Total Synthesis and Antimicrobial Activity of a Natural Cycloheptapeptide of Marine Origin

Abstract
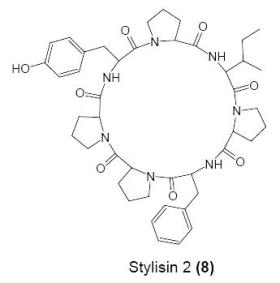
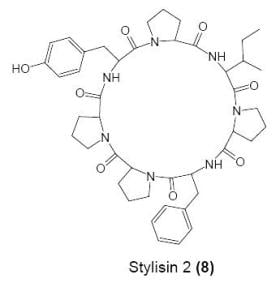
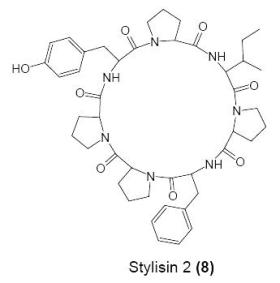
:
1. Introduction
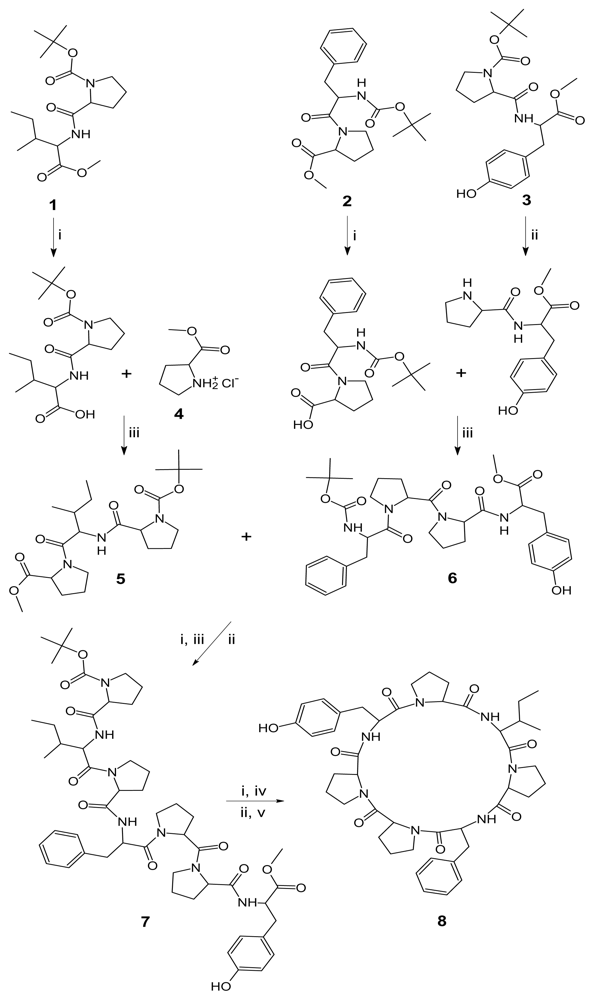
2. Results and Discussion
Reagents and conditions
3. Experimental
3.1. General procedure for the preparation of linear tri/tetrapeptide segments
tert-Butyloxycarbonyl-l-prolyl-l-isoleucyl-l-proline methyl ester (5)
tert-Butyloxycarbonyl-l-phenylalanyl-l-prolyl-l-prolyl-l-tyrosine methyl ester (6)
3.2. Deprotection of tripeptide unit at the carboxyl terminal
3.3. Deprotection of tetrapeptide unit at amino terminal
3.4. Procedure for the synthesis of the linear heptapeptide unit
tert-Butyloxycarbonyl-l-prolyl-l-isoleucyl-l-prolyl-l-phenylalanyl-l-prolyl-l-prolyl-l-tyrosine methyl ester (7)
3.5. Synthesis of the cyclic heptapeptide 8
Cyclo(l-prolyl-l-isoleucyl-l-prolyl-l-phenylalanyl-l-prolyl-l-prolyl-l-tyrosinyl) (8)
3.6. Biological activity studies
3.6.1. Antibacterial screening
3.6.2. Antifungal screening
4. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References
- Fusetani, N; Matsunaga, S. Bioactive Sponge Peptides. Chem. Rev 1993, 93, 1793–1806. [Google Scholar]
- Zainuddin, EN; Jansen, R; Nimtz, M; Wray, V; Preisitsch, M; Lalk, M; Mundt, S. Lyngbyazothrins A–D, Antimicrobial Cyclic Undecapeptides from the Cultured Cyanobacterium Lyngbya sp. J. Nat. Prod 2009, 72, 1373–1378. [Google Scholar]
- Williams, DE; Yu, K; Behrisch, HW; van Soest, R; Andersen, RJ. Rolloamides A and B, Cytotoxic Cyclic Heptapeptides isolated from the Caribbean Marine Sponge Eurypon laughlini. J. Nat. Prod 2009, 72, 1253–1257. [Google Scholar]
- Ford, PW; Gustafson, KR; McKee, TC; Shigematsu, N; Maurizi, LK; Pannell, LK; Williams, DE; de Silva, ED; Lassota, P; Allen, TM; van Soest, R; Andersen, RJ; Boyd, MR. Papuamides A–D, HIV-Inhibitory and Cytotoxic Depsipeptides from the Sponges Theonella mirabilis and Theonella swinhoei Collected in Papua New Guinea. J. Am. Chem. Soc 1999, 121, 5899–5909. [Google Scholar]
- Capon, RJ; Ford, J; Lacey, E; Gill, JH; Heiland, K; Friedel, T. Phoriospongin A and B: Two New Nematocidal Depsipeptides from the Australian Marine Sponges Phoriospongia sp. and Callyspongia bilamellata. J. Nat. Prod 2002, 65, 358–363. [Google Scholar]
- Randazzo, A; Bifulco, G; Giannini, C; Bucci, M; Debitus, C; Cirino, G; Gomez-Paloma, L. Halipeptins A and B: Two Novel Potent Anti-inflammatory Cyclic Depsipeptides from the Vanuatu Marine Sponge Haliclona species. J. Am. Chem. Soc 2001, 123, 10870–10876. [Google Scholar]
- Gunasekera, SP; Miller, MW; Kwan, JC; Luesch, H; Paul, VJ. Molassamide, A Depsipeptide Serine Protease Inhibitor from the Marine Cyanobacterium Dichothrix utahensis. J. Nat. Prod 2010, 73, 459–462. [Google Scholar]
- Muller, D; Krick, A; Kehraus, S; Mehner, C; Hart, M; Kupper, FC; Saxena, K; Prinz, H; Schwalbe, H; Janning, P; Waldmann, H; Konig, GM. Brunsvicamides A–C: Sponge-Related Cyanobacterial Peptides with Mycobacterium tuberculosis Protein Tyrosine Phosphatase Inhibitory Activity. J. Med. Chem 2006, 49, 4871–4878. [Google Scholar]
- Mohammed, R; Peng, J; Kelly, M; Hamann, MT. Cyclic Heptapeptides from the Jamaican Sponge Stylissa caribica. J. Nat. Prod 2006, 69, 1739–1744. [Google Scholar]
- Kobayashi, J; Tsuda, M; Nakamura, T; Mikami, Y; Shigemori, H. Hymenamides A and B, New Proline-Rich Cyclic Heptapeptides from the Okinawan Marine Sponge Hymeniacidon sp. Tetrahedron 1993, 49, 2391–2402. [Google Scholar]
- Dahiya, R; Pathak, D; Himaja, M; Bhatt, S. First Total Synthesis and Biological Screening of Hymenamide E. Acta Pharm 2006, 56, 399–415. [Google Scholar]
- Wele, A; Zhang, Y; Dubost, L; Pousset, J-L; Bodo, B. Cyclic Peptides from the Seeds of Annona glauca and A. cherimola. Chem. Pharm. Bull 2006, 54, 690–692. [Google Scholar]
- Dahiya, R; Kumar, A; Gupta, R. Synthesis, Cytotoxic and Antimicrobial Screening of a Proline- Rich Cyclopolypeptide. Chem. Pharm. Bull 2009, 57, 214–217. [Google Scholar]
- Dahiya, R. Synthesis and in vitro Cytotoxic Activity of a Natural Peptide of Plant Origin. J. Iran. Chem. Soc 2008, 5, 445–452. [Google Scholar]
- Dahiya, R. Synthetic Studies on a Cyclic Hexapeptide from Dianthus Superbus. Chem. Pap 2008, 62, 527–535. [Google Scholar]
- Dahiya, R. Total Synthesis and Biological Potential of Psammosilenin A. Arch. Pharm. Chem. Life Sci 2008, 341, 502–509. [Google Scholar]
- Dahiya, R. Synthesis, Spectroscopic and Biological Investigation of Cyclic Octapeptide: Cherimolacyclopeptide G. Turk. J. Chem 2008, 32, 205–215. [Google Scholar]
- Dahiya, R. Synthetic and Pharmacological Studies on Longicalycinin A. Pak. J. Pharm. Sci 2007, 20, 317–323. [Google Scholar]
- Dahiya, R. Synthesis of a Phenylalanine-Rich Peptide as Potential Anthelmintic and Cytotoxic Agent. Acta Pol. Pharm 2007, 64, 509–516. [Google Scholar]
- Dahiya, R. Synthesis, Characterization and Biological Evaluation of a Glycine-Rich Peptide—Cherimolacyclopeptide E. J. Chil. Chem. Soc 2007, 52, 1224–1229. [Google Scholar]
- Dahiya, R; Kaur, K. Synthetic and Biological Studies on Natural Cyclic Heptapeptide: Segetalin E. Arch. Pharm. Res 2007, 30, 1380–1386. [Google Scholar]
- Dahiya, R; Pathak, D. First Total Synthesis and Biological Evaluation of Halolitoralin A. J. Serb. Chem. Soc 2007, 72, 101–107. [Google Scholar]
- Dahiya, R; Kaur, K. Synthetic and Pharmacological Investigation of Segetalin C as a Novel Antifungal and Cytotoxic Agent. Arzneim. Forsch 2008, 58, 29–34. [Google Scholar]
- Dahiya, R; Kumar, A. Synthetic and Biological Studies on a Cyclopolypeptide of Plant Origin. J. Zhejiang Univ. Sci. B 2008, 9, 391–400. [Google Scholar]
- Dahiya, R; Sharma, RD. Synthesis and Bioactivity of a Novel Cyclic Hexapeptide from Stellaria delavayi. Eur. J. Sci. Res 2008, 21, 277–287. [Google Scholar]
- Dahiya, R; Maheshwari, M; Kumar, A. Toward the Synthesis and Biological Evaluation of Hirsutide. Monatsh. Chem 2009, 140, 121–127. [Google Scholar]
- Dahiya, R; Maheshwari, M; Yadav, R. Synthetic, Cytotoxic and Antimicrobial Activity Studies on Annomuricatin B. Z. Naturforsch 2009, 64b, 237–244. [Google Scholar]
- Bodanzsky, M; Bodanzsky, A. The Practice of Peptide Synthesis; Springer: New York, NY, USA, 1984; pp. 68–143. [Google Scholar]
- Bauer, AW; Kirby, WM; Sherris, JC; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol 1966, 45, 493–496. [Google Scholar]
- Khan, ZK; Gyanchandani, A. Verma, RS, Khan, IK, Singh, AP, Eds.; Trends in antifungals: past, present and future. In Antifungal Agents: Past, Present, Future Prospects; National Academy of Chemistry and Biology: Lucknow, India, 1998; pp. 55–128. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Compound | Diameter of zone of inhibition (mm) | |||||||
|---|---|---|---|---|---|---|---|---|
| Bacterial strains | Fungal strains | |||||||
| B. sub. | S. aur. | P. aeru. | K. pneu. | C. alb. | M. audo. | A. niger | T. menta. | |
| 7 | 10(25)† | 14(12.5) | 22(6) | 23(6) | 21(6) | 11(6) | – | 14(6) |
| 8 | 13(25) | 16(12.5) | 26(6) | 28(6) | 27(6) | 15(6) | – | 18(6) |
| Control * | – | – | – | – | – | – | – | – |
| Gatifloxacin | 18(12.5) | 28(6) | 22(6) | 25(6) | – | – | – | – |
| Griseofulvin | – | – | – | – | 20(6) | 17(6) | 18(12.5) | 20(6) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dahiya, R.; Gautam, H. Total Synthesis and Antimicrobial Activity of a Natural Cycloheptapeptide of Marine Origin. Mar. Drugs 2010, 8, 2384-2394. https://doi.org/10.3390/md8082384
Dahiya R, Gautam H. Total Synthesis and Antimicrobial Activity of a Natural Cycloheptapeptide of Marine Origin. Marine Drugs. 2010; 8(8):2384-2394. https://doi.org/10.3390/md8082384
Chicago/Turabian StyleDahiya, Rajiv, and Hemendra Gautam. 2010. "Total Synthesis and Antimicrobial Activity of a Natural Cycloheptapeptide of Marine Origin" Marine Drugs 8, no. 8: 2384-2394. https://doi.org/10.3390/md8082384