The Outer Vestibule of the Na+ Channel–Toxin Receptor and Modulator of Permeation as Well as Gating

Abstract
:1. Introduction
2. Use-dependent Block by Guanidinium Toxins
3. Partial Blockers of the Outer Vestibule Allow Assessment of Changes in Gating Behavior during Drug Binding
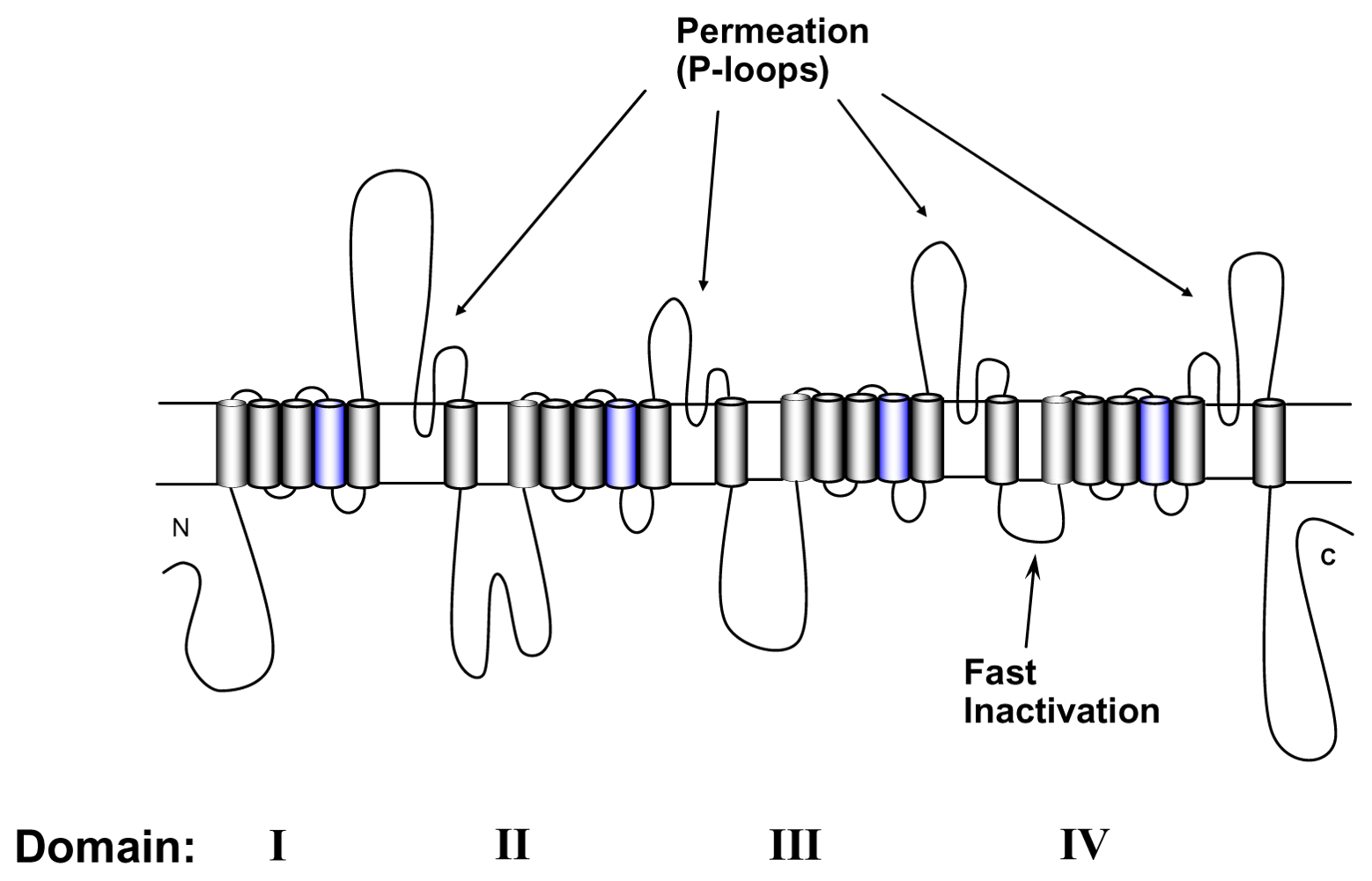
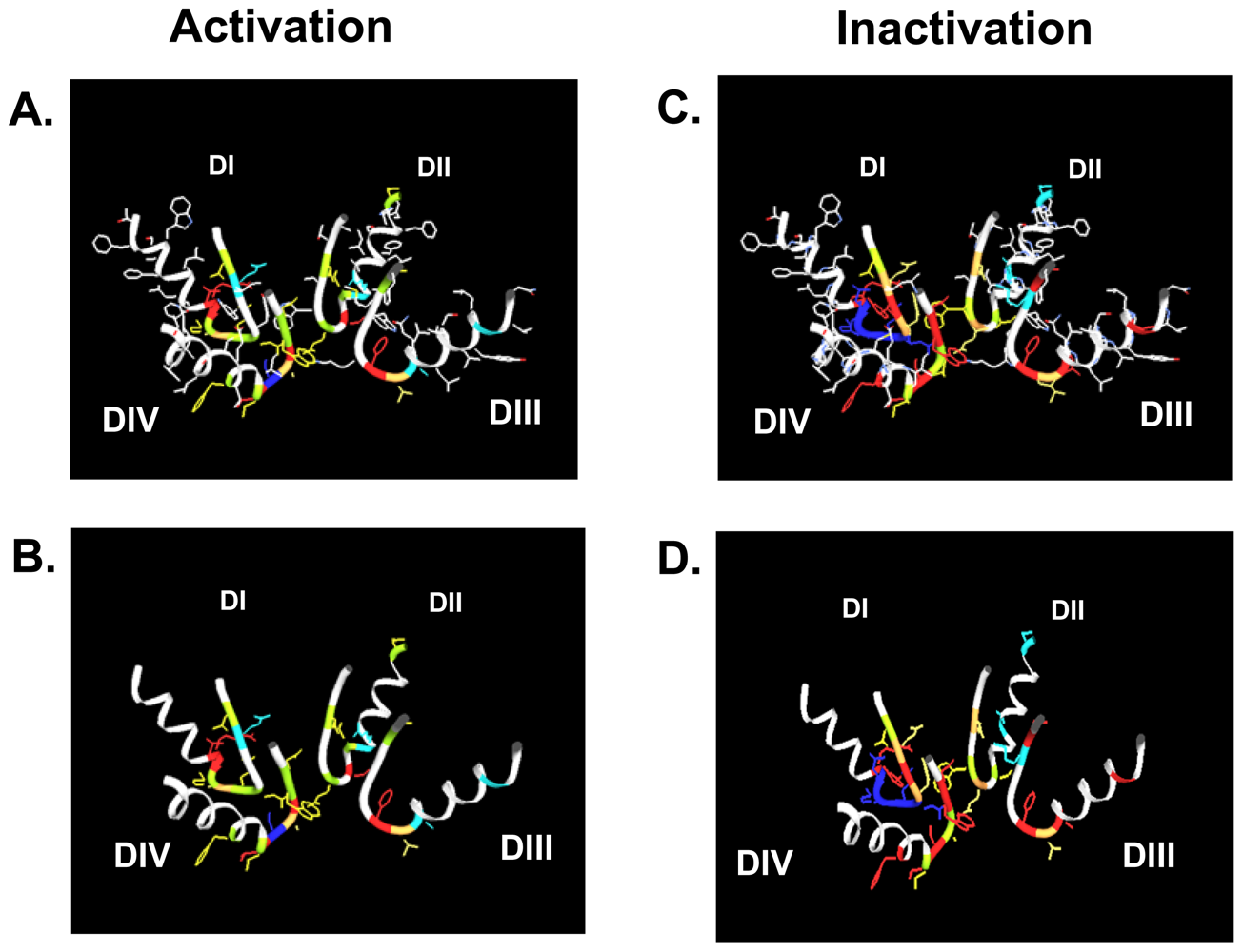
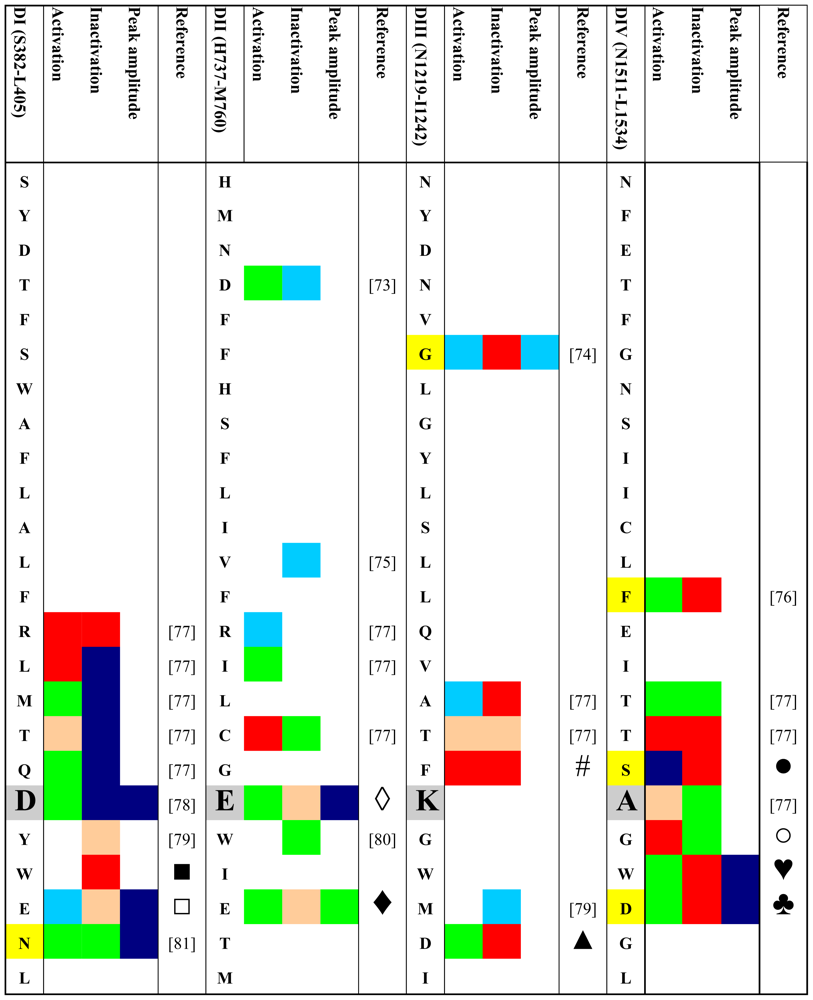
4. Gating Changes by Mutations in the Outer Vestibule—A Topological Overview
- Fast and slow inactivated states were lumped together.
- Effects on the half point of steady state availability were categorized as described for the conductance versus voltage curve. Steady state probability of an inactivated state was considered “major” if the change was >50%. Effects on time constants of development and/or recovery were considered “minor” if the change was 1.5–2 fold. >2-fold changes were considered “major”. The same criteria were applied to changes in the relative amplitude of the respective state(s).
- If changes in several parameters were reported for a given state, and/or if several inactivated states were reported we considered the respective value/state with the greatest change.
5. Physiological Relevance of Gating Changes by Mutations in the Outer Vestibule
6. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References and Notes
- Sutkowski, EM; Catterall, WA. Beta 1 subunits of sodium channels. Studies with subunit-specific antibodies. J Biol Chem 1990, 265, 12393–12399. [Google Scholar]
- Isom, LL; De Jongh, KS; Patton, DE; Reber, BF; Offord, J; Charbonneau, H; Walsh, K; Goldin, AL; Catterall, WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 1992, 256, 839–842. [Google Scholar]
- Goldin, AL; Snutch, T; Lubbert, H; Dowsett, A; Marshall, J; Auld, V; Downey, W; Fritz, LC; Lester, HA; Dunn, R; et al. Messenger RNA coding for only the alpha subunit of the rat brain Na+ channel is sufficient for expression of functional channels in Xenopus oocytes. Proc Natl Acad Sci USA 1986, 83, 7503–7507. [Google Scholar]
- Noda, M; Shimizu, S; Tanabe, T; Takai, T; Kayano, T; Ikeda, T; Takahashi, H; Nakayama, H; Kanaoka, Y; Minamino, N; et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 1984, 312, 121–127. [Google Scholar]
- Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000, 80, 555–592. [Google Scholar]
- Catterall, WA; Goldin, AL; Waxman, SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005, 57, 397–409. [Google Scholar]
- Pusch, M; Noda, M; Stühmer, W; Numa, S; Conti, F. Single point mutations of the sodium channel drastically reduce the pore permeability without preventing its gating. Eur Biophys J 1991, 20, 127–133. [Google Scholar]
- Heinemann, SH; Terlau, H; Stühmer, W; Imoto, K; Numa, S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 1992, 356, 441–443. [Google Scholar]
- Lee, CH; Ruben, PC. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels (Austin) 2008, 2, 407–412. [Google Scholar]
- Noda, M; Suzuki, H; Numa, S; Stühmer, W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett 1989, 259, 213–216. [Google Scholar]
- Terlau, H; Heinemann, SH; Stuhmer, W; Pusch, M; Conti, F; Imoto, K; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett 1991, 293, 93–96. [Google Scholar]
- Lipkind, GM; Fozzard, HA. A Structural Model of the Tetrodotoxin and Saxitoxin Binding-Site of the Na+ Channel. Biophys J 1994, 66, 1–13. [Google Scholar]
- Penzotti, JL; Fozzard, HA; Lipkind, GM; Dudley, SC, Jr. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J 1998, 75, 2647–2657. [Google Scholar]
- Lipkind, GM; Fozzard, HA. KcsA crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 2000, 39, 8161–8170. [Google Scholar]
- Hille, B. Ionic channels in nerve membranes. Prog Biophys Mol Biol 1970, 21, 1–32. [Google Scholar]
- Narahashi, T. Chemicals as tools in the study of excitable membranes. Physiol Rev 1974, 54, 813–889. [Google Scholar]
- Ulbricht, W. Ionic channels and gating currents in excitable membranes. Annu Rev Biophys Bioeng 1977, 6, 7–31. [Google Scholar]
- Benitah, JP; Chen, Z; Balser, JR; Tomaselli, GF; Marban, E. Molecular dynamics of the sodium channel pore vary with gating: interactions between P-segment motions and inactivation. J Neurosci 1999, 19, 1577–1585. [Google Scholar]
- Benitah, JP; Ranjan, R; Yamagishi, T; Janecki, M; Tomaselli, GF; Marban, E. Molecular motions within the pore of voltage-dependent sodium channels. Biophys J 1997, 73, 603–613. [Google Scholar]
- Eriksson, MA; Roux, B. Modeling the structure of agitoxin in complex with the ShakerK+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys J 2002, 83, 2595–2609. [Google Scholar]
- Huang, X; Dong, F; Zhou, HX. Electrostatic recognition and induced fit in the κ-PVIIA toxin binding to Shaker potassium channel. J Am Chem Soc 2005, 127, 6836–6849. [Google Scholar]
- Lange, A; Giller, K; Hornig, S; Martin-Eauclaire, MF; Pongs, O; Becker, S; Baldus, M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature 2006, 440, 959–962. [Google Scholar]
- Szendroedi, J; Sandtner, W; Zarrabi, T; Zebedin, E; Hilber, K; Dudley, SC, Jr; Fozzard, HA; Todt, H. Speeding the recovery from ultraslow inactivation of voltage-gated Na+ channels by metal ion binding to the selectivity filter: a foot-on-the-door? Biophys J 2007, 93, 4209–4224. [Google Scholar]
- Courtney, KR. Mechanism of frequency-dependent inhibition of sodium currents in frog myelinated nerve by the lidocaine derivative GEA. J Pharmacol Exp Ther 1975, 195, 225–236. [Google Scholar]
- Starmer, CF. Theoretical characterization of ion channel blockade: ligand binding to periodically accessible receptors. J Theor Biol 1986, 119, 235–249. [Google Scholar]
- Hondeghem, LM; Katzung, BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta 1977, 472, 373–398. [Google Scholar]
- Hille, B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 1977, 69, 497–515. [Google Scholar]
- Baer, M; Best, PM; Reuter, H. Voltage-Dependent Action of Tetrodotoxin in Mammalian Cardiac-Muscle. Nature 1976, 263, 344–345. [Google Scholar]
- Cohen, CJ; Bean, BP; Colatsky, TJ; Tsien, RW. Tetrodotoxin Block of Sodium-Channels in Rabbit Purkinje-Fibers-Interactions between Toxin Binding and Channel Gating. J Gen Physiol 1981, 78, 383–411. [Google Scholar]
- Gonoi, T; Sherman, SJ; Catterall, WA. Voltage clamp analysis of tetrodotoxin-sensitive and -insensitive sodium channels in rat muscle cells developing in vitro. J Neurosci 1985, 5, 2559–2564. [Google Scholar]
- Vassilev, PM; Hadley, RW; Lee, KS; Hume, JR. Voltage-Dependent Action of Tetrodotoxin in Mammalian Cardiac Myocytes. Am J Physiol 1986, 251, 475–480. [Google Scholar]
- Salgado, VL; Yeh, JZ; Narahashi, T. Use-Dependent and Voltage-Dependent Block of the Sodium-Channel by Saxitoxin. Ann NY Acad Sci 1986, 479, 84–95. [Google Scholar]
- Makielski, JC; Satin, J; Fan, Z. Post-repolarization block of cardiac sodium channels by saxitoxin. Biophys J 1993, 65, 790–798. [Google Scholar]
- Carmeliet, E. Voltage-Dependent Block by Tetrodotoxin of the Sodium-Channel in Rabbit Cardiac Purkinje-Fibers. Biophys J 1987, 51, 109–114. [Google Scholar]
- Clarkson, CW; Matsubara, T; Hondeghem, LM. Evidence for voltage-dependent block of cardiac sodium channels by tetrodotoxin. J Mol Cell Cardiol 1988, 20, 1119–1131. [Google Scholar]
- Lonnendonker, U. Use-Dependent Block of Sodium-Channels in Frog Myelinated Nerve by Tetrodotoxin and Saxitoxin at Negative Holding Potentials. Biochim Biophys Acta 1989, 985, 153–160. [Google Scholar]
- Eickhorn, R; Weirich, J; Hornung, D; Antoni, H. Use Dependence of Sodium Current Inhibition by Tetrodotoxin in Rat Cardiac-Muscle - Influence of Channel State. Pflug Arch Eur J Phy 1990, 416, 398–405. [Google Scholar]
- Patton, DE; Goldin, AL. A voltage-dependent gating transition induces use-dependent block by tetrodotoxin of rat IIA sodium channels expressed in Xenopus oocytes. Neuron 1991, 7, 637–647. [Google Scholar]
- Lonnendonker, U. Use-Dependent Block with Tetrodotoxin and Saxitoxin at Frog Ranvier Nodes.1. Intrinsic Channel and Toxin Parameters. Eur Biophys J 1991, 20, 135–141. [Google Scholar]
- Lonnendonker, U. Use-Dependent Block with Tetrodotoxin and Saxitoxin at Frog Ranvier Nodes. 2. Extrinsic Influence of Cations. Eur Biophys J 1991, 20, 143–149. [Google Scholar]
- Satin, J; Kyle, JW; Chen, M; Bell, P; Cribbs, LL; Fozzard, HA; Rogart, RB. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 1992, 256, 1202–1205. [Google Scholar]
- Satin, J; Kyle, JW; Fan, Z; Rogart, R; Fozzard, HA; Makielski, JC. Post-repolarization block of cloned sodium channels by saxitoxin: the contribution of pore-region amino acids. Biophys J 1994, 66, 1353–1363. [Google Scholar]
- West, JW; Patton, DE; Scheuer, T; Wang, Y; Goldin, AL; Catterall, WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA 1992, 89, 10910–10914. [Google Scholar]
- Hartmann, HA; Tiedeman, AA; Chen, SF; Brown, AM; Kirsch, GE. Effects of III–IV linker mutations on human heart Na+ channel inactivation gating. Circ Res 1994, 75, 114–122. [Google Scholar]
- Dumaine, R; Hartmann, H. Two conformational states involved in the use-dependent TTX blockade of human cardiac Na+ channel. Am J Physiol-Heart C 1996, 39, 2029–2037. [Google Scholar]
- Richmond, JE; Featherstone, DE; Hartmann, HA; Ruben, PC. Slow inactivation in human cardiac sodium channels. Biophys J 1998, 74, 2945–2952. [Google Scholar]
- Conti, F; Gheri, A; Pusch, M; Moran, O. Use dependence of tetrodotoxin block of sodium channels: a revival of the trapped-ion mechanism. Biophys J 1996, 71, 1295–1312. [Google Scholar]
- Moran, O; Picollo, A; Conti, F. Tonic and phasic guanidinium toxin-block of skeletal muscle Na+ channels expressed in Mammalian cells. Biophys J 2003, 84, 2999–3006. [Google Scholar]
- Boccaccio, A; Moran, O; Imoto, K; Conti, F. Tonic and phasic tetrodotoxin block of sodium channels with point mutations in the outer pore region. Biophys J 1999, 77, 229–240. [Google Scholar]
- Rosker, C; Lohberger, B; Hofer, D; Steinecker, B; Quasthoff, S; Schreibmayer, W. The TTX metabolite 4,9-anhydro-TTX is a highly specific blocker of the NaV1.6 voltage-dependent sodium channel. Am J Physiol-Cell Ph 2007, 293, 783–789. [Google Scholar]
- Zhou, Y; Morais-Cabral, JH; Kaufman, A; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001, 414, 43–48. [Google Scholar]
- Boccaccio, A; Conti, F; Olivera, BM; Terlau, H. Binding of κ-conotoxin PVIIA to ShakerK+ channels reveals different K+ and Rb+ occupancies within the ion channel pore. J Gen Physiol 2004, 124, 71–81. [Google Scholar]
- Heggeness, ST; Starkus, JG. Saxitoxin and Tetrodotoxin - Electrostatic Effects on Sodium-Channel Gating Current in Crayfish Axons. Biophys J 1986, 49, 629–643. [Google Scholar]
- Keynes, RD; Greeff, NG; Forster, IC; Bekkers, JM. The effect of tetrodotoxin on the sodium gating current in the squid giant axon. Proc Biol Sci 1991, 246, 135–140. [Google Scholar]
- Ulbricht, W. Sodium channel inactivation: molecular determinants and modulation. Physiol Rev 2005, 85, 1271–1301. [Google Scholar]
- Choi, KL; Aldrich, RW; Yellen, G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci USA 1991, 88, 5092–5095. [Google Scholar]
- Moczydlowski, E; Olivera, BM; Gray, WR; Strichartz, GR. Discrimination of muscle and neuronal Na+-channel subtypes by binding competition between [3H]saxitoxin and μ–conotoxins. Proc Natl Acad Sci USA 1986, 83, 5321–5325. [Google Scholar]
- Chang, NS; French, RJ; Lipkind, GM; Fozzard, HA; Dudley, S. Predominant interactions between μ–conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 1998, 37, 4407–4419. [Google Scholar]
- Chang, NS; Dudley, SC, Jr; Lipkind, G; French, RJ; Fozzard, H. μ-Conotoxin binding to the voltage-gated Na+ channel: structural implications for the outer vestibule. Biophys J 1997, 72, A361. [Google Scholar]
- Dudley, SC, Jr; Chang, N; Hall, J; Lipkind, G; Fozzard, HA; French, RJ. μ–conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J Gen Physiol 2000, 116, 679–689. [Google Scholar]
- Dudley, SC, Jr; Todt, H; Lipkind, G; Fozzard, HA. A μ–conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophys J 1995, 69, 1657–1665. [Google Scholar]
- Fozzard, HA; Lipkind, G. The guanidinium toxin binding site on the sodium channel. Jpn Heart J 1996, 37, 683–692. [Google Scholar]
- Penzotti, JL; Fozzard, HA; Lipkind, GM; Dudley, SC, Jr. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J 1998, 75, 2647–2657. [Google Scholar]
- Li, RA; Ennis, II; French, RJ; Dudley, SC, Jr; Tomaselli, GF; Marban, E. Clockwise domain arrangement of the sodium channel revealed by β-conotoxin (GIIIA) docking orientation. J Biol Chem 2001, 276, 11072–11077. [Google Scholar]
- Becker, S; Prusak-Sochaczewski, E; Zamponi, G; Beck-Sickinger, AG; Gordon, RD; French, RJ. Action of derivatives of μ–conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar]
- French, RJ; Prusak-Sochaczewski, E; Zamponi, GW; Becker, S; Kularatna, AS; Horn, R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron 1996, 16, 407–413. [Google Scholar]
- Todt, H; Dudley, SC, Jr; Kyle, JW; French, RJ; Fozzard, HA. Ultra-slow inactivation in μ1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys J 1999, 76, 1335–1345. [Google Scholar]
- Hilber, K; Sandtner, W; Kudlacek, O; Glaaser, IW; Weisz, E; Kyle, JW; French, RJ; Fozzard, HA; Dudley, SC, Jr; Todt, H. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. J Biol Chem 2001, 276, 27831–27839. [Google Scholar]
- Hilber, K; Sandtner, W; Kudlacek, O; Schreiner, B; Glaaser, I; Schutz, W; Fozzard, HA; Dudley, SC, Jr; Todt, H. Interaction between fast and ultra-slow inactivation in the voltage-gated sodium channel. Does the inactivation gate stabilize the channel structure. J Biol Chem 2002, 277, 37105–37115. [Google Scholar]
- Sandtner, W; Szendroedi, J; Hilber, K; Zarrabi, T; Fozzard, HA; Dudley, SC, Jr; Todt, H. Lidocaine: a foot in the door of the inner pore of the voltage-gated Na+ channel. Biophys J 2004, 86, 118a. [Google Scholar]
- Cuello, LG; Jogini, V; Cortes, DM; Pan, AC; Gagnon, DG; Cordero-Morales, JF; Chakrapani, S; Roux, B; Perozo, E. Structural basis for the coupling between activation and inactivation gating in potassium channels. Biophys J 2009, 96, 381a. [Google Scholar]
- Doyle, DA; Morais, CJ; Pfuetzner, RA; Kuo, A; Gulbis, JM; Cohen, SL; Chait, BT; MacKinnon, R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar]
- Kontis, KJ; Goldin, AL. Site-directed mutagenesis of the putative pore region of the rat IIA sodium channel. Mol Pharmacol 1993, 43, 635–644. [Google Scholar]
- Tan, BH; Valdivia, CR; Song, CH; Makielski, JC. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am J Physiol-Heart C 2006, 291, 1822–1828. [Google Scholar]
- Vilin, YY; Fujimoto, E; Ruben, PC. A single residue differentiates between human cardiac and skeletal muscle Na+ channel slow inactivation. Biophys J 2001, 80, 2221–2230. [Google Scholar]
- Otagiri, T; Kijima, K; Osawa, M; Ishii, K; Makita, N; Matoba, R; Umetsu, K; Hayasaka, K. Cardiac Ion Channel Gene Mutations in Sudden Infant Death Syndrome. Pediatr Res 2008, 64, 482–487. [Google Scholar]
- Yamagishi, T; Xiong, W; Kondratiev, A; Velez, P; Mendez-Fitzwilliam, A; Balser, JR; Marban, E; Tomaselli, GF. Novel molecular determinants in the pore region of sodium channels regulate local anesthetic binding. Mol Pharmacol 2009, 76, 861–871. [Google Scholar]
- Kuhn, FJ; Greeff, NG. Mutation D384N Alters Recovery of the Immobilized Gating Charge in Rat Brain IIA Sodium Channels. J Membr Biol 2002, 185, 145–155. [Google Scholar]
- Struyk, AF; Cannon, SC. Slow inactivation does not block the aqueous accessibility to the outer pore of voltage-gated Na+ channels. J Gen Physiol 2002, 120, 509–516. [Google Scholar]
- Balser, JR; Nuss, HB; Chiamvimonvat, N; Perez-Garcia, MT; Marban, E; Tomaselli, GF. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol (Lond) 1996, 494, 431–442. [Google Scholar]
- Rossenbacker, T; Carroll, SJ; Liu, H; Kuiperi, C; de Ravel, TJ; Devriendt, K; Carmeliet, P; Kass, RS; Heidbuchel, H. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm 2004, 1, 610–615. [Google Scholar]
- Kambouris, NG; Hastings, LA; Stepanovic, S; Marban, E; Tomaselli, GF; Balser, JR. Mechanistic link between lidocaine block and inactivation probed by outer pore mutations in the rat μ1 skeletal muscle sodium channel. J Physiol 1998, 512, 693–705. [Google Scholar]
- Tomaselli, GF; Chiamvimonvat, N; Nuss, HB; Balser, JR; Perez-Garcia, MT; Xu, RH; Orias, DW; Backx, PH; Marban, E. A mutation in the pore of the sodium channel alters gating. Biophys J 1995, 68, 1814–1827. [Google Scholar]
- Xiong, W; Farukhi, YZ; Tian, Y; DiSilvestre, D; Li, RA; Tomaselli, GF. A conserved ring of charge in mammalian Na+ channels: a molecular regulator of the outer pore conformation during slow inactivation. J Physiol 2006, 576, 739–754. [Google Scholar]
- Khan, A; Romantseva, L; Lam, A; Lipkind, G; Fozzard, HA. Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J Physiol (Lond) 2002, 543, 71–84. [Google Scholar]
- Shirai, N; Makita, N; Sasaki, K; Yokoi, H; Sakuma, I; Sakurada, H; Akai, J; Kimura, A; Hiraoka, M; Kitabatake, A. A mutant cardiac sodium channel with multiple biophysical defects associated with overlapping clinical features of Brugada syndrome and cardiac conduction disease. Cardiovasc Res 2002, 53, 348–354. [Google Scholar]
- Akai, J; Makita, N; Sakurada, H; Shirai, N; Ueda, K; Kitabatake, A; Nakazawa, K; Kimura, A; Hiraoka, M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett 2000, 479, 29–34. [Google Scholar]
- Yang, YC; Hsieh, JY; Kuo, CC. The external pore loop interacts with S6 and S3-S4 linker in domain 4 to assume an essential role in gating control and anticonvulsant action in the Na+ channel. J Gen Physiol 2009, 134, 95–113. [Google Scholar]
- Tsang, SY; Tsushima, RG; Tomaselli, GF; Li, RA; Backx, PH. A multifunctional aromatic residue in the external pore vestibule of Na+ channels contributes to the local anesthetic receptor. Mol Pharmacol 2005, 67, 424–434. [Google Scholar]
- Amin, AS; Verkerk, AO; Bhuiyan, ZA; Wilde, AAM; Tan, HL. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol Scand 2005, 185, 291–301. [Google Scholar]
- Wang, SY; Mitchell, J; Tikhonov, DB; Zhorov, BS; Wang, GK. How batrachotoxin modifies the sodium channel permeation pathway: computer modeling and site-directed mutagenesis. Mol Pharmacol 2006, 69, 788–795. [Google Scholar]
- Lipkind, GM; Fozzard, HA. KcsA crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 2000, 39, 8161–8170. [Google Scholar]
- Sheets, MF; Kyle, JW; Kallen, RG; Hanck, DA. The Na+ channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J 1999, 77, 747–757. [Google Scholar]
- Chahine, M; George, AL, Jr; Zhou, M; Ji, S; Sun, W; Barchi, RL; Horn, R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron 1994, 12, 281–294. [Google Scholar]
- Chen, LQ; Santarelli, V; Horn, R; Kallen, RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol 1996, 108, 549–556. [Google Scholar]
- Kontis, KJ; Rounaghi, A; Goldin, AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol 1997, 110, 391–401. [Google Scholar]
- Krafte, DS; Goldin, AL; Auld, VJ; Dunn, RJ; Davidson, N; Lester, HA. Inactivation of cloned Na+ channels expressed in Xenopus oocytes. J Gen Physiol 1990, 96, 689–706. [Google Scholar]
- Stuhmer, W; Conti, F; Suzuki, H; Wang, XD; Noda, M; Yahagi, N; Kubo, H; Numa, S. Structural parts involved in activation and inactivation of the sodium channel. Nature 1989, 339, 597–603. [Google Scholar]
- Wang, SY; Wang, GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J 1997, 72, 1633–1640. [Google Scholar]
- Takahashi, MP; Cannon, SC. Enhanced slow inactivation by V445M: a sodium channel mutation associated with myotonia. Biophys J 1999, 76, 861–868. [Google Scholar]
- O’Reilly, JP; Wang, SY; Wang, GK. A point mutation in domain 4-segment 6 of the skeletal muscle sodium channel produces an atypical inactivation state. Biophys J 2000, 78, 773–784. [Google Scholar]
- O’Reilly, JP; Wang, SY; Wang, GK. Residue-specific effects on slow inactivation at V787 in D2-S6 of NaV1.4 sodium channels. Biophys J 2001, 81, 2100–2111. [Google Scholar]
- Wang, SY; Russell, C; Wang, GK. Tryptophan Substitution of a Putative D4S6 Gating Hinge Alters Slow Inactivation in Cardiac Sodium Channels. Biophys J 2005, 88, 3991–3999. [Google Scholar]
- Vedantham, V; Cannon, SC. Rapid and slow voltage-dependent conformational changes in segment IVS6 of voltage-gated Na+ channels. Biophys J 2000, 78, 2943–2958. [Google Scholar]
- Chen, Y; Yu, FH; Surmeier, DJ; Scheuer, T; Catterall, WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron 2006, 49, 409–420. [Google Scholar]
- Ragsdale, DS; McPhee, JC; Scheuer, T; Catterall, WA. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc Natl Acad Sci USA 1996, 93, 9270–9275. [Google Scholar]
- Yarov-Yarovoy, V; McPhee, JC; Idsvoog, D; Pate, C; Scheuer, T; Catterall, WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem 2002, 277, 35393–35401. [Google Scholar]
- Sunami, A; Dudley, SC, Jr; Fozzard, HA. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc Natl Acad Sci USA 1997, 94, 14126–14131. [Google Scholar]
- Ong, BH; Tomaselli, GF; Balser, JR. A Structural Rearrangement in the Sodium Channel Pore Linked to Slow Inactivation and Use Dependence. J Gen Physiol 2000, 116, 653–662. [Google Scholar]
- Yifrach, O; MacKinnon, R. Energetics of pore opening in a voltage-gated K+ channel. Cell 2002, 111, 231–239. [Google Scholar]
- Sadovsky, E; Yifrach, O. Principles underlying energetic coupling along an allosteric communication trajectory of a voltage-activated K+ channel. Proc Natl Acad Sci USA 2007, 104, 19813–19818. [Google Scholar]
- Ruff, RL; Simoncini, L; Stühmer, W. Slow sodium channel inactivation in mammalian muscle: a possible role in regulating excitability. Muscle Nerve 1988, 11, 502–510. [Google Scholar]
- George, AL, Jr. Inherited disorders of voltage-gated sodium channels. J Clin Invest 2005, 115, 1990–1999. [Google Scholar]
- Antzelevitch, C; Brugada, P; Borggrefe, M; Brugada, J; Brugada, R; Corrado, D; Gussak, I; LeMarec, H; Nademanee, K; Perez, Riera AR; Shimizu, W; Schulze-Bahr, E; Tan, H; Wilde, A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005, 111, 659–670. [Google Scholar]
- Kapplinger, JD; Tester, DJ; Alders, M; Benito, B; Berthet, M; Brugada, J; Brugada, P; Fressart, V; Guerchicoff, A; Harris-Kerr, C; Kamakura, S; Kyndt, F; Koopmann, TT; Miyamoto, Y; Pfeiffer, R; Pollevick, GD; Probst, V; Zumhagen, S; Vatta, M; Towbin, JA; Shimizu, W; Schulze-Bahr, E; Antzelevitch, C; Salisbury, BA; Guicheney, P; Wilde, AA; Brugada, R; Schott, JJ; Ackerman, MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar]
- Akai, J; Makita, N; Sakurada, H; Shirai, N; Ueda, K; Kitabatake, A; Nakazawa, K; Kimura, A; Hiraoka, M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett 2000, 479, 29–34. [Google Scholar]
- Krous, HF; Beckwith, JB; Byard, RW; Rognum, TO; Bajanowski, T; Corey, T; Cutz, E; Hanzlick, R; Keens, TG; Mitchell, EA. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 2004, 114, 234–238. [Google Scholar]
- Rasmusson, RL; Morales, MJ; Wang, SM; Liu, SG; Campbell, DL; Brahmajothi, MV; Strauss, HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res 1998, 82, 739–750. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cervenka, R.; Zarrabi, T.; Lukacs, P.; Todt, H. The Outer Vestibule of the Na+ Channel–Toxin Receptor and Modulator of Permeation as Well as Gating. Mar. Drugs 2010, 8, 1373-1393. https://doi.org/10.3390/md8041373
Cervenka R, Zarrabi T, Lukacs P, Todt H. The Outer Vestibule of the Na+ Channel–Toxin Receptor and Modulator of Permeation as Well as Gating. Marine Drugs. 2010; 8(4):1373-1393. https://doi.org/10.3390/md8041373
Chicago/Turabian StyleCervenka, René, Touran Zarrabi, Peter Lukacs, and Hannes Todt. 2010. "The Outer Vestibule of the Na+ Channel–Toxin Receptor and Modulator of Permeation as Well as Gating" Marine Drugs 8, no. 4: 1373-1393. https://doi.org/10.3390/md8041373