3.1. Chemistry
All the chemicals were obtained from commercial sources. The NMR spectra were measured on Bruker Avance spectrometers (400 MHz for 1H; Avance III 400, and 125 MHz for 13C; Avance III 500, Bruker Biospin AG, Uster, Switzerland). Chemical shifts were expressed in δ (ppm) and coupling constants (Ј) in Hz. Commercial Silica gel (200–300 mesh, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) was used for column chromatography and pre-coated Silica gel plates (HSGF254, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) were used for analytical TLC. ESI-MS spectra were recorded on a Q-TOF Micromass spectrometer (1290-6545 UHPLC-QTOF, Micromass, Wythenshawe, UK).
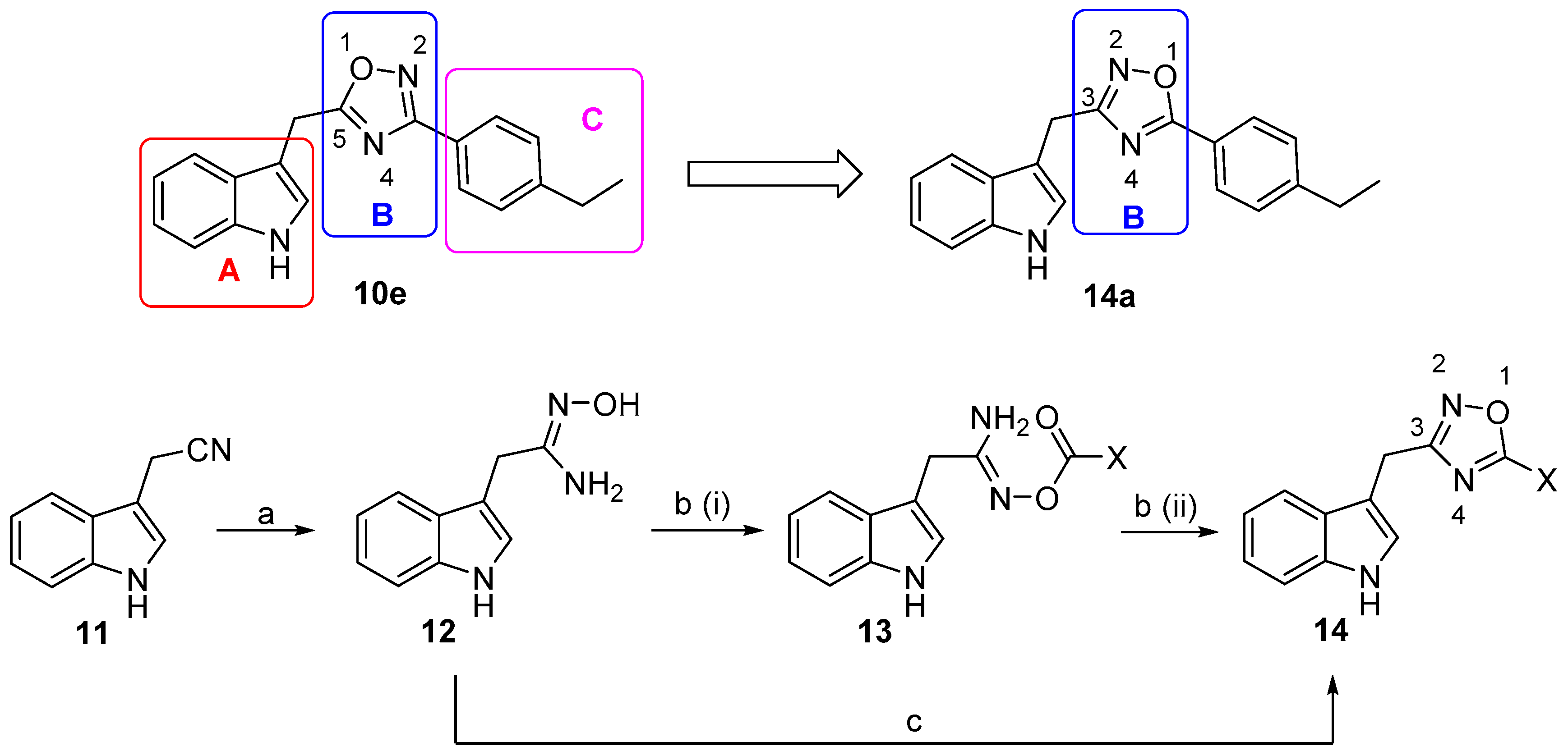
3.1.1. General Synthetic Procedure of Oxime 5 and Nitrile 6
To a solution of aldehyde 4 (9.4 mmol, 1.0 equiv) in EtOH (30 mL) was added hydroxylamine hydrochloride (11.3 mmol, 1.2 equiv) and sodium hydroxide (18.8 mmol, 2.0 equiv). The mixture was stirred at room temperature overnight. EtOH was removed in vacuo. The residue was added water and extracted with ethyl acetate (3 × 30 mL), washed with brine (3 × 30 mL), dried over anhydrous MgSO4, and concentrated. The residue was subjected to silica gel chromatography with petroleum ether/CH2Cl2 (3:2) to make oxime 5. To a solution of oxime 5 (6.0 mmol, 1.0 equiv) in acetonitrile was added [Ru2(p-PriC6H4Me)2(μ-Cl)Cl]2 (0.3 mmol, 0.05 equiv) and refluxed for 4 h. The mixture was filtered, and the filtrate was concentrated in vacuo. The residue was subjected to silica gel chromatography with petroleum ether/CH2Cl2 (2:1) to make nitrile 6.
3.1.2. Synthesis of Carboxamidine 7
To a solution of nitrile 6 (3.3 mmol, 1.0 equiv) in EtOH (15 mL) was added hydroxylamine hydrochloride (4.0 mmol, 1.2 equiv) and NaHCO3 (6.6 mmol, 2.0 equiv). The mixture was refluxed for 4 h. The reaction mixture was diluted with EtOAc, filtered, and concentrated in vacuo. Water was added to the residue and extracted with ethyl acetate (3 × 30 mL), washed with brine (1 × 30 mL), dried over anhydrous MgSO4, and concentrated. The residue was subjected to silica gel chromatography with EtOAc/MeOH (9:1) to make carboxamidine 7.
3.1.3. Synthesis of Carboxamidine 9
To a solution of 3-indoleacetic acid (3.6 mmol, 1.0 equiv) in CH2Cl2 (20 mL), DIPEA (4.7 mmol, 1.3 equiv) and HATU (3.6 mmol, 1.0 equiv) were added, and the reaction mixture was stirred for 30 min, then carboxamidine 7 (3.6 mmol, 1.0 equiv) dissolved in CH2Cl2 (10 mL) was added and stirred for 2 h. The mixture was filtered and the residue was washed with CH2Cl2, after which the solution was combined and concentrated. The residue was subjected to silica gel chromatography with petroleum ether/EtOAc (2:1) to give carboxamidine 9.
9a: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 8.13 (d, J = 7.15 Hz, 1H),7.70 (d, J = 7.78 Hz, 1H), 7.61 (d, J = 7.45 Hz, 1H), 7.50 (t, J = 7.66 Hz, 2H), 7.35 (d, J = 8.12 Hz, 1H), 7.27 (s, 1H), 7.11 (t, 1H), 7.03 (t, 1H), 3.73(s, 2H); 13C NMR (125 MHz, CD3OD): δ 166.42, 161.34, 138.25, 134.25, 130.70, 130.55, 129.64, 128.49, 125.03, 122.67, 120.01, 119.63, 112.29, 109.59, 28.25; HR-ESIMS: [M + H]+ calcd. for C17H16N3O2 294.1237, found: 294.1231.
9b: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 7.75 (m, 2H), 7.64 (d, 1H), 7.37 (d, 1H), 7.25 (s, 1H), 7.15 (m, 3H), 7.05 (t, 1H), 3.96(s, 2H); 13C NMR (125 MHz, CD3OD): δ 172.08, 164.41, 158.89, 137.69, 130.55, 130.46, 128.58, 127.79, 124.95, 122.60, 120.03, 119.51, 116.54, 116.32, 112.41, 108.40, 30.80; HR-ESIMS: [M + H]+ calcd. for C17H15FN3O2 312.1143, found: 312.1144.
9c: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 7.70 (d, J = 8.79 Hz, 2H), 7.62 (d, J = 6.72 Hz, 1H), 7.43 (d, J = 8.79 Hz, 2H), 7.36 (d, J = 8.10 Hz, 1H), 7.25 (s, 1H), 7.11 (t, 1H), 7.06 (t, 1H), 3.96 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 172.08, 158.85, 138.08, 137.85, 131.44, 129.81, 129.74, 129.57, 128.81, 128.61, 124.84, 122.58, 120.02, 119.42, 112.38, 108.37, 30.91; HR-ESIMS: [M − H]− calcd. for C17H13ClN3O2 326.0702, found: 326.0705.
9d: White solid, Yield 90%; 1H NMR (400 MHz, CD3OD): δ 8.28 (d, J = 9.07 Hz, 2H), 7.96 (d, J = 9.07 Hz, 2H), 7.64 (d, J = 7.92 Hz, 1H), 7.37 (d, J = 8.06 Hz, 1H), 7.26 (s, 1H), 7.12 (t, 1H), 7.06 (t, 1H), 3.98 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 172.00, 157.92, 150.69, 138.90, 138.09, 129.53, 124.87, 124.56, 122.60, 120.03, 119.41, 112.40, 108.29, 30.85; HR-ESIMS: [M − H]− calcd. for C17H13N4O4 337.0942, found: 337.0952.
9e: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 7.63 (m, 3H), 7.36 (d, 1H), 7.26 (m, 3H), 7.12 (t, 1H), 7.05 (t, 1H), 3.96 (s, 2H), 2.67 (m, 2H), 1.23 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 172.15, 159.95, 148.71, 138.09, 130.05, 129.03, 128.63, 128.21, 124.82, 122.58, 120.02, 119.43, 112.37, 108.46, 30.97, 29.68, 15.89; HR-ESIMS: [M + H]+ calcd. for C19H20N3O2 322.1550, found: 322.1543.
3.1.4. Synthesis of Compound 10
A solution of carboxamidine 9 (3.2 mmol, 1.0 equiv) and sodium acetate (6.4 mmol, 2.0 equiv) in 30% EtOH/H2O (10 mL) was refluxed overnight. The EtOH was removed in vacuo, and the residue was added to water and extracted with ethyl acetate (3 × 30 mL), then washed with brine (1 × 30 mL), dried over anhydrous MgSO4, and concentrated. The residue was subjected to silica gel chromatographic with petroleum ether/EtOAc (5:1) to make compound 10.
10a: White solid, Yield 94%; 1H NMR (400 MHz, CD3OD): δ 8.10 (d, J = 7.05 Hz, 1H), 7.63 (d, 2H), 7.56 (d, 2H), 7.35(d, J = 8.13 Hz, 1H), 7.23 (s, 1H), 7.10 (t, 1H), 7.01 (t, 1H), 4.27(s, 2H); 13C NMR (125 MHz, CD3OD): δ 177.04, 172.0, 138.17, 134.06, 130.36, 129.02, 128.41, 125.39, 124.55, 122.59, 119.91, 119.37, 112.33, 109.82, 23.39; HR-ESIMS: [M + H]+ calcd. for C17H14N3O 276.1131, found: 276.1125.
10b: White solid, Yield 92%; 1H NMR (400 MHz, CD3OD): δ 8.08 (d, 1H), 8.06 (d, 1H), 7. 58 (d, J = 7.92 Hz, 1H), 7.37 (d, J = 8.14 Hz, 1H), 7.28 (s, 1H), 7.23 (t, 2H), 7.12 (t, 1H), 7.03 (t, 1H),4.46(s, 2H); 13C NMR (125 MHz, CD3OD): δ 181.15, 168.75, 164.99, 138.12, 130.73, 130.66, 128.16, 124.80, 124.60, 122.79, 120.19, 119.16, 117.09, 116.91, 112.46, 108.06, 24.01; HR-ESIMS: [M − H]− calcd. for C17H11FN3O 292.0892, found: 292.0886.
10c: White solid, Yield 92%; 1H NMR (400 MHz, CD3OD): δ 8.01 (d, J = 8.75 Hz, 2H), 7.92 (d, J = 7.92 Hz, 1H), 7. 50 (d, J = 8.74 Hz, 2H), 7.37 (d, J = 8.14 Hz, 1H), 7.28 (s, 1H), 7.12 (t, 1H), 7.04 (t, 1H), 4.47 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 181.27, 168.77, 164.99, 138.34, 138.12, 130.27, 129.86, 128.16, 126.92, 124.79, 122.79, 120.19, 119.15, 112.46, 108.03, 24.02; HR-ESIMS: [M − H]− calcd. for C17H11ClN3O 308.0596, found: 308.0596.
10d: White solid, Yield 90%; 1H NMR (400 MHz, dimethyl sulfoxide (DMSO)-d6, not soluble in MeOH or CHCl3): δ 8.38 (d, J = 8.98 Hz, 2H), 8.24 (d, J = 8.98 Hz, 2H), 7.57 (d, J = 7.80 Hz, 1H), 7.43 (s, 1H), 7.39 (d, J = 8.11 Hz, 1H), 7.11 (t, 1H), 7.02 (t, 1H), 4.56 (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ 180.31, 166.43, 149.13, 136.17, 132.05, 128.39, 126.61, 124.49, 124.44, 121.37, 118.86, 118.19, 111.63, 106.21, 22.76; HR-ESIMS: [M − H]− calcd. for C17H11N4O3 319.0837, found: 319.0835.
10e: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 8.55 (t, 2H), 8.20 (t, 1H), 7.94 (m, 4H), 7.73 (t, 1H), 7.66 (t, 1H), 5.07 (m, 2H), 3.93 (s, 2H), 1.87 (m, 3H); 13C NMR (125 MHz, CD3OD): δ 180.87, 169.54, 149.26, 138.12, 129.47, 128.40, 128.17, 125.52, 124.78, 122.78, 120.18, 118.17, 112.45, 108.11, 29.79, 24.03, 15.82; HR-ESIMS: [M + H]+ calcd. for C19H18N3O 304.1444, found: 304.1450.
3.1.5. Synthesis of Carboxamidine 12
To a solution of compound 11 (64.1 mmol, 1.0 equiv) in EtOH (60 mL) was added hydroxylamine hydrochloride (96.2 mmol, 1.5 equiv) and NaHCO3 (192.3 mmol, 3.0 equiv). The mixture was stirred for 4 h at 65 °C. The reaction mixture was diluted with EtOAc, filtered and concentrated in vacuo. Water was added to the residue and extracted with ethyl acetate (3 × 100 mL), washed with brine (1 × 100 mL), dried over anhydrous MgSO4, and concentrated. The residue was subjected to silica gel chromatography with EtOAc /MeOH (9:1) to make carboxamidine 12.
3.1.6. Synthesis of Carboxamidine 13
To a solution of carboxylic acid (0.8 mmol, 1.0 equiv) in CH2Cl2 (5 mL), DIPEA (1.0 mmol, 1.3 equiv) and HATU (0.8 mmol, 1.0 equiv) were added, and the reaction mixture was stirred for 30 min, after which compound 7 (0.8 mmol, 1.0 equiv) in CH2Cl2 (2 mL) was added and stirred for 2 h. The mixture was filtered, the residue was washed with CH2Cl2, and then the solution was combined and concentrated. The residue was subjected to silica gel chromatography with petroleum ether/EtOAc (2:1) to give 13.
13a: White solid, Yield 88%; 1H NMR (400 MHz, CD3OD): δ 8.04 (d, 2H), 7.70 (d, 1H), 7.34 (m, 3H), 7.27 (s, 1H), 7.11 (t, 1H), 7.03 (t, 1H), 3.72 (s, 2H), 2.72 (q, 2H), 1.25 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 166.44, 161.22, 151.46, 138.26, 130.74, 129.13, 125.01, 122.66, 120.01, 119.66, 112.28, 109.65, 29.89, 28.26, 15.73; HR-ESIMS: [M + H]+ calcd. for C19H20N3O2 322.1550, found: 322.1555.
13b: White solid, Yield 82%; 1H NMR (400 MHz, CD3OD): δ 8.03 (d, 2H), 7.70 (d, 1H), 7.35 (d, 1H), 7.31 (d, 2H), 7.26 (s, 1H), 7.11 (t, 1H), 7.02 (t, 1H), 3.72 (s, 2H), 2.69 (t, 2H), 1.61 (m, 2H), 1.37 (m, 2H), 0.94 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 166.45, 161.22, 150.08, 138.25, 130.64, 129.69, 125.01, 122.66, 120.01, 119.65, 112.28, 109.64, 36.62, 34.54, 28.26, 23.32, 14.20; HR-ESIMS: [M + H]+ calcd. for C21H24N3O2 350.1863, found: 350.1870.
13c: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 8.03 (d, 2H), 7.70 (d, 1H), 7.35 (d, 1H), 7.30 (d, 2H), 7.26 (s, 1H), 7.11 (t, 1H), 7.02 (t, 1H), 3.72 (s, 2H), 2.68 (t, 2H), 1.64 (m, 2H), 1.32 (m, 6H), 0.89 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 166.46, 161.22, 150.10, 138.25, 130.64, 129.69, 125.01, 122.66, 120.01, 119.65, 112.28, 109.64, 36.92, 32.81, 32.31, 29.99, 28.26, 23.63, 14.37; HR-ESIMS: [M + H]+ calcd. for C23H28N3O2 378.2176, found: 378.2182.
13d: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 8.01 (t, 1H), 7.69 (d, 1H), 7.61 (m, 1H), 7.35 (d, 1H), 7.29 (t, 2H), 7.26 (s, 1H), 7.22 (t, 1H), 7.11 (t, 1H), 7.04 (t, 1H), 3.72 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 164.04, 164.01, 161.96, 161.55, 138.23, 136.05, 135.98, 133.15, 128.46, 125.57, 125.54, 125.03, 122.67, 120.02, 119.60, 119.11, 119.03, 118.03, 117.85, 112.30, 109.45, 28.02; HR-ESIMS: [M + H]+ calcd. for C17H15FN3O2 312.1143, found: 312.1144.
13e: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 8.32 (d, J = 8.14 Hz, 2H), 7.81 (d, J = 8.28 Hz, 2H), 7.70 (d, J = 7.91 Hz,1H), 7.35 (d, J = 8.14 Hz, 1H), 7.27 (s, 1H), 7.11 (t, 1H), 7.02 (t, 1H), 3.73 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 165.01, 161.67, 138.26, 135.23, 134.42, 131.27, 131.17, 128.48, 126.60, 126.57, 125.03, 122.67, 120.01, 119.65, 112.29, 109.59, 28.25; HR-ESIMS: [M + H]+ calcd. for C18H15F3N3O2 362.1111, found: 362.1112.
13f: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 8.08 (d, J = 8.97 Hz, 2H), 7.70 (d, J = 7.92 Hz, 1H), 7.35 (d, J = 8.12 Hz,1H), 7.26 (s, 1H), 7.11 (t, J = 7.56 Hz, 1H), 7.04 (s, 1H), 6.99 (t, J = 9.00 Hz, 1H), 3.85 (s, 2H), 3.71 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 166.23, 165.19, 161.09, 138.25, 132.63, 128.50, 125.00, 122.78, 122.66, 120.00, 119.66, 114.88, 112.28, 109.66, 55.99, 28.27; HR-ESIMS: [M + H]+ calcd. for C18H18N3O3 324.1343, found: 324.1347.
13g: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 7.69 (d, 1H), 7.56 (d, 1H), 7.35 (d, 1H), 7.33 (d, 1H), 7.26 (s, 1H), 7.11 (t, 1H), 7.01 (m, 2H), 3.81 (s, 3H), 3.71 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 166.11, 161.55, 160.37, 138.24, 135.87, 134.49, 128.47, 125.04, 122.67, 120.03, 119.80, 119.63, 117.40, 112.29, 111.77 109.46, 56.24, 28.05; HR-ESIMS: [M + H]+ calcd. for C18H17BrN3O3 402.0448, found: 402.0445.
13h: White solid, Yield 85%; 1H NMR (400 MHz, CD3OD): δ 7.64 (d, 1H), 7.34 (d, 1H), 7.21 (s, 1H), 7.10 (t, 1H), 7.01 (t, 1H), 3.63 (s, 2H), 2.43 (t, 2H), 1.65 (m, 2H), 1.40 (m, 2H), 0.94 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 173.57, 160.85, 138.21, 128.45, 124.93, 122.62, 119.93, 119.63, 112.25, 109.63, 33.38, 28.23, 28.12, 23.30, 14.07; HR-ESIMS: [M + H]+ calcd. for C15H20N3O2 274.1550, found: 274.1553.
13i: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 7.64 (d, 1H), 7.34 (d, 1H), 7.22 (s, 1H), 7.10 (t, 1H), 7.00 (t, 1H), 3.63 (s, 2H), 3.47 (t, 2H), 2.47 (t, 2H), 1.91 (m, 2H), 1.82 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 173.07, 160.95, 138.21, 128.44, 124.93, 122.62, 119.94, 119.63, 112.25, 109.63, 33.72, 33.23, 32.63, 28.12, 24.65; HR-ESIMS: [M + H]+ calcd. for C15H19BrN3O2 352.0655, found: 352.0663.
3.1.7. Synthesis of Compound 14
A solution of compound 9 (0.4 mmol, 1.0 equiv) and sodium acetate (0.8 mmol, 2.0 equiv) in 30% EtOH/H2O (5 mL), was refluxed overnight. The EtOH was removed in vacuo, the residue was added with water and extracted with ethyl acetate (3 × 5 mL), washed with brine (1 × 5 mL), dried over anhydrous MgSO4, and concentrated. The residue was subjected to silica gel chromatography with petroleum ether/EtOAc (5:1) to give compound 14.
14a: White solid, Yield 84%; 1H NMR (400 MHz, CD3OD): δ 7.99 (d, 2H), 7.59 (d, 1H), 7.37 (d, 2H), 7.34 (d, 1H), 7.22 (s, 1H), 7.09 (t, 1H), 7.01 (t, 1H), 4.24 (s, 2H), 2.71 (q, 2H), 1.25 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 177.14, 171.85, 151.36, 138.15, 129.83, 129.13, 124.53, 122.57, 119.90, 119.37, 112.32, 109.84, 29.89, 23.38, 16.63; HR-ESI: [M + H]+ calcd. for C19H18N3O 304.1444, found: 304.1447.
14b: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 7.98 (d, 2H), 7.59 (d, 1H), 7.34 (m, 3H), 7.22 (s, 1H), 7.09 (t, 1H), 7.02 (t, 1H), 4.25 (s, 2H), 2.68 (t, 2H), 1.61 (m, 2H), 1.36 (m, 2H), 0.94 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 177.15, 171.85, 150.01, 138.15, 130.39, 129.05, 124.53, 122.58, 119.90, 119.37, 112.32, 109.84, 36.62, 34.47, 23.39, 23.32, 14.19; HR-ESI: [M + H]+ calcd. for C21H22N3O 332.1757, found: 332.1754.
14c: White solid, Yield 86%; 1H NMR (400 MHz, CD3OD): δ 7.97 (d, 2H), 7.59 (d, 1H), 7.34 (m, 3H), 7.21 (s, 1H), 7.09 (t, 1H), 7.00 (t, 1H), 4.24 (s, 2H), 2.65 (t, 2H), 1.61 (m, 2H), 1.31 (m, 6H), 0.88 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 177.13, 171.83, 150.00, 138.14, 130.36, 129.03, 124.53, 122.57, 119.90, 119.37, 112.32, 109.84, 36.91, 32.79, 32.23, 29.98, 23.61, 23.29, 14.37; HR-ESI: [M + H]+ calcd. for C23H26N3O 360.2070, found: 360.2073.
14d: White solid, Yield 82%; 1H NMR (400 MHz, CD3OD): δ 8.09 (t, 1H), 7.63 (m, 2H), 7.35 (m, 3H), 7.23 (s, 1H), 7.09 (t, 1H), 7.01 (t, 1H), 4.29 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 174.00, 171.73, 163.05, 160.99, 138.14, 136.20, 136.13, 131.93, 128.39, 126.10, 126.08, 124.57, 122.58, 119.91, 119.39, 118.19, 118.02, 113.76, 112.32, 109.79, 23.32; HR-ESI: [M + H]+ calcd. for C17H13FN3O 294.1037, found: 294.1035.
14e: White solid, Yield 91%; 1H NMR (400 MHz, CD3OD): δ 8.27 (d, J = 8.16 Hz, 2H), 7.86 (d, J = 8.27 Hz, 2H), 7.60 (s, 2H), 7.34 (d, J = 8.13 Hz, 1H), 7.23 (s, 1H), 7.09 (t, J = 7.58 Hz, 1H), 7.01 (t, J = 7.50 Hz, 1H), 4.28 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 175.67, 172.34, 138.15, 135.23, 134.97, 129.72, 128.88, 128.38, 127.32, 127.29, 126.17, 124.59, 124.00, 122.60, 119.93, 119.36, 112.34, 109.72, 23.36; HR-ESIMS: [M + H]+ calcd. for C18H13F3N3O 344.1005, found: 344.1010.
14f: White solid, Yield 90%; 1H NMR (400 MHz, CD3OD): δ 8.04 (d, 2H), 7.59 (d, 1H), 7.34 (d, 1H), 7.22 (s, 1H), 7.09 (m, 2H), 7.01 (t, 2H), 4.23 (s, 2H), 3.88 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 176.99, 171.76, 165.01, 138.17, 131.00, 128.42, 124.53, 122.57, 119.90, 119.37, 117.66, 115.75, 112.32, 109.88, 56.10, 23.38; HR-ESI: [M + H]+ calcd. for C18H16N3O2 306.1237, found: 306.1234.
14g: White solid, Yield 90%; 1H NMR (400 MHz, CD3Cl, in methanol dissolved is not good): δ 7.76 (d, 1H), 7.60 (d, 1H), 7.46 (d, 1H), 7.36 (m, 1H), 7.21 (m, 2H), 7.14 (t, 1H), 6.94 (dd, 1H), 4.34 (s, 2H), 3.82 (s, 3H); 13C NMR (125 MHz, CD3Cl): δ 174.77, 170.16, 158.86, 136.37, 135.62, 127.25, 126.40, 123.14, 122.46, 120.00, 119.84, 119.23, 116.71, 112.51, 111.31, 110.04, 55.89, 22.91; HR-ESI: [M + H]+ calcd. for C18H15BrN3O2 384.0342, found: 384.0338.
14h: White solid, Yield 80%; 1H NMR (400 MHz, CD3OD): δ 7.51 (d, 1H), 7.33 (d, 1H), 7.16 (s, 1H), 7.08 (t, 1H), 6.98 (t, 1H), 4.15 (s, 2H), 2.84 (t, 2H), 1.72 (m, 2H), 1.35 (m, 2H), 0.91 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 181.73, 171.02, 138.12, 128.32, 124.46, 122.54, 119.85, 119.30, 112.29, 109.79, 29.59, 26.83, 23.18, 23.06, 13.80; HR-ESI: [M + H]+ calcd. for C15H18N3O 256.1372, found: 256.1377.
14i: White solid, Yield 65%; 1H NMR (400 MHz, CD3OD): δ 7.51 (d, 1H), 7.33 (d, 1H), 7.17 (s, 1H), 7.09 (t, 1H), 6.98 (t, 1H), 4.17 (s, 2H), 4.06 (t, 2H), 2.91 (t, 2H), 2.00 (s, 3H), 1.85 (m, 2H), 1.69 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 181.33, 172.92, 171.12, 138.14, 128.34, 124.48, 122.55, 119.86, 119.32, 112.30, 109.79, 64.90, 28.92, 26.70, 24.09, 23.20, 20.74; HR-ESI: [M + H]+ calcd. for C17H20N3O3 314.1499, found: 314.1500.
14j: White solid, Yield 70%; 1H NMR (400 MHz, CD3OD): δ 7.53 (d, 1H), 7.33 (d, 1H), 7.16 (s, 1H), 7.08 (t, 1H), 6.99 (t, 1H), 4.15 (s, 2H), 3.17 (m, 1H), 1.32 (d, 6H), 1.35 (m, 2H), 0.91 (t, 3H); 13C NMR (125 MHz, CD3OD): δ 185.49, 170.96, 138.10, 128.33, 124.47, 122.54, 119.86, 119.31, 112.29, 109.77, 28.54, 23.22, 20.32; HR-ESI: [M + H]+ calcd. for C14H16N3O 242.1215, found: 242.1212.
14k: White solid, Yield 65%; 1H NMR (400 MHz, CD3OD): δ 7.55 (d, 1H), 7.35 (m, 2H), 7.19 (s, 1H), 7.10 (t, 1H), 7.00 (t, 1H), 4.24 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 175.37, 172.18, 138.08, 128.24, 124.62, 122.62, 119.96, 119.26, 112.34, 109.19, 59.86, 23.21; HR-ESI: [M + H]+ calcd. for C12H10Cl2N3O 282.0123, found: 282.0127.
14p: White solid, Yield 65%; 1H NMR (400 MHz, CD3OD): δ 7.50 (d, 1H), 7.45 (d, 1H), 7.33 (dd, 2H), 7.17 (s, 1H), 7.13 (s, 1H), 7.09 (m, 2H), 6.97 (t, tH), 4.32 (s, 2H), 4.14 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 178.67, 169.88, 136.35, 136.24, 123.12, 122.56, 122.35, 120.02, 119.76, 119.76, 119.06, 118.76, 111.42, 111.32, 109.82, 107.95, 23.47, 22.78; HR-ESI: [M + H]+ calcd. for C20H17N4O 329.1397, found: 329.1388.
14q: White solid, Yield 74%; 1H NMR (400 MHz, CD3OD): δ 7.50 (d, 1H), 7.29 (m, 2H), 7.20 (s, 1H), 7.16 (d, 1H), 7.12 (s, 1H), 7.07 (t, 1H), 6.97 (t, 1H), 6.87 (t, 1H) 4.25 (s, 2H), 4.13 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 180.53, 171.21, 159.80, 158.26, 138.07, 134.57, 128.43, 128.36, 128.29, 126.73, 124.46, 122.54, 119.89, 119.31, 113.29, 113.22, 112.27, 110.98, 110.80, 109.74, 108.29, 108.25, 103.96, 103.80, 23.74, 23.17; HR-ESIMS: [M − H]− calcd. for C20H14FN4O 345.1157, found: 345.1161.
14r: White solid, Yield 78%; 1H NMR (400 MHz, CD3OD): δ 7.51 (m, 2H), 7.31 (m, 2H), 7.24 (s, 1H), 7.14 (s, 1H), 7.08 (m, 2H), 6.97 (t, 1H), 4.31 (s, 2H), 4.16 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 180.47, 171.27, 138.11, 136.45, 129.22, 128.33, 126.51, 125.99, 124.46, 122.94, 122.54, 119.90, 119.31, 118.69, 113.67, 112.28, 109.77, 108.00, 23.69, 23.19; HR-ESIMS: [M − H]− calcd. for C20H14ClN4O 361.0862, found: 361.0858.
14s: White solid, Yield 82%; 1H NMR (400 MHz, CD3OD): δ 7.66 (d, 1H), 7.51 (d, 1H), 7.32 (d, 1H), 7.20 (m, 2H), 7.14 (s, 1H), 7.08 (t, 1H), 6.98 (t, 1H), 4.30 (s, 2H), 4.15 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 180.44, 171.26, 138.11, 136.69, 129.88, 128.33, 126.35, 125.54, 124.46, 122.54, 121.84, 119.91, 119.31, 114.10, 113.38, 112.28, 109.76, 107.90, 23.67, 23.18; HR-ESIMS: [M − H]− calcd. for C20H14BrN4O 405.0356, found: 405.0356.
14t: White solid, Yield 88%; 1H NMR (400 MHz, CD3OD): δ 7.51 (m, 2H), 7.31 (m, 2H), 7.24 (s, 1H), 7.14 (s, 1H), 7.08 (m, 2H), 6.97 (t, 1H), 4.31 (s, 2H), 4.16 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 180.47, 171.27, 138.11, 136.45, 129.22, 128.33, 126.51, 125.99, 124.46, 122.94, 122.54, 119.90, 119.31, 118.69, 113.67, 112.28, 109.77, 108.00, 23.69, 23.19; HR-ESIMS: [M − H]− calcd. for C20H14ClN4O 361.0862, found: 361.0870.
14u: White solid, Yield 82%; 1H NMR (400 MHz, CD3OD): δ 7.49 (d, 1H), 7.29 (t, 2H), 7.21 (s, 1H), 7.10 (s, 1H), 7.08 (t, 1H), 7.03 (t, 1H), 6.96 (m, 2H), 4.54 (s, 2H), 4.13 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 181.46, 171.12, 139.70, 138.08, 123.37, 122.51, 120.86, 119.85, 119.33, 112.24, 111.62, 109.78, 107.89, 25.42, 23.21; HR-ESIMS: [M − H]− calcd. for C20H14ClN4O 361.0862, found: 361.0864.
3.1.8. Synthesis of Compound 14l−14o
To a solution of acyl chloride (2.7 mmol, 1.0 equiv) and potassium carbonate (3.2 mmol, 1.2 equiv) in toluene (10 mL), compound 12 (2.7 mmol, 1.0 equiv) was added and under reflux for 4h. The mixture was filtered, and the residue was washed with CH2Cl2; then, the solution was combined and concentrated. The residue was subjected to silica gel chromatography with petroleum ether/EtOAc (5:1) to give compounds 14l−14o.
14l: White solid, Yield 60%; 1H NMR (400 MHz, CD3OD): δ 7.53 (d, 1H), 7.33 (d, 1H), 7.17 (s, 1H), 7.09 (t, 1H), 6.98 (t, 1H), 4.16 (s, 2H), 3.75 (m, 1H), 2.39 (m, 4H), 2.08 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 183.61, 171.03, 138.09, 128.33, 124.48, 122.54, 119.86, 119.30, 112.29, 109.77, 32.52, 27.98, 23.22, 19.57; HR-ESIMS: [M + H]+ calcd. for C15H16N3O 254.1288, found: 254.1285.
14m: White solid, Yield 65%; 1H NMR (400 MHz, CD3OD): δ 7.53 (d, 1H), 7.33 (d, 1H), 7.17 (s, 1H), 7.09 (t, 1H), 6.98 (t, 1H), 4.16 (s, 2H), 3.75 (m, 1H), 2.39 (m, 4H), 2.08 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 183.61, 171.03, 138.09, 128.33, 124.48, 122.54, 119.86, 119.30, 112.29, 109.77, 32.52, 27.98, 23.22, 19.57; HR-ESIMS: [M + H]+ calcd. for C16H18N3O 268.1382, found: 268.1391.
14n: White solid, Yield 60%; 1H NMR (400 MHz, CD3OD): δ 7.53 (d, 1H), 7.33 (d, 1H), 7.16 (s, 1H), 7.09 (t, 1H), 6.99 (t, 1H), 4.16 (s, 2H), 2.95 (m, 1H), 2.03 (m, 2H), 1.80 (m, 2H); 1.59 (m, 2H); 1.42 (m, 2H); 1.32 (m, 2H); 13C NMR (125 MHz, CD3OD): δ 184.49, 170.88, 138.12, 128.34, 124.46, 122.54, 119.85, 119.31, 112.29, 109.77, 37.42, 31.25, 30.76, 26.62, 23.34; HR-ESIMS: [M + H]+ calcd. for C17H20N3O 282.1601, found: 282.1603.
14o: White solid, Yield 63%; 1H NMR (400 MHz, CD3OD): δ 8.72 (d, 1H), 8.24 (d, J = 7.92 Hz, 1H), 8.03 (t, 1H), 7.60 (m, 2H), 7.34 (d, J = 8.13 Hz 1H), 7.24 (s, 1H), 7.09 (t, 1H), 7.02 (t, 1H), 4.31 (s, 2H); 13C NMR (125 MHz, CD3OD): δ 175.36, 172.34, 151.26, 144.35, 139.54, 138.14, 128.45, 125.58, 124.62, 122.59, 119.93, 119.33, 112.33, 109.70, 23.38; HR-ESIMS: [M + H]+ calcd. for C16H13N4O 277.1084, found: 277.1089.

 and
and 

{kind=link}
{kind=link}
{kind=link}
{kind=link}























