A Dereplication and Bioguided Discovery Approach to Reveal New Compounds from a Marine-Derived Fungus Stilbella fimetaria

 , ,
, ,
Abstract
:1. Introduction
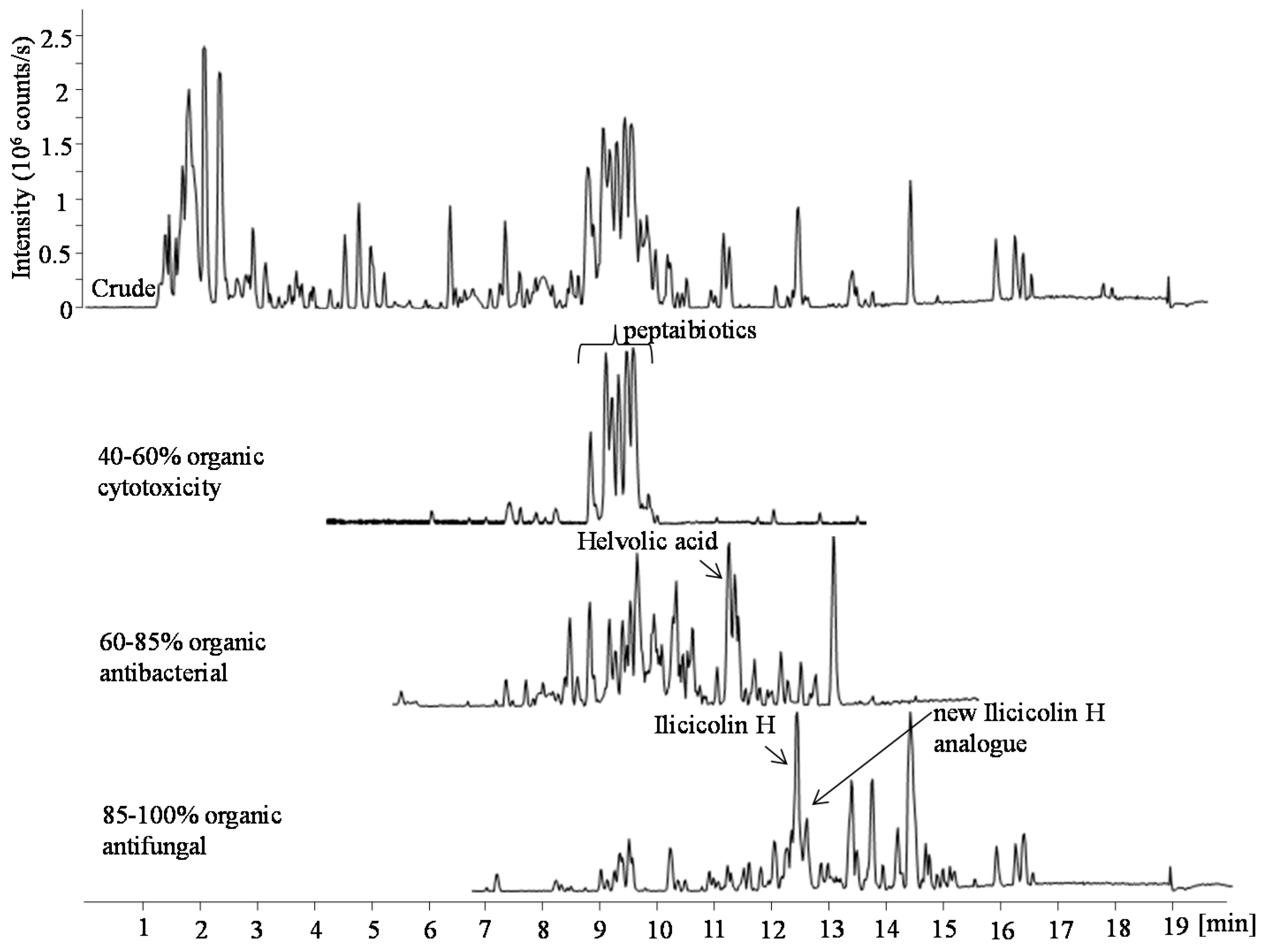
2. Results and Discussion
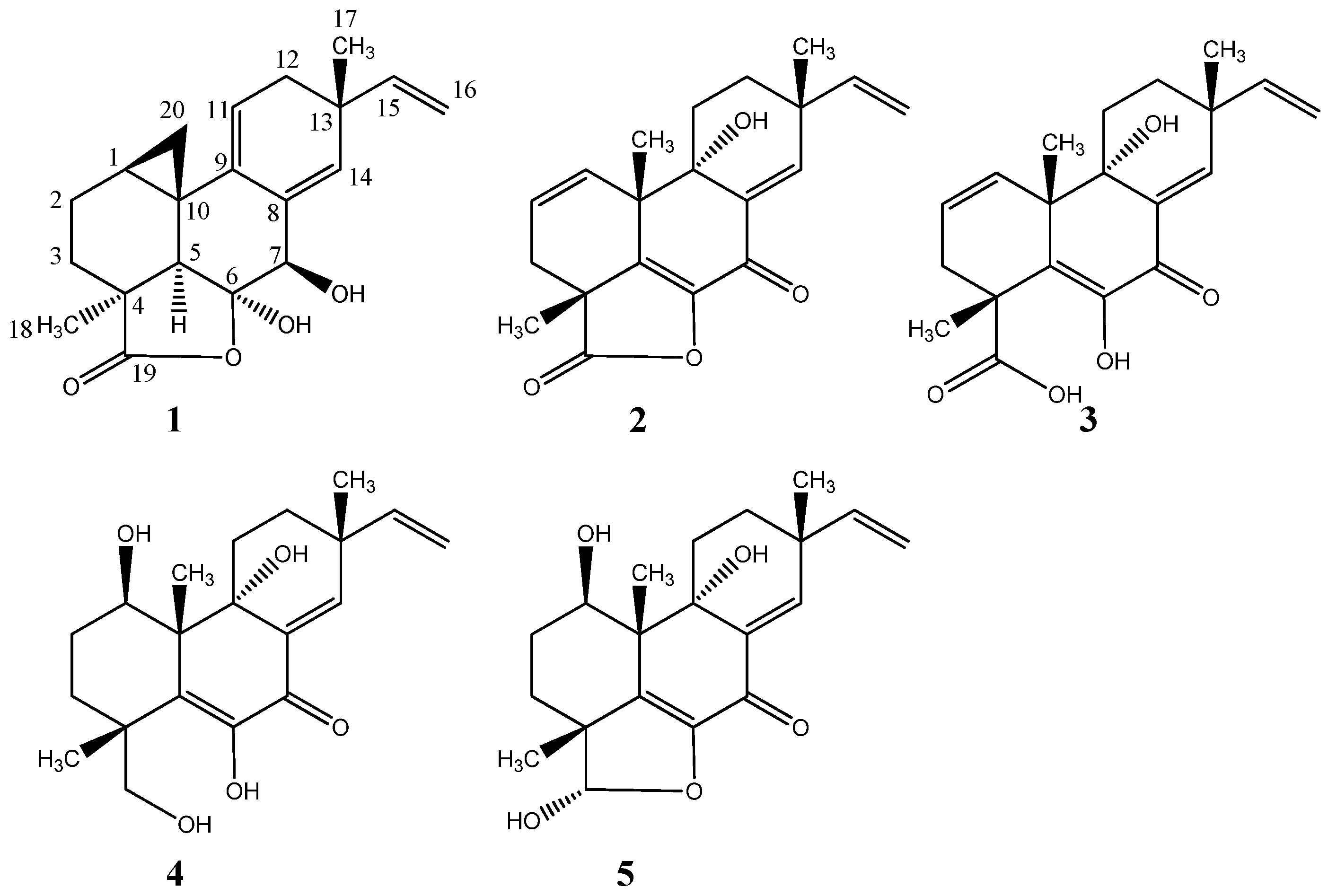
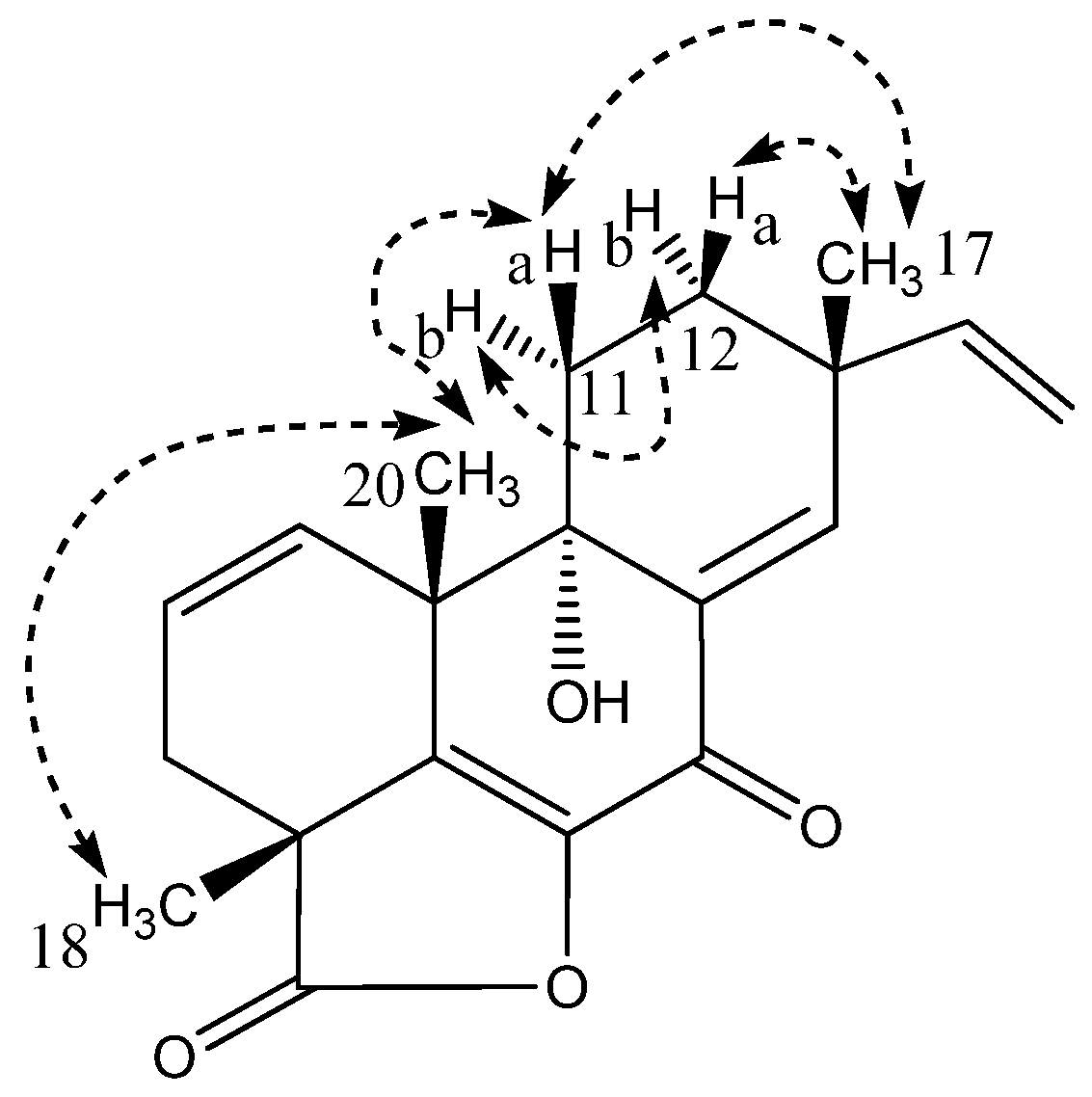
2.1. Pimarane-Type Diterpenoids Exhibiting Cytotoxicity
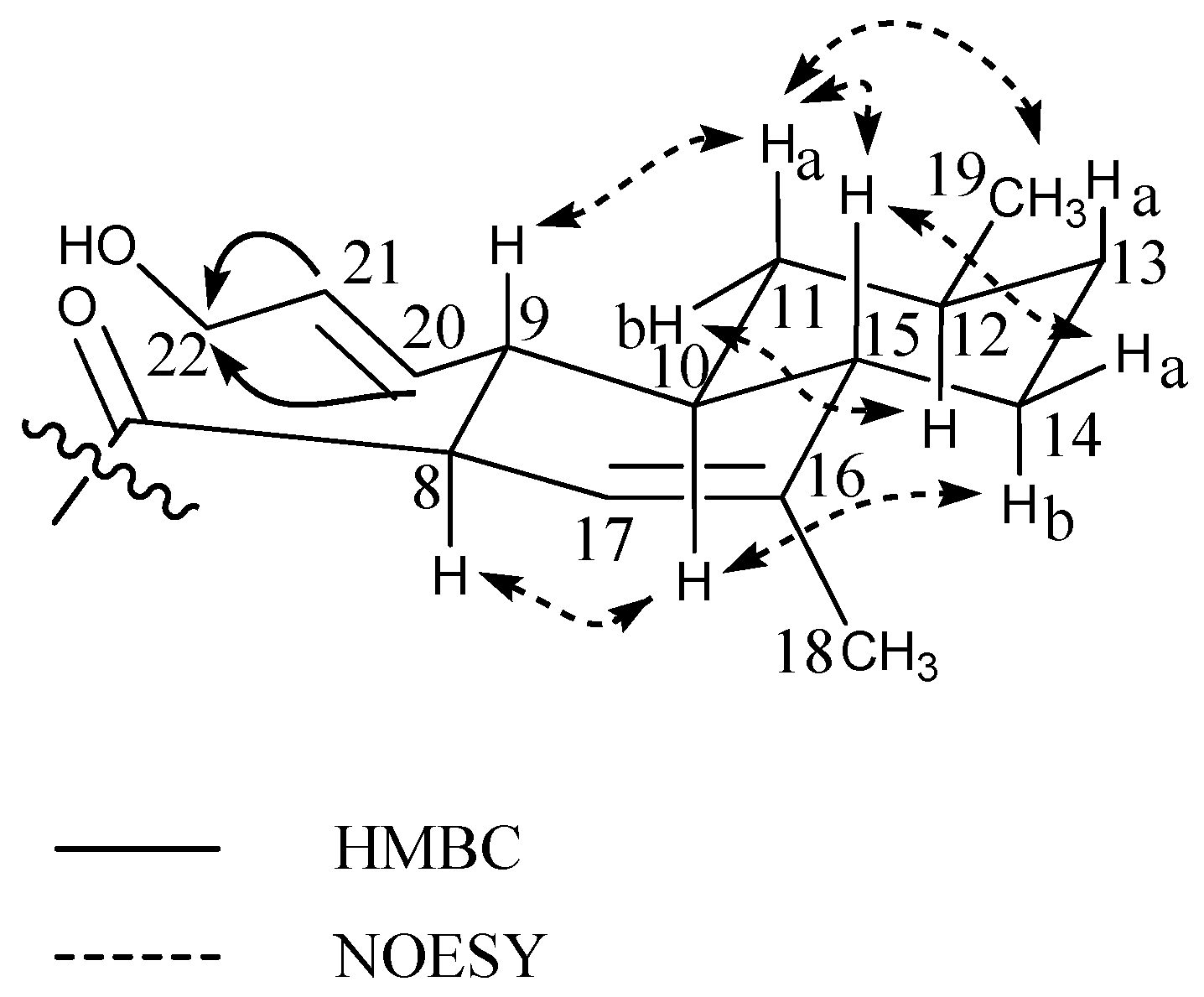
2.2. Ilicicolin H, A Broad-Spectrum Antifungal, and New Analogues
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Strain and Identification
3.3. Cultivation
3.4. Extraction and Isolation
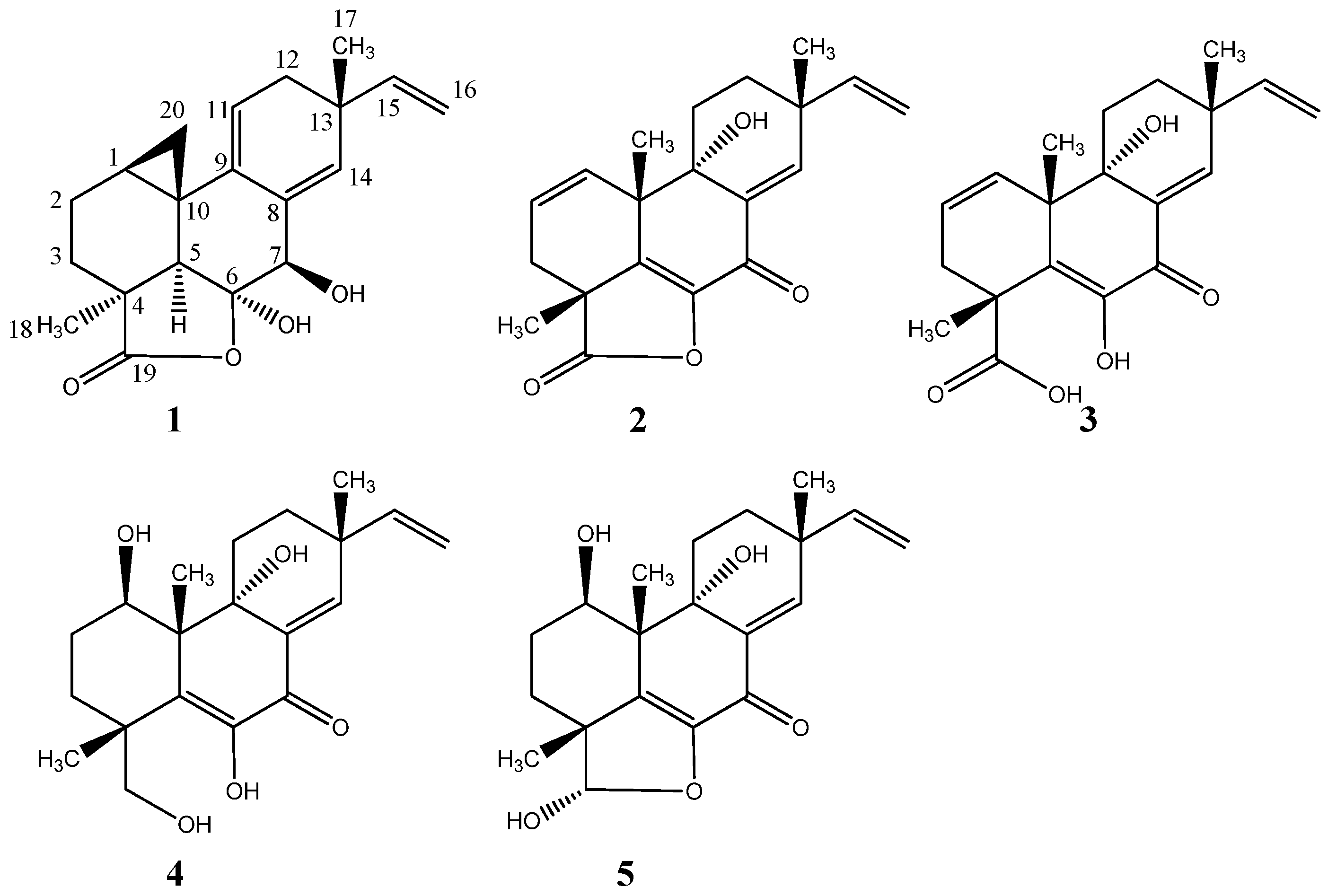
- Helvolic acid: white solid; UV (MeCN) λmax: 234 nm; 13C NMR see Figure S12 and Table 1; HRESIMS m/z 591.2932 ([M + Na]+ calculated for C33H44O8Na, m/z 591.2922)
- Myrocin F: white solid; UV (MeCN) λmax: 215 nm, 270 nm; 13C- and 1H-NMR see Table 1; HRESIMS m/z 329.1745 ([M + H]+ calculated for C20H25O4, m/z 329.1746)
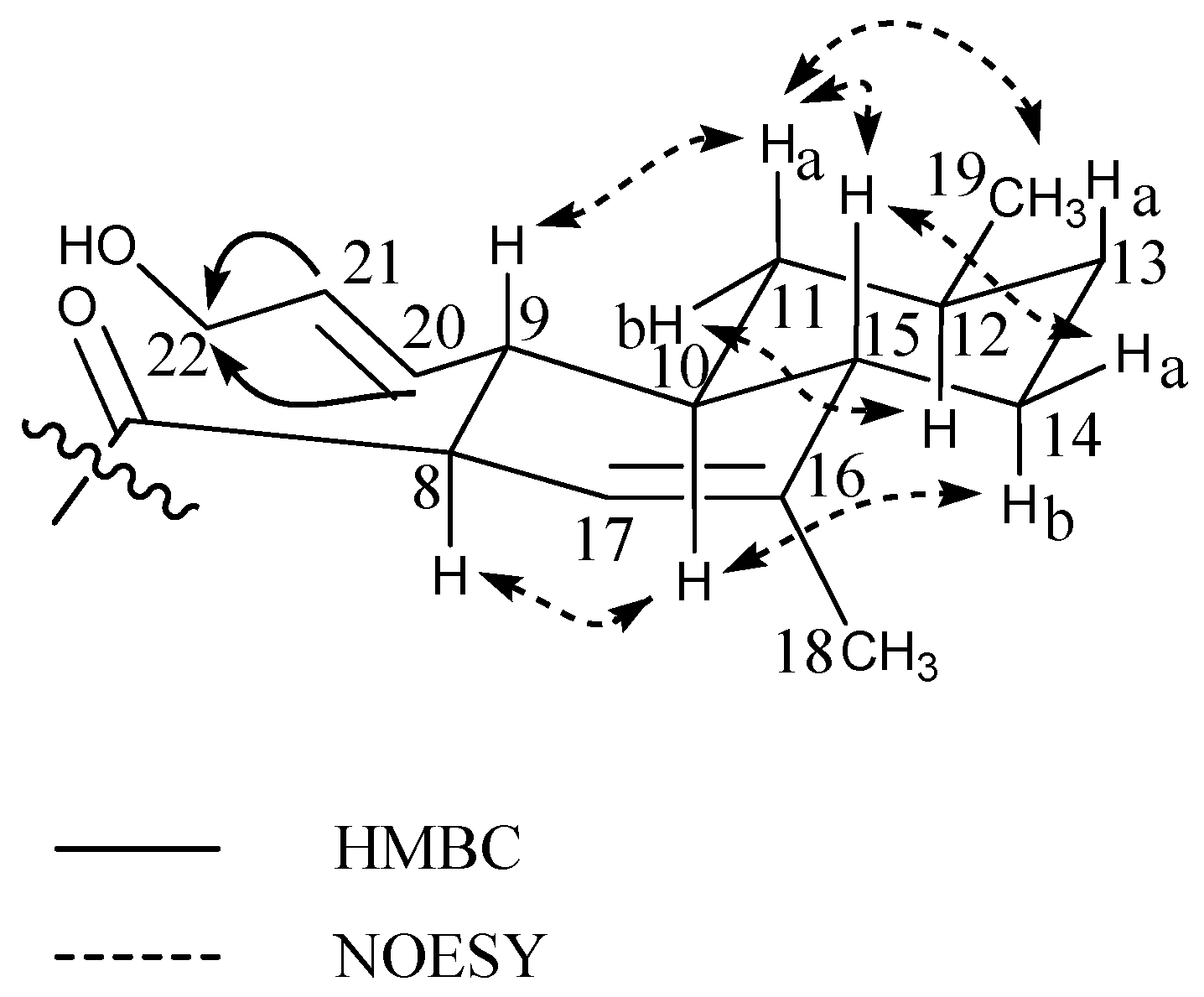
- Libertellenone M: white solid; −81° (c 0.10, MeOH); UV (MeCN) λmax: 220 sh nm, 270 sh nm, 290 nm; 13C- and 1H-NMR see Table 2; HRESIMS m/z 327.1592 ([M + H]+ calculated for C20H23O4, m/z 327.1590)
- Opened γ-lactone ring of libertellenone M: white solid; UV (MeCN) λmax: 220 sh nm, 270 nm, 315 nm; 13C- and 1H-NMR see Table 2; HRESIMS m/z 345.1692 ([M + H]+ calculated for C20H25O5, m/z 345.1695)
- Libertellenone C: white solid; −98° (c 0.11, MeOH); UV (MeCN) λmax: 218 nm, 270 nm, 325 nm; 13C- and 1H-NMR see Figure S31 and Table 2; HRESIMS m/z 349.2012 ([M + H]+ calculated for C20H29O5, m/z 349.2007)
- Libertellenone E: white solid; +24.6° (c 0.13, MeOH); UV (MeCN) λmax: 214 nm, 268 nm, 314 nm; 13C- and 1H-NMR see Figure S31 and Table 2; HRESIMS m/z 347.1858 ([M + H]+ calculated for C20H27O5, m/z 347.1851)
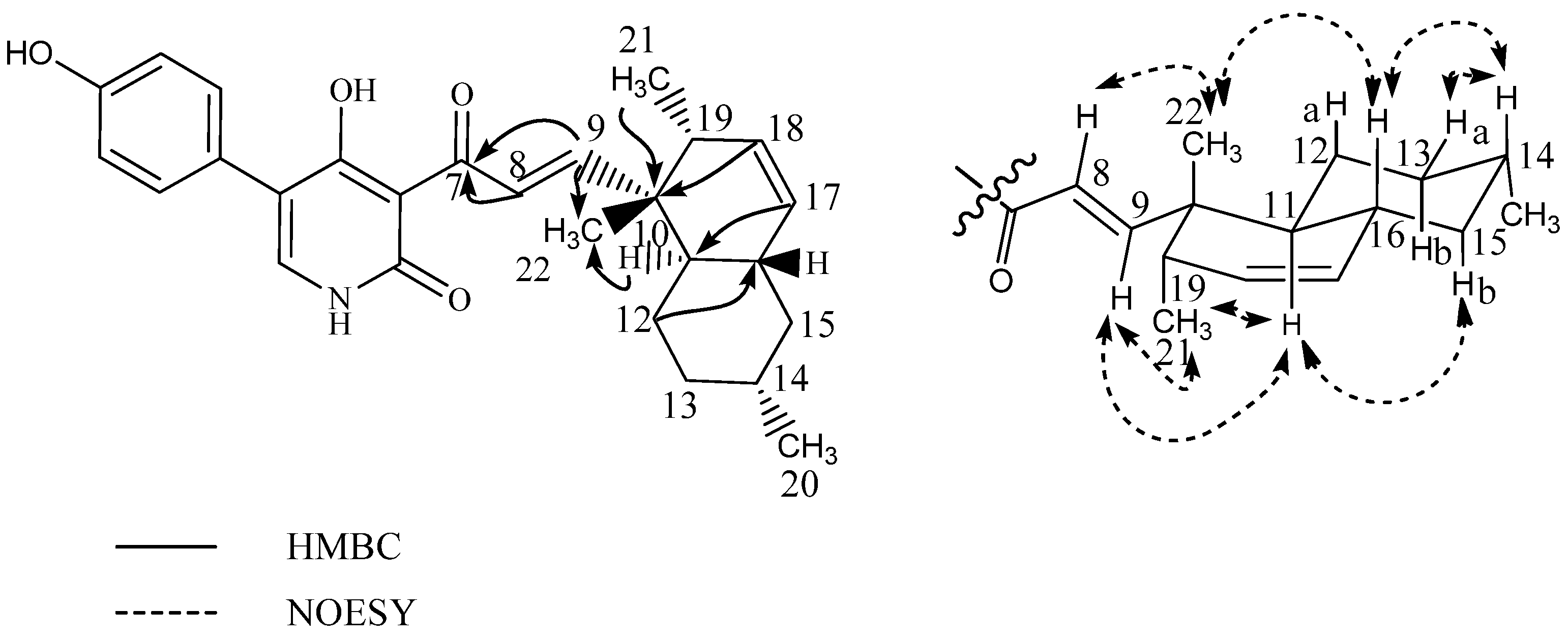
- Ilicicolin H: yellow solid; −159° (c 0.11, MeOH); UV (MeCN) λmax: 250 nm, 295 nm, 350 nm; 13C- and 1H-NMR see Table 3; HRESIMS m/z 434.2325 ([M + H]+ calculated for C27H32NO4, m/z 434.2323)
- Hydroxyl-ilicicolin H: yellow solid; UV (MeCN) λmax: 250 nm, 295 nm, 350 nm; 13C- and 1H-NMR see Table 3; HRESIMS m/z 450.2278 ([M + H]+ calculated for C27H32NO5, m/z 450.2272)
- Ilicicolin I: yellow solid; UV (MeCN) λmax: 254 nm, 308 nm, 365 nm; 13C- and 1H-NMR see Table 3; HRESIMS m/z 434.2325 ([M + H]+ calculated for C27H32NO4, m/z 434.2323
3.5. Cytotoxicity Assay
3.6. Antibacterial and Antifungal Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Debbab, A.; Aly, A.H.; Lin, W.H.; Proksch, P. Bioactive compounds from marine bacteria and fungi. Microb. Biotechnol. 2010, 3, 544–563. [Google Scholar] [CrossRef] [PubMed]
- Duarte, K.; Rocha-Santos, T.A.P.; Freitas, A.C.; Duarte, A.C. Analytical techniques for discovery of bioactive compounds from marine fungi. Trends Anal. Chem. 2012, 34, 97–109. [Google Scholar] [CrossRef]
- Overy, D.P.; Bayman, P.; Kerr, R.G.; Bills, G.F. An assessment of natural product discovery from marine (sensu strictu) and marine-derived fungi. Mycology 2014, 5, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Burgaud, G.; Le Calvez, T.; Arzur, D.; Vadenkoornhuyse, P.; Barbier, G. Diversity of culturable marine filamentous fungi from deep-sea hydrothermal vents. Environ. Microbiol. 2009, 11, 1588–1600. [Google Scholar] [CrossRef] [PubMed]
- Jones, E. Are there more marine fungi to be described? Bot. Mar. 2011, 54, 343–354. [Google Scholar] [CrossRef]
- Jones, E. Fifty years of marine mycology. Fungal Divers. 2011, 50, 73–112. [Google Scholar] [CrossRef]
- Richards, T.A.; Jones, M.D.M.; Leonard, G.; Bass, D. Marine fungi: Their ecology and molecular diversity. Annu. Rev. Mar. Sci. 2012, 4, 495–522. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Bladt, T.B.; Dürr, C.; Knudsen, P.B.; Kildgaard, S.; Frisvad, J.C.; Gotfredsen, C.H.; Seiffert, M.; Larsen, T.O. Bio-activity and dereplication-based discovery of ophiobolins and other fungal secondary metabolites targeting leukemia cells. Molecules 2013, 18, 14629–14650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kildgaard, S.; Mansson, M.; Dosen, I.; Klitgaard, A.; Frisvad, J.C.; Larsen, T.O.; Nielsen, K.F. Accurate dereplication of bioactive secondary metabolites from marine-derived fungi by UHPLC-DAD-QTOFMS and a MS/HRMS library. Mar. Drugs 2014, 12, 3681–3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, K.F.; Månsson, M.; Rank, C.; Frisvad, J.C.; Larsen, T.O. Dereplication of microbial natural products by LC-DAD-TOFMS. J. Nat. Prod. 2011, 74, 2338–2348. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, A.; Iversen, A.; Andersen, M.R.; Larsen, T.O.; Frisvad, J.C.; Nielsen, K.F. Aggressive dereplication using UHPLC-DAD-QTOF—Screening extracts for up to 3000 fungal secondary metabolites. Anal. Bioanal. Chem. 2014, 406, 1933–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthals, A.; Watrous, J.D.; Dorrestein, P.C.; Bandeira, N. The spectral networks paradigm in high throughput mass spectrometry. Mol. BioSyst. 2012, 8, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [PubMed]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A.; et al. Integrating molecular networking and biological assays to target the isolation of a cytotoxic cyclic octapeptide, samoamide A, from an American Samoan marine cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Appleton, D.R.; Buss, A.D.; Butler, M.S. A simple method for high-throughput extract prefractionation for biological screening. Chimia 2007, 61, 327–331. [Google Scholar] [CrossRef]
- Wagenaar, M.M. Pre-fractionated microbial samples—The second generation natural products library at Wyeth. Molecules 2008, 13, 1406–1426. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Fontaine, F.; Cooper, M.A. Natural product libraries: Assembly, maintenance, and screening. Planta Med. 2014, 80, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Månsson, M.; Phipps, R.K.; Gram, L.; Munro, M.H.G.; Larsen, T.O.; Nielsen, K.F. Explorative solid-phase extraction (E-SPE) for accelerated microbial natural product discovery, dereplication, and purification. J. Nat. Prod. 2010, 73, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Ratnaweera, P.B.; Williams, D.E.; de Silva, E.D.; Wijesundera, R.L.C.; Dalisay, D.S.; Andersen, R.J. Helvolic acid, an antibacterial nortriterpenoid from a fungal endophyte, Xylaria sp. of orchid Anoectochilus setaceus endemic to Sri Lanka. Mycology 2014, 5, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, B.; Guan, J.; Zhang, G. In vitro synergistic antibacterial activities of helvolic acid on multi-drug resistant Staphylococcus aureus. Nat. Prod. Res. 2009, 23, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, H.; Negishi, E.; Yamaguchi, K.; Nishi, N.; Yamazaki, M. Isolation of new tremorgenic metabolites from an Ascomycete, Corynascus setosus. Chem. Pharm. Bull. 1996, 44, 1843–1848. [Google Scholar] [CrossRef]
- Matsumoto, M.; Minato, H. Structure of ilicicolin H, an antifungal. Tetrahedron Lett. 1976, 42, 3827–3838. [Google Scholar] [CrossRef]
- Tanaba, M.; Uranot, S. Biosynthetic studies with 13C the antifungal antibiotic ilicicolin H. Tetrahedron 1983, 39, 3569–3574. [Google Scholar] [CrossRef]
- Singh, B.S.; Liu, W.; Li, X.; Chen, T.; Shafiee, A.; Card, D.; Abruzzo, G.; Flattery, A.; Gill, C.; Thompson, J.R.; et al. Antifungal spectrum, in vivo efficacy, and structure-activity relationship of ilicicolin H. ACS Med. Chem. Lett. 2012, 3, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Liu, W.; Li, X.; Chen, T.; Shafiee, A.; Dreikorn, S.; Hornak, V.; Meinz, M.; Onishi, J.C. Structure—activity relationship of cytochrome bc1 reductase inhibitor broad spectrum antifungal ilicicolin H. Bioorg. Med. Chem. Let. 2013, 23, 3018–3022. [Google Scholar] [CrossRef] [PubMed]
- Shervington, A.; Lu, C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Investig. 2008, 26, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Stoppacher, N.; Neumann, N.K.N.; Burgstaller, L.; Zeilinger, S.; Degenkolb, T.; Bruckner, H.; Schuhmacher, R. The Comprehensive Peptaibiotics Database. Chem. Biodivers. 2013, 10, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Thirumalachar, M.J. Antiamoebin anti parasit a new anti protozoal anti helminthic antibiotic I production and biological studies Emericellopsis-Poonensis Emericellopsis-Synnematicola Cephalosporium-Pimprina. Hindustan Antibiot. Bull. 1968, 10, 287–289. [Google Scholar] [PubMed]
- Lehr, N.-A.; Meffert, A.; Antelo, L.; Sterner, O.; Anke, H.; Weber, R.W.S. Antiamoebins, myrocin B and the basis of antifungal antibiosis in the coprophilous fungus Stilbella erythrocephala (syn. S. fimetaria). FEMS Microbiol. Ecol. 2006, 55, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, A.; Bruckner, H. New sequences and new fungal producers of peptaibol antibiotics antiamoebins. Pept. Sci. 2000, 6, 149–167. [Google Scholar] [CrossRef]
- Klemke, C.; Kehraus, S.; Wright, A.D.; König, G.M. New secondary metabolites from the marine endophytic fungus Apiospora montagnei. J. Nat. Prod. 2004, 67, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Nakagawa, M.; Hirota, A.; Shima, S.; Nakayama, M. Structure of myrocin B, a new diterpene antibiotic produced by Myrothecium verrucaria. Agric. Biol. Chem. 1988, 52, 1305–1307. [Google Scholar] [CrossRef]
- Tsukada, M.; Fukai, M.; Miki, K.; Shiraishi, T.; Suzuki, T.; Nishio, K.; Sugita, T.; Ishino, M.; Kinoshita, K.; Takahashi, K.; et al. Chemical constituents of a marine fungus, Arthrinium sacchari. J. Nat. Prod. 2011, 74, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Shiono, Y.; Matsui, N.; Imaizumi, T.; Koseki, T.; Murayama, T.; Kwon, E.; Abe, T.; Kimura, K.-I. An unusual spirocyclic isopimarane diterpenoid and other isopimarane diterpenoids from fruiting bodies of Xylaria polymorpha. Phytochem. Lett. 2013, 6, 439–443. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Hirota, A.; Shima, S.; Nakagawa, M.; Adachi, T.; Nozaki, H.; Nakayama, M. Myrocin C, a new diterpene antitumor antibiotic from Myrothecium verrucaria. J. Antibiot. 1989, 42, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-C.; Jensen, P.R.; Kauffman, C.A.; Fenical, W. Libertellenones A–D: Induction of cytotoxic diterpenoid biosynthesis by marine microbial competition. Bioorg. Med. Chem. 2005, 13, 5267–5273. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Wei, W.; Jiang, R.; Zhao, G. Yi Zhong Er Tie Libertellenone G Ji Qi Zhi Bei Fang Fa Yu Yong Tu. CN Patent 103073527 A, 1 May 2013. [Google Scholar]
- Laatsch, H. AntiBase 2012; Wiley-VCH: Weinheim, Germany, 2012; Available online: http://www.wileyvch.de/stmdata/antibase.php (accessed on 1 July 2017).
- Hayakawa, S.; Minato, H.; Katagiri, K. The ilicicolins, antibiotics from Cylindrocladium ilicicola. J. Antibiot. 1971, 24, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Bremmer, M.L.; Brown, D.L.; D’Antuono, J. Total Synthesis of (f)-Ilicicolin H. J. Org. Chem. 1985, 50, 2807–2809. [Google Scholar] [CrossRef]
- Singh, S.B.; Li, X.; Chen, T. Biotransformation of antifungal ilicicolin H. Tetrahedron Lett. 2011, 52, 6190–6191. [Google Scholar] [CrossRef]
- Andersen, A.J.C.; Hansen, P.J.; Jørgensen, K.; Nielsen, K.F. Dynamic cluster analysis: An unbiased method for identifying A + 2 element containing compounds in liquid chromatographic high-resolution time of flight mass spectrometric data. Anal. Chem. 2016, 88, 12461–12469. [Google Scholar] [CrossRef] [PubMed]
- Samson, R.A.; Houbraken, J.; Thrane, U.; Frisvad, J.C.; Andersen, B. Food and Indoor Fungi; CBS-KNAW Fungal Biodiversity Centre: Utrecht, The Netherlands, 2010. [Google Scholar]
- Smedsgaard, J. Micro-Scale extraction procedure for standardized screening of fungal metabolite production in cultures. J. Chromatogr. A 1997, 760, 264–270. [Google Scholar] [CrossRef]
- Audoin, C.; Bonhomme, D.; Ivanisevic, J.; de la Cruz, M.; Cautain, B.; Monteiro, M.C.; Reyes, F.; Rios, L.; Perez, T.; Thomas, O.P. Balibalosides, an original family of glucosylated sesterterpenes produced by the Mediterranean sponge Oscarella balibaloi. Mar. Drugs 2013, 11, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.C.; de la Cruz, M.; Cantizani, J.; Moreno, C.; Tormo, J.R.; Mellado, E.; De Lucas, J.R.; Asensio, F.; Valiante, V.; Brakhage, A.A.; et al. A new approach to drug discovery: High-throughput screening of microbial natural extracts against Aspergillus fumigatus using resazurin. J. Biomol. Screen. 2012, 17, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
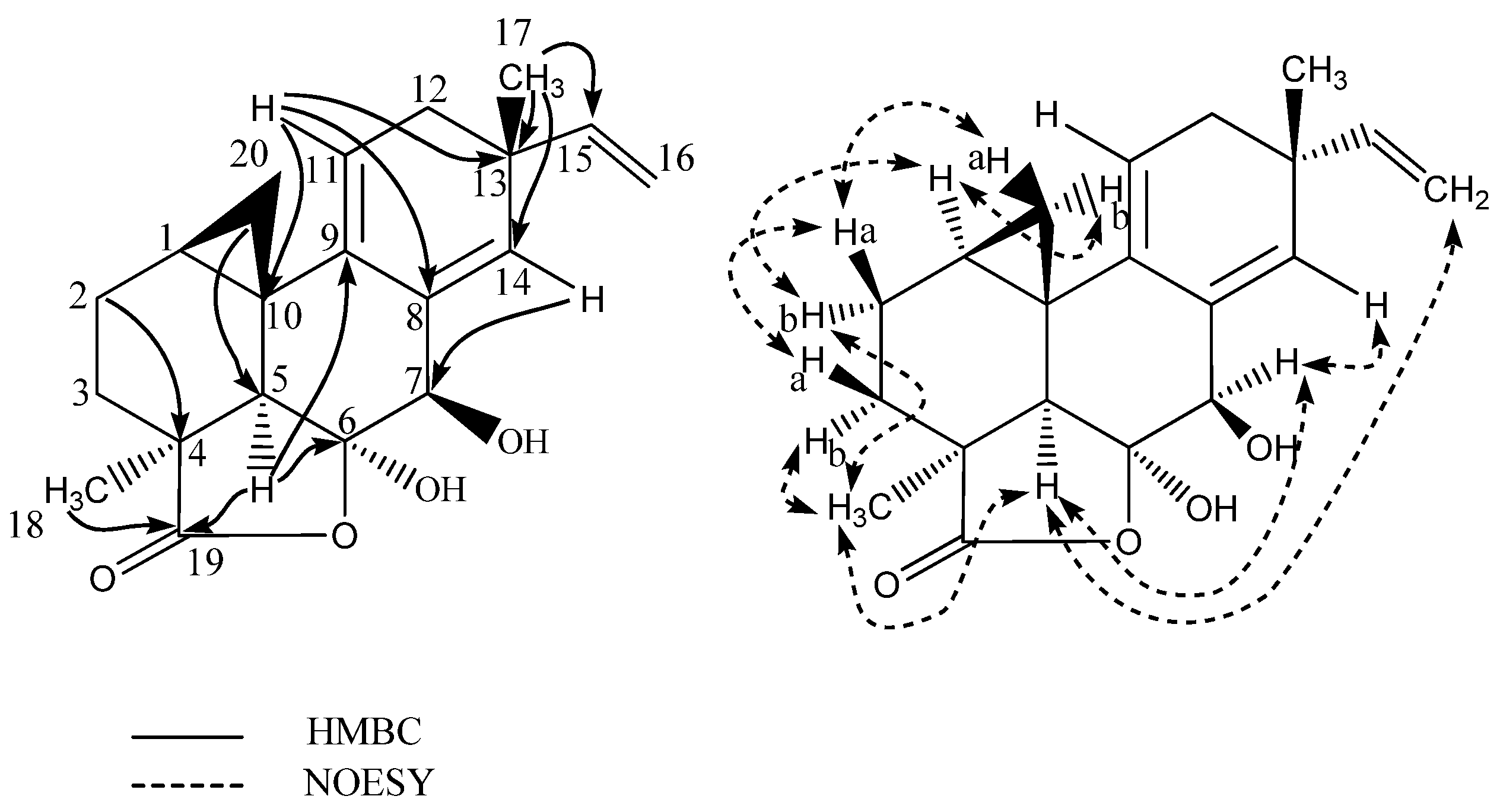
| Position | δ13C | δ1H (Mult, J) | HMBC | NOESY |
|---|---|---|---|---|
| 1 | 14.0 | 1.63 m | 3 | 2b,5,11,20b |
| 2a | 19.8 | 1.78 m | 3,4 | 3a,20a |
| 2b | 1.81 m | 3,4 | 1,5,18 | |
| 3a | 28.8 | 1.44 m | 1,4,5,19 | 2a,20a |
| 3b | 1.74 m | 4,19 | 18 | |
| 4 | 42.8 | - | ||
| 5 | 52.5 | 2.11 s | 4,6,9,10,18–20 | 1,2b,7,16a,18 |
| 6 | 107.6 | - | ||
| 7 | 77.4 | 4.24 s | 5,6,8,9,14 | 5,14 |
| 8 | 135.5 | - | ||
| 9 | 138.6 | - | ||
| 10 | 20.5 | - | ||
| 11 | 114.6 | 5.24 t(4.5) | 8,10,12–14 | 1,12,20b |
| 12 | 37.0 | 2.19 m | 9,11,13–15,17 | 11,15,17 |
| 13 | 38.9 | - | ||
| 14 | 135.8 | 5.56 s | 7,9,11–13,15,17 | 7,16a,17 |
| 15 | 144.0 | 5.67 dd(17.4/10.4) | 13,14,17 | 12,16a/b,17 |
| 16a | 112.3 | 5.03 dd (17.4/1.5) | 13 | 5,14,15,17 |
| 16b | 4.89 dd(10.4/1.5) | 13 | 15 | |
| 17 | 28.0 | 1.15 s | 12–15 | 12,14,15,16a |
| 18 | 29.6 | 1.42 s | 3–5,19 | 2b,3b,5 |
| 19 | 185.6 | - | ||
| 20a | 17.1 | 0.85 t(5.2) | 1,5,9 | 2a,3a |
| 20b | 0.25 dd(8.2/5.7) | 2,9 | 1,11 |
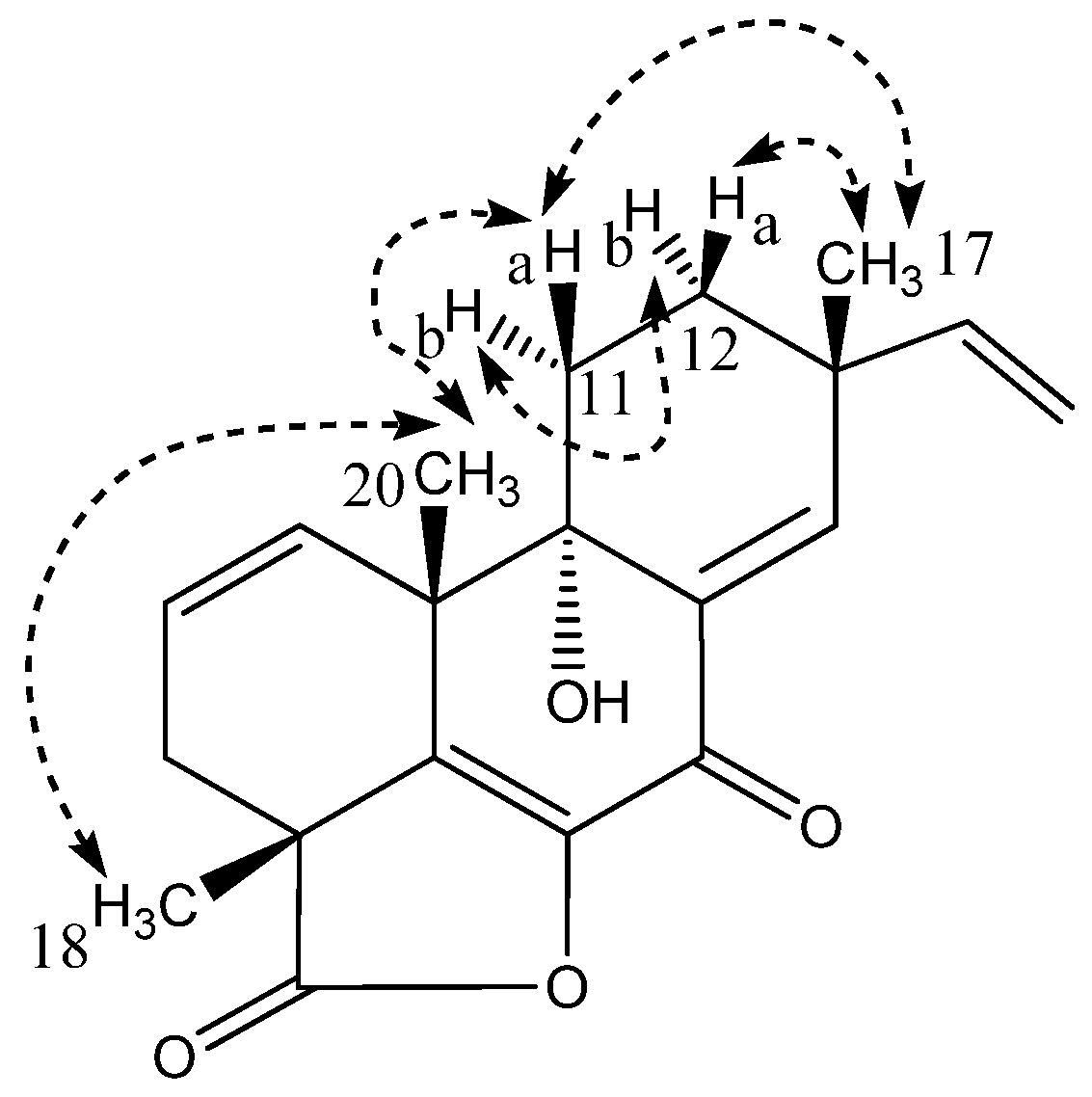
| Libertellenone M (2) | Opened γ-lactam libertellenone M (3) | |||||
|---|---|---|---|---|---|---|
| Position | δ13C | δ1H (Mult, J) | HMBC | δ13C | δ1H (Mult, J) | HMBC |
| 1 | 130.7 | 5.78 dd(9.9,3.0) | 3,5,6,10,20 | 130.7 | 5.94 m | 3,10 |
| 2 | 127.4 | 5.91 m | 3,4,10 | 126.7 | 5.98 m | 3,10 |
| 3a | 34.3 | 2.36 dt(16.5,2.5) | 1,2,4,5,18 | 36.2 | 2.16 m | - |
| 3b | 2.43 dd(16.5,5.9) | 1,2,4,18,19 | 2.64 m | 1,2 | ||
| 4 | 46.2 | - | 46.8 | - | - | |
| 5 | 146.9 | - | 137.0 | - | - | |
| 6 | 143.1 | - | * | - | - | |
| 7 | 177.3 | - | 183.6 | - | - | |
| 8 | 137.6 | - | * | - | - | |
| 9 | 76.6 | - | 76.0 | - | - | |
| 10 | 45.5 | - | 46.6 | - | - | |
| 11a | 27.5 | 2.24 m | 9,10,12,13 | 27.4 | 2.16 m | - |
| 11b | 1.72 ddd(14.0,5.0,3.5) | 8–10,12,13 | 1.93 m | - | ||
| 12a | 30.9 | 1.59 m | 9,11,14,17 | 30.6 | 1.60 m | - |
| 12b | 1.78 td(13.0,3.5) | 9,11,13–15,17 | 1.92 m | 17 | ||
| 13 | 39.8 | - | 40.0 | - | - | |
| 14 | 148.8 | 6.90 s | 7–9,12,13,15,17 | 148.8 | 6.98 s | 7,9,12,15 |
| 15 | 147.0 | 5.93 m | 12–14,17 | 147.0 | 5.92 m | - |
| 16a | 113.5 | 5.09 d(17.5) | 13,15 | 113.0 | 5.12 d(17.2) | 13 |
| 16b | 5.07 d(10.5) | 13,15 | 5.06 d(10.5) | 13 | ||
| 17 | 24.8 | 1.17 s | 12–15 | 23.8 | 1.16 s | 12–15 |
| 18 | 23.4 | 1.48 s | 3–5,19 | 24.1 | 1.55 s | 3–5,19 |
| 19 | 181.2 | - | 181.1 | - | ||
| 20 | 24.1 | 1.29 s | 1,5,9,10 | 28.3 | 1.23 s | 1,5,9,10 |
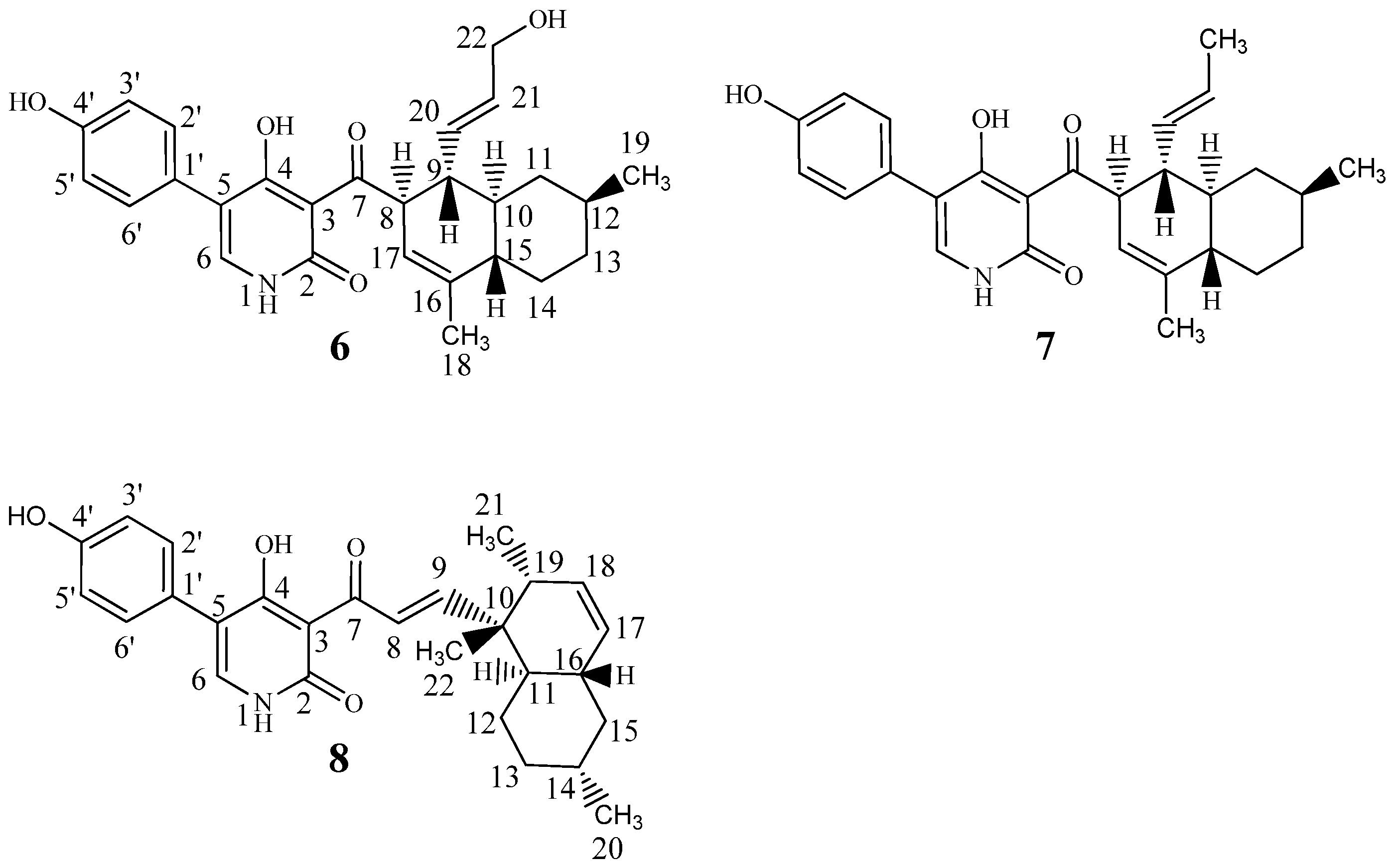
| Hydroxyl-ilicicolin H (6) | Ilicicolin H (7) | Ilicicolin I (8) | ||||
|---|---|---|---|---|---|---|
| Position | δ13C | δ1H (Mult, J) | δ13C | δ1H (Mult, J) | δ13C | δ1H (Mult, J) |
| 1′ | 125.9 | - | 125.8 | - | 126.0 | - |
| 3′5′ | 116.4 | 6.83 d(8.6) | 116.6 | 6.83 d(8.6) | 116.4 | 6.84 d(8.6) |
| 2′6′ | 131.8 | 7.27 d(8.6) | 131.7 | 7.26 d(8.6) | 131.8 | 7.29 d(8.6) |
| 4′ | 157.8 | - | 157.8 | - | 157.9 | - |
| 4′OH | - | 16.7 br.s. | - | 17.6 br.s. | - | - |
| 1NH | - | 9.46 br.s. | - | 9.56 br.s. | - | 9.44 br.s. |
| 2 | 163.0 | - | 162.9 | - | 163.3 | - |
| 3 | 108.7* | - | 108.1 | - | 107.5 | - |
| 4 | 178.2 | - | 178.0 | - | 179.5 | - |
| 5 | 114.9 | - | 114.8 | - | 115.1 | - |
| 6 | 141.3 | 7.40 s | 141.4 | 7.40 s | 141.2 | 7.42 s |
| 7 | 210.8 | - | 211.0 | - | 195.7 | - |
| 8 | 54.1 | 4.98 m | 54.1 | 4.97 m | 127.5 | 7.98 d(16.0) |
| 9 | 45.7 | 2.56 q(10.4) | 46.2 | 2.48 q(10.4) | 160.2 | 7.26 d(16.0) |
| 10 | 44.5 | 1.28 m | 44.5 | 1.23 m | 42.6 | - |
| 11a | 40.6 | 0.61 q(11.8) | 40.6 | 0.58 q(11.8) | 43.2 | 1.41 m |
| 11b | 1.78 m | 1.77 m | ||||
| 12a | 33.8 | 1.40 m | 33.8 | 1.38 m | 28.5 | 1.07 dq(12.4,3.4) |
| 12b | 1.40 m | |||||
| 13a | 36.6 | 0.97 m | 36.6 | 0.97 m | 36.8 | 1.73 m |
| 13b | 1.76 m | 1.77 m | 1.00 dq(12.5,3.4) | |||
| 14a | 31.0 | 2.07 m | 31.0 | 2.04 m | 34.3 | 1.47 m |
| 14b | 0.99 m | 0.99 m | ||||
| 15a | 45.6 | 1.70 m | 45.4 | 1.68 m | 43.1 | 1.80 m |
| 15b | 0.80 q(12.5) | |||||
| 16 | 139.5 | - | 139.5 | - | 39.3 | 1.81 m |
| 17 | 121.0 | 5.22 s | 120.9 | 5.21 m | 131.1 | 5.41 d(10.0) |
| 18 | 21.4 | 1.65 s | 21.5 | 1.63 s | 132.5 | 5.58 ddd(10.0,4.7,2.6) |
| 19 | 23.3 | 0.90 d(6.5) | 23.4 | 0.89 d(6.5) | 44.5 | 1.91 m |
| 20 | 134.1 | 5.41 dd(15.5,8.2) | 134.8 | 5.21 m | 23.2 | 0.90 d(6.5) |
| 21 | 132.6 | 5.47 dt(15.5,5.1) | 127.3 | 5.32 m | 18.6 | 0.98 d(7.0) |
| 22 | 63.5 | 3.85 d(4.8) | 18.5 | 1.53 d(6.5) | 18.5 | 1.10 s |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kildgaard, S.; Subko, K.; Phillips, E.; Goidts, V.; De la Cruz, M.; Díaz, C.; Gotfredsen, C.H.; Andersen, B.; Frisvad, J.C.; Nielsen, K.F.; et al. A Dereplication and Bioguided Discovery Approach to Reveal New Compounds from a Marine-Derived Fungus Stilbella fimetaria. Mar. Drugs 2017, 15, 253. https://doi.org/10.3390/md15080253
Kildgaard S, Subko K, Phillips E, Goidts V, De la Cruz M, Díaz C, Gotfredsen CH, Andersen B, Frisvad JC, Nielsen KF, et al. A Dereplication and Bioguided Discovery Approach to Reveal New Compounds from a Marine-Derived Fungus Stilbella fimetaria. Marine Drugs. 2017; 15(8):253. https://doi.org/10.3390/md15080253
Chicago/Turabian StyleKildgaard, Sara, Karolina Subko, Emma Phillips, Violaine Goidts, Mercedes De la Cruz, Caridad Díaz, Charlotte H. Gotfredsen, Birgitte Andersen, Jens C. Frisvad, Kristian F. Nielsen, and et al. 2017. "A Dereplication and Bioguided Discovery Approach to Reveal New Compounds from a Marine-Derived Fungus Stilbella fimetaria" Marine Drugs 15, no. 8: 253. https://doi.org/10.3390/md15080253