
Induction of p53-Independent Apoptosis and G1 Cell Cycle Arrest by Fucoidan in HCT116 Human Colorectal Carcinoma Cells

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
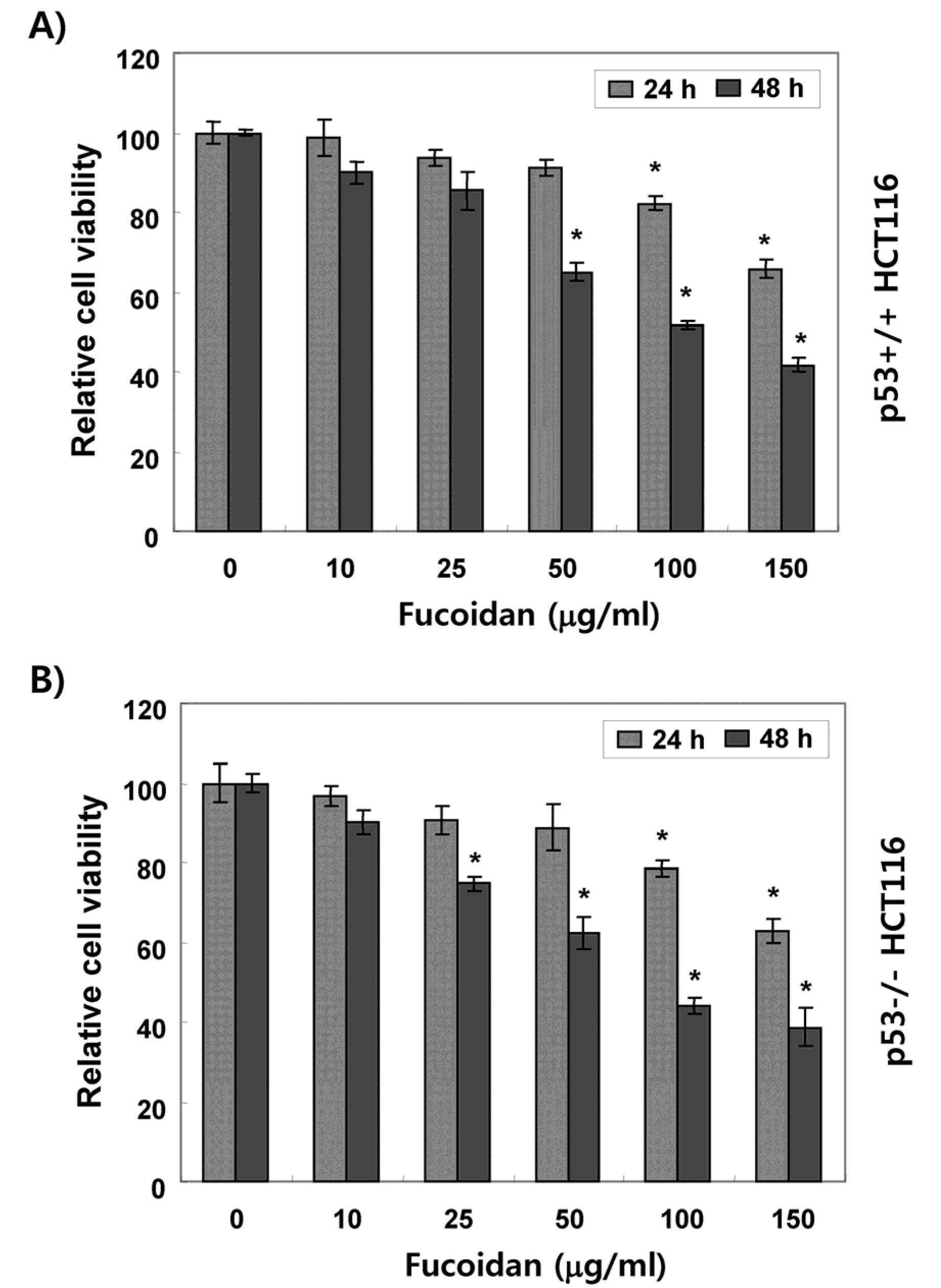
2.1. Fucoidan Suppresses Cell Survival in HCT116 Cells
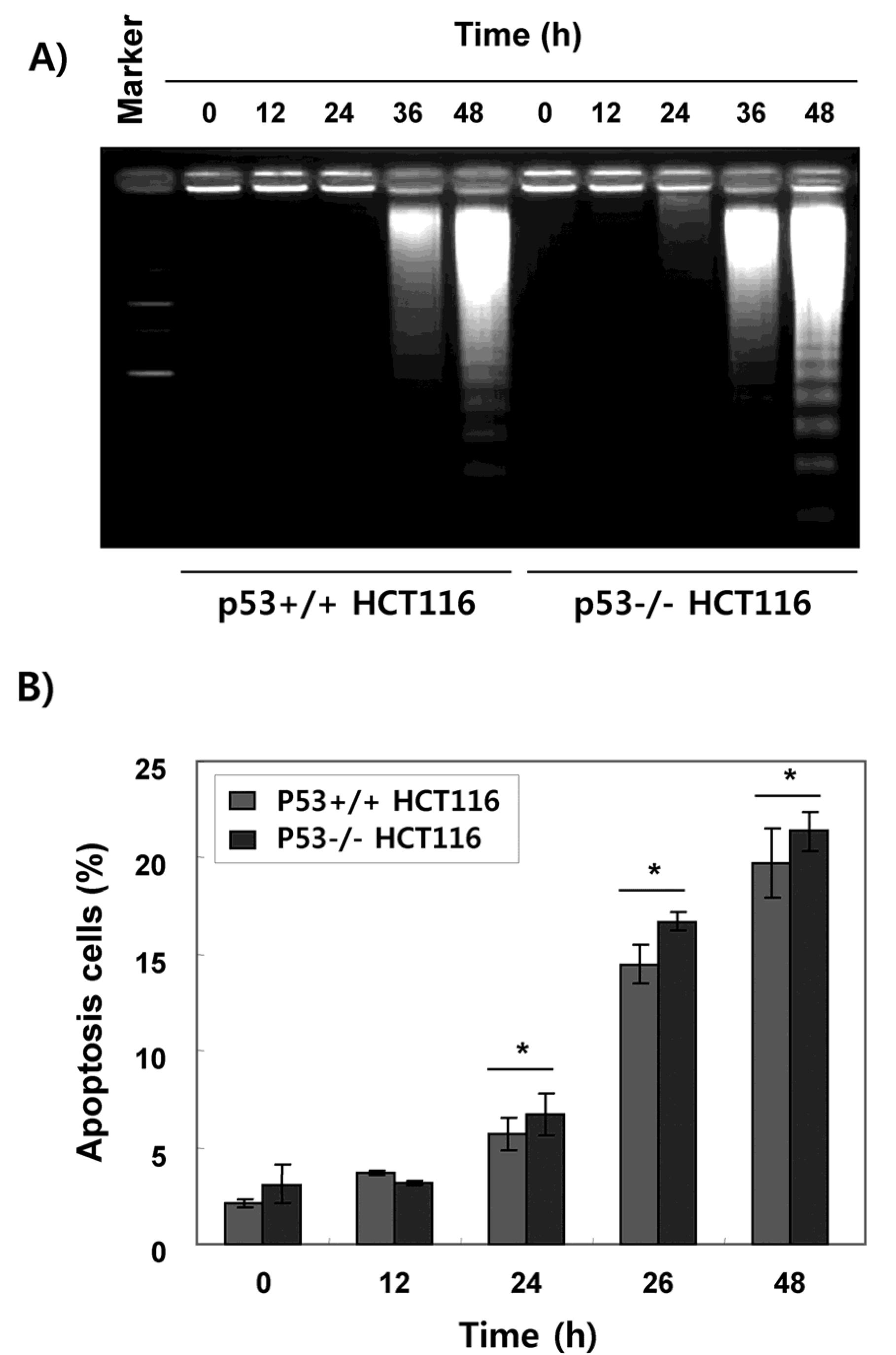
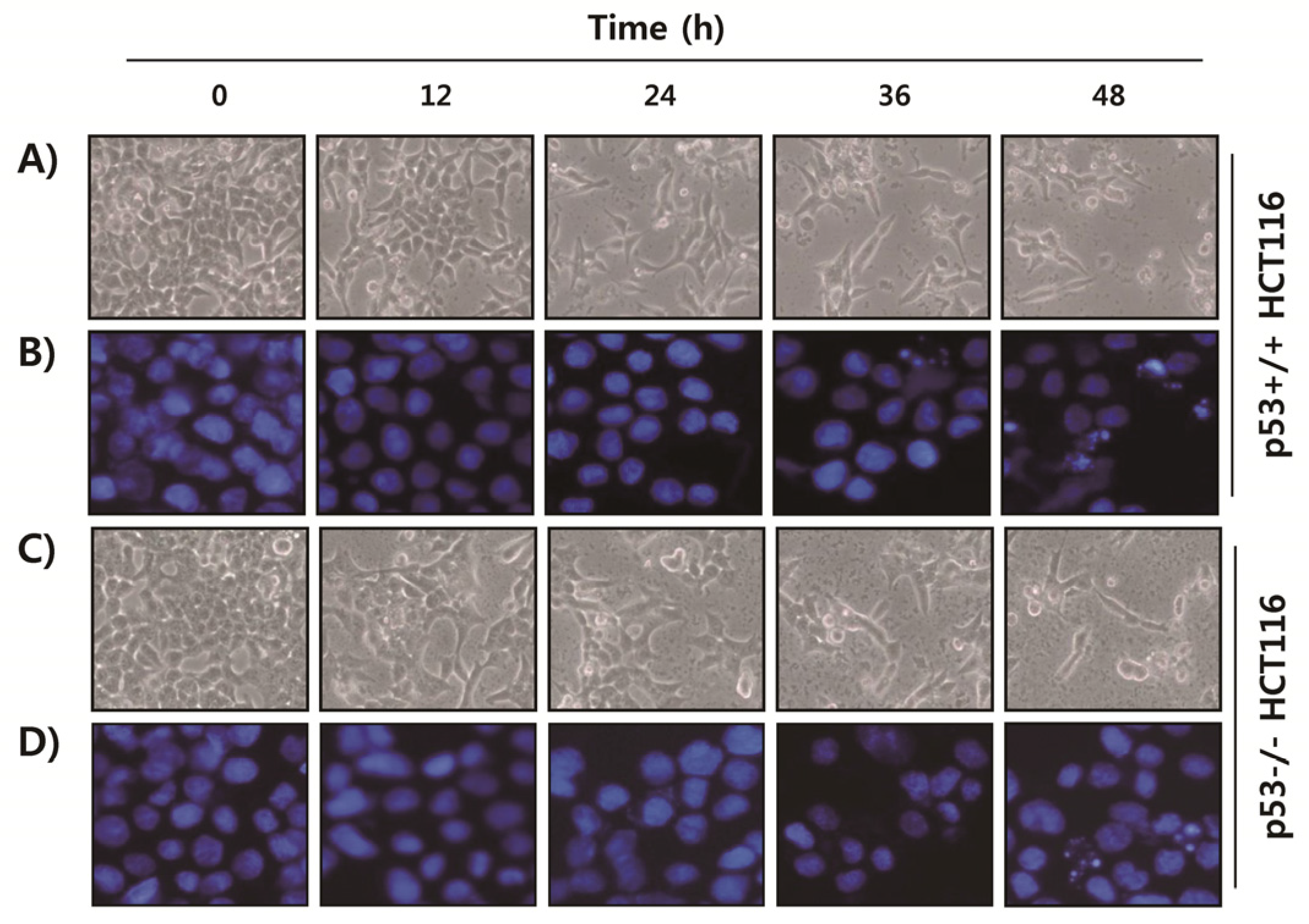
2.2. Fucoidan Induces Apoptotic Cell Death in HCT116 Cells
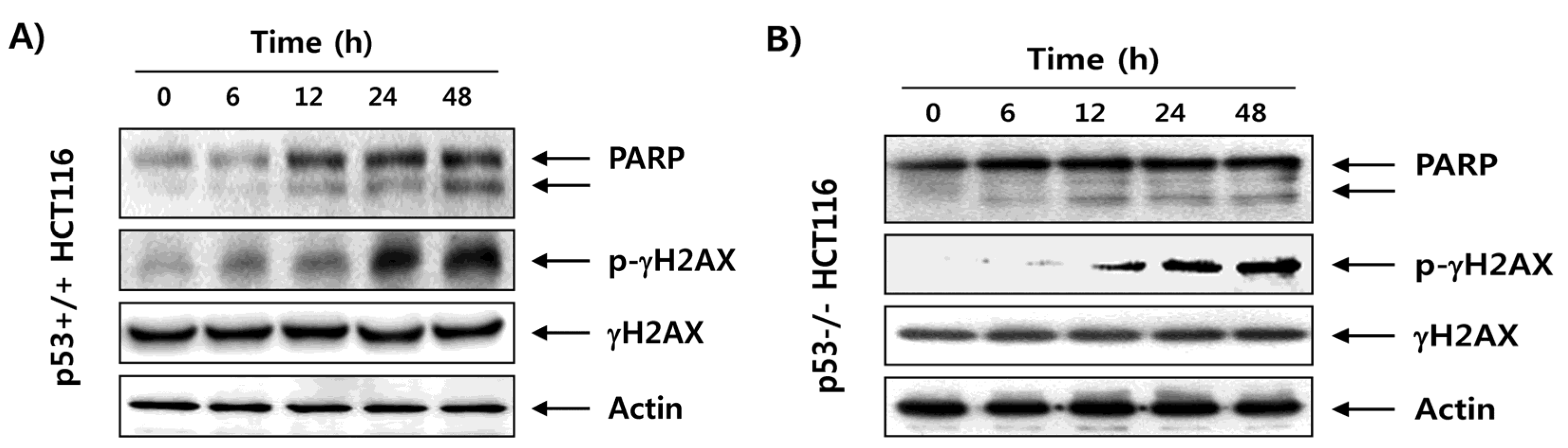
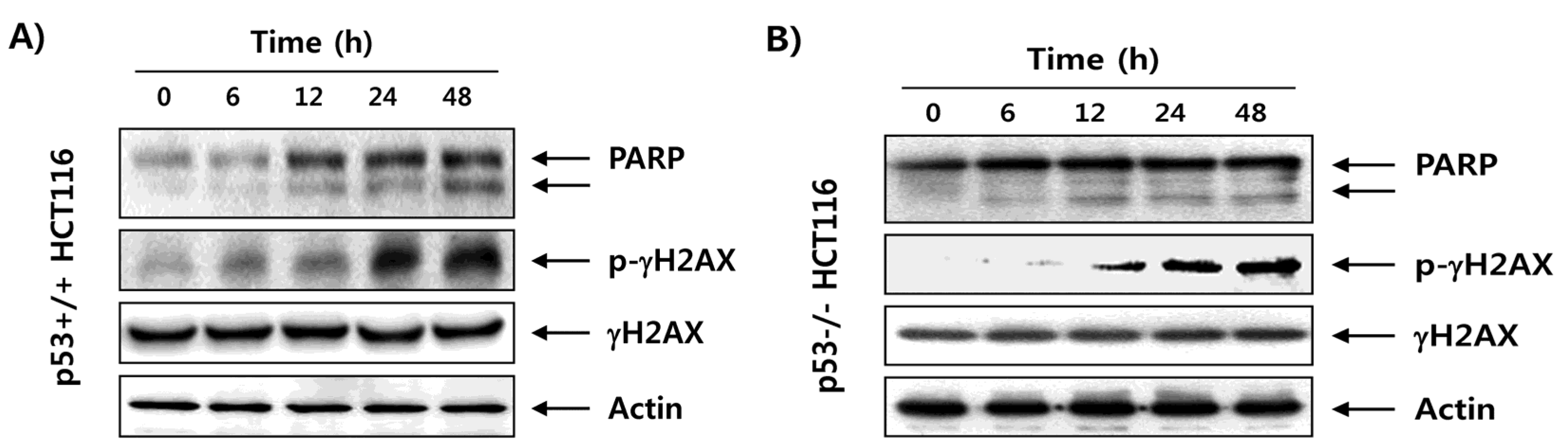
2.3. Fucoidan Enhances Degradation of Poly(ADP-ribose) Polymerase (PARP) and Phosphorylation of γH2AX in HCT116 Cells
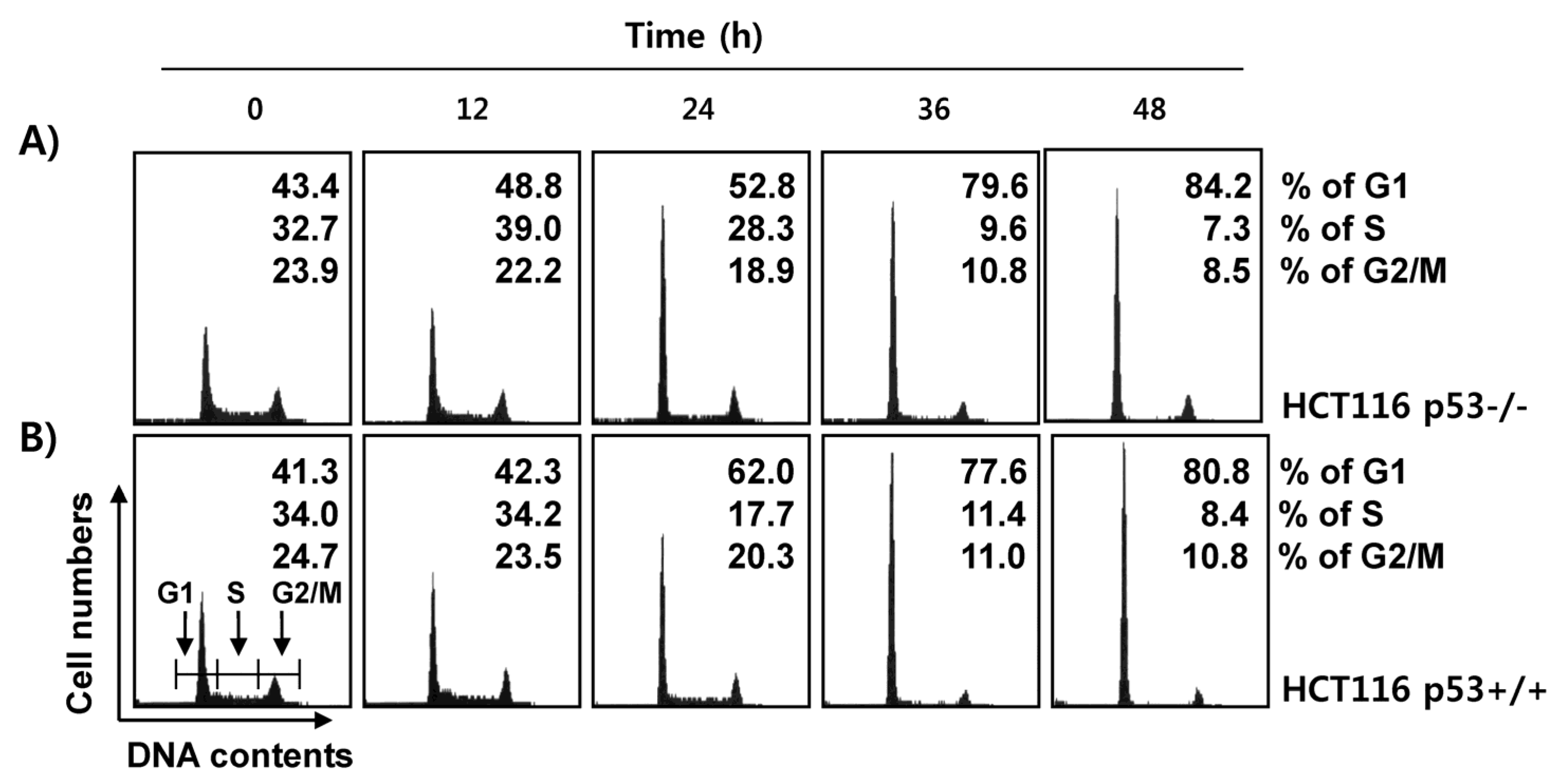
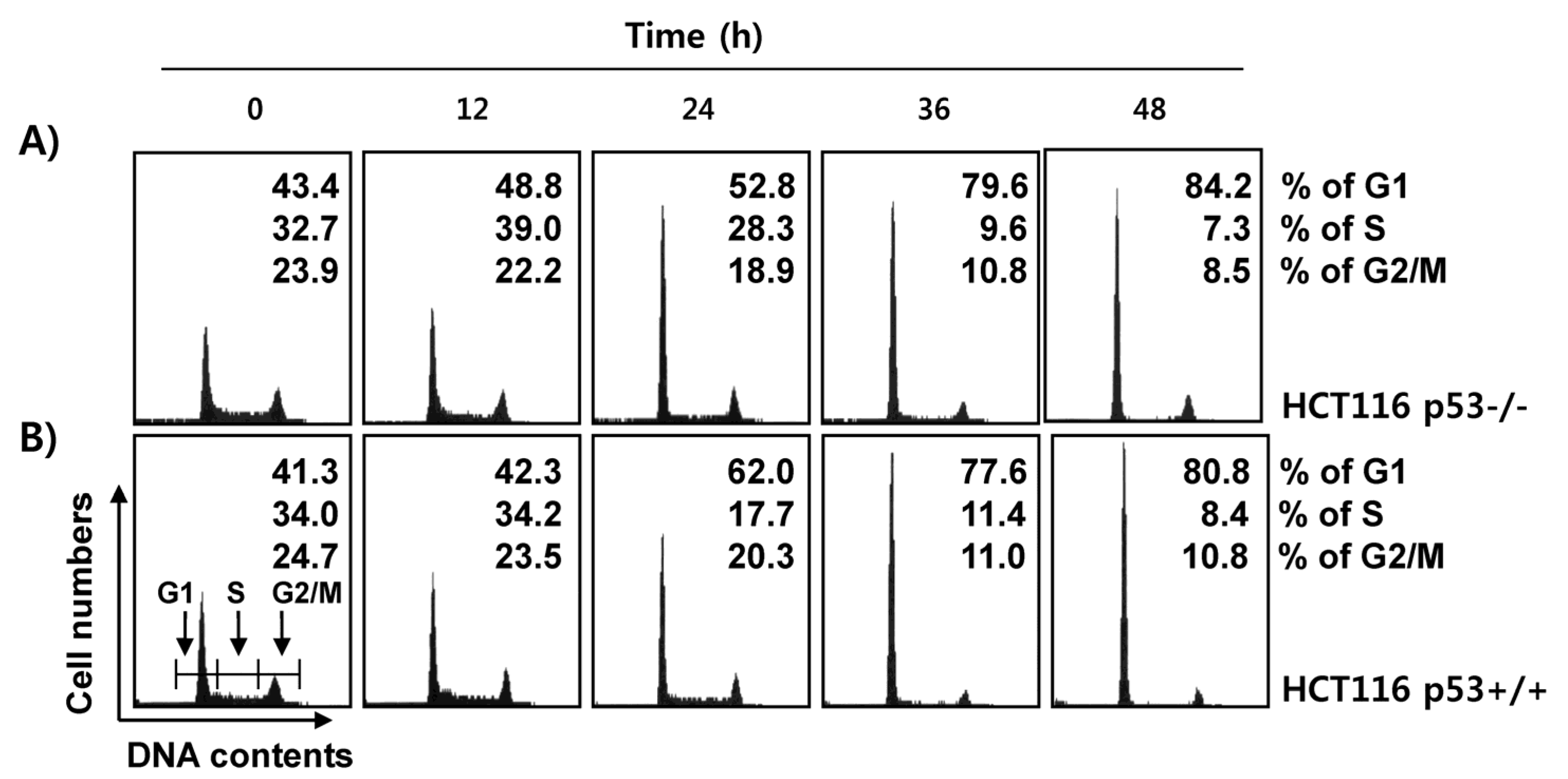
2.4. Fucoidan Induces G1 Cell Cycle Arrest in HCT116 Cells
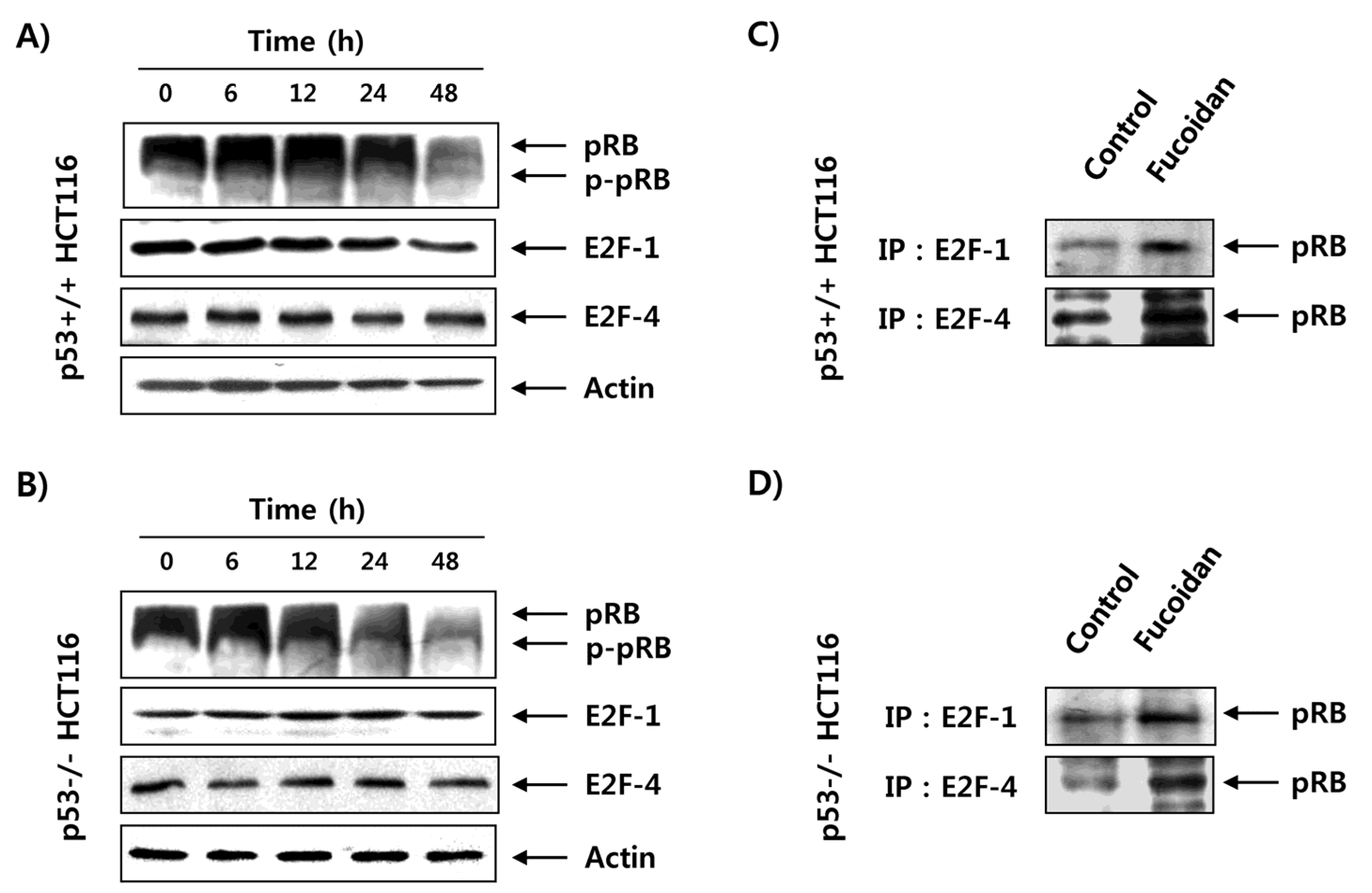
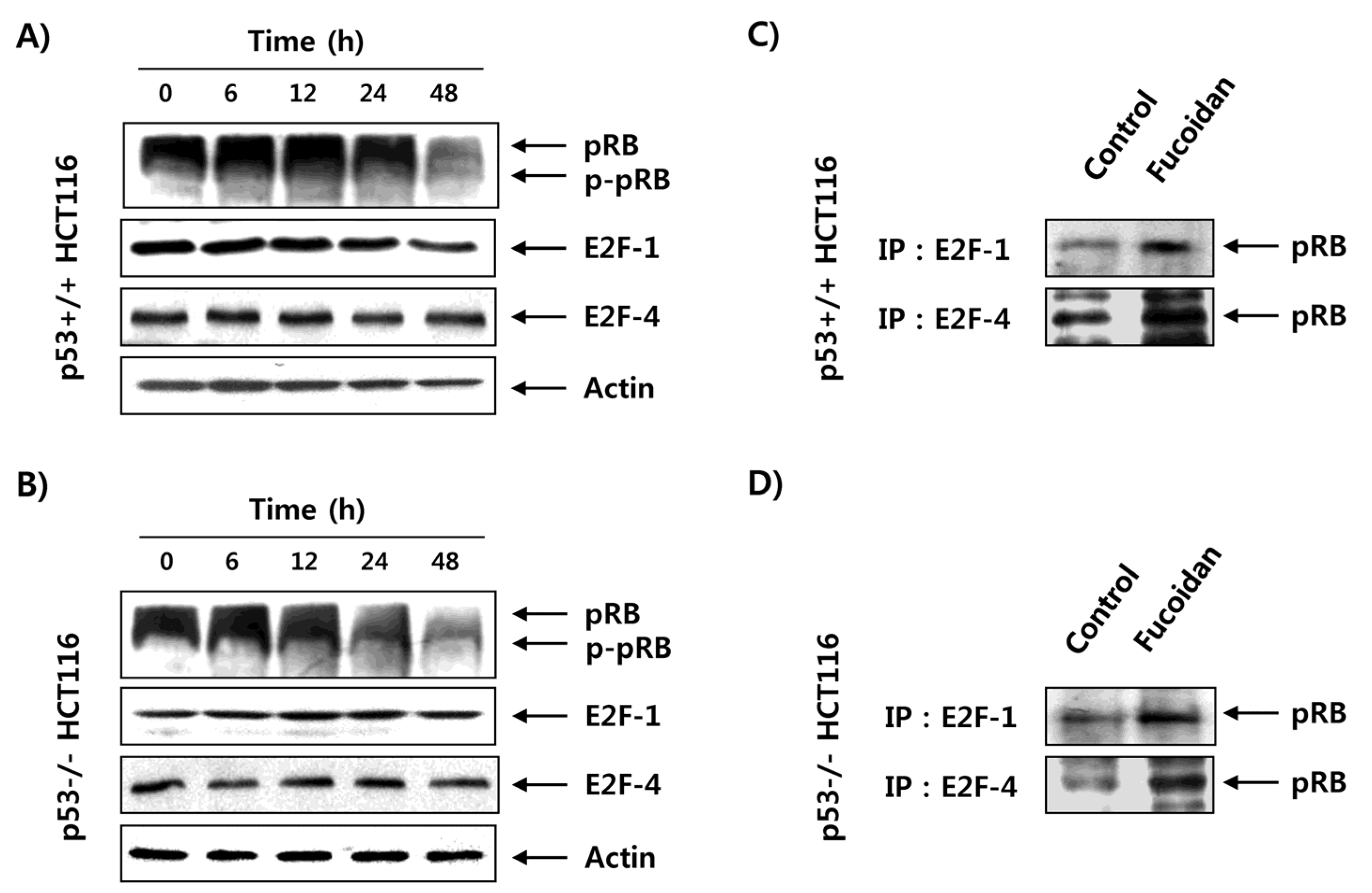
2.5. Fucoidan Inhibits pRB Phosphorylation and Enhances Binding pRB with E2Fs in HCT116 Cells
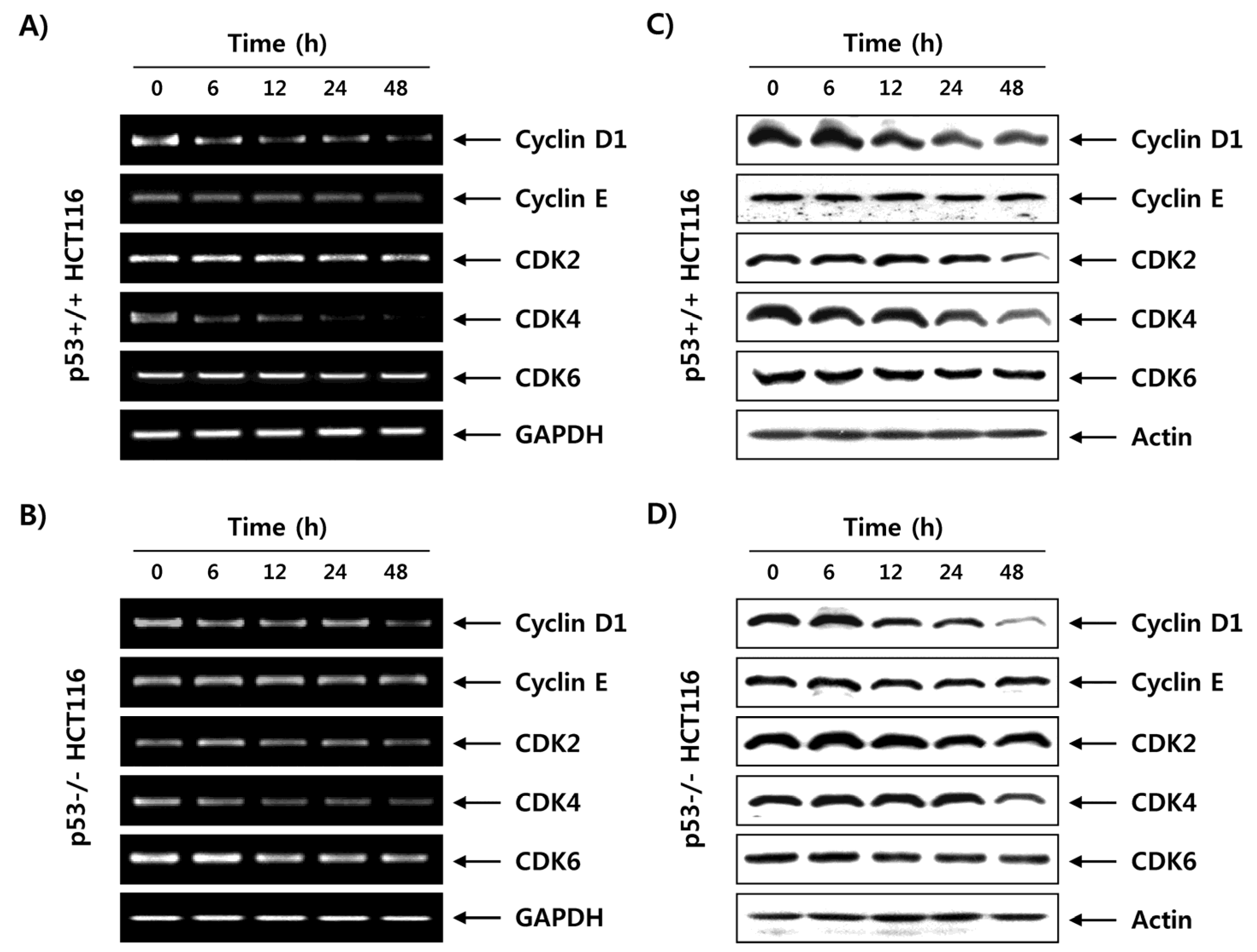
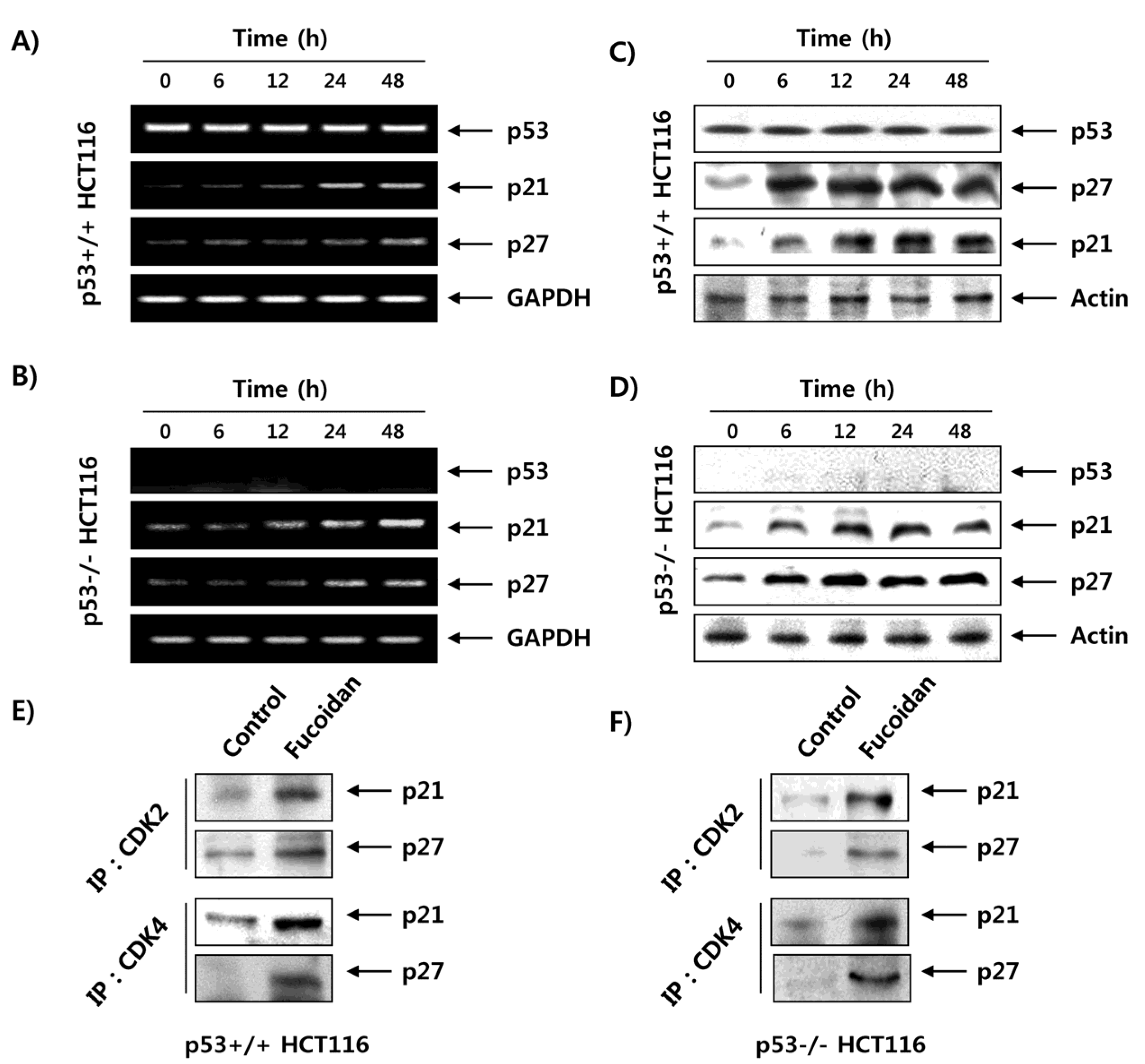
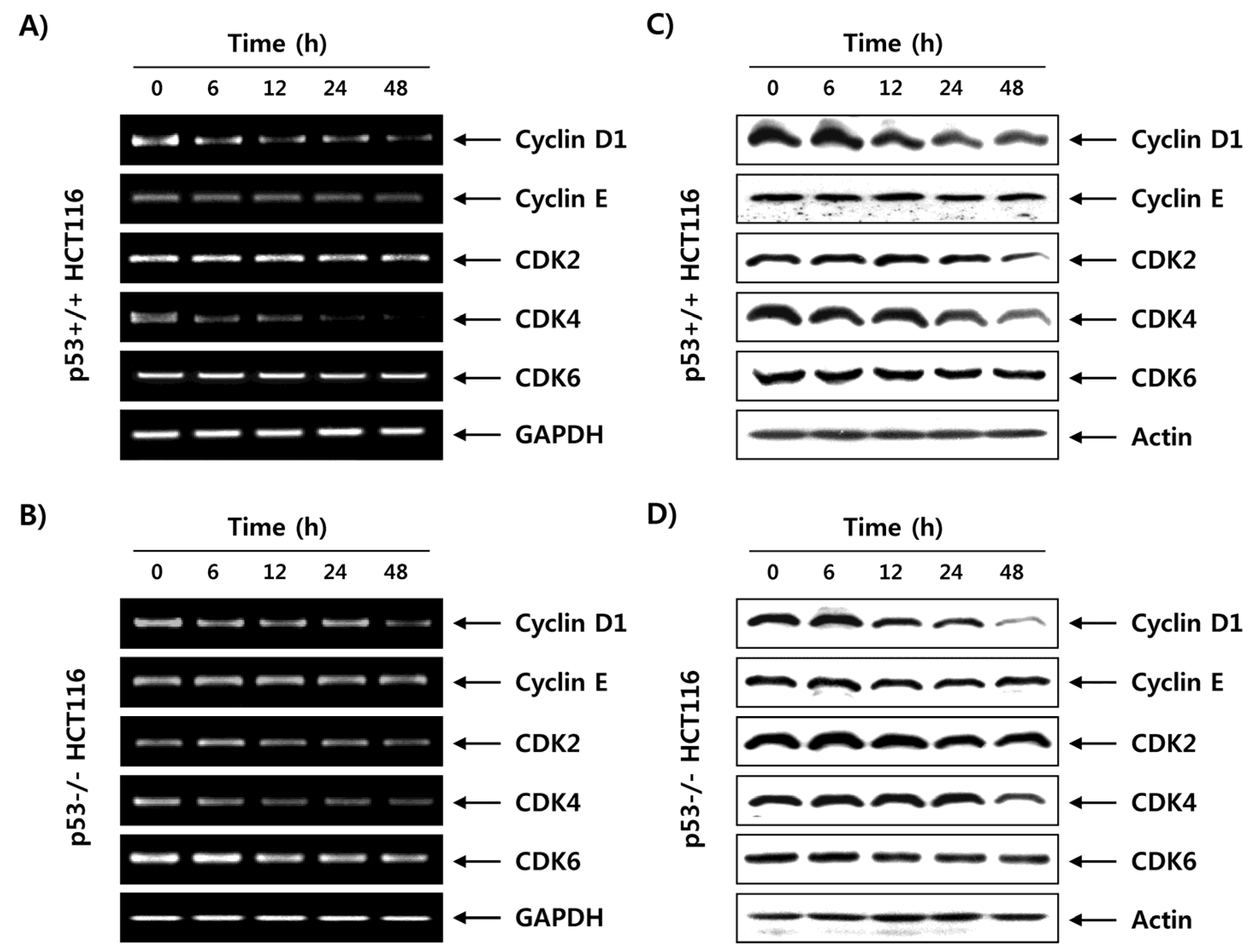
2.6. Fucoidan Regulates the Expression of G1 Phase-Associated Cyclins and CDKs in HCT116 Cells
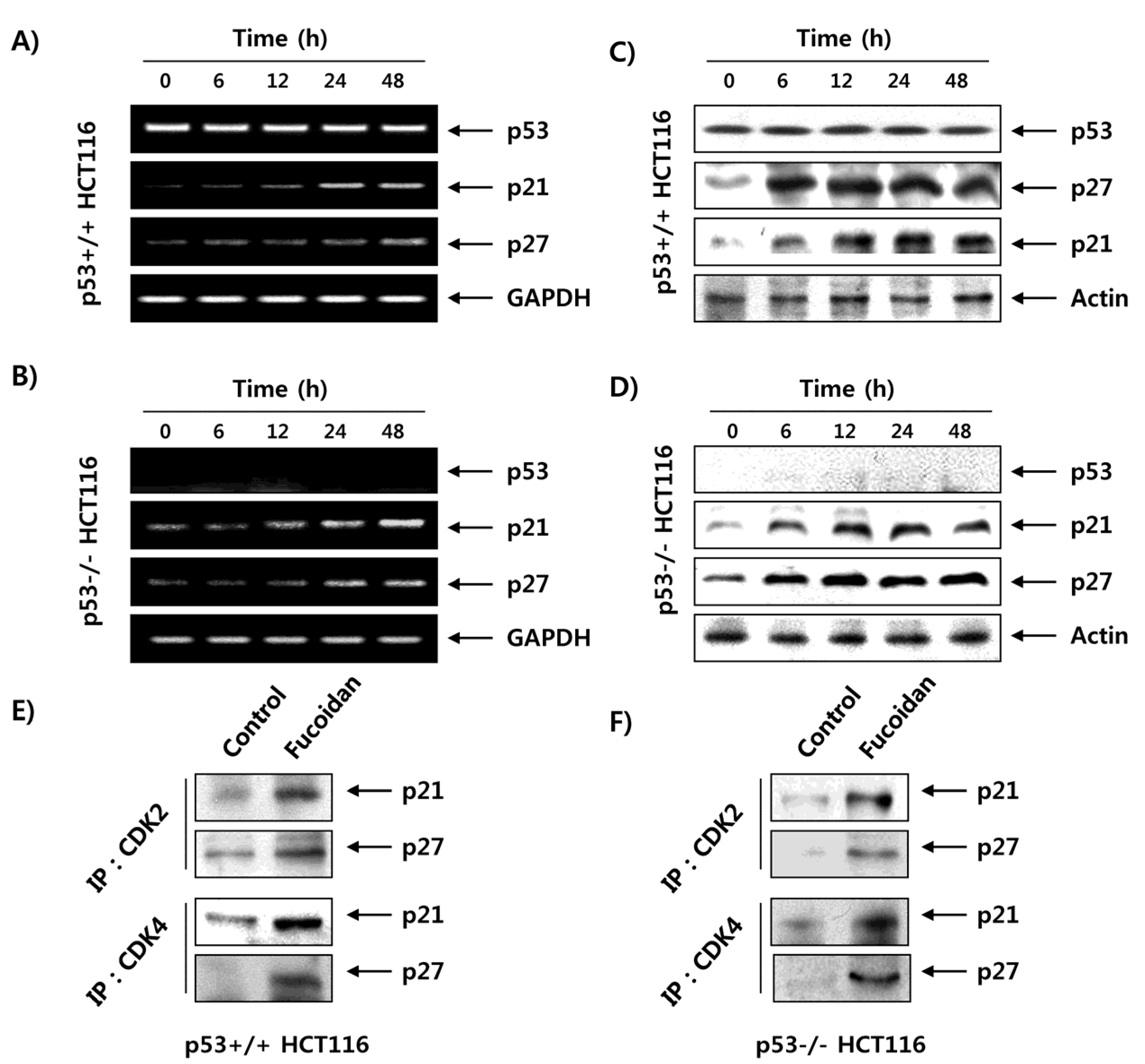
2.7. Fucoidan Increases the Expression of CDK Inhibitors (CDKI) in HCT116 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Fucoidan Treatment
4.2. Cell Viability Assay
4.3. Cell Cycle Analysis
4.4. Nuclear Staining with DAPI
4.5. DNA Fragmentation Assay
4.6. Protein Isolation, Immunoprecipitation, and Western Blot Analysis
4.7. RNA Isolation and RT–PCR Analysis
4.8. Coimmunoprecipitation Analysis
4.9. Statistical Analysis
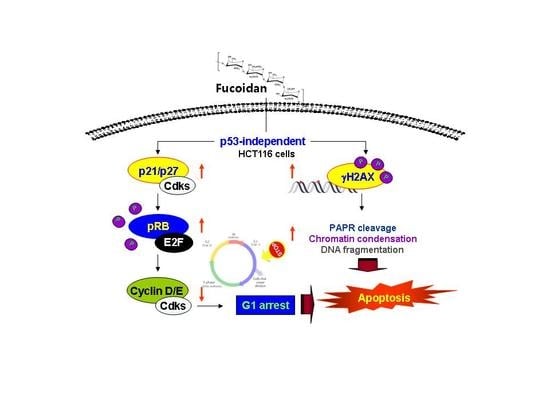
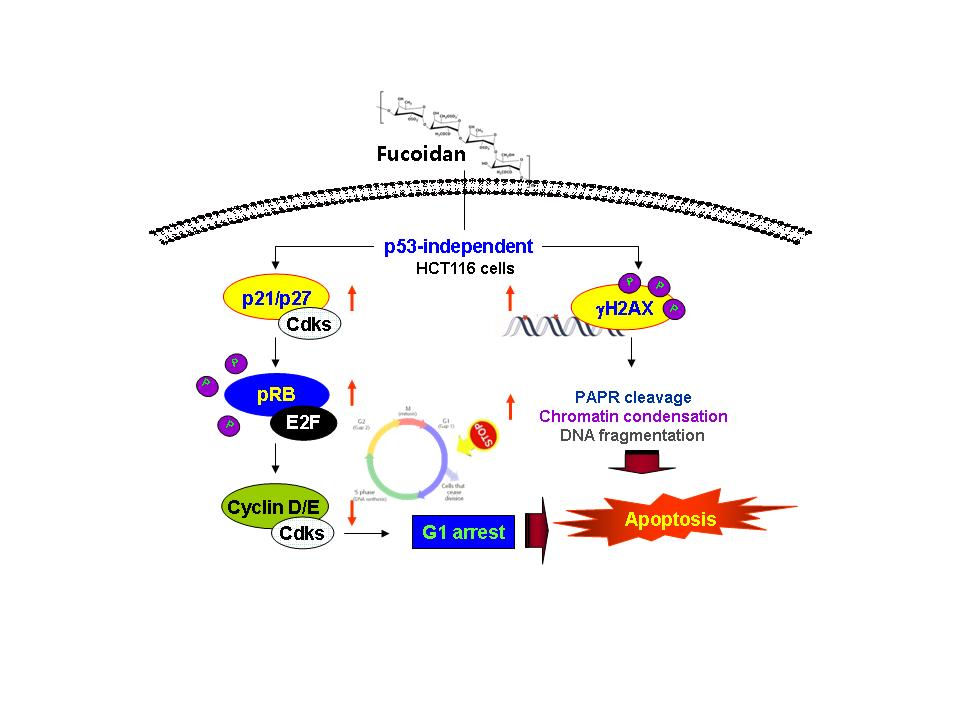
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Okręglicka, K. Health effects of changes in the structure of dietary macronutrients intake in western societies. Rocz. Panstw. Zakl. Hig. 2015, 66, 97–105. [Google Scholar] [PubMed]
- Williams, T.G.; Cubiella, J.; Griffin, S.J.; Walter, F.M.; Usher-Smith, J.A. Risk prediction models for colorectal cancer in people with symptoms: A systematic review. BMC Gastroenterol. 2016, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 154. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.A.; Manan, A.A.; Chow, K.Y.; Cornain, S.F.; Devi, C.R.; Triningsih, F.X.; Laudico, A.; Mapua, C.A.; Mirasol-Lumague, M.R.; Noorwati, S. Cancer epidemiology and control in peninsular and island South-east Asia-past, present and future. Asian. Pac. J. Cancer Prev. 2010, 11, 81–98. [Google Scholar] [PubMed]
- Brody, H. Colorectal cancer. Nature 2015, 521, S1. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Sullivan, A.; Lu, X. ASPP: A new family of oncogenes and tumour suppressor genes. Br. J. Cancer 2007, 96, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Beckta, J.M.; Ahmad, S.F.; Yang, H.; Valerie, K. Revisiting p53 for cancer-specific chemo- and radiotherapy: Ten years after. Cell Cycle 2014, 13, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, 026104. [Google Scholar] [CrossRef] [PubMed]
- Tokino, T.; Nakamura, Y. The role of p53-target genes in human cancer. Crit. Rev. Oncol. Hematol. 2000, 33, 1–6. [Google Scholar] [CrossRef]
- Yang, B.; Eshleman, J.R.; Berger, N.A.; Markowitz, S.D. Wild-type p53 protein potentiates cytotoxicity of therapeutic agents in human colon cancer cells. Clin. Cancer Res. 1996, 2, 1649–1657. [Google Scholar] [PubMed]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Speidel, D. The role of DNA damage responses in p53 biology. Arch. Toxicol. 2015, 89, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.; Koff, A. Cell-cycle inhibitors: Three families united by a common cause. Gene 2000, 247, 1–15. [Google Scholar] [CrossRef]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, K.; Manivasagan, P.; Venkatesan, J.; Kim, S.K. Brown seaweed fucoidan: Biological activity and apoptosis, growth signaling mechanism in cancer. Int. J. Biol. Macromol. 2013, 60, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Takai, M.; Miyazaki, Y.; Tachibana, H.; Yamada, K. The enhancing effect of fucoidan derived from Undaria pinnatifida on immunoglobulin production by mouse spleen lymphocytes. Biosci. Biotechnol. Biochem. 2014, 78, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Hsu, W.L.; Hwang, P.A.; Chou, T.C. Low molecular weight fucoidan inhibits tumor angiogenesis through downregulation of HIF-1/VEGF signaling under hypoxia. Mar. Drugs 2015, 13, 4436–4451. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Ge, Y.; Zhang, J.; Liu, Y.; Wang, Q.; Hou, L.; Zheng, Z. Fucoidan inhibited 4T1 mouse breast cancer cell growth in vivo and in vitro via downregulation of Wnt/β-catenin signaling. Nutr. Cancer 2013, 65, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kim, G.Y.; Moon, S.K.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. Fucoidan inhibits the proliferation of human urinary bladder cancer T24 cells by blocking cell cycle progression and inducing apoptosis. Molecules 2014, 19, 5981–5998. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Lee, J.H.; Lee, S.H. Antitumor effects of fucoidan on human colon cancer cells via activation of Akt signaling. Biomol. Ther. (Seoul) 2015, 23, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhang, Q.; Kong, Y.; Xie, B.; Gao, M.; Tao, Y.; Xu, H.; Zhan, F.; Dai, B.; Shi, J.; Wu, X. Antitumor activity of fucoidan against diffuse large B cell lymphoma in vitro and in vivo. Acta. Biochim. Biophys. Sin. (Shanghai) 2015, 47, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Lowenthal, R.M.; Woods, G.M.; Holloway, A.F.; Karpiniec, S.S.; Dickinson, J.L. Fucoidan suppresses the growth of human acute promyelocytic leukemia cells in vitro and in vivo. J. Cell. Physiol. 2016, 231, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Alwarsamy, M.; Gooneratne, R.; Ravichandran, R. Effect of fucoidan from Turbinaria conoides on human lung adenocarcinoma epithelial (A549) cells. Carbohydr. Polym. 2016, 152, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Seville, L.L.; Shah, N.; Westwell, A.D.; Chan, W.C. Modulation of pRB/E2F functions in the regulation of cell cycle and in cancer. Curr. Cancer Drug Targets 2005, 5, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Dyson, N.J. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011, 21, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.K.; Mitrea, D.M.; Ou, L.; Kriwacki, R.W. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem. Soc. Trans. 2012, 40, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Chari, N.S.; Pinaire, N.L.; Thorpe, L.; Medeiros, L.J.; Routbort, M.J.; McDonnell, T.J. The p53 tumor suppressor network in cancer and the therapeutic modulation of cell death. Apoptosis 2009, 14, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yu, X.; Liu, S.; Deng, Q.; Liu, X.; Peng, S.; Li, H.; Liu, J.; Cao, Y. The role of targeting kinase activity by natural products in cancer chemoprevention and chemotherapy (Review). Oncol. Rep. 2015, 34, 547–554. [Google Scholar] [PubMed]
- Lee, M.H.; Yang, H.Y. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell. Mol. Life Sci. 2001, 58, 1907–1922. [Google Scholar] [CrossRef] [PubMed]
- Haneji, K.; Matsuda, T.; Tomita, M.; Kawakami, H.; Ohshiro, K.; Uchihara, J.N.; Masuda, M.; Takasu, N.; Tanaka, Y.; Ohta, T.; Mori, N. Fucoidan extracted from Cladosiphon okamuranus Tokida induces apoptosis of human T-cell leukemia virus type 1-infected T-cell lines and primary adult T-cell leukemia cells. Nutr. Cancer 2005, 52, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.M.; Kim, W.J.; Moon, S.K. Akt signaling is involved in fucoidan-induced inhibition of growth and migration of human bladder cancer cells. Food Chem. Toxicol. 2014, 64, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell. Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Safi, W.; Kuehnl, A.; Nüssler, A.; Eckstein, H.H.; Pelisek, J. Differentiation of human CD14+ monocytes: An experimental investigation of the optimal culture medium and evidence of a lack of differentiation along the endothelial line. Exp. Mol. Med. 2016, 48, e227. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Lee, S.M.; Ko, J.W.; Park, S.H.; Shin, I.S.; Moon, C.; Kim, S.H.; Kim, J.C. Role of mitogen-activated protein kinases and nuclear factor-kappa B in 1,3-dichloro-2-propanol-induced hepatic injury. Lab. Anim. Res. 2016, 32, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, C.M.; Kim, Y.C. Anti-melanogenic effect of Oenothera laciniata methanol extract in melan-a cells. Toxicol. Res. 2017, 33, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Koo, J.; Wang, S.; Hur, S.J.; Bahk, Y.Y. A systematic study of nuclear interactome of C-terminal domain small phosphatase-like 2 using inducible expression system and shotgun proteomics. BMB Rep. 2016, 49, 319–324. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.Y.; Park, S.-H.; Jeong, J.-W.; Yoon, D.; Han, M.H.; Lee, D.-S.; Choi, G.; Yim, M.-J.; Lee, J.M.; Kim, D.-H.; et al. Induction of p53-Independent Apoptosis and G1 Cell Cycle Arrest by Fucoidan in HCT116 Human Colorectal Carcinoma Cells. Mar. Drugs 2017, 15, 154. https://doi.org/10.3390/md15060154
Park HY, Park S-H, Jeong J-W, Yoon D, Han MH, Lee D-S, Choi G, Yim M-J, Lee JM, Kim D-H, et al. Induction of p53-Independent Apoptosis and G1 Cell Cycle Arrest by Fucoidan in HCT116 Human Colorectal Carcinoma Cells. Marine Drugs. 2017; 15(6):154. https://doi.org/10.3390/md15060154
Chicago/Turabian StylePark, Hye Young, Shin-Hyung Park, Jin-Woo Jeong, Dahye Yoon, Min Ho Han, Dae-Sung Lee, Grace Choi, Mi-Jin Yim, Jeong Min Lee, Do-Hyung Kim, and et al. 2017. "Induction of p53-Independent Apoptosis and G1 Cell Cycle Arrest by Fucoidan in HCT116 Human Colorectal Carcinoma Cells" Marine Drugs 15, no. 6: 154. https://doi.org/10.3390/md15060154