24-Methyl-Cholesta-5,24(28)-Diene-3β,19-diol-7β-Monoacetate Inhibits Human Small Cell Lung Cancer Growth In Vitro and In Vivo via Apoptosis Induction

 ,
,
Abstract
:1. Introduction
2. Results
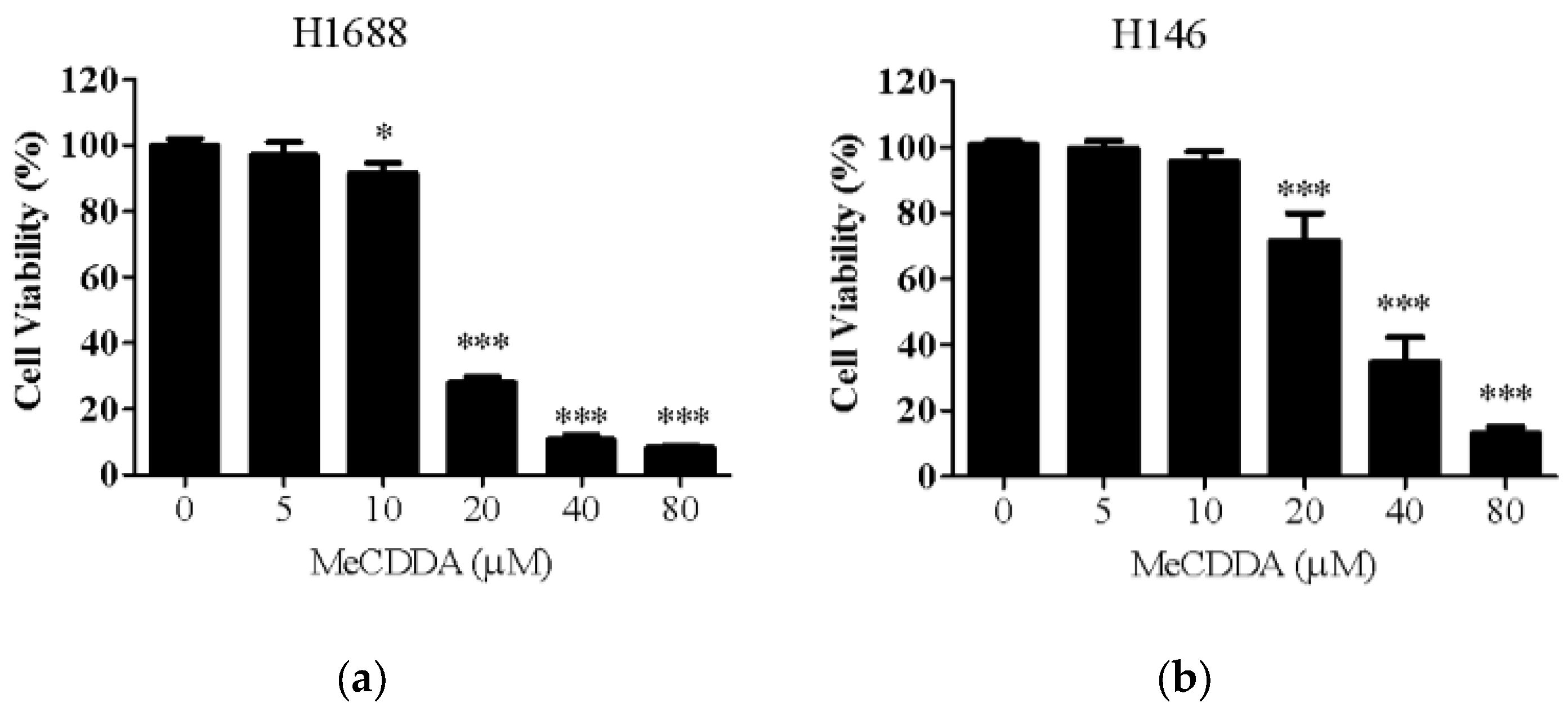
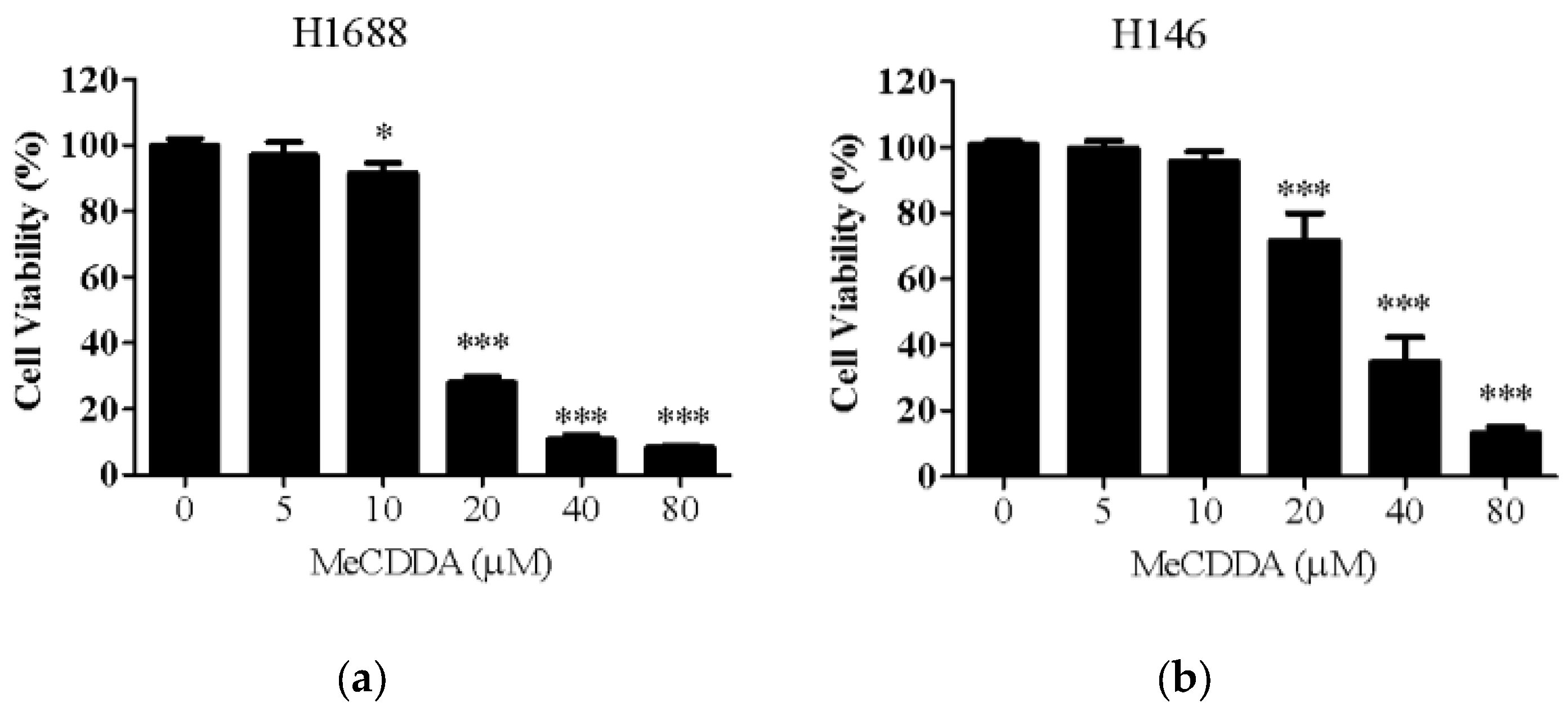
2.1. MeCDDA Decreased the Cell Viability of H1688 and H146 in vitro
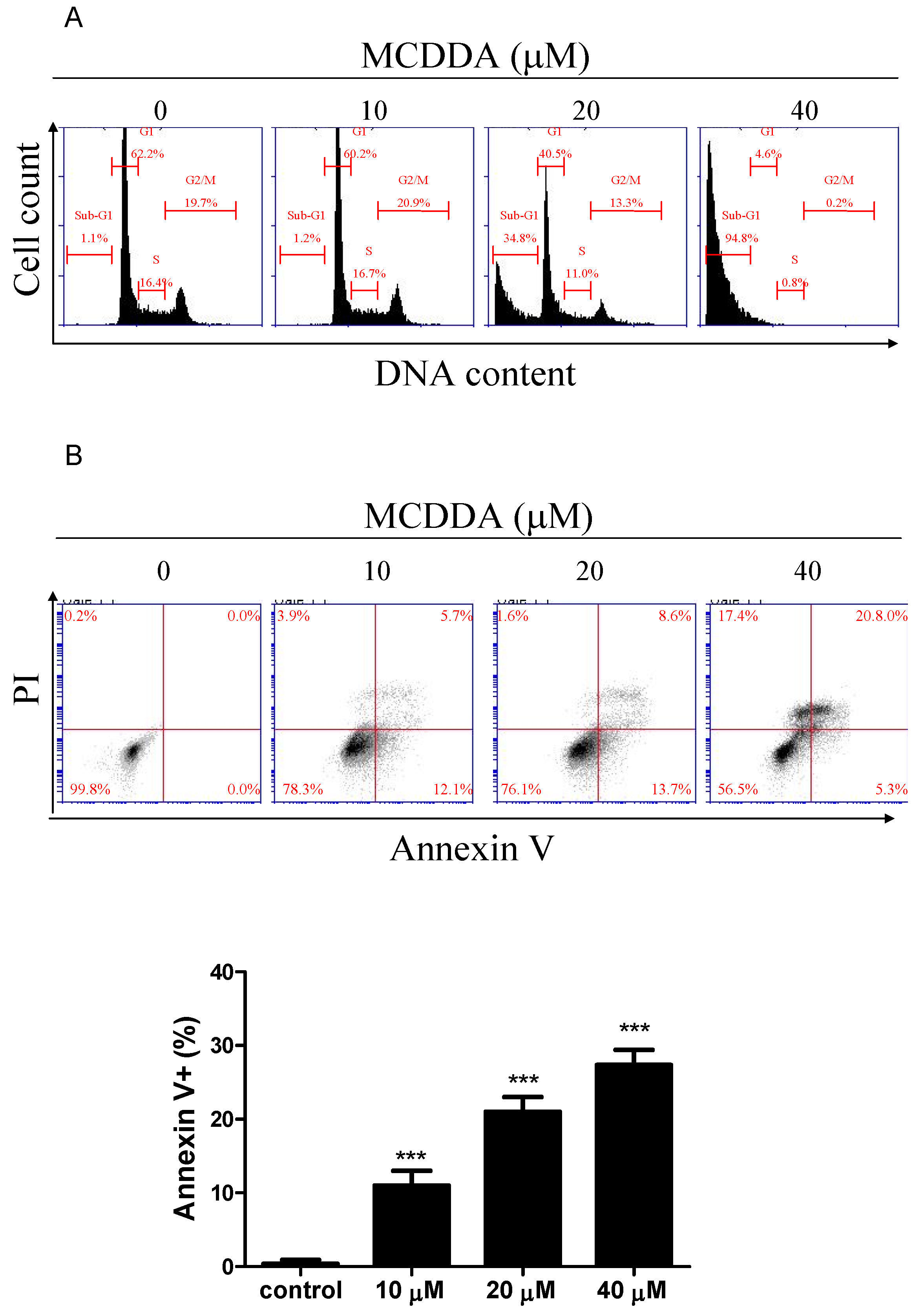
2.2. MeCDDA Induced Apoptosis of H1688
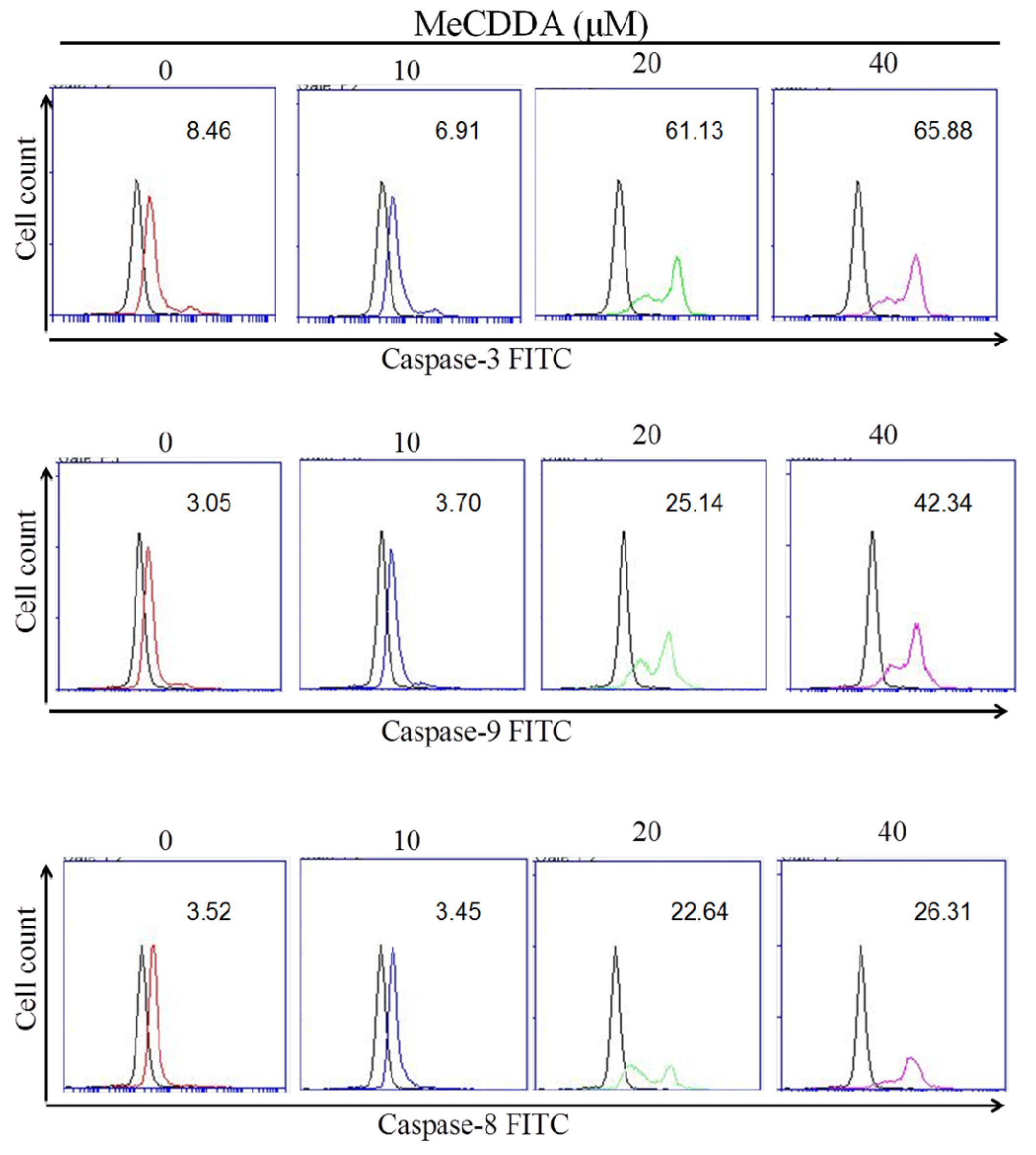
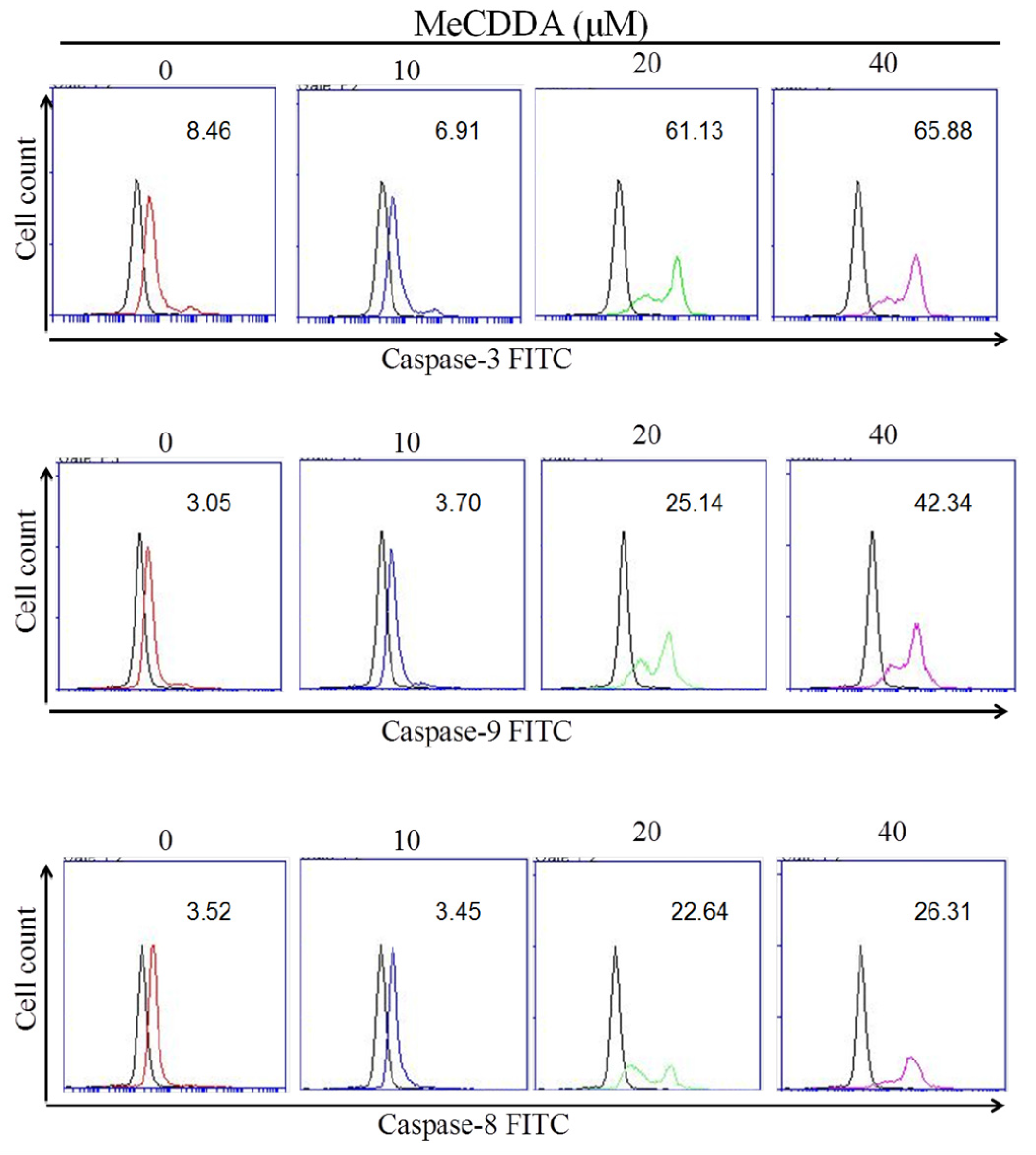
2.3. MeCDDA Induced H1688 Cells Apoptosis through a Caspase-Dependent Pathway
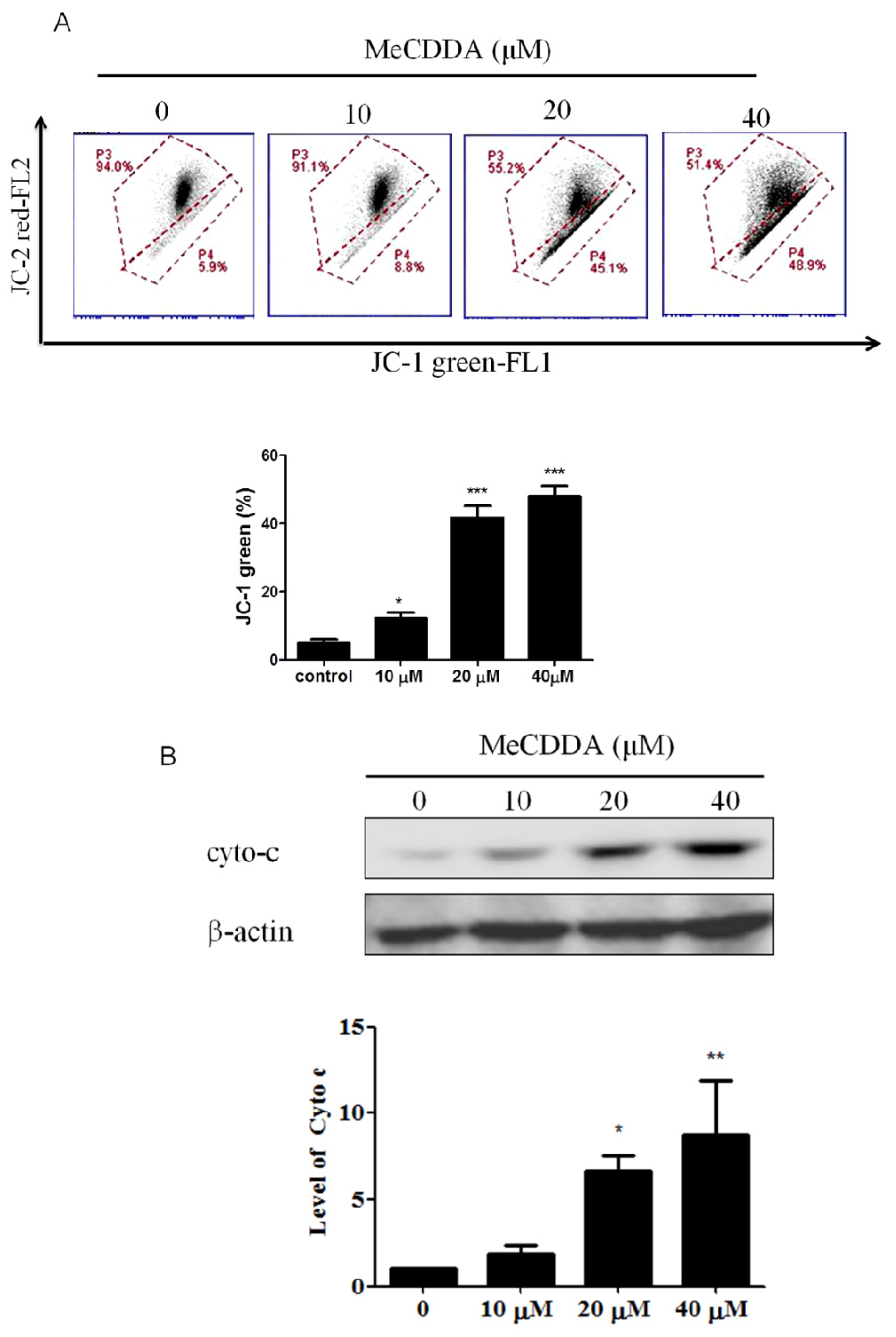
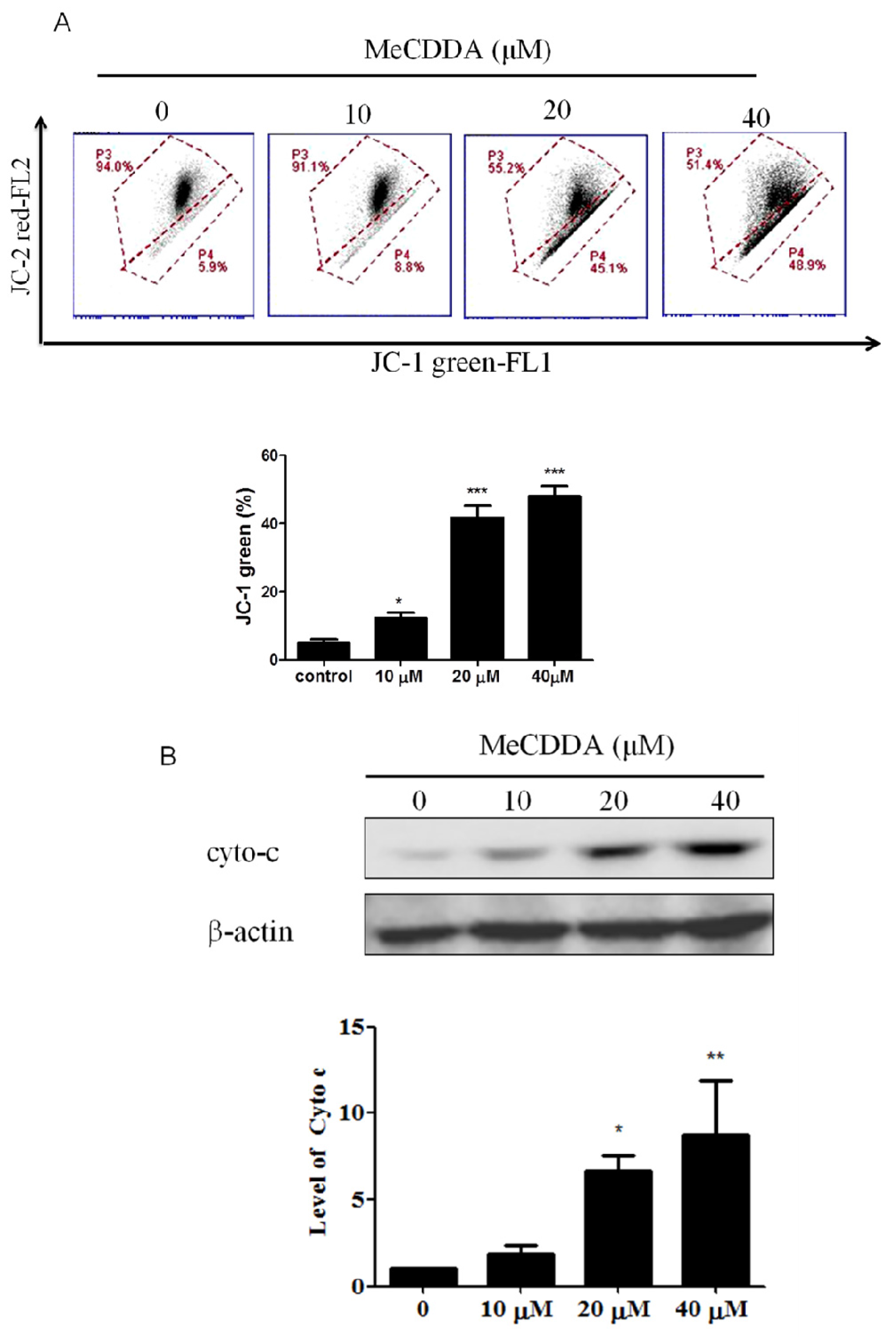
2.4. DA-Induced Loss of Mitochondrial Membrane Potential and Facilitated Cytochrome C Release
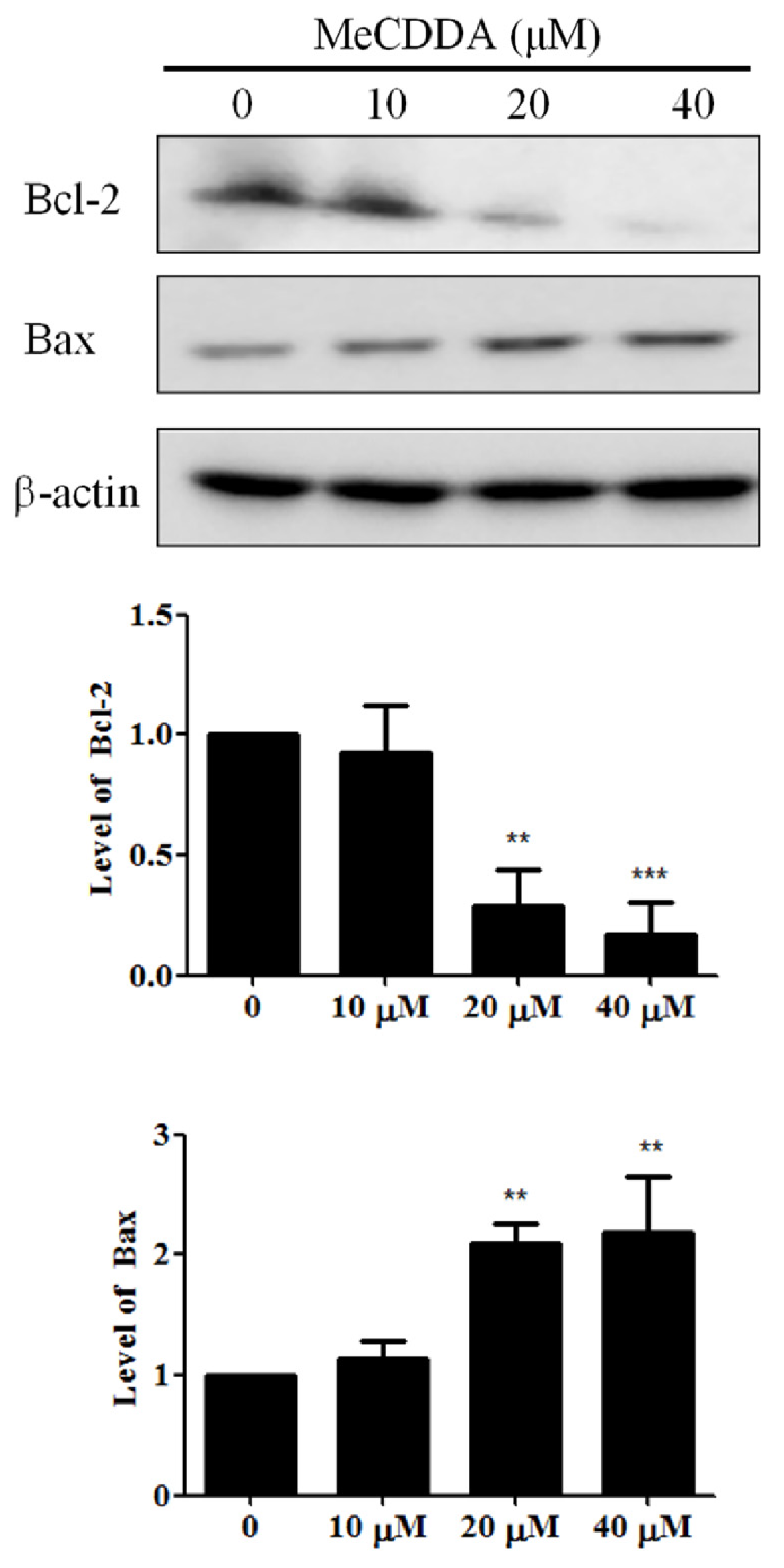
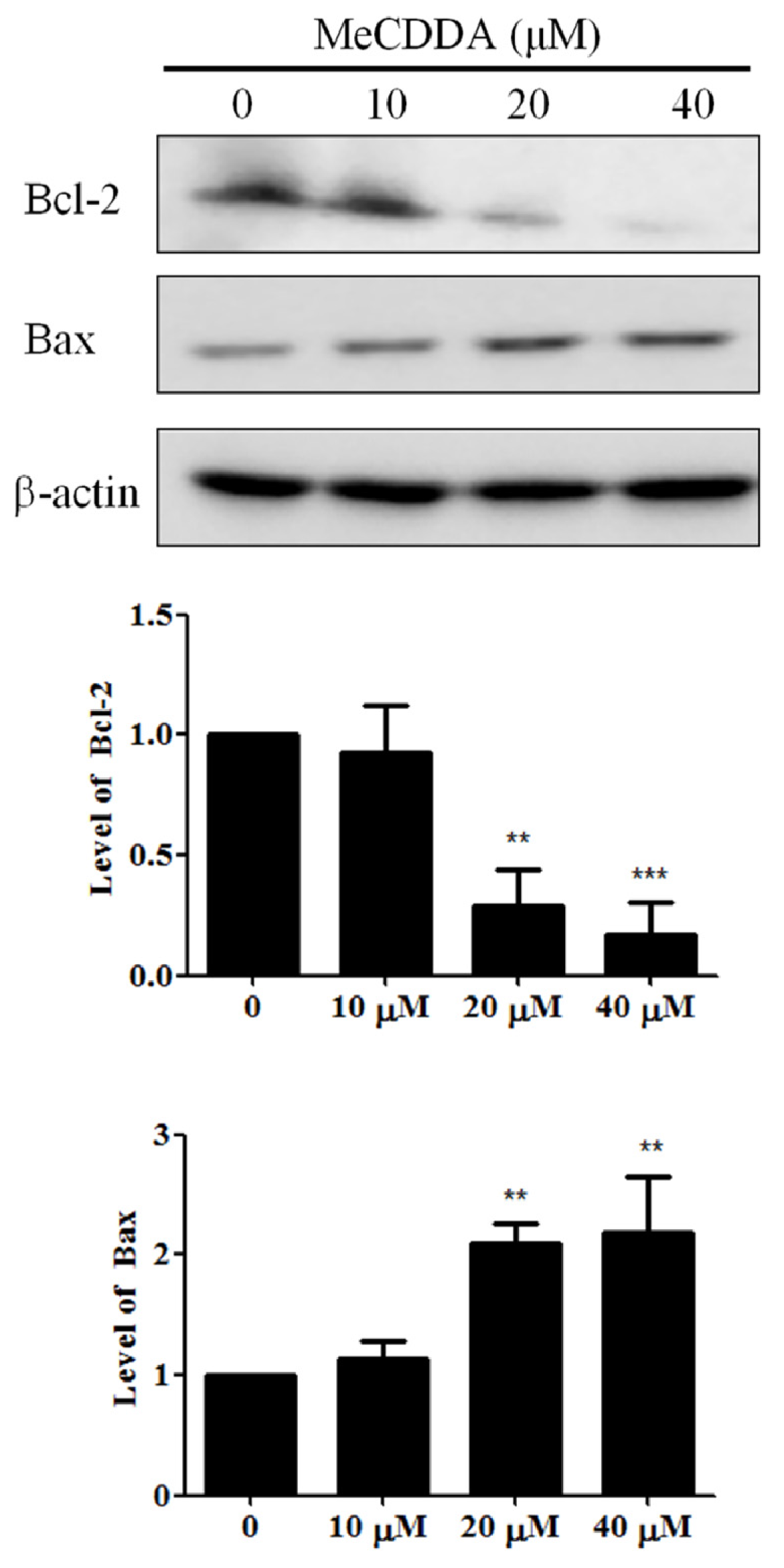
2.5. MeCDDA Modulated Bcl-2 Family Protein Expression in H1688 Cells
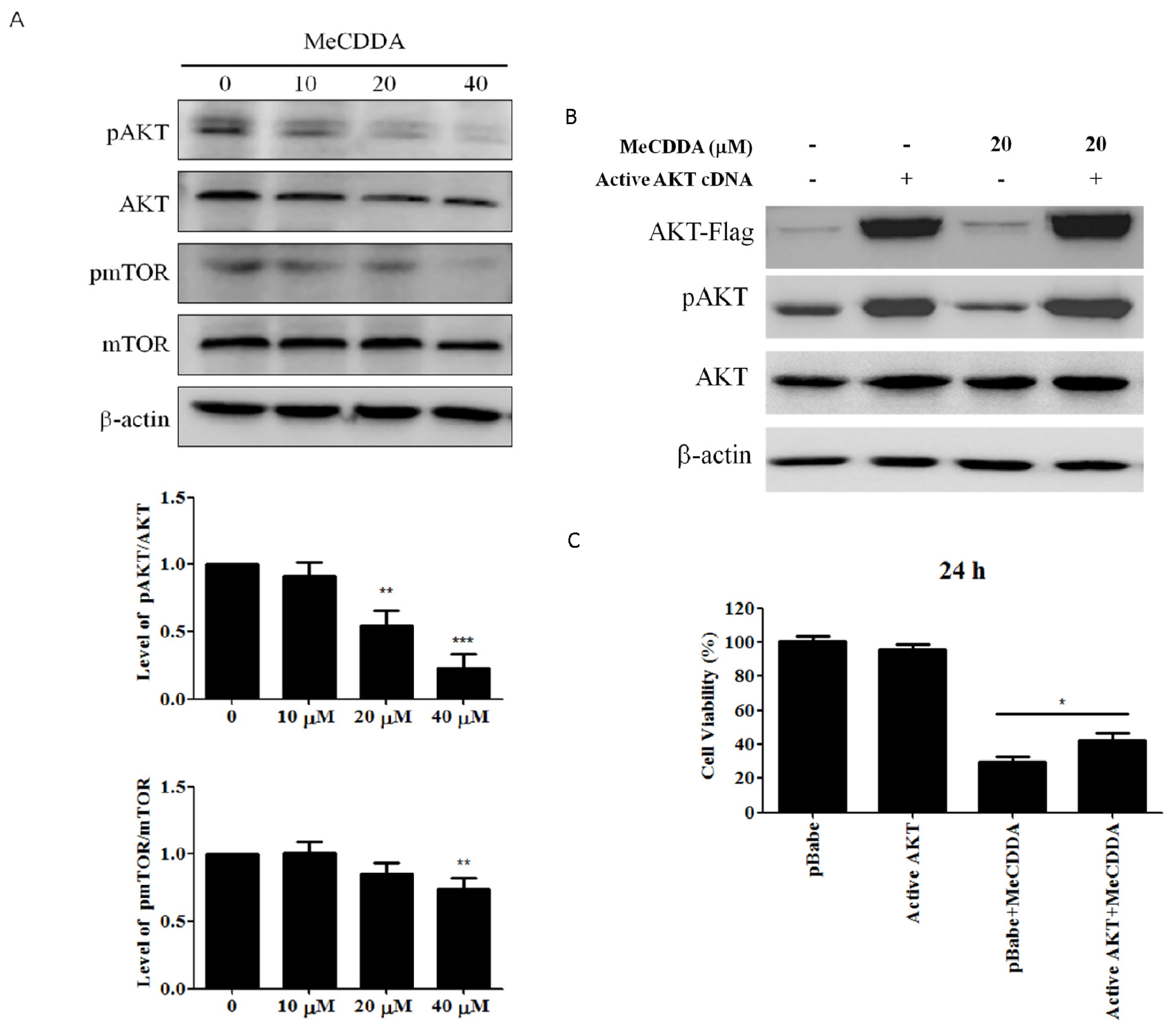
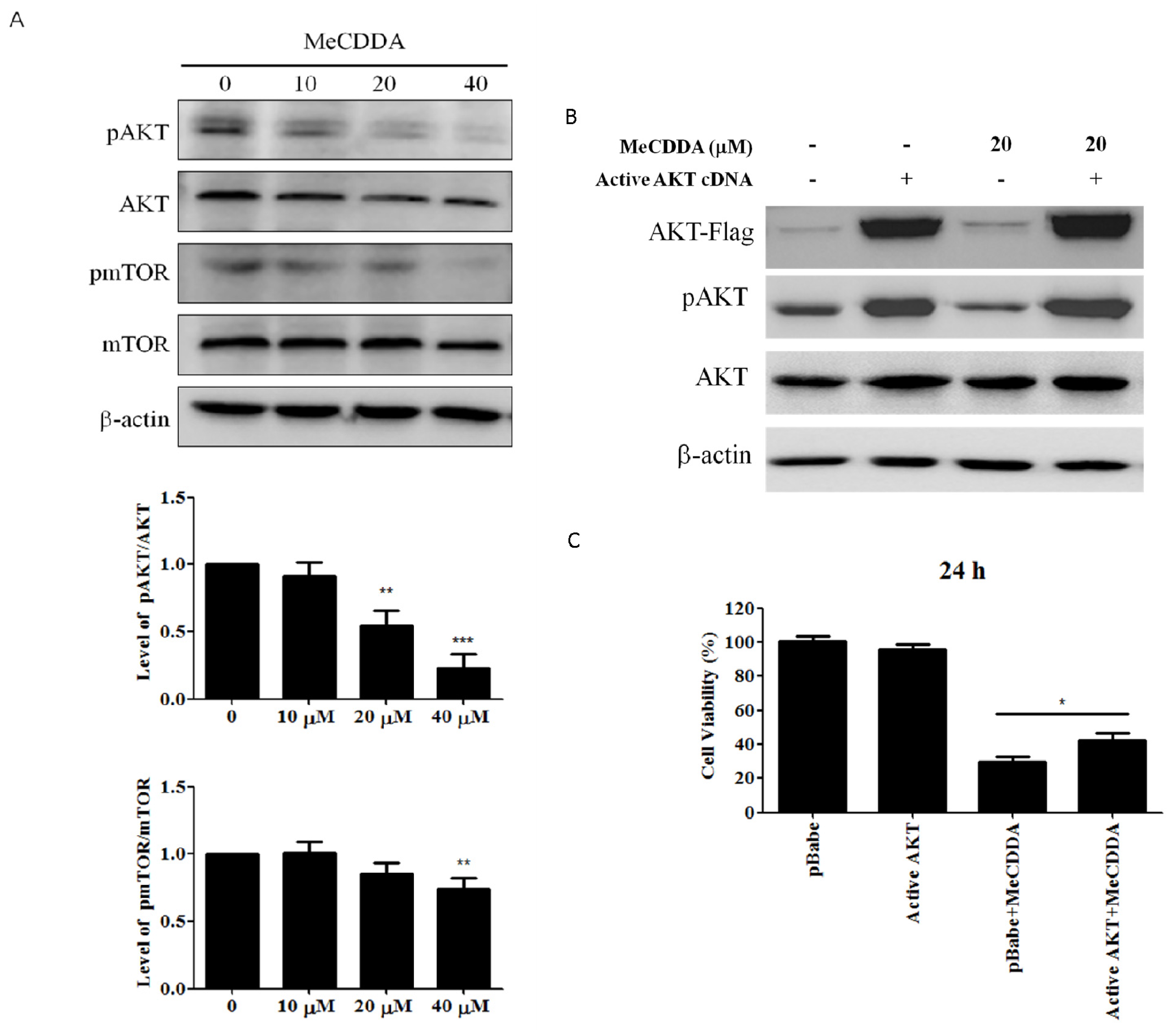
2.6. MeCDDA Inactivated the PI3K/Akt Pathway in H1688 Cells
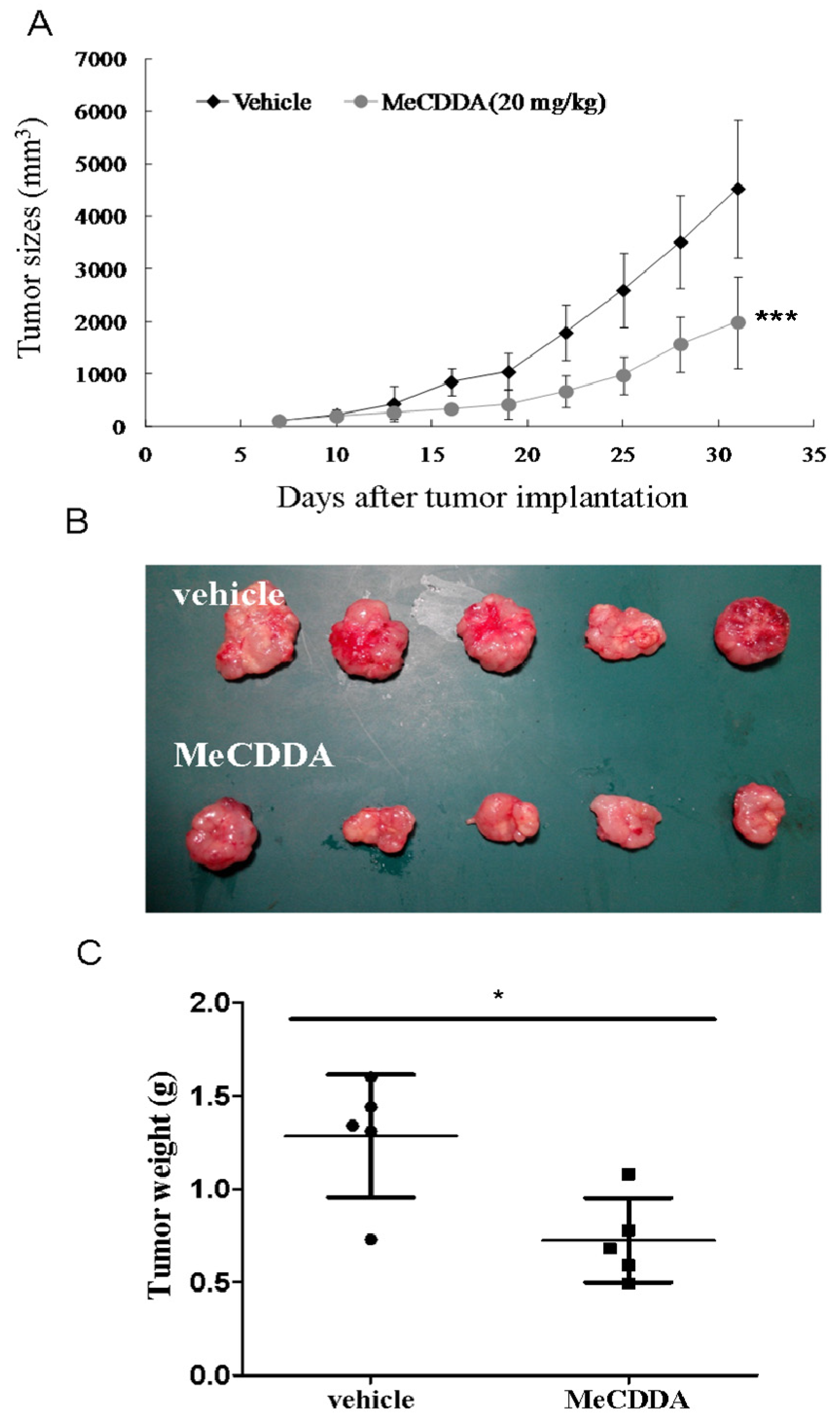
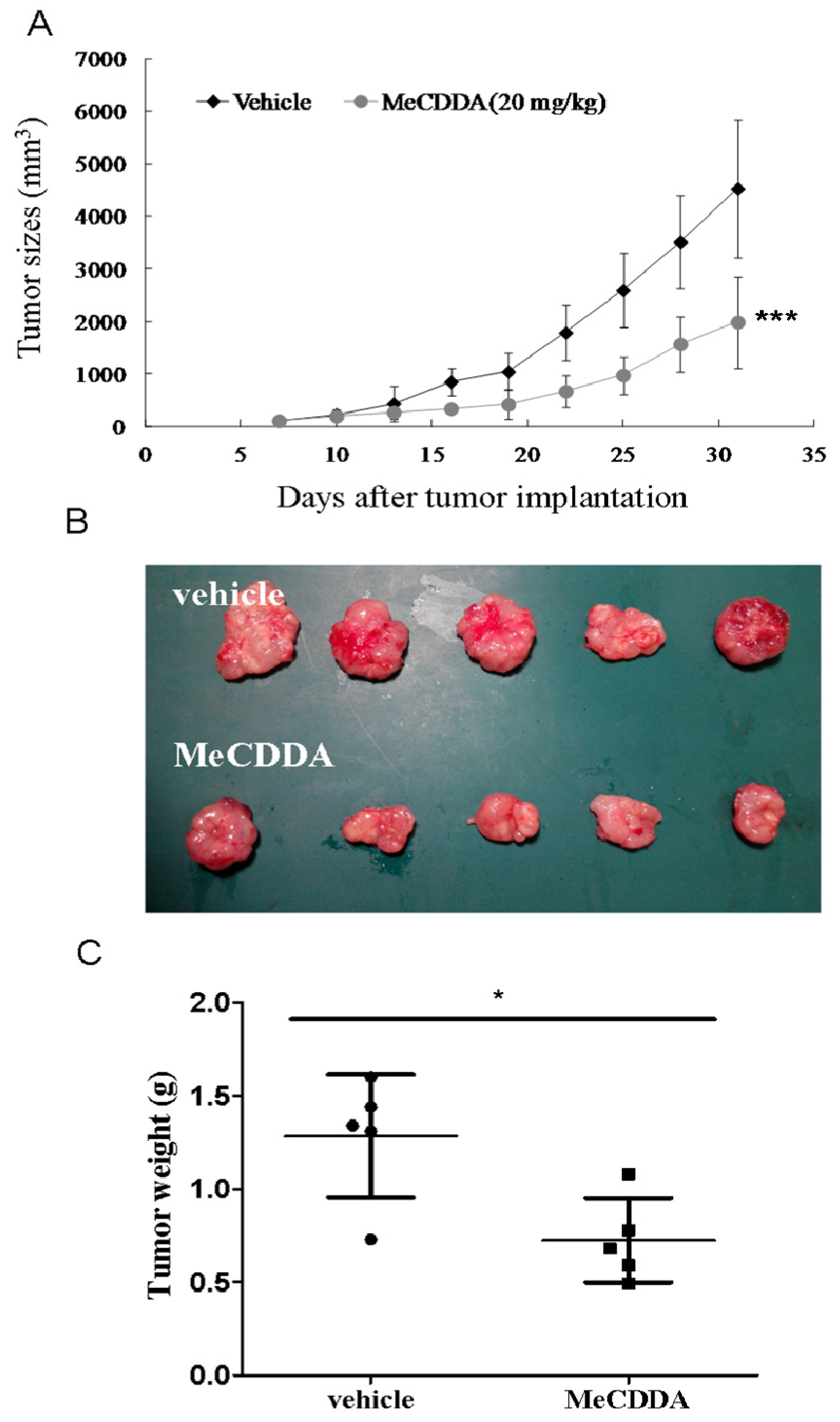
2.7. MeCDDA-Inhibited H1688 Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines
4.3. Over-Expression of AKT Protein in Transfected H1688 Cells
4.4. DNA Profile Analysis and Apoptosis Dectection
4.5. Caspase-3, 8, and 9 Activities Assay
4.6. Mitochondrial Membrane Potential Assay
4.7. Cytochrome C Measurement
4.8. Western Blotting Analysis
4.9. Animals
4.10. MeCDDA Treatment of Xenograft Tumors
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Toh, C.K.; Lim, W.T. Lung cancer in never-smokers. J. Clin. Pathol. 2007, 60, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Abeloff, M.D.; Goodwin, G.; Carney, D.N.; Gazdar, A.F. Activities of L-dopa decarboxylase and diamine oxidase (histaminase) in human lung cancers and decarboxylase as a marker for small (oat) cell cancer in cell culture. Cancer Res. 1980, 40, 1990–1994. [Google Scholar] [PubMed]
- Vescio, R.A.; Connors, K.M.; Bordin, G.M.; Robb, J.A.; Youngkin, T.; Umbreit, J.N.; Hoffman, R.M. The distinction of small cell and non-small cell lung cancer by growth in native-state histoculture. Cancer Res. 1990, 50, 6095–6099. [Google Scholar] [PubMed]
- Byers, L.A.; Rudin, C.M. Small cell lung cancer: Where do we go from here? Cancer 2015, 121, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.; Turrisi, A.T., III. A review of first-line treatment for small-cell lung cancer. J. Thorac. Oncol. 2006, 1, 270–278. [Google Scholar] [CrossRef]
- American Cancer Society. Small Cell Lung Cancer; ACS: Atlanta, GA, USA, 2016; Available online: https://www.cancer.org/cancer/small-cell-lung-cancer.html (accessed on 16 May 2016).
- Lu, B.; Zhang, J.Q.; Yang, H. Nonphospholipid vesicles of Carboplatin for lung targeting. Drug Deliv. 2003, 10, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sarma, N.S.; Krishna, M.S.; Pasha, S.G.; Rao, T.S.; Venkateswarlu, Y.; Parameswaran, P.S. Marine metabolites: The sterols of soft coral. Chem. Rev. 2009, 109, 2803–2828. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Hsu, C.H.; Chao, C.H.; Wen, Z.H.; Wu, Y.C.; Dai, C.F.; Sheu, J.H. Cytotoxic and anti-inflammatory metabolites from the soft coral Scleronephthya gracillimum. Mar. Drugs 2013, 11, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.C.; Wang, K.L.; Liao, X.J.; Deng, Z.; Xu, S.H. Pentacyclic hemiacetal sterol with antifouling and cytotoxic activities from the soft coral Nephthea sp. Bioorg. Med. Chem. Lett. 2013, 23, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chang, C.W.; Tseng, Y.J.; Lee, J.; Sung, P.J.; Su, J.H.; Hwang, T.L.; Dai, C.F.; Wang, H.C.; Sheu, J.H. Bioactive Steroids from the Formosan Soft Coral Umbellulifera petasites. Mar. Drugs 2016, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.C.; Huang, Y.T.; Chou, S.K.; Shih, M.C.; Chiang, C.Y.; Su, J.H. Cytotoxic Oxygenated Steroids from the Soft Coral Nephthea erecta. Chem. Pharm. Bull. (Tokyo) 2016, 64, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Mignotte, B.; Vayssiere, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Levy, M.A.; Claxton, D.F. Therapeutic inhibition of BCL-2 and related family members. Expert Opin. Investig. Drugs 2017, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Synthetic lethality by co-targeting mitochondrial apoptosis and PI3K/Akt/mTOR signaling. Mitochondrion 2014, 19, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.; Honecker, F. Marine Compounds and Cancer: Where Do We Stand? Mar. Drugs 2015, 13, 5657–5665. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.R.; Huang, C.Y.; Tsai, Y.Y.; Lin, Y.S.; Hwang, T.L.; Su, J.H.; Sung, P.J.; Dai, C.F.; Sheu, J.H. New cytotoxic and anti-inflammatory steroids from the soft coral Klyxum flaccidum. Bioorg. Med. Chem. Lett. 2016, 26, 3253–3257. [Google Scholar] [CrossRef] [PubMed]
- Urda, C.; Fernandez, R.; Perez, M.; Rodriguez, J.; Jimenez, C.; Cuevas, C. Protoxenicins A and B, cytotoxic long-chain acylated xenicanes from the soft coral Protodendron repens. J. Nat. Prod. 2017, 80, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; Wong, B.S.; Yea, S.H.; Lu, C.I.; Weng, S.H. Sinularin induces apoptosis through mitochondria dysfunction and inactivation of the pI3K/Akt/mTOR pathway in gastric carcinoma cells. Mar. Drugs 2016, 14, E142. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Puu, S.Y.; Duh, C.Y. New 19-oxygenated steroids from the soft coral Nephthea chabrolii. Mar. Drugs 2012, 10, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Chien, Y.C.; Pan, C.H.; Sheu, J.H.; Chen, C.Y.; Wu, C.H. Inhibitory effect of dihydroaustrasulfone alcohol on the migration of human non-small cell lung carcinoma A549 cells and the antitumor effect on a lewis lung carcinoma-bearing tumor model in C57BL/6J mice. Mar. Drugs 2014, 12, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Araki, J.; Suto, R.; Shimada, M.; Nakagawa, K.; Fukuoka, M. EGFR mutation in gefitinib-responsive small-cell lung cancer. Ann. Oncol. 2006, 17, 1028–1029. [Google Scholar] [CrossRef] [PubMed]
- Shiao, T.H.; Chang, Y.L.; Yu, C.J.; Chang, Y.C.; Hsu, Y.C.; Chang, S.H.; Shih, J.Y.; Yang, P.C. Epidermal growth factor receptor mutations in small cell lung cancer: A brief report. J. Thorac. Oncol. 2011, 6, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, L.; Gao, J.; Hooten, K.G.; Wang, E.; Thonhoff, J.R.; Dunn, T.J.; Gao, T.; Wu, P. Critical role of PI3K/Akt/GSK3β in motoneuron specification from human neural stem cells in response to FGF2 and EGF. PLoS ONE 2011, 6, e23414. [Google Scholar] [CrossRef] [PubMed]
- Hung, D.T.; Chen, J.; Schreiber, S.L. (+)-Discodermolide binds to microtubules in stoichiometric ratio to tubulin dimers, blocks taxol binding and results in mitotic arrest. Chem. Biol. 1996, 3, 287–293. [Google Scholar] [CrossRef]
- ter Haar, E.; Kowalski, R.J.; Hamel, E.; Lin, C.M.; Longley, R.E.; Gunasekera, S.P.; Rosenkranz, H.S.; Day, B.W. Discodermolide, a cytotoxic marine agent that stabilizes microtubules more potently than taxol. Biochemistry 1996, 35, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Su, T.R.; Lin, J.J.; Chiu, C.C.; Chen, J.Y.; Su, J.H.; Cheng, Z.J.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. Proteomic investigation of anti-tumor activities exerted by sinularin against A2058 melanoma cells. Electrophoresis 2012, 33, 1139–1152. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sub-G1 | G0/G1 | S | G2/M |
|---|---|---|---|---|
| Control | 0.9 ± 0.2 | 62.0 ± 0.4 | 16.8 ± 0.4 | 19.9 ± 0.2 |
| MeCDDA 10 μM | 1.2 ± 0.2 | 62.0 ± 1.2 | 17.1 ± 0.6 | 20.4 ± 0.4 |
| MeCDDA 20 μM | 32.5 ± 2.7 *** | 39.4 ± 1.4 *** | 12.8 ± 1.6 * | 15.0 ± 1.6 * |
| MeCDDA 40 μM | 89.9 ± 5.8 *** | 4.9 ± 0.4 *** | 2.7 ± 2.4 *** | 2.8 ± 3.1 * |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, T.-W.; Su, J.-H.; Lin, C.-C.; Li, Y.-R.; Chao, Y.-H.; Lin, S.-H.; Chan, H.-L. 24-Methyl-Cholesta-5,24(28)-Diene-3β,19-diol-7β-Monoacetate Inhibits Human Small Cell Lung Cancer Growth In Vitro and In Vivo via Apoptosis Induction. Mar. Drugs 2017, 15, 210. https://doi.org/10.3390/md15070210
Chung T-W, Su J-H, Lin C-C, Li Y-R, Chao Y-H, Lin S-H, Chan H-L. 24-Methyl-Cholesta-5,24(28)-Diene-3β,19-diol-7β-Monoacetate Inhibits Human Small Cell Lung Cancer Growth In Vitro and In Vivo via Apoptosis Induction. Marine Drugs. 2017; 15(7):210. https://doi.org/10.3390/md15070210
Chicago/Turabian StyleChung, Ting-Wen, Jui-Hsin Su, Chi-Chen Lin, Yi-Rong Li, Ya-Hsuan Chao, Sheng-Hao Lin, and Hong-Lin Chan. 2017. "24-Methyl-Cholesta-5,24(28)-Diene-3β,19-diol-7β-Monoacetate Inhibits Human Small Cell Lung Cancer Growth In Vitro and In Vivo via Apoptosis Induction" Marine Drugs 15, no. 7: 210. https://doi.org/10.3390/md15070210