 10, 18 |
| 10 | 28.3, CH2 | 1.36, 2H, m | |
| 11 | 29.4, CH2 | 1.26, 2H, m | |
| 12–14 | 29.6–30.0, CH2 | 1.26 × 3, 6H, m | |
| 15 | 32.1, CH2 | 1.26, 2H, m | 16, 17 |
| 16 | 22.8, CH2 | 1.26, 2H, m | 15, 17 |
| 17 | 14.3, CH3 | 0.89, 3H, t (6.8) | 15, 16 |
| 18 | 16.2, CH3 | 1.57, 3H, s | 6, 8, 9 |
| 1′ | 174.8, C | ||
| 2′ | 73.1, CH | 4.56, 1H, d (5.3) | |
| 3′ | 126.6, CH | 5.49, 1H, dd (15.8, 4.2) | |
| 4′ | 134.3, CH | 5.85, 1H, dt (15.8, 5.8) | 2′ |
| 5′ | 32.9, CH2 | 2.00, 2H, m | 6′ |
| 6′ | 32.7, CH2 | 1.35, 2H, m | |
| 7′–12′ | 29.6–30.0, CH2 | 1.26 × 6, 12H, m | |
| 13′ | 29.4, CH2 | 1.26, 2H, m | |
| 14′–16′ | 29.6–30.0, CH2 | 1.26 × 3, 6H, m | |
| 17′ | 32.1, CH2 | 1.26, 2H, m | 18′, 19′ |
| 18′ | 22.8, CH2 | 1.26, 2H, m | 17′, 19′ |
| 19′ | 14.3, CH3 | 0.89, 3H, t (6.8) | 17′, 18′ |
| 1″ | 103.1, CH | 4.37, 1H, s | |
| 2″ | 73.4, CH | 3.35, 1H, m | |
| 3″ | 76.1, CH | 3.36, 1H, m | |
| 4″ | 69.6, CH | 3.52, 1H, m | |
| 5″ | 76.1, CH | 3.53, 1H, m | |
| 6″ | 61.1, CH2 | 3.85, 2H, m | |
| NH | 7.39, 1H, s | ||
| Compounds | Inhibition Rate | |
|---|---|---|
| 10 µM | IC50 | |
| 1 | 57.7% | |
| 2 | 88.1% | |
| 3–24 | <35.0% | |
| JQ1 b | 239.6 nM | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Zhang, D.; Zhou, B.; Ma, Z. Inhibitors of BRD4 Protein from a Marine-Derived Fungus Alternaria sp. NH-F6. Mar. Drugs 2017, 15, 76. https://doi.org/10.3390/md15030076
Ding H, Zhang D, Zhou B, Ma Z. Inhibitors of BRD4 Protein from a Marine-Derived Fungus Alternaria sp. NH-F6. Marine Drugs. 2017; 15(3):76. https://doi.org/10.3390/md15030076
Chicago/Turabian StyleDing, Hui, Dashan Zhang, Biao Zhou, and Zhongjun Ma. 2017. "Inhibitors of BRD4 Protein from a Marine-Derived Fungus Alternaria sp. NH-F6" Marine Drugs 15, no. 3: 76. https://doi.org/10.3390/md15030076