Four New C9 Metabolites from the Sponge-Associated Fungus Gliomastix sp. ZSDS1-F7-2
by
 ,
,
Jun Zhang
1,3,* ,
,
Zhiqiang Yang
1,
Yan Liang
1,
Liping Zhong
1,
Huiting Lin
1,
Balian Zhong
1,
Liangchun Li
2,*,
Shihai Xu
3,* and
Yonghong Liu
4 1
National Engineering Research Center of Navel Orange, Gannan Normal University, Ganzhou 341000, China
2
School of Life Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, China
3
Department of Chemistry, Jinan University, Guangzhou 510000, China
4
Center for Marine Microbiology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou 510000, China
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2018, 16(7), 231; https://doi.org/10.3390/md16070231
Submission received: 26 May 2018
/
Revised: 6 July 2018
/
Accepted: 6 July 2018
/
Published: 9 July 2018
(This article belongs to the Special Issue Marine Natural Products from Symbiotic Ecosystems)
Abstract
:Four new structurally related metabolites, one γ-lactone named gliomasolide F (1), one δ-lactone named gliomasolide G (2), and two medium-chain fatty acids named gliomacids A–B (3–4), each containing nine carbons in total, were identified from the sponge-associated fungus Gliomastix sp. ZSDS1-F7-2. The planar chemical structures of these novel C9 metabolites were elucidated by nuclear magnetic resonance (NMR) spectroscopic methods, in connection with the analysis of high-resolution mass spectrometry (HRMS) and infrared (IR) data. The absolute configuration of 1, was determined by comparisons of experimental circular dichroism (CD) and optical rotation (OR) value with corresponding ones computed by quantum chemistry. The relative configuration of 2 was determined by the Nuclear Overhauser effect spectroscopy (NOESY) spectrum, while its absolute configuration was tentatively determined in view of the biogenetic and biosynthetic relationships between 1 and 2. Compounds 3–4, originally as an inseparable mixture, were successfully isolated after chemical modifications. The stereo-chemistries of compounds 3–4 were assumed by comparison of 13C NMR with those of the similar moiety reported in literature, in addition to the biogenetic and biosynthetic relationships with 1. The plausible biosynthetic relationships among these four C9 metabolites were supposed. Biologically, compounds 1–4 showed no cytotoxic effect against HeLa cell line at concentrations up to 25 μg/mL, while 1 exhibited moderate antifouling activity against the settlement of Balanus amphitrite larvae with IC50 being 12.8 μg/mL and LC50 > 25 μg/mL. The co-occurrence of macrolides gliomasolides A—E and four C9 metabolites in the same fermentation culture made us assume that these C9 metabolites might be biosynthetic building blocks toward the construction of more complex macrolides such as gliomasolides A—E or other unidentified polyketides.
1. Introduction
Marine sponge-associated microorganisms, known as one of the most prolific resources for the discovery of chemical and biological diversified secondary metabolites, have been and continue to be among the research interests of chemists and biologists worldwide [1,2,3,4,5,6,7]. During the past decades, a myriad of structurally unique and pharmacologically interesting drug leads have been identified from the microorganisms derived from varieties of marine sponges, involving diterpenes inhibiting the growth of a panel of cancer cell lines from a fungus Trichoderma harzianum OUPS-111D-4 separated from a marine sponge Halichondria okadai [8], antibactetial and antifungal isocoumarins from a culture of the fungus Pestalotiopsis heterocornis associated with sponge Phakellia fusca [9], cytotoxic lactones from the sponge-derived fungus Talaromyces rugulosus [10], and cyclopentenones with β-amyloid fibrillization inhibitory effects from a sponge-derived fungus Trichoderma sp. HPQJ-34 [11].
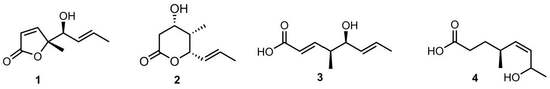
As part of our continuous effort to identify structurally and biologically interesting natural products from marine resources [12,13,14], the fungus Gliomastix sp. ZSDS1-F7-2 from the marine sponge Phakellia fusca Thiele, which was collected from the South China Sea in March 2012, has been investigated with the discovery of a series of unusual macrolides gliomasolides A—E with cytotoxic effect against the growth of HeLa cell line [15]. To further dig out bioactive metabolites from the remaining fractions of the same crude extract, we carried out systematically purification work, resulting in the discovery of four new metabolites (1–4), each having nine carbons in total, briefly named as C9 metabolites (Figure 1).
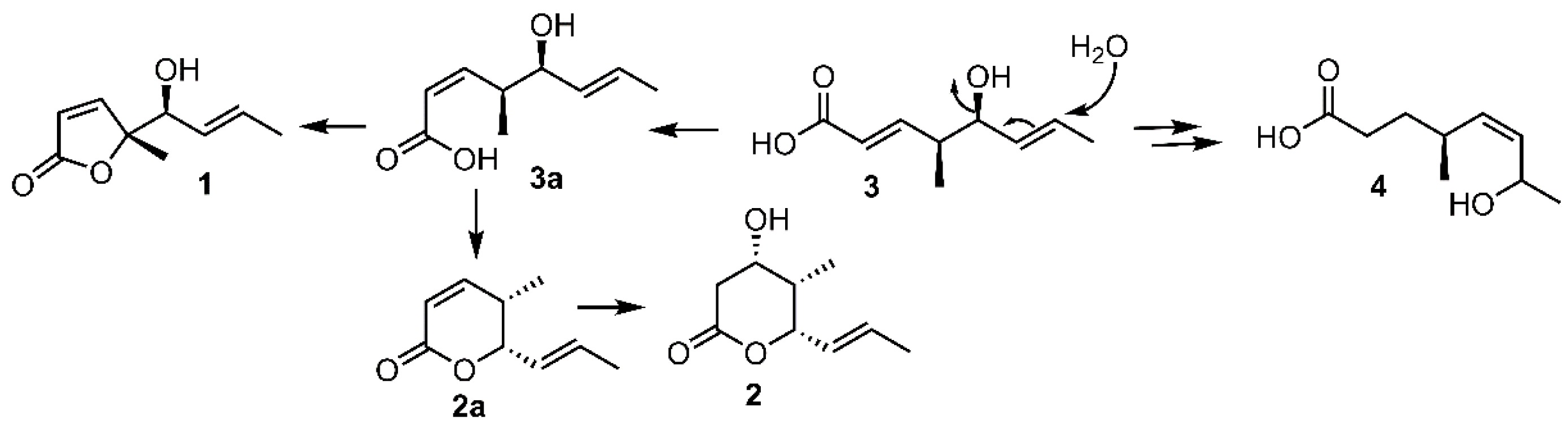
Interestingly, these four C9 metabolites represented three kinds of structure-related skeletons. Compound 1 was an unusual new γ-lactone which differed from other γ-lactones mainly by the presence of a methyl group in the γ position of the ester carbonyl. 2 represented a new δ-lactone, while compounds 3–4 were two new medium-chain fatty acids. In view of their structure similarity, a plausible biosynthetic relationship was assumed. As depicted in Scheme 1, compound 3 might shift the double bond and hydroxyl group by allylic alcohol rearrangement, followed by hydrogenation of the conjugated double bond to give 4. The intramolecular lactonisation of 3a which was formed by trans-cis isomerization of 3, at the γ position furnished 1, while cyclization of 3a at the δ position giving the δ-lactone 2a, which might undergo Michael addition by water to give 2. The co-occurrence of macrolides gliomasolides A—E and four C9 metabolites in the same fermentation culture made us assume that these C9 metabolites might be biosynthetic building blocks toward the construction of more complex macrolides such as gliomasolides A—E or other unidentified polyketides. This assumption was consistent with the widely accepted hypothesis of step by step functionalization of growing polyketide chains [16,17]. Biologically, these novel C9 metabolites exhibited no cytotoxic effect against HeLa cell line at concentrations up to 25 μg/mL, while 1 exhibited moderate antifouling activity against the settlement of Balanus amphitrite larvae with IC50 being 12.8 μg/mL and LC50 > 25 μg/mL. This paper reported the isolation, structural elucidation, biosynthetic relationship, and biological effects of four new C9 metabolites from the fungus Gliomastix sp. ZSDS1-F7-2.
2. Results and Discussion
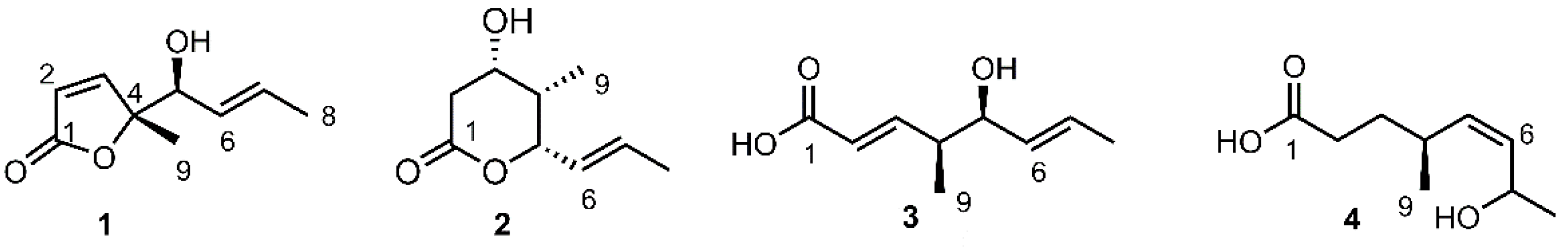
Compound 1 was obtained as a brown oil. The molecular formula of 1 was determined to be C9H12O3 by high-resolution time-of-flight mass spectrometry (HR-TOF-MS), displaying ion peak [M + H]+ at m/z 169.0872 (calcd. for C9H13O3+ at m/z 169.0865). The infrared (IR) spectrum of compound 1 showed absorption bands for hydroxyl group (3487 cm−1) and carbonyl group (1746 cm−1). Preliminary inspection of the 1H nuclear magnetic resonance (NMR) spectrum of 1 led to the clear identification of a singlet methyl group at δH 1.40 (3H, s, H-9), a doublet methyl at δH 1.70 (3H, d, J = 6.0 Hz, H-8) in the upfield area (Table 1). The presence of an oxygenated methine in 1, was clearly suggested by the chemical shifts of δH 4.14 (1H, d, J = 6.9 Hz, H-5) and the corresponding δC 75.8 in the 1H and 13C NMR, respectively. In the downfield region of the 1H NMR, the signals at δH 6.05 (1H, d, J = 5.4 Hz, H-2), 7.40 (1H, d, J = 5.4 Hz, H-3), 5.43 (1H, dd, J = 15.0, 6.9 Hz, H-6) and 5.78 (1H, dq, J = 15.0, 6.0 Hz, H-7), combined with the corresponding 13C NMR signals at δC 121.8 (C-2), 158.5 (C-3), 127.6 (C-6), 131.0 (C-7), supported the presence of two pairs of olefinic groups in 1. The linear fragment extending from C-5 to C-8, was established by the 1H-1H correlation spectroscopy (COSY) showing sequential correlations of between H-5 with H-6, H-6 with H-7, and H-7 with H-8, which was further supported by the correlations of from H-5 to C-7, H-8 to C-6/C-7 in heteronuclear multiple-bond correlation spectroscopy (HMBC) (Figure 2). The fragment extending from C-1 to C-4 in 1, was established by the HMBC correlations of from both H-2/H-3 to C-1/C-4, as shown in Figure 2. Whereas, the connection between C-1 with C-4 via an oxygen atom to form the butenolide moiety was deduced by the high-resolution mass spectrometry (HRMS) spectrum referring the molecular formula of C9H12O3 to 1, requiring the cyclization to satisfy the unsaturated degrees. This assignment was also in agreement with the IR absorbance of carbonyl carbon at 1746 cm−1 which was found at a much higher wavenumber than the α,β unsaturated acid (around 1700 cm−1). The arrangement of a singlet methyl group connected to the C-4, was determined by the correlations of from H-9 to C-4/C-3/C-5 in HMBC (Figure 2). Furthermore, the connection of the moiety extending from C-5 to C-8, and the butenolide moiety extending from C-1 to C-4, to form the complete planar structure of 1, as shown in Figure 1, was established by the HMBC correlations between H-5 and C-3/C-4/C-9, and between H-3 and C-4/C-5/C-9, respectively.
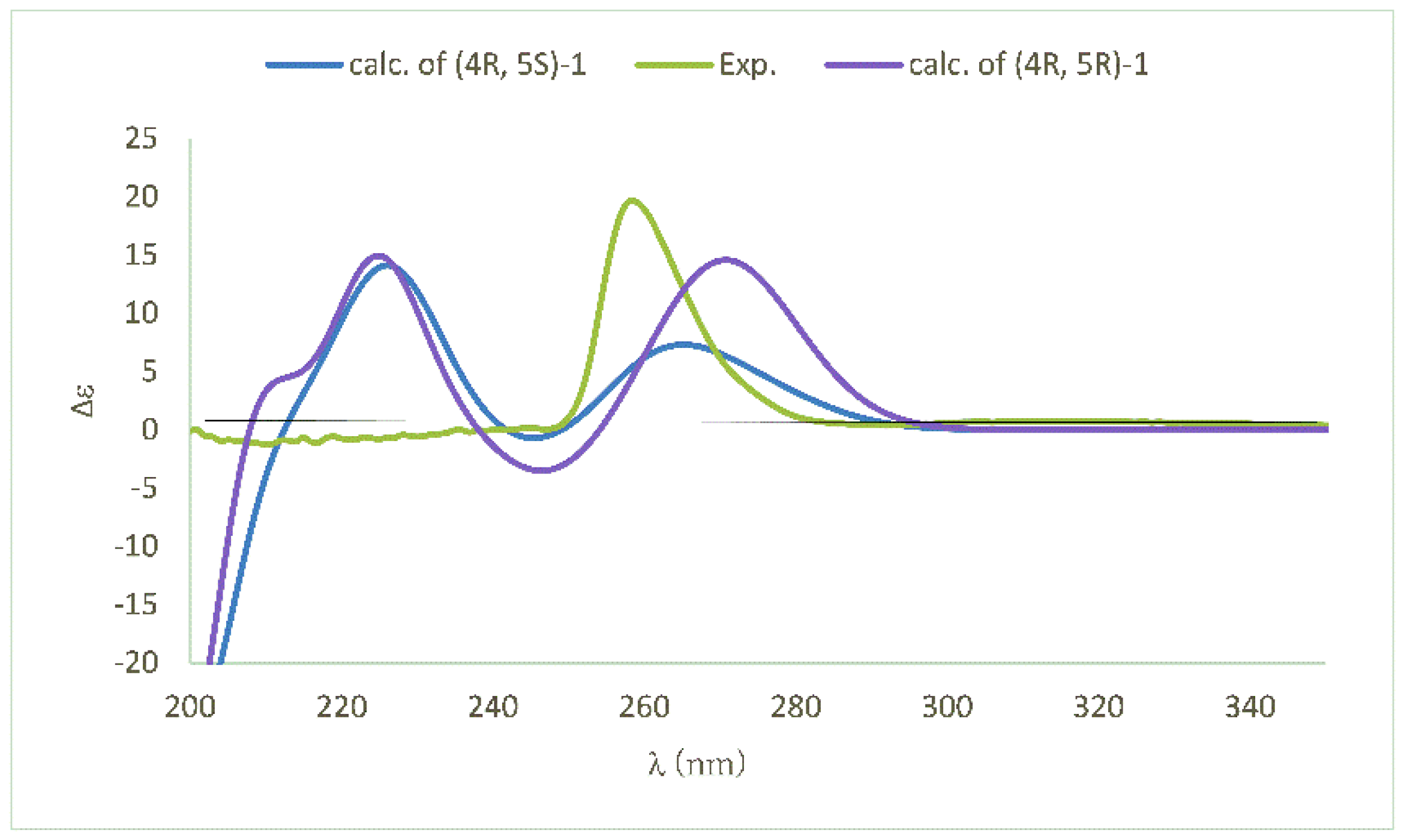
The stereochemistry of the double bond Δ6,7 was determined to be trans in view of the coupling constant of J6,7 = 15.0 Hz. To determine the stereo-orientation of the hydroxyl group at C-5, the Mosher ester method was employed. According to the procedure previously reported by our group, the (S)-MTPA (methoxy(trifluoromethyl)phenylacetic acid) ester was successfully obtained, unfortunately, in spite of modifying the reaction conditions several times, we failed to obtain the (R)-MTPA ester, which made our effort using the Mosher ester method in vain. Pleasingly, the absolute configurations of both chiral carbons C-4/C-5, were successfully determined by the quantum chemical method using TDDFT (time-dependent density functional theory) calculations. Theoretically, there are four configurations for the structure of 1, being (4R, 5S)-1, (4R, 5R)-1 and their enantiomers. The absolute configuration of 1 was determined by comparisons of the calculated CD (circular dichroism) and OR (optical rotation) value with those of the corresponding experimental data. The calculated CD spectra of both (4R, 5S)-1 and (4R, 5R)-1 were similar to the experimental spectrum of 1 (Figure 3), excluding the possibility of assigning their enantiomers to 1. Furthermore, the OR values of (4R, 5S)-1 and (4R, 5R)-1 were calculated at the B3LYP/6-311++g (2d, p) level using methanol as solvent. The computed OR values of (4R, 5S)-1 and (4R, 5R)-1 were +99.6 and +241.6, respectively, which supported the assignment of (4R, 5S) stereochemistry to 1 in view of the information that the computed OR value of (4R, 5S)-1 was more close to the experimental value (+88.3) than that of (4R, 5R)-1. Thus, based on both the electronic circular dichroism (ECD) and OR calculations, the structure with absolute configuration of 1 was determined to be (R)-5-((S,E)-1-hydroxybut-2-en-1-yl)-5-methylfuran-2(5H)-one, trivially named gliomasolide F.
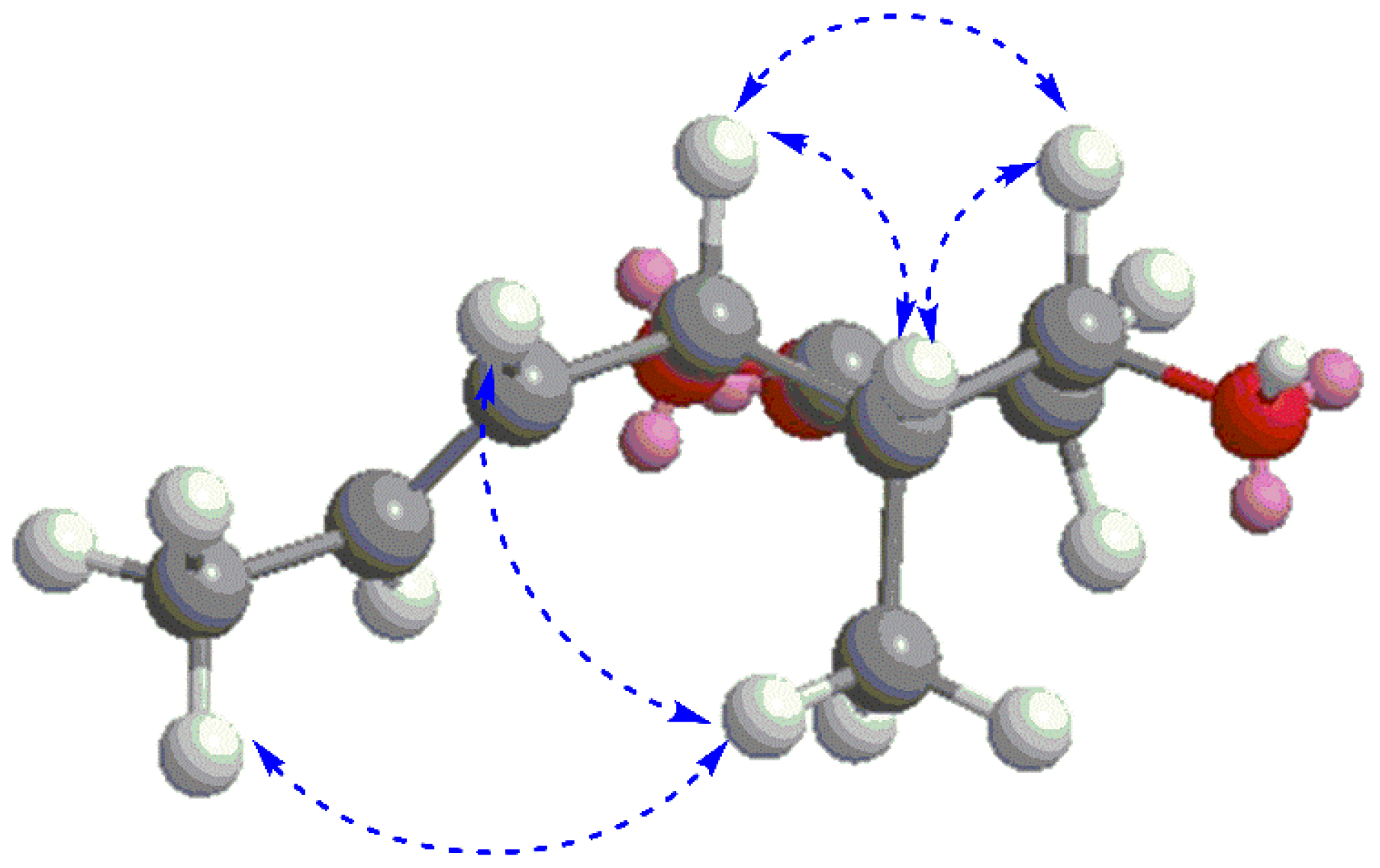
Compound 2 was obtained as a brown oil. Its molecular formula C9H14O3 was determined by HR-TOF-MS displaying ion peaks at m/z 171.1029 [M + H]+ (calcd. for C9H15O3+: 171.1021) and 153.0932 [M − H2O + H]+ (calcd. for C9H13O2+: 153.0916). The IR spectrum suggested the presence of hydroxyl (3503 cm−1) and conjugated ester groups (1718 cm−1). The NMR data of 2 was similar to that of prelactone C first isolated from the Streptomyces sp. [17], with the major difference arising from the signals of CH-3, CH-4, CH-5, and CH3-9, as indicated in Table S1. The planar structure of 2 was unambiguously determined to be the same as that of prelactone C based on the comprehensive elucidation of the 2D NMR involving 1H–1H COSY, heteronuclear single quantum correlation (HSQC) and HMBC. The relative stereochemistry of chiral carbons C-3/C-4/C-5 was established according to the nuclear Overhauser effect spectroscopy (NOESY) spectrum in which the correlations of H-3 with both H-4 and H-5, H-9 with both H-6 and H-8 were clearly identified (Figure 4), allowing the assignment of all-cis configurations for hydroxyl at C-3, methyl at C-4, and propenyl at C-5, as shown in Figure 1. The absolute configurations of 3S, 4S and 5S were tentatively assigned to 2 based on the biogenetic point of view, in combination with the plausible biosynthetic relationship between 1 and 2 (Scheme 1). Worthy of note, the carbon chemical shift of CH3-9 in 2 resonated at δC 5.9, significantly upfield shifted compared to that of its isomer prelactone C at δC 13.7 [17]. We assumed that, due to the hydroxyl, propenyl, and methyl groups all being in cis-relationship, they were close to each other in spatial distance, making both the hydroxyl and propenyl groups exhibit strong gamma gauche effect which was generally observed causing an upfield shift of the resonating carbon [18,19], resulting in significant shield effect on methyl. Therefore, the chemical structure of 2 was tentatively elucidated, trivially named gliomasolide G.
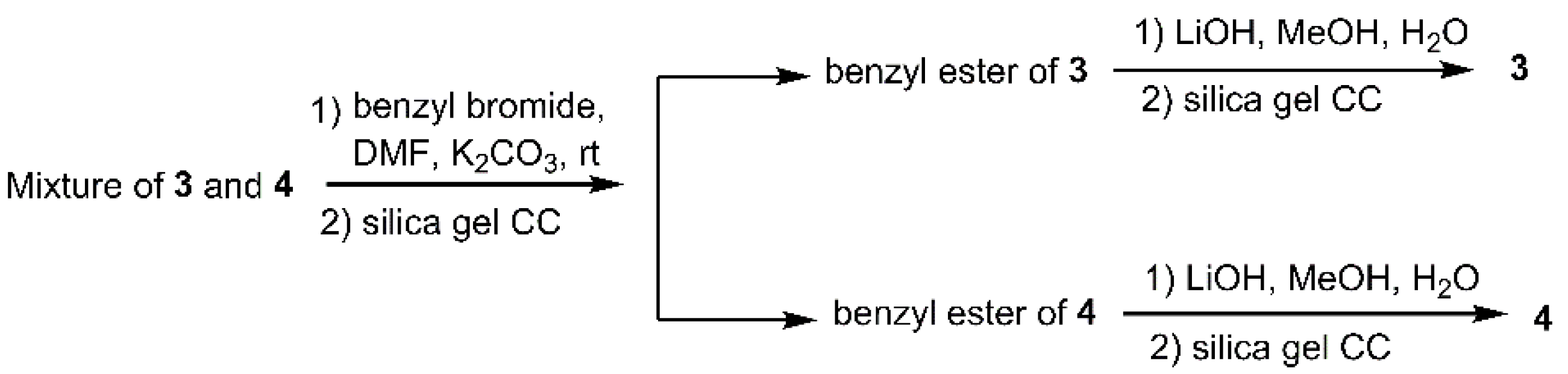
Based on NMR analysis, compounds 3 and 4 were found to be mixed in one thin-layer chromatography (TLC) fraction, with a rough 1:1 ratio, which were then subjected to prep-high-performance liquid chromatography (HPLC) for purification. However, in spite of trying varieties of HPLC conditions, these two compounds could not be separated well. After carefully examining the NMR spectra and the HRMS data, we suspected that this mixture should consist of a pair of medium-chain fatty acids, inspiring us to esterify them before processing separation. Fortunately, after esterification using benzyl bromide, their corresponding esters were successfully separated by silica gel column chromatography, which were then subjected to LiOH solution to remove the benzyl group, giving the pure compounds 3 and 4 (Scheme 2).
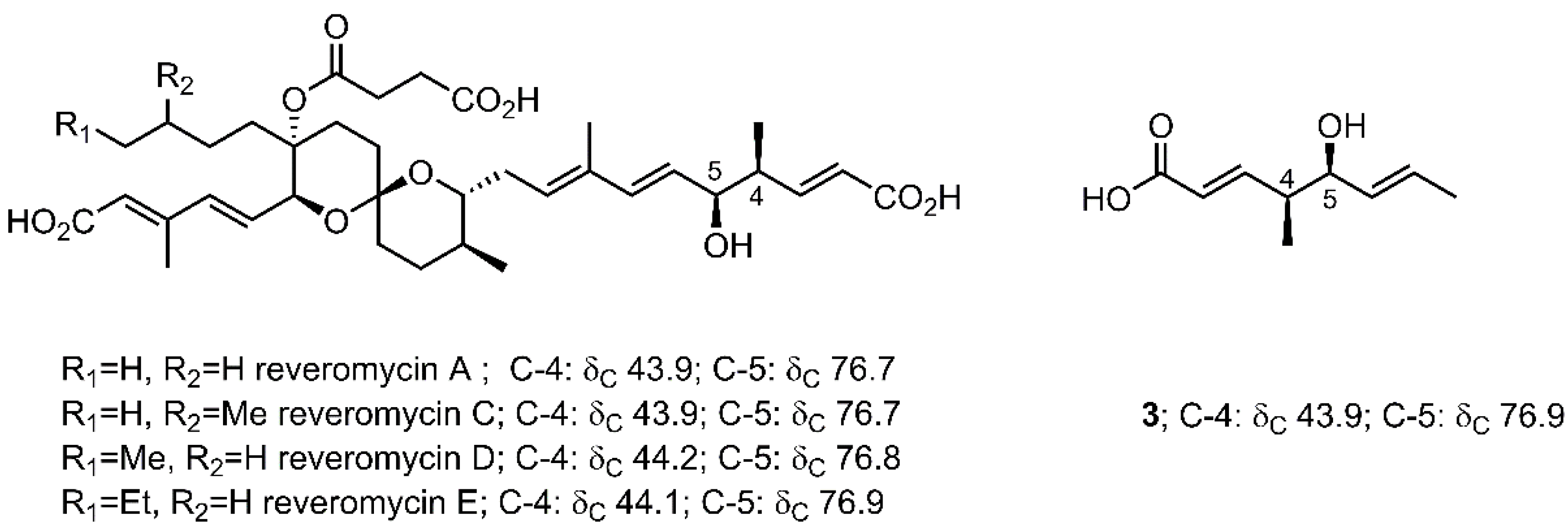
Compound 3 was obtained as a yellow oil, and its molecular formula C9H14O3 was established by the 1H, 13C NMR data, in combination with the HRMS displaying ion peaks at m/z 153.0924 [M − H2O + H]+ (calcd. for C9H13O2+: 153.0916). The IR displayed typical linear α,β-unsaturated carbonyl carbon absorption at 1697 cm−1, in agreement with the observation of chemical shift of δC 171.2 (C-1) in the 13C NMR spectrum. The 1H, 13C NMR data, indicated the presence of four olefinic protons and carbons, one oxygenated methine, and two doublet methyls (Table 2). The 1H–1H COSY correlations sequentially extending from H-2 to H-9, combined with the correlations of from H-2/H-3 with C-1, H-9 with C-3/C-5, H-5 with C-9/C-7, and H-8 with C-6/C-7 in HMBC, clearly constructed the planar structure of 3, shown in Figure 1. The geometric configurations of both the double bonds Δ2,3, Δ6,7 were determined to be trans by the coupling constants, being 15.0 Hz for both J2,3 and J6,7. The relative configurations, with hydroxyl and methyl CH3-9 being assigned as cis, was established by comparison of 13C NMR data between 3 and the similar moiety of reveromycins, a series of polyketide spiroketals from Streptomyces sp. [20]. The reveromycins contained a medium-chain acid, being very similar to 3, as its structure moiety. As shown in Figure 5, the carbon chemical shifts of C-4 and C-5 in all reveromycins resonated at ~44.0 and ~76.9 ppm in CD3OD, respectively, almost the same as those corresponding signals in 3 with C-4 and C-5 at δC 43.9 and 76.9, respectively. The absolute configurations 4S/5S, were tentatively assigned in view of the biogenetic and biosynthetic relationships between 1 and 3. Therefore, the chemical structure of compound 3 was achieved, being (2E,4S,5S,6E)-5-hydroxy-4-methylocta-2,6-dienoic acid, trivially named gliomacid A.
Compound 4, structurally similar to 3, has nine carbons in total, involving one carbonyl, two olefinic carbons and one oxygenated methine, as revealed by 13C NMR. The HRMS displayed ion peaks at m/z 155.1073 [M − H2O + H]+ (calcd. for C9H15O2+: 155.1072), giving a molecular formula C9H16O3 to 4. Corresponding to the 13C NMR and HRMS, there are two more protons in the 1H NMR compared with 3. According to the detailed 2D NMR analysis (Figure 2), the planar structure of 4 was established as shown in Figure 1. The stereochemistry of C-4 was assumed to be the same as that of 3 based on the biogenetic and biosynthetic viewpoint, whereas the stereochemistry of C-7 remained unknown currently. The cis geometric configuration of the double bond Δ5,6 was determined by the peak pattern of the olefinic protons H-5/H-6 which overlapped to form what looks like a broad singlet with coupling constant of J5,6 obviously less than 15 Hz. Compound 4, therefore, was elucidated to be (4S,Z)-7-hydroxy-4-methyloct-5-enoic acid, trivially named gliomacid B.
All the isolates were evaluated for cytotoxic activity against the proliferation of HeLa cell line and the antifouling effect against the settlement of Balanus amphitrite larvae. Unfortunately, none of the tested compounds 1–4 showed cytotoxicity at the concentrations up to 25 μg/mL. Pleasingly, compound 1 exhibited a non-toxic moderate inhibitory effect against the settlement of Balanus amphitrite larvae with IC50 being 12.8 μg/mL and LC50 > 25 μg/mL, whereas 2–4 displayed no anti-fouling effect at the concentrations up to 25 μg/mL.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotation was measured on a JASCO P-2000 digital polarimeter (Jasco, Tokyo, Japan). The ultraviolet (UV) spectrum was recorded on a Shimadzu UV2401PC spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were recorded on a Perkin-Elmer 16 PC Fourier transform–infrared (FT–IR) spectrometer (Perkin-Elmer, Boston, MA, USA). NMR spectra were recorded on Varian Inova-500 NMR spectrometer (Toso, Tokyo, Japan) and Varian Mercury VX 300 spectrometer (Varian, Japan) using TMS (Tetramethylsilane) as internal standard, the chemical shifts were reported in ppm relative to CD3OD (δH 3.31 and δC 49.00) or CDCl3 (δH 7.26 and δC 77.16). High-resolution mass spectra (HRMS) were performed on QSTAR XL mass spectrometry system (Applied Biosystems, Foster City, CA. USA). The preparative HPLC was performed on Waters systems (Waters, Manchester, UK) involving Waters 2545 binary gradient module, Waters 2996 PDA detector, MassLynx V4.1 workstation, and XBridge RP18 column (19 × 300 mm, 5 µm). Column chromatography (CC) was performed with silica gel (230–400 mesh, Merck, Darmstadt, Hessen, Germany) or octadecyl silane (ODS) (Fuji Silysia Chemical Ltd. Kasugai, Aichi, Japan). TLC was performed on aluminium sheet precoated with silica gel G60 (Merck, Darmstadt, Hessen, Germany). Spots were visualized under UV light or by spraying with 5% H2SO4 in EtOH followed by heating.
3.2. Fungal Material
The fungal strain, Gliomastix sp. ZSDS1-F7-2, was isolated from the sponge Phakellia fusca Thiele collected from the Xisha Islands of China in 2012. The sponge was identified by Prof. Jinhe Li at the Institute of Oceanology, Chinese Academy of Sciences. A voucher specimen of the fungus (No. 12071301) was deposited in the Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences. The fungus was identified using a molecular biological protocol by DNA amplification and sequencing of the 18S rRNA gene (GenBank Accession No. KR054959). By comparison with the GenBank database, the most similar strain was Gliomastix murorum YNS1116-4, with a sequence identity of 99% (95% query coverage). So the fungus strain ZSDS1-F7-2 belonged to the genus Gliomastix, and was designated as Gliomastix sp.
3.3. Fermentation, Extraction and Isolation
The strain Gliomastix sp. ZSDS1-F7-2 was cultured in 100 mL Erlenmeyer flasks each containing 25 mL of malt medium (20 g/L of malt extract (Oxoid), 1 g/L of peptone (BD Difco), and 30 g/L of sea salt (Anhui Hongsifang Co., Ltd., CNSG, Anhui, China), to give the seed culture (20 × 100 mL flasks), which was transferred to the 1000 mL Erlenmeyer flask containing solid rice medium (200 g of rice (North-eastern rice, COFCO), 200 g water, 6 g sea salt (Anhui Hongsifang Co., Ltd., CNSG, Anhui, China, 20 × 1 L flasks). After incubation at 25 °C under daylight for 2 months, the culture was broken up with a spatula and extracted with 95% EtOH (3 × 10 L). The combined extract was filtered and evaporated under vacuum to give 95% EtOH extract (105.1 g), which was suspended in water (1 L) and extracted successively with petroleum ether (1 L × 3), EtOAc (1 L × 3), and n-BuOH (1 L × 3). The EtOAc fraction (70 g) was subjected to a chromatography column (CC) on silica gel (400 g; 9.0 cm diam × 50 cm height), eluted with a step gradient of petroleum ether (PE) in EtOAc (10–100%), and then with MeOH–EtOAc (10–30%), to yield 10 fractions (Fr. A-J). Fr. B (5.0 g) was further subjected to octadecyl silane (ODS) CC (60 g; 3.0 cm diam. × 16 cm height), eluted with a gradient solution of methanol in water (10–100%) to furnish 10 fractions (Fr. B1-Fr. B10). Then, Fr. B4 (0.2 g) was purified by preparative high-performance liquid chromatography (HPLC) (25% acetonitrile–H2O, 15.0 mL/min) to yield compounds 1 (19.5 mg), 2 (5.3 mg). Fr. B6 (0.3 g) was purified by preparative HPLC (30% acetonitrile-H2O, 15.0 mL/min) to yield a mixture (18.9 mg) containing both compounds 3 and 4. To the solution of this mixture (15.0 mg) in dried DMF (1 mL), was added K2CO3 (40 mg), followed by benzyl bromide (16 μL), then this was stirred at room temperature for 8 h, then quenched with saturated NaHCO3 aqueous (2 mL), extracted with DCM (3 mL × 2), washed sequentially by brine (5 mL × 2) and water (5 mL); the residue obtained after evaporation was purified by silica gel CC, eluting with 15% EtOAc/PE, to give the benzyl ester of 3 (6.5 mg, 1H NMR recorded at 300 MHz in CDCl3: δH 1.06 (3H, d, J = 6.9), 1.70 (3H, d, J = 6.3), 2.52 (1H, m), 4.01 (1H, m), 5.18 (2H, s), 5.47 (1H, m), 5.70 (1H, m), 5.90 (1H, d, J = 15.9), 7.03 (1H, dd, J = 15.9, 6.9), 7.38 (5H, m)), and the benzyl ester of 4 (3.0 mg, 1H NMR recorded at 300 MHz in CDCl3: δH 0.99 (3H, d, J = 6.6), 1.24 (3H, d, J = 6.3), 1.66 (2H, m), 2.13 (1H, m), 2.34 (2H, t, J = 7.5), 4.23 (1H, m), 5.11 (2H, s), 5.46 (2H, m), 7.36 (5H, m)). The separated benzyl esters 3 (3.0 mg), was dissolved in methanol/water (1 mL/0.5 mL), followed by the addition of LiOH (2.1 mg), the mixture was stirred at room temperature for 4 h, then quenched with 1 M HCl aqueous (2 mL), extracted with DCM (3 mL × 2), washed with brine (3 mL × 2) and water (3 mL), and the combined DCM layer was evaporated to obtain the residue which was subjected to CC, eluted with 30% EtOAc/PE to give 3 (2.0 mg). The compound 4 (1.2 mg) was obtained from the separated benzyl esters 4 by the same procedure.
Gliomasolide F (1): brown oil; +88.1 (c = 5.53, CH3OH); IR (film) νmax 3487, 2938, 1746, 1450, 1377, 1111, 1027, 820 cm−1; UV (CH3OH) λmax (log ε) 210 (3.75) nm; CD λmax (Δε) 260 (18.9); 1H and 13C NMR data see Table 1; HR-TOF-MS m/z 169.0872 [M + H]+ (calcd. for C9H13O3+: 169.0865).
Gliomasolide G (2): brown oil; −38.7 (c = 0.87, CH3OH); IR (film) νmax: 3503, 2966, 1718, 1459, 1249, 1051, 966, 844 cm−1; UV (CH3OH) λmax (log ε): 244 (3.32) nm; 1H and 13C NMR data see Table 1; HR-TOF-MS m/z 171.1029 [M + H]+ (calcd. for C9H15O3+: 171.1021), 153.0932 [M − H2O + H]+ (calcd. for C9H13O2+: 153.0916).
Gliomacid A (3): brown oil; −409.7 (c 0.08, MeOH); IR (film) νmax 3394, 2917, 1697, 1652, 1398, 967 cm–1; UV (CH3OH) λmax(log ε) 232 (3.86) nm; 1H and 13C NMR data see Table 2; HR-TOF-MS m/z 153.0924 [M − H2O + H]+ (calcd. for C9H13O2+: 153.0916).
Gliomacid B (4): brown oil; +76.8 (c 0.09, MeOH); IR (film) νmax 3382, 2923, 1707, 1651, 1379, 1059 cm–1; UV (CH3OH) λmax(log ε) 228 (2.95) nm; 1H and 13C NMR data see Table 2; HR-TOF-MS m/z 155.1073 [M − H2O + H]+ (calcd. for C9H15O2+: 155.1072).
3.4. Computation Section
Conformational studies were performed using the Amber and MMFF94S force field, respectively. The geometries with relative energy less than 5.5 kcal/mol were selected and calculated at B3LYP/6-31G(d)//B3LYP/3-21G(d) level to obtain global minima. Conformers within an energy range of 3 kcal/mol from the global minima were subjected to geometrical optimization (DFT/B3LYP/6-31G(d)) in the gas phase, combined with calculation of vibrational modes to confirm these minima. No imaginary frequencies were found. After conformational search, 10 conformations for (4R, 5S)-1, and 8 conformations for (4R, 5R)-1 with low energy were found. For these conformations, ECD spectra were calculated at the B3LYP/6-311+G(d,p) level in MeOH (SCRF/IEFPCM). OR values were calculated at the B3LYP/6-311++G(2d,p) level. Each calculated ECD spectra and OR values was assigned a Boltzmann weight according to the energy of the minimized conformers at 298.15 K and overlaid prior to comparison with the experimental results. All of the DFT (density functional theory) calculations reported in this study were performed with the Gaussian 09 package [21].
3.5. Cytotoxicity Test
The cytotoxicity against HeLa cervical cancer cell line was determined using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] method. Briefly, the HeLa cells were cultured in Dulbecco’s modified Eagle's medium (DEME) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FCS (fetal calf serum) (Invitrogen, Carlsbad, CA, USA) in a 96-well plate at a density of 1 × 104 cells per well and incubated for 20 hours, then the cells were treated with testing samples at various concentrations and then incubated at 37 °C for 48 hours in a 5% CO2 atmosphere. The supernatant was removed, followed by the addition of 25 μL of MTT (2.5 mg mL−1 in PBS (phosphate buffered saline)) to each well. After incubation at 37 °C for 4 h, 100 μL of dimethyl sulfoxide (DMSO) was added to each well and incubated for another 20 min. The absorbance of each well was then measured at 570 nm using a Thermo scientific Multiskan FC multiplate photometer (Waltham, MA, USA).
3.6. Antifouling Effect
The antifouling assay was evaluated according to the procedure reported previously [13]. Briefly, fresh cyprids of the barnacle Balanus amphitrite were used in the testing. Larval settlement assays were examined using 24-well polystyrene plates with Sea-Nine 211TM (4,5-dichloro-2-n-octyl-4-iso-thiazolin-3-one) being served as positive control. The tested samples and Sea-Nine 211TM were dissolved with a small amount of dimethyl sulfoxide (DMSO) and then diluted with 0.22 μm filtered seawater (FSW) to achieve the final concentrations of 25.0, 12.5, 6.25, 3.13, and 1.56 µg/mL. About 15−20 competent larvae were added to each well containing 1 mL of test solution in triplicate, with wells having only FSW, DMSO, and larvae being served as negative control. The plates were incubated at 25 °C for 48 h, followed by examination under a microscope to count settled, unsettled larvae, and where appropriate, potential toxic effects were recorded. The number of settled larvae was expressed as a percentage of the total number of larvae per well. The EC50 was calculated as the concentration where 50% of the larval individuals were inhibited to settle as compared to the control, while LC50 was calculated as the concentration where 50% of the larval population was dead.
4. Conclusions
As part of our continuous effort to explore the biologically and structurally interesting metabolites from marine resources, we carried out a systematical investigation on the secondary metabolites from the sponge-associated fungus Gliomastix sp. ZSDS1-F7-2, leading to the discovery of four new structurally related metabolites, each having nine carbons in total, therefore briefly named as C9 metabolites. Their structures involving absolute configurations were successfully elucidated by the detailed analyses of NMR, HRMS, quantum chemistry calculations, biosynthetic relationship, and by comparison with reported literature. Biological assay revealed that these C9 metabolites showed no cytotoxic effect against the HeLa cell line, whereas one γ-lactone named gliomasolide F (1) exhibited moderate anti-fouling activity against the settlement of the barnacle Balanus Amphitrite. Furthermore, based on their structure similarity, a plausible biosynthetic relationship between these C9 metabolites was assumed. Interestingly, the co-isolation of unusual macrolides gliomasolides A—E and these four look like simple C9 metabolites, from the same fermentation extract of the marine fungus Gliomastix sp. ZSDS1-F7-2, and strongly inspired our assumption that these C9 metabolites might represent the early steps of the polyketide pathway toward gliomasolides A—E or other macrolides that remained unknown within the fermentation extract. For a more detailed understanding of the role of these C9 metabolites in the macrolides biosynthesis, incorporation experiments will be carried out in our laboratory in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/16/7/231/s1, Table S1: 1H and 13C NMR data of compounds 2 and prelactone C; Computational data, HRMS, IR and NMR spectra of compounds 1–4.
Author Contributions
J.Z. designed the research plan, elucidated the chemical structures and wrote the paper; Z.Y. carried out the isolation and purification process; Y.L. performed the biological activities test; L.Z. assisted in the purification work; H.L. assisted in the biological activities test; B.Z. and Y.L. contributed to the revision of the paper; L.L. contributed to the quantum chemistry calculation, compounds isolation, and wrote partial sections of the paper; S.X. assisted in the chemical structures elucidation, prepared the draft paper, and conceived the project.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 41506156), Natural Science Fundation of Guangdong Province (No. 2016A030310095), and Natural Science Fundation of Jiangxi Province (No. 20171BCB24011).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, Y.M.; Debbab, A.; Wray, V.; Lin, W.H.; Schulz, B.; Trepos, R.; Pile, C.; Hellio, C.; Proksch, P.; Aly, A.H. Marine bacterial inhibitors from the sponge-derived fungus Aspergillus sp. Tetrahedron Lett. 2014, 55, 2789–2792. [Google Scholar] [CrossRef] [Green Version]
- Xin, Z.H.; Fang, Y.C.; Du, L.; Zhu, T.J.; Duan, L.; Chen, J.; Gu, Q.Q.; Zhu, W.M. Aurantiomides A–C, quinazoline alkaloids from the sponge-derived fungus Penicillium aurantiogriseum SP0-19. J. Nat. Prod. 2007, 70, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Jiao, J.; Li, J.; Wang, W.; Gu, Q.; Zhu, T.; Li, D. Pyronepolyene C-glucosides with NF-κB inhibitory and anti-influenza A viral (H1N1) activities from the sponge-associated fungus Epicoccum sp. JJY40. Bioorg. Med. Chem. Lett. 2012, 22, 3188–3190. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Sun, Y.L.; Zhang, X.Y.; Han, Z.; Gao, H.C.; He, F.; Qian, P.Y.; Qi, S.H. Antifouling and antibacterial polyketides from marine gorgonian coral-associated fungus Penicillium sp. SCSGAF 0023. J. Antibiot. 2013, 66, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Umebayashi, Y.; Kawashima, M.; Sugiura, Y.; Kikuchi, T.; Tanaka, R. Determination of the chemical structures of Tandyukisins B–D, isolated from a marine sponge-derived fungus. Mar. Drugs 2015, 13, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, H.; Lin, X.; Han, L.; Ma, J.; Dong, K.; Wang, X.; Zhang, J.; Mu, Y.; Liu, Y.; Huang, X. Polyketide derivatives from a marine-sponge-associated fungus Pestalotiopsis heterocornis. Phytochemistry 2017, 142, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Suzue, M.; Arai, T.; Kikuchi, T.; Tanaka, R. Trichodermanins C–E, new diterpenes with a fused 6-5-6-6 ring system produced by a marine sponge-derived fungus. Mar. Drugs 2017, 15, 169. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Lin, X.; Han, L.; Ma, J.; Ma, Q.; Zhong, J.; Liu, Y.; Sun, T.; Wang, J.; Huang, X. New metabolites and bioactive chlorinated benzophenone derivatives produced by a marine-derived fungus Pestalotiopsis heterocornis. Mar. Drugs 2017, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Küppers, L.; Ebrahim, W.; El-Neketi, M.; Özkaya, F.C.; Mándi, A.; Kurtán, T.; Orfali, R.S.; Müller, W.E.G.; Hartmann, R.; Lin, W.; et al. Lactones from the sponge-derived fungus Talaromyces rugulosus. Mar. Drugs 2017, 15, 359. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Zhao, J.; Ding, L.; Huang, C.; Naman, C.B.; He, S.; Wu, B.; Zhu, P.; Luo, Q.; Gerwick, W.H.; et al. 5-Hydroxycyclopenicillone, a new β-amyloid fibrillization inhibitor from a sponge-derived fungus Trichoderma sp. HPQJ-34. Mar. Drugs 2017, 15, 260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Zhong, B.; Liao, X.; Xu, S. 9,11-Secosteroids with cytotoxic activity from the South China Sea gorgonian coral Subergorgia suberosa. Steroids 2015, 98, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Wang, K.; Liao, X.; Deng, Z.; Xu, S. Antifouling steroids from the South China Sea gorgonian coral Subergorgia suberosa. Steroids 2014, 79, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, L.; Wang, K.; Liao, X.; Deng, Z.; Xu, S. Pentacyclic hemiacetal sterol with antifouling and cytotoxic activities from the soft coral Nephthea sp. Bioorg. Med. Chem. Lett. 2013, 23, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lin, X.; Li, L.; Zhong, B.; Liao, X.; Liu, Y.; Xu, S. Gliomasolides A—E, unusual macrolides from a sponge-derived fungus Gliomastix sp. ZSDS1-F7-2. RSC Adv. 2015, 5, 54645–54648. [Google Scholar] [CrossRef]
- Cane, D.E.; Yang, C.C. Macrolide biosynthesis. 4. Intact incorporation of a chain-elongation intermediate into erythromycin. J. Am. Chem. Soc. 1987, 109, 1255–1257. [Google Scholar] [CrossRef]
- Bindseil, K.U.; Zeeck, A. Metabolic products of microorganisms. Part 265. Prelactones C and B, oligoketides from Streptomyces producing concanamycins and bafilomycins. Helv. Chim. Acta 1993, 76, 150–157. [Google Scholar] [CrossRef]
- Clemans, G.B.; Alemayehu, M. The gamma gauche substituent effect in 13C NMR. Tetrahedron Lett. 1993, 34, 1563–1566. [Google Scholar] [CrossRef]
- Beierbeck, H.; Saunders, J.K. A reinterpretation of beta, gamma, and delta substituent effects on 13C chemical shifts. Can. J. Chem. 1976, 54, 2985–2995. [Google Scholar] [CrossRef]
- Fremlin, L.; Farrugia, M.; Piggott, A.M.; Khalil, Z.; Lacey, E.; Capon, R.J. Reveromycins revealed: New polyketide spiroketals from Australian marine-derived and terrestrial Streptomyces spp. A case of natural products vs. artifacts. Org. Biomol. Chem. 2011, 9, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision D.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
Figure 1.
Structures of compounds 1–4.

Scheme 1.
Assumed biosynthetic relationships of 1–4.

Figure 2.
Key 1H–1H correlation spectroscopy (COSY) and heteronuclear multiple-bond correlation spectroscopy (HMBC) correlations of 1–4.
Figure 2.
Key 1H–1H correlation spectroscopy (COSY) and heteronuclear multiple-bond correlation spectroscopy (HMBC) correlations of 1–4.

Figure 3.
Experimental and calculated circular dichroism (CD) spectra of 1.

Figure 4.
Key nuclear Overhauser effect (NOE) correlations of 2.

Scheme 2.
Chemical modifications of 3 and 4.

Figure 5.
Comparison of 13C NMR data of reveromycins and 3.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
1H (300 MHz) and 13C (75 MHz) nuclear magnetic resonance (NMR) data of compounds 1 in CDCl3 and 2 in CD3OD.
Table 1.
1H (300 MHz) and 13C (75 MHz) nuclear magnetic resonance (NMR) data of compounds 1 in CDCl3 and 2 in CD3OD.
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (Mult., J in Hz) | δC | δH (Mult., J in Hz) | δC | |
| 1 | - | 172.8 | - | 173.4 |
| 2 | 6.05 (d, J = 5.4) | 121.8 | 2.42 (dd, J = 18.3, 9.0) 2.81 (dd, J = 18.3, 6.6) | 36.4 |
| 3 | 7.40 (d, J = 5.4) | 158.5 | 4.20 (m) | 67.4 |
| 4 | - | 90.9 | 2.16 (m) | 38.9 |
| 5 | 4.14 (d, J = 6.9) | 75.8 | 4.81 (dd, J = 6.9, 2.1) | 83.2 |
| 6 | 5.43 (dd, J = 15.0, 6.9) | 127.6 | 5.66 (dd, J = 15.3, 6.9) | 128.6 |
| 7 | 5.78 (dq, J = 15.0, 6.0) | 131.0 | 5.84 (dq, J = 15.3, 6.3) | 130.6 |
| 8 | 1.70 (d, J = 6.0) | 17.9 | 1.75 (d, J = 6.3) | 17.9 |
| 9 | 1.40 (s) | 20.1 | 0.92 (d, J = 7.2) | 5.9 |
Table 2.
1H (500 MHz) and 13C (75 MHz) NMR data of compounds 3–4 in CD3OD.
| No | 3 | 4 | ||
|---|---|---|---|---|
| δH (Mult., J in Hz) | δC | δH (Mult., J in Hz) | δC | |
| 1 | - | 171.2 | - | 179.1 |
| 2 | 5.79 (d, J = 15.5) | 123.7 | 2.25 (m) | 33.9 |
| 3 | 6.89 (dd, J = 15.5,7.5) | 151.5 | 1.57 (m), 1.62 (m) | 33.4 |
| 4 | 2.43 (m) | 43.9 | 2.13 (m) | 37.4 |
| 5 | 3.93 (m) | 76.9 | 5.47 (m) | 136.2 |
| 6 | 5.44 (dd, J = 15.5, 7.5) | 132.8 | 5.47 (m) | 134.8 |
| 7 | 5.68 (dq, J = 15.5, 6.5) | 128.8 | 4.18 (m) | 69.3 |
| 8 | 1.70 (d, J = 6.5) | 17.9 | 1.20 (d, J = 6.5) | 23.7 |
| 9 | 1.06 (d, J = 7.0) | 15.4 | 1.01 (d, J = 6.5) | 20.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, J.; Yang, Z.; Liang, Y.; Zhong, L.; Lin, H.; Zhong, B.; Li, L.; Xu, S.; Liu, Y. Four New C9 Metabolites from the Sponge-Associated Fungus Gliomastix sp. ZSDS1-F7-2. Mar. Drugs 2018, 16, 231. https://doi.org/10.3390/md16070231
AMA Style
Zhang J, Yang Z, Liang Y, Zhong L, Lin H, Zhong B, Li L, Xu S, Liu Y. Four New C9 Metabolites from the Sponge-Associated Fungus Gliomastix sp. ZSDS1-F7-2. Marine Drugs. 2018; 16(7):231. https://doi.org/10.3390/md16070231
Chicago/Turabian StyleZhang, Jun, Zhiqiang Yang, Yan Liang, Liping Zhong, Huiting Lin, Balian Zhong, Liangchun Li, Shihai Xu, and Yonghong Liu. 2018. "Four New C9 Metabolites from the Sponge-Associated Fungus Gliomastix sp. ZSDS1-F7-2" Marine Drugs 16, no. 7: 231. https://doi.org/10.3390/md16070231
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.