New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation

Abstract
:1. Introduction
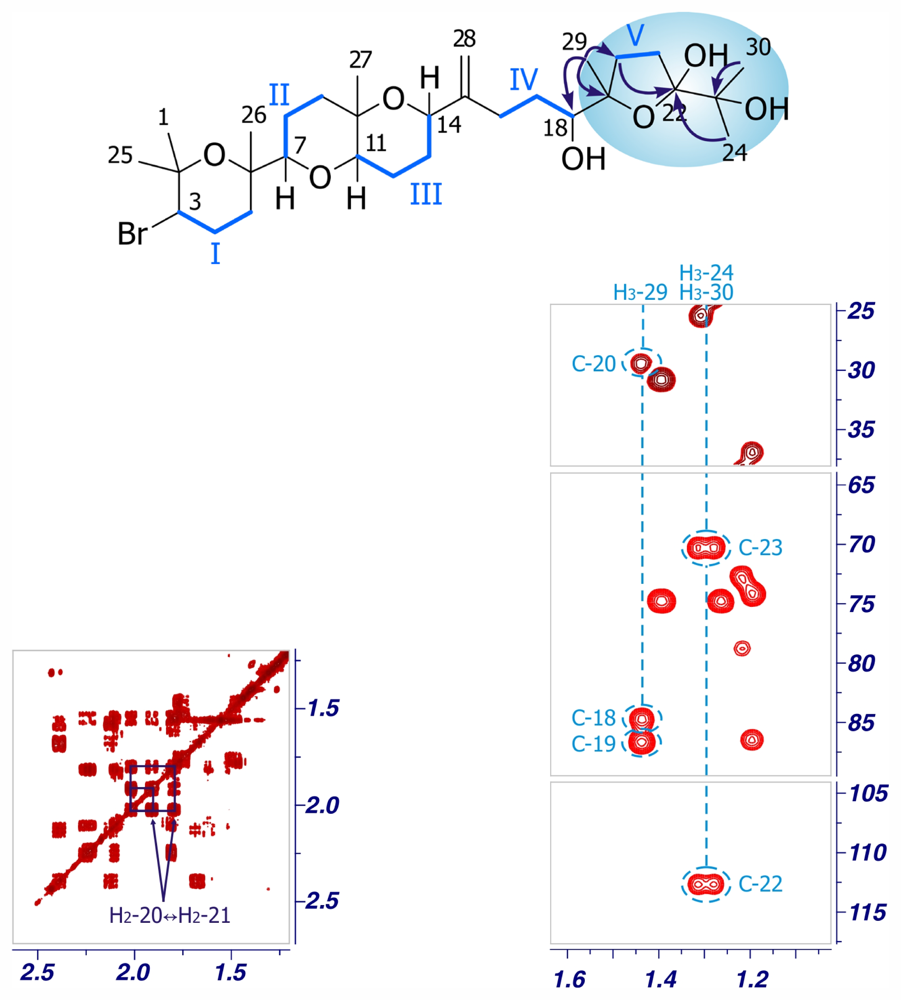
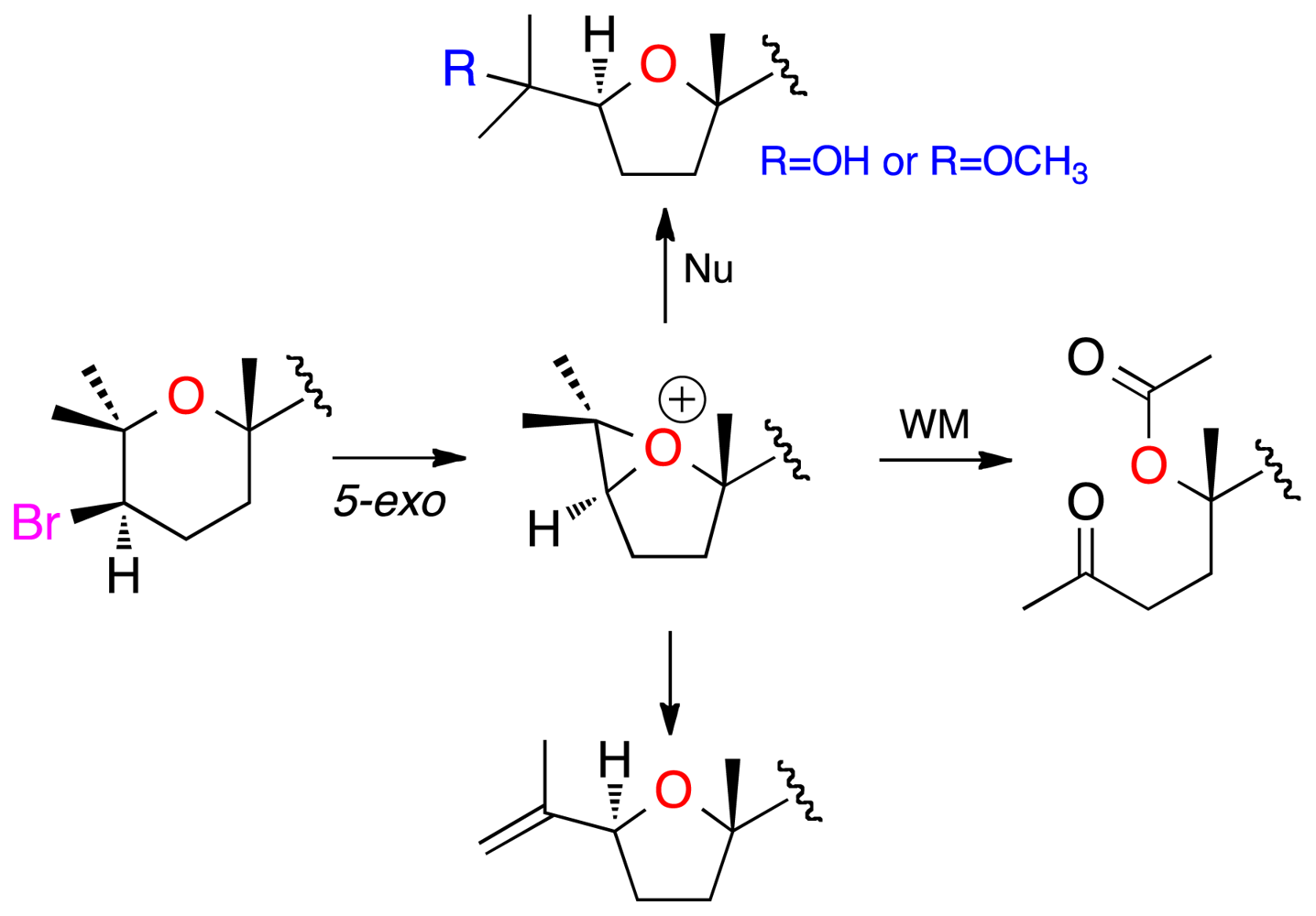
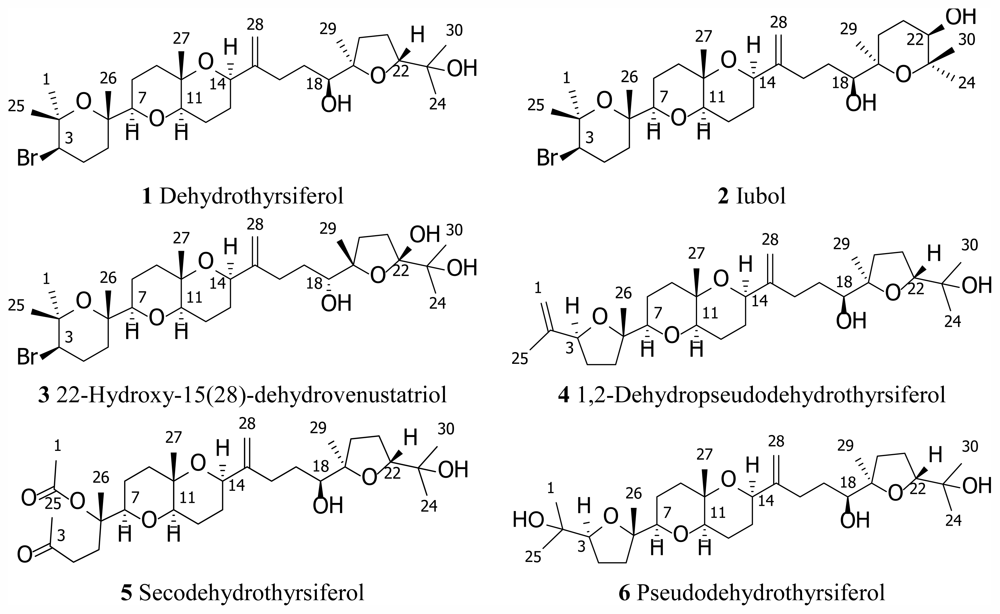
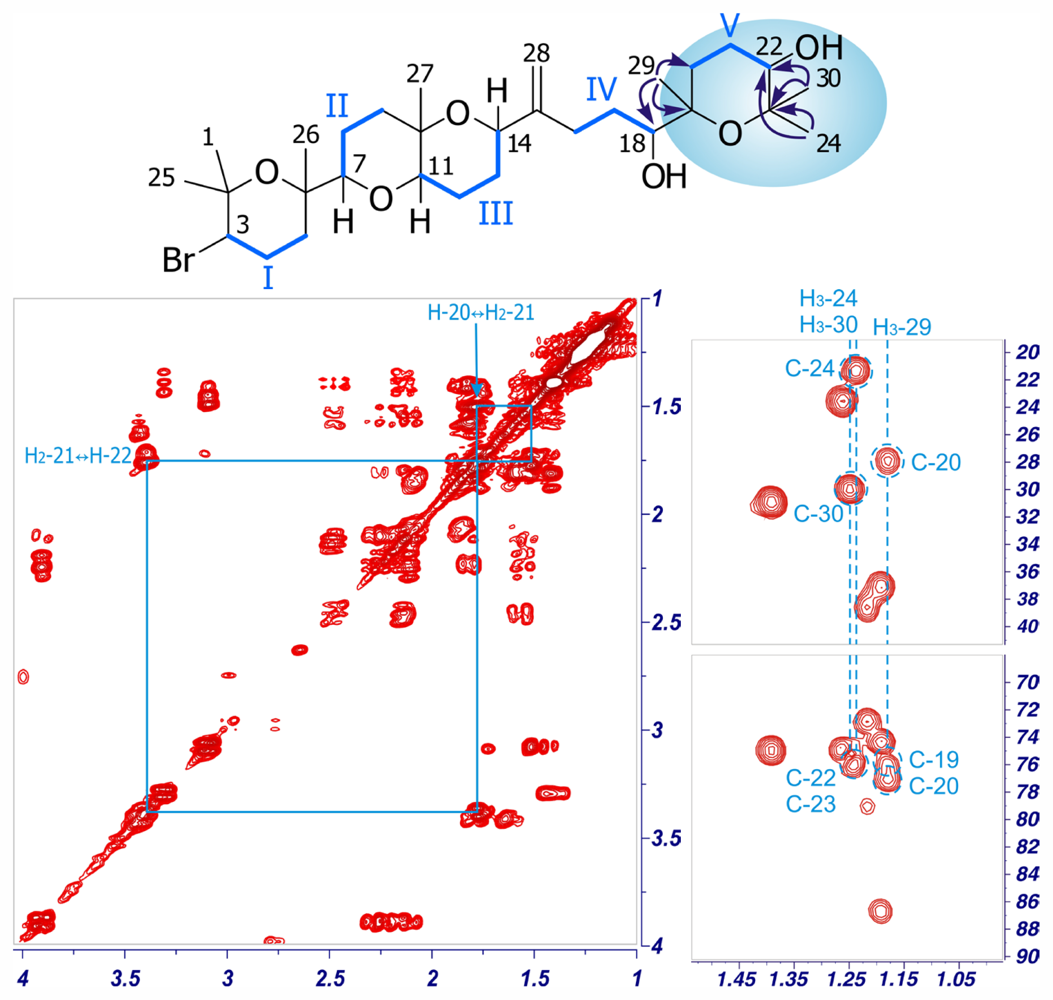
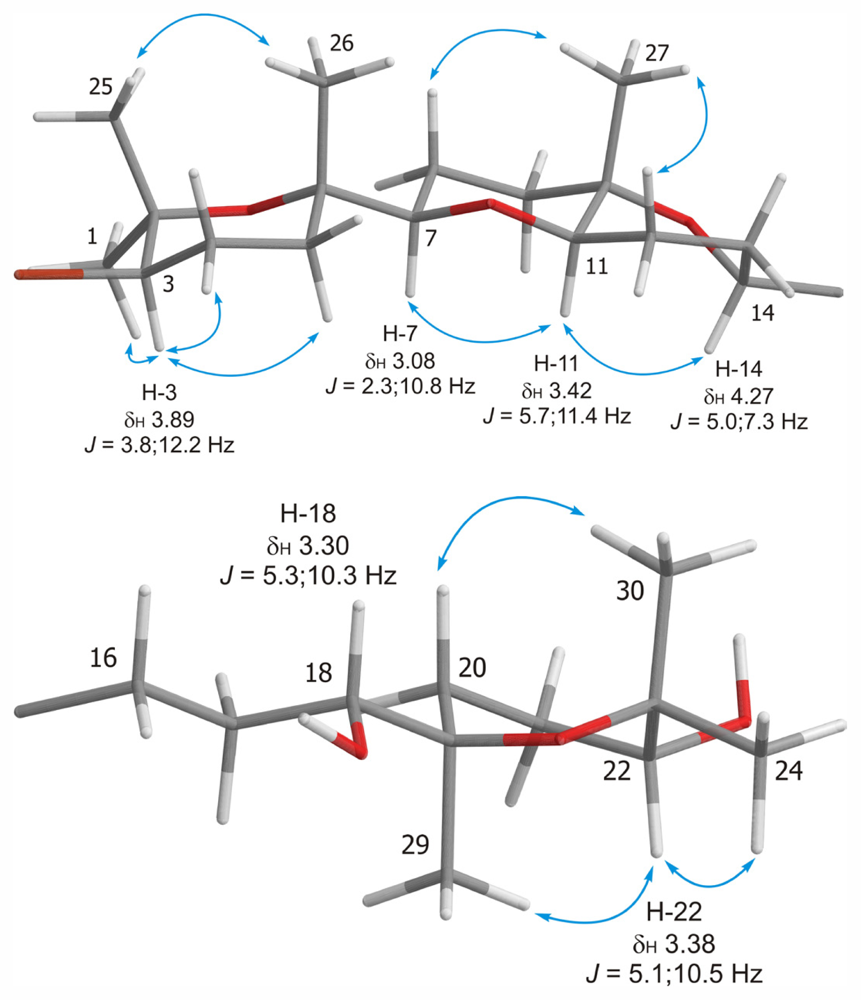
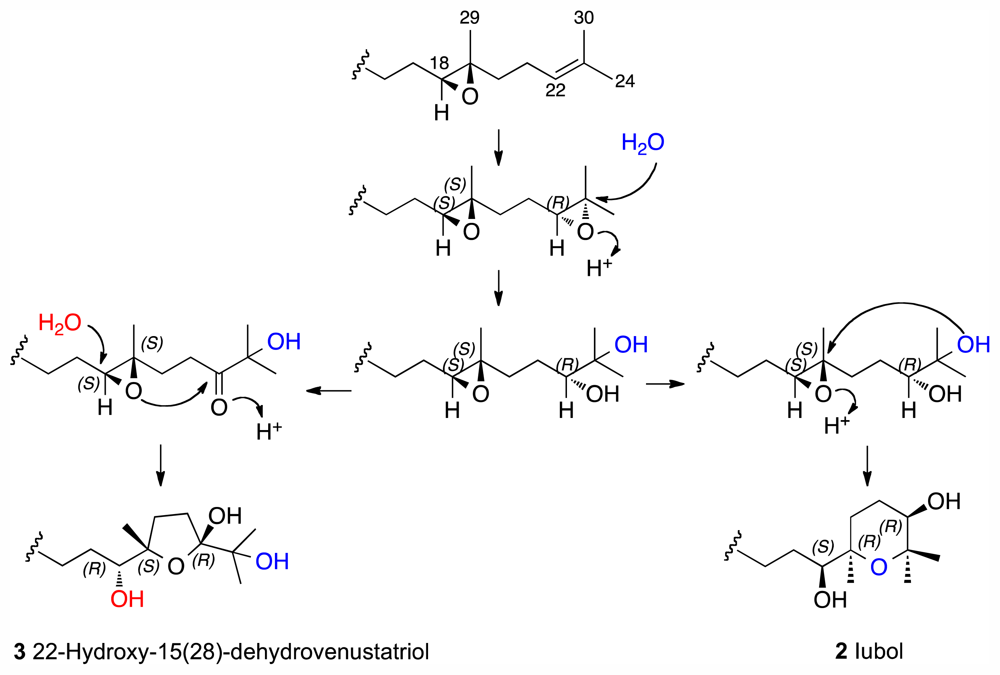
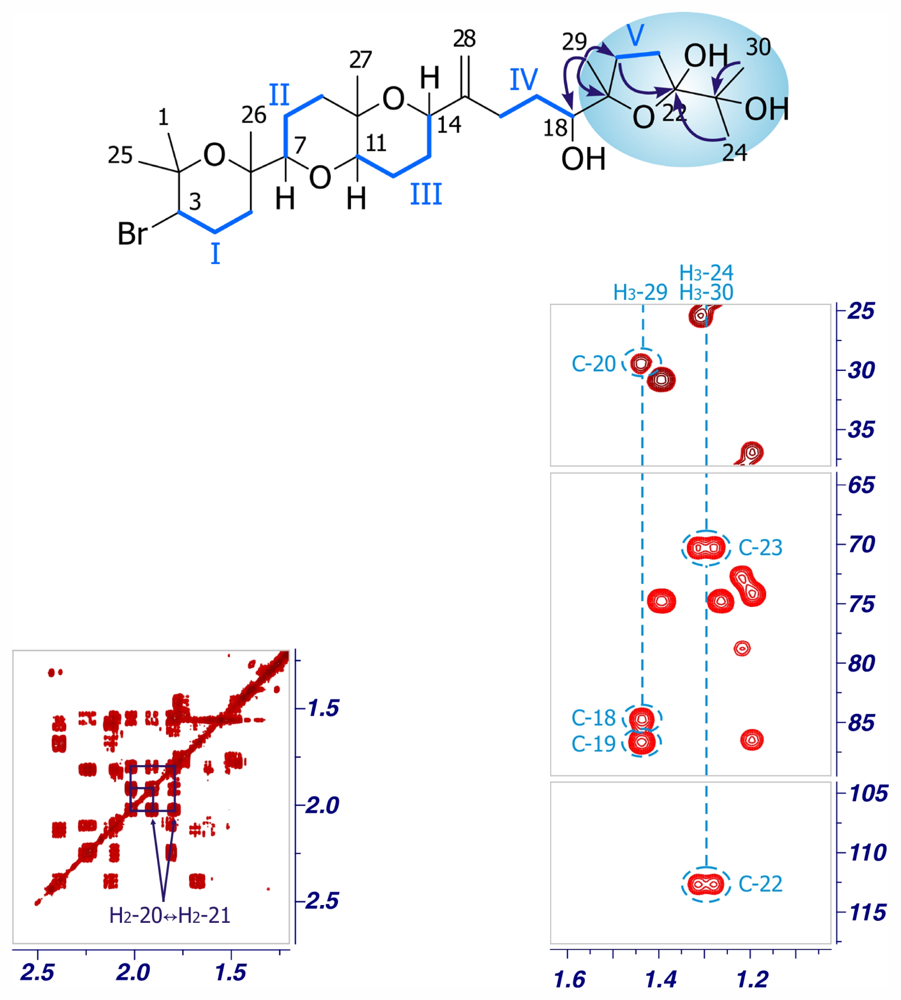
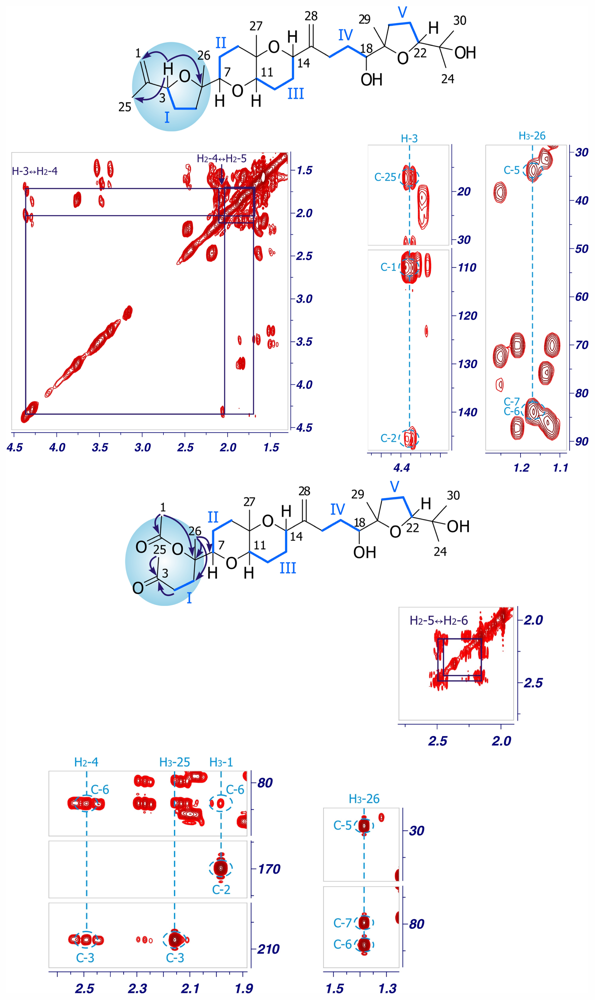
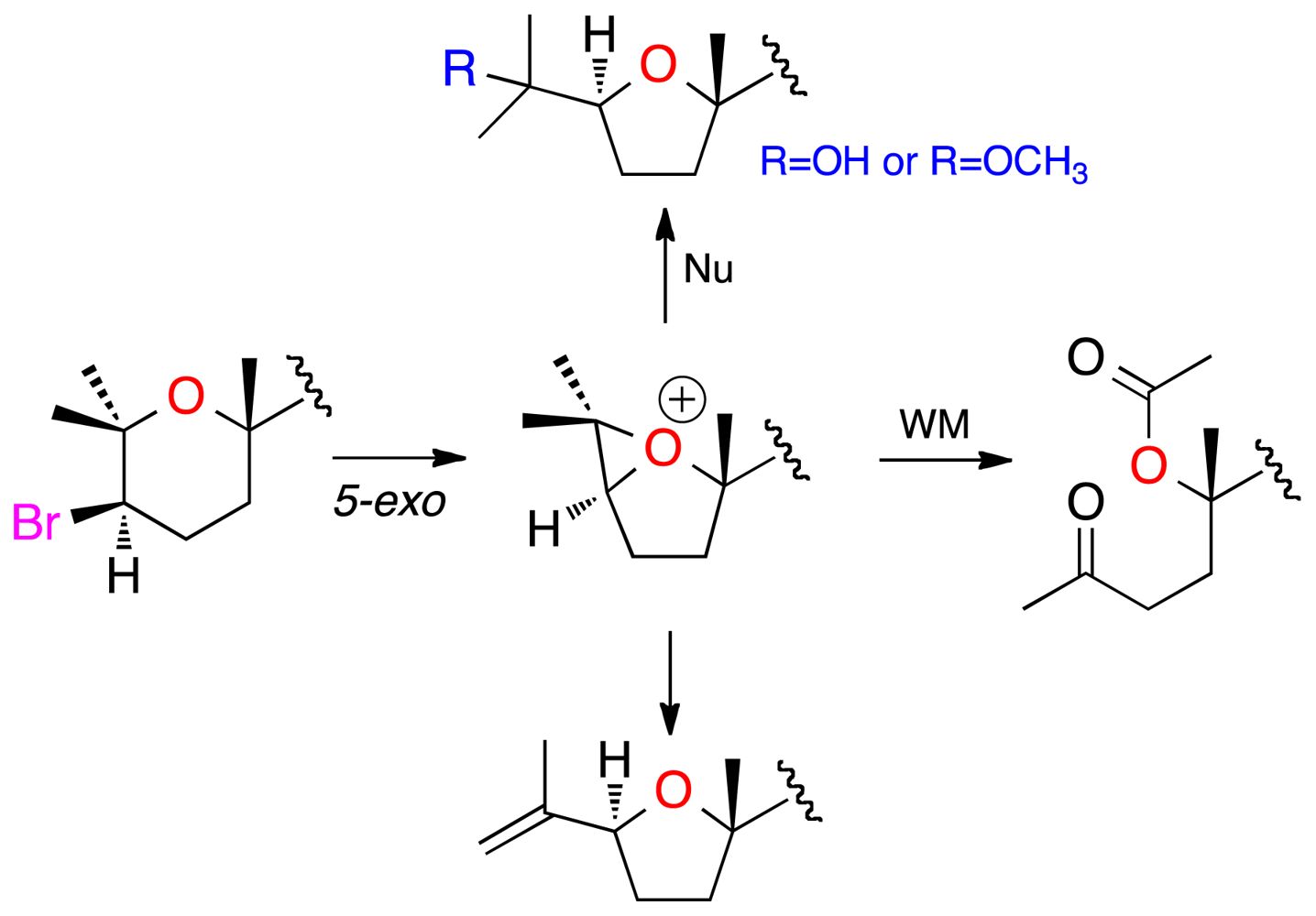
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Chromatographic Separation
Iubol (2)
22-Hydroxy-15(28)-dehydrovenustatriol (3)
1,2-Dehydropseudodehydrothyrsiferol (4)
Secodehydrothyrsiferol (5)
3.4. Chemical Correlations
3.5. Cell Culture
3.6. Cell Growth Inhibition Assay
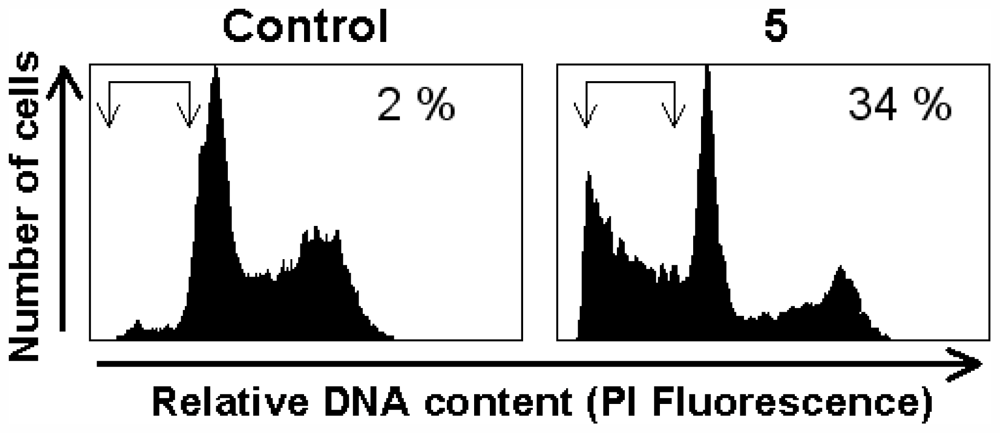
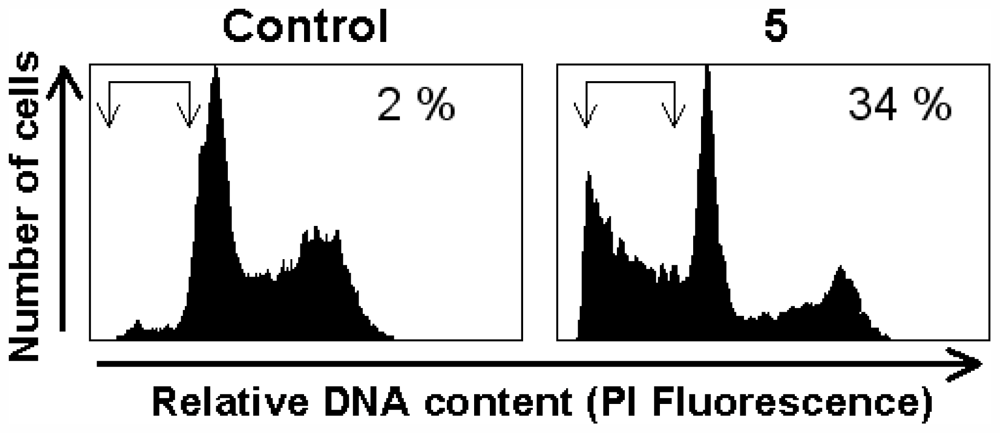
3.7. Apoptosis Assay
Supplementary Material
marinedrugs-09-02220-s001.pdfAcknowledgments
- Samples Availability: Available from the authors.
References
- Cragg, G.M.; Newman, D.J. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod 2007, 70, 461–477. [Google Scholar]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev 2009, 109, 3012–3043. [Google Scholar]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep 2002, 19, 1–48. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep 2011, 28, 196–268. [Google Scholar]
- Fernández, J.J.; Souto, M.L.; Norte, M. Marine polyether triterpenes. Nat. Prod. Rep 2000, 17, 235–246. [Google Scholar]
- Gil-Rodríguez, M.C.; Haroun, R. Laurencia viridis sp. Nov. (Ceramiales, Rhodomelacea) from the macaronesian archipielagos. Bot. Mar 1992, 35, 227–237. [Google Scholar]
- Norte, M.; Fernández, J.J.; Souto, M.L. Evaluation of the cytotoxic activity of polyethers isolated from Laurencia. Bioorg. Med. Chem 1998, 6, 2237–2243. [Google Scholar]
- Souto, M.L.; Manríquez, C.P.; Norte, M.; Leira, F.; Fernández, J.J. The inhibitory effects of squalene-derived triterpenes on protein phosphatase PP2A. Bioorg. Med. Chem. Lett 2003, 13, 1261–1264. [Google Scholar]
- Pec, M.K.; Aguirre, A.; Moser-Thier, K.; Fernández, J.J.; Souto, M.L.; Dorta, J.F.; Díaz-González, F.; Villar, J. Induction of apoptosis in estrogen dependent and independent breast cancer cells by the marine terpenoid dehydrothyrsiferol. Biochem. Pharm 2003, 65, 1451–1461. [Google Scholar]
- Pec, M.K.; Artwohl, M.; Fernández, J.J.; Souto, M.L.; Giraldes, T.; de la Rosa, A.D.; Valenzuela-Fernández, A.; Díaz-González, F. Chemical modulation of VLA integrin affinity in human breast cancer cells. Exp. Cell Res 2007, 313, 1121–1134. [Google Scholar]
- Cen-Pacheco, F.; Villa-Pulgarin, J.A.; Mollinedo, F.; Norte, M.; Daranas, A.; Fernández, J.J. Cytotoxic oxasqualenoids from the red alga Laurencia viridis. Eur. J. Med. Chem 2011, 46, 3302–3308. [Google Scholar]
- Gonzalez, A.G.; Arteaga, J.M.; Fernandez, J.J.; Martín, J.D.; Norte, M.; Ruano, J.Z. Terpenoids of the red alga Laurencia pinnatifida. Tetrahedron 1984, 40, 2751–2755. [Google Scholar]
- Dewick, P.M. Secondary Metabolism: The Building Blocks and Construction Mechanisms. In Medicinal Natural Products: A Biosinthetic Approach, 3rd ed; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 7–38. [Google Scholar]
- Braddock, D.C. A hypothesis concerning the biosynthesis of the obsutallene family of marine natural products via electrophilic bromination. Org. Lett 2006, 8, 6055–6058. [Google Scholar]
- Souto, M.L.; Manríquez, C.P.; Norte, M.; Fernández, J.J. Novel marine polyethers. Tetrahedron 2002, 58, 8119–8125. [Google Scholar]
- Manríquez, C.P.; Souto, M.L.; Gavín, J.A.; Norte, M.; Fernández, J.J. Several new squalene-derived triterpenes from Laurencia. Tetrahedron 2001, 57, 3117–3123. [Google Scholar]
- Pec, M.K.; Aguirre, A.; Fernández, J.J.; Souto, M.L.; Dorta, J.; Villar, J. Dehydrothyrsiferol does not modulate multidrug resistance-associated protein 1 resistance: A functional screening system for MRP1 substrates. Int. J. Mol. Med 2002, 10, 605–608. [Google Scholar]
- David-Cordonnier, M.H.; Gajate, C.; Olmea, O.; Laine, W.; de la Iglesia-Vicente, J.; Perez, C.; Cuevas, C.; Otero, G.; Manzanares, I.; Bailly, C.; et al. DNA and non-DNA targets in the mechanism of action of the antitumor drug trabectedin. Chem. Biol 2005, 12, 1201–1210. [Google Scholar]
- Gajate, C.; Santos-Beneit, A.M.; Macho, A.; Lazaro, M.; Hernandez-De Rojas, A.; Modolell, M.; Munoz, E.; Mollinedo, F. Involvement of mitochondria and caspase-3 in ET-18-OCH3-induced apoptosis of human leukemic cells. Int. J. Cancer 2000, 86, 208–218. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dehydrothyrsiferol (1) | Iubol (2) | |||||||
|---|---|---|---|---|---|---|---|---|
| Position | δC, mult. | δH (J in Hz) | HMBC a | δC, mult. | δH (J in Hz) | HMBC a | ||
| 1 | 31.0, | CH3 | 1.27, s | 2, 3, 25 | 31.0, | CH3 | 1.26, s | 2, 3, 25 |
| 2 | 74.9, | C | 74.9, | C | ||||
| 3 | 59.0, | CH | 3.89, dd (4.1, 12.6) | 1, 2, 4, 5, 25 | 59.1, | CH | 3.89, dd (3.8, 12.2) | 1, 2, 5, 25 |
| 4 | 28.2, | CH2 | 2.13(α), m 2.26(β), m | 2, 3, 5, 6 | 28.3, | CH2 | 2.10(α), m 2.24(β), m | 2, 3, 5, 6 |
| 5 | 37.1, | CH2 | 1.52(α), m 1.80(β), m | 3, 4, 6, 7 | 37.1, | CH2 | 1.53(α), m 1.80(β), m | 3, 4, 6, 7 |
| 6 | 74.4, | C | 74.3, | C | ||||
| 7 | 86.7, | CH | 3.08, dd (2.5, 11.0) | 6, 8, 9, 11 | 86.6, | CH | 3.08, dd (2.3, 10.8) | 6, 8, 9, 11 |
| 8 | 22.9, | CH2 | 1.49(β), m 1.76(α), m | 7, 10 | 23.0, | CH2 | 1.45(β), m 1.72(α), m | 7, 10 |
| 9 | 38.7, | CH2 | 1.44(α), m 1.77(β), m | 7, 10, 11 | 38.7, | CH2 | 1.50(α), m 1.77(β), m | 7, 10, 11 |
| 10 | 72.9, | C | 72.9, | C | ||||
| 11 | 78.9, | CH | 3.43, dd (5.7, 11.3) | 7, 9, 10, 12, 13 | 79.1, | CH | 3.42, dd (5.7, 11.4) | 7, 10, 13 |
| 12 | 21.8, | CH2 | 1.60(β), m 1.81(α), m | 10, 11, 14 | 21.8, | CH2 | 1.63(β), m 1.78(α), m | 10, 11, 14 |
| 13 | 26.6, | CH2 | 1.86(β), m 2.11(α), m | 11, 14 | 26.3, | CH2 | 1.82(β), m 2.03(α), m | 11, 14 |
| 14 | 72.5, | CH | 4.28, dd (4.0, 8.1) | 10, 15, 16, 28 | 72.4, | CH | 4.27, dd (4.8, 7.3) | 15, 16, 28 |
| 15 | 151.3, | C | 151.3, | C | ||||
| 16 | 29.9, | CH2 | 2.12, m 2.43, m | 14, 15, 28, 18 | 29.9, | CH2 | 2.12, m 2.46, m | 14, 15, 28 |
| 17 | 30.3, | CH2 | 1.45, m 1.63, m | 18, 19 | 28.2, | CH2 | 1.38 | 18, 19 |
| 18 | 76.2, | CH | 3.52, dd (1.7, 10.4) | 16, 17, 19, 20 | 77.0, | CH | 3.30, dd (1.3, 10.5) | 16, 19, 20 |
| 19 | 86.0, | C | 76.1, | C | ||||
| 20 | 31.7, | CH2 | 1.60, m 2.12, m | 18, 19, 22 | 27.9, | CH2 | 1.50, m 1.79, m | 18, 19 |
| 21 | 26.3, | CH2 | 1.84, m | 19, 22 | 24.7, | CH2 | 1.75, m | 22 |
| 22 | 87.6, | CH | 3.76, dd (6.0, 9.9) | 19, 23, 24, 30 | 75.2, | CH | 3.38, dd (5.1, 10.5) | 23, 24, 30 |
| 23 | 70.5, | C | 75.7, | C | ||||
| 24 | 24.0, | CH3 | 1.12, s | 22, 23, 30 | 21.4, | CH3 | 1.24, s | 22, 23, 30 |
| 25 | 23.6, | CH3 | 1.39, s | 1, 2, 3 | 23.6, | CH3 | 1.39, s | 1, 2, 3 |
| 26 | 20.1, | CH3 | 1.20, s | 5, 6, 7 | 20.0, | CH3 | 1.20, s | 5, 6, 7 |
| 27 | 19.4, | CH3 | 1.23, s | 9, 10, 11 | 19.3, | CH3 | 1.22, s | 9, 10, 11 |
| 28 | 109.8, | CH2 | 4.88, bs 5.05, bs | 14, 15, 16 | 109.7, | CH2 | 4.86, bs 5.03, bs | 14, 15, 16 |
| 29 | 23.7, | CH3 | 1.14, s | 18, 19, 20 | 22.4, | CH3 | 1.18, s | 18, 19, 20 |
| 30 | 27.7, | CH3 | 1.21, s | 22, 23, 24 | 30.0, | CH3 | 1.25, s | 22, 23, 24 |
| 22-Hydroxy-15(28)dehydrovenustatriol (3) | 1,2-Dehydropseudodehydrothyrsiferol (4) | |||||||
|---|---|---|---|---|---|---|---|---|
| Position | δC, mult. | δH (J in Hz) | HMBC a | δC, mult. | δH (J in Hz) | HMBC a | ||
| 1 | 31.0, | CH3 | 1.27, s | 2, 3, 25 | 110.0, | CH2 | 4.77, bs 4.99, bs | 2, 3, 25 |
| 2 | 74.7, | C | 145.3, | C | ||||
| 3 | 59.0, | CH | 3.89, dd (3.9, 12.5) | 1, 2, 5, 25 | 83.2, | CH | 4.36, dd (6.1,8.7) | 1, 2, 4, 25 |
| 4 | 28.2, | CH2 | 2.11(α), m 2.25(β), m | 2, 3, 5, 6 | 30.9, | CH2 | 1.70, m 2.04, m | 3, 6 |
| 5 | 36.9, | CH2 | 1.52(α), m 1.81(β), m | 3, 4, 6, 7, 26 | 34.2, | CH2 | 1.62, m 2.10, m | 6, 7, 26 |
| 6 | 74.3, | C | 84.1, | C | ||||
| 7 | 86.4, | CH | 3.08, dd (2.2, 11.2) | 6, 9, 11, 26 | 83.7, | CH | 3.37, dd (2.7,11.0) | 6, 9, 11, 26 |
| 8 | 22.7, | CH2 | 1.47(β), m 1.75(α), m | 7, 10 | 24.6, | CH2 | 1.47(β), m 1.65(α), m | 6, 7, 10 |
| 9 | 38.5, | CH2 | 1.53(α), m 1.78(β), m | 7, 10, 11 | 38.4, | CH2 | 1.56(β), m 1.80(α), m | 10, 11 |
| 10 | 73.0, | C | 72.4, | C | ||||
| 11 | 78.8, | CH | 3.42, dd (5.7, 11.3) | 7, 10, 13, 27 | 78.6, | CH | 3.48, dd (5.5,11.6) | 9, 10, 13, 27 |
| 12 | 21.7, | CH2 | 1.61(β), m 1.81(α), m | 10, 11, 14 | 21.4, | CH2 | 1.66(β), m 1.82(α), m | 10, 11 |
| 13 | 26.6, | CH2 | 1.85(β), m 2.10(α), m | 11, 14 | 26.4, | CH2 | 1.85(β), m 2.09(α), m | 11, 14, 15 |
| 14 | 72.5, | CH | 4.26, dd (4.4, 7.4) | 15, 16, 28 | 72.3, | CH | 4.28, dd (4.0,7.7) | 12, 15, 16, 28 |
| 15 | 151.0, | C | 151.0, | C | ||||
| 16 | 29.9, | CH2 | 2.12, m 2.39, m | 14, 15, 28 | 29.3, | CH2 | 2.18, m 2.46, m | 15, 28 |
| 17 | 29.5, | CH2 | 1.57, m 1.68, m | 18, 19 | 29.7, | CH2 | 1.48, m 1.64, m | 15, 18 |
| 18 | 84.8, | CH | 3.55, m | 16, 19, 20, 29 | 76.0, | CH | 3.52, d (9.2) | 17, 19, 20, 29 |
| 19 | 86.9, | C | 86.1, | C | ||||
| 20 | 29.3, | CH2 | 2.00, m | 18, 19 | 31.3, | CH2 | 1.57, m 2.08, m | 18, 19 |
| 21 | 32.0, | CH2 | 1.80, m 1.91, m | 19, 22 | 26.3, | CH2 | 1.84, m | 19, 22 |
| 22 | 112.5, | C | 87.4, | CH | 3.76, dd (5.8,10.2) | 21, 23, 24, 30 | ||
| 23 | 70.4, | C | 70.3, | C | ||||
| 24 | 25.2, | CH3 | 1.29, s | 22, 23, 24 | 23.7, | CH3 | 1.12, s | 22, 23, 30 |
| 25 | 23.3, | CH3 | 1.40, s | 1, 2, 3 | 17.2, | CH3 | 1.70, s | 1, 2, 3 |
| 26 | 19.7, | CH3 | 1.20, s | 5, 6, 7 | 22.7, | CH3 | 1.17, s | 5, 6, 7 |
| 27 | 19.1, | CH3 | 1.22, s | 9, 10, 11 | 19.5, | CH3 | 1.26, s | 9, 10, 11 |
| 28 | 109.4, | CH2 | 4.86, bs 5.05, bs | 14, 15, 16 | 109.6, | CH2 | 4.88, bs 5.05, bs | 14, 15, 16 |
| 29 | 17.5, | CH3 | 1.44, s | 18, 19, 20 | 23.6, | CH3 | 1.14, s | 18, 19, 20 |
| 30 | 23.6, | CH3 | 1.31, s | 22, 23, 30 | 27.5, | CH3 | 1.21, s | 22, 23, 24 |
| Secodehydrothyrsiferol (5) | Pseudodehydrothyrsiferol (6) | |||||||
|---|---|---|---|---|---|---|---|---|
| Position | δC, mult. | δH (J in Hz) | HMBC a | δC, mult. | δH (J in Hz) | HMBC a | ||
| 1 | 22.2, | CH3 | 1.98, s | 6, 25 | 24.0, | CH3 | 1.11, s | 2, 3, 25 |
| 2 | 170.1, | C | 70.6, | C | ||||
| 3 | 208.5, | C | 86.7, | CH | 3.76, dd (5.8, 9.1) | 1, 2, 4, 5, 25 | ||
| 4 | 38.1, | CH2 | 2.50, m | 3, 5, 6 | 26.3, | CH2 | 1.84, m | 3, 6 |
| 5 | 29.3, | CH2 | 2.16, m 2.26, m | 3, 4, 6, 7, 26 | 35.2, | CH2 | 1.66, m 2.04, m | 6, 7, 26 |
| 6 | 84.1, | C | 84.0, | C | ||||
| 7 | 80.0, | CH | 3.93, dd (3.9, 9.9) | 5, 6, 9, 11, 26 | 84.0, | CH | 3.32, dd (2.6, 11.4) | 6, 9, 11, 26 |
| 8 | 23.6, | CH2 | 1.56, m | 7, 10 | 24.5, | CH2 | 1.51(β), m 1.66(α), m | 7, 10 |
| 9 | 38.6, | CH2 | 1.57(β), m 1.81(α), m | 10, 11, 27 | 38.7, | CH2 | 1.57(β), m 1.81(α), m | 10, 11, 27 |
| 10 | 72.5, | C | 72.8, | C | ||||
| 11 | 79.0, | CH | 3.46, dd (5.6, 11.4) | 9, 10, 13, 27 | 78.9, | CH | 3.46, dd (5.6, 11.7) | 9, 10, 13, 27 |
| 12 | 21.7, | CH2 | 1.63(β), m 1.83(α), m | 10, 11 | 21.8, | CH2 | 1.65(β), m 1.84(α), m | 10, 11 |
| 13 | 26.2, | CH2 | 1.84(β), m 2.05(α), m | 11, 14, 15 | 26.4, | CH2 | 1.85(β), m 2.08(α), m | 11, 14, 15 |
| 14 | 72.5, | CH | 4.28, dd (3.8, 7.5) | 13, 15, 16, 28 | 72.5, | CH | 4.29, dd (4.2, 7.1) | 13, 15, 16, 28 |
| 15 | 151.1, | C | 151.3, | C | ||||
| 16 | 29.5, | CH2 | 2.16, m 2.45, m | 15, 28 | 29.7, | CH2 | 2.20, m 2.46, m | 15, 28 |
| 17 | 29.9, | CH2 | 1.46, m 1.64, m | 15, 18 | 29.9, | CH2 | 1.48, m 1.64, m | 15, 18 |
| 18 | 76.2, | CH | 3.52, dd (1.4, 10.4) | 19, 20, 29 | 76.2, | CH | 3.53, dd (1.5, 10.8) | 17, 19, 20, 29 |
| 19 | 86.1, | C | 86.1, | C | ||||
| 20 | 31.5, | CH2 | 1.57, m 2.10, m | 18, 19 | 31.6, | CH2 | 1.58, m 2.10, m | 18, 19 |
| 21 | 26.6, | CH2 | 1.84, m | 19, 22 | 26.5, | CH2 | 1.83, m | 19, 22 |
| 22 | 87.6, | CH | 3.76, dd (5.8, 10.1) | 23, 24, 30 | 87.6, | CH | 3.76, dd (6.5, 9.8) | 21, 23, 24, 30 |
| 23 | 70.4, | C | 70.4, | C | ||||
| 24 | 23.9, | CH3 | 1.13, s | 22, 23, 30 | 23.9, | CH3 | 1.13, s | 22, 23, 30 |
| 25 | 29.9, | CH3 | 2.15, s | 3 | 27.5, | CH3 | 1.19, s | 1, 2, 3 |
| 26 | 20.0, | CH3 | 1.39, s | 5, 6, 7 | 22.7, | CH3 | 1.14, s | 5, 6, 7 |
| 27 | 19.5, | CH3 | 1.25, s | 9, 10, 11 | 19.4, | CH3 | 1.25, s | 9, 10, 11 |
| 28 | 110.0, | CH2 | 4.89, bs 5.05, bs | 14, 15, 16 | 109.9, | CH2 | 4.89, bs 5.05, bs | 14, 15, 16 |
| 29 | 23.8, | CH3 | 1.15, s | 18, 19, 20 | 23.7, | CH3 | 1.14, s | 18, 19, 20 |
| 30 | 27.7, | CH3 | 1.22, s | 22, 23, 24 | 27.7, | CH3 | 1.21, s | 22, 23, 24 |
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| Jurkat | MM144 | HeLa | CADO-ES1 | |
| 1 | 13.5 ± 1.8 | 21.5 ± 2.1 | 34.5 ± 3.2 | 12.0 ± 1.4 |
| 2 | 3.5 ± 0.4 | 13.0 ± 1.9 | 27.0 ± 2.6 | 11.0 ± 1.5 |
| 3 | 2.0 ± 0.2 | ND | 2.9 ± 0.5 | ND |
| 4 | 15.5 ± 2.8 | 16.5 ± 2.5 | 24.0 ± 3.5 | 10.6 ± 1.5 |
| 5 | 2.5 ± 0.3 | 12.0 ± 1.7 | 30.0 ± 3.5 | 12.2 ± 1.6 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pacheco, F.C.; Villa-Pulgarin, J.A.; Mollinedo, F.; Martín, M.N.; Fernández, J.J.; Daranas, A.H. New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation. Mar. Drugs 2011, 9, 2220-2235. https://doi.org/10.3390/md9112220
Pacheco FC, Villa-Pulgarin JA, Mollinedo F, Martín MN, Fernández JJ, Daranas AH. New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation. Marine Drugs. 2011; 9(11):2220-2235. https://doi.org/10.3390/md9112220
Chicago/Turabian StylePacheco, Francisco Cen, Janny A. Villa-Pulgarin, Faustino Mollinedo, Manuel Norte Martín, José Javier Fernández, and Antonio Hernández Daranas. 2011. "New Polyether Triterpenoids from Laurencia viridis and Their Biological Evaluation" Marine Drugs 9, no. 11: 2220-2235. https://doi.org/10.3390/md9112220