Marine Polysaccharides in Microencapsulation and Application to Aquaculture: “From Sea to Sea”


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Polysaccharides
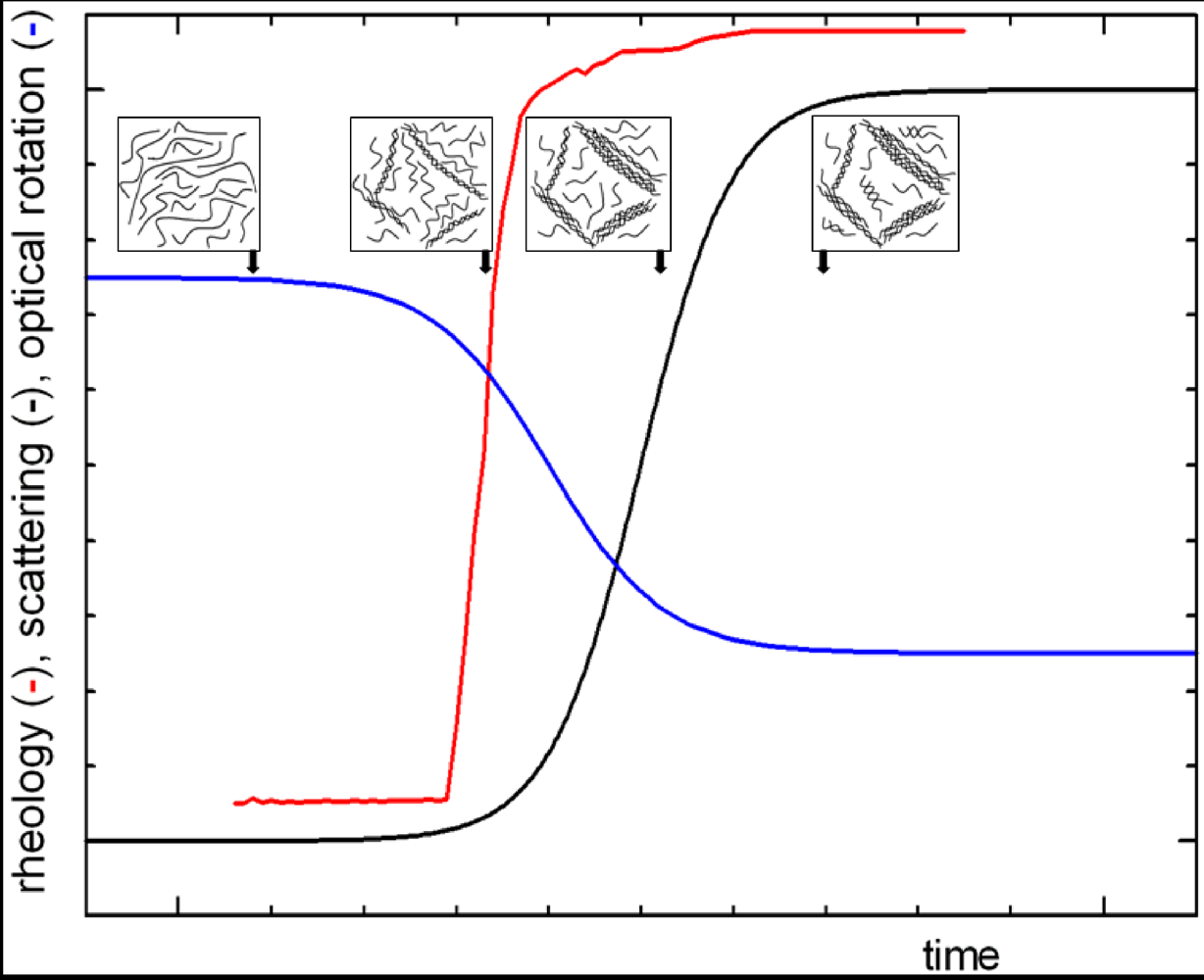
2.1. Structure and Shape Determinants for Aggregation and Gelation

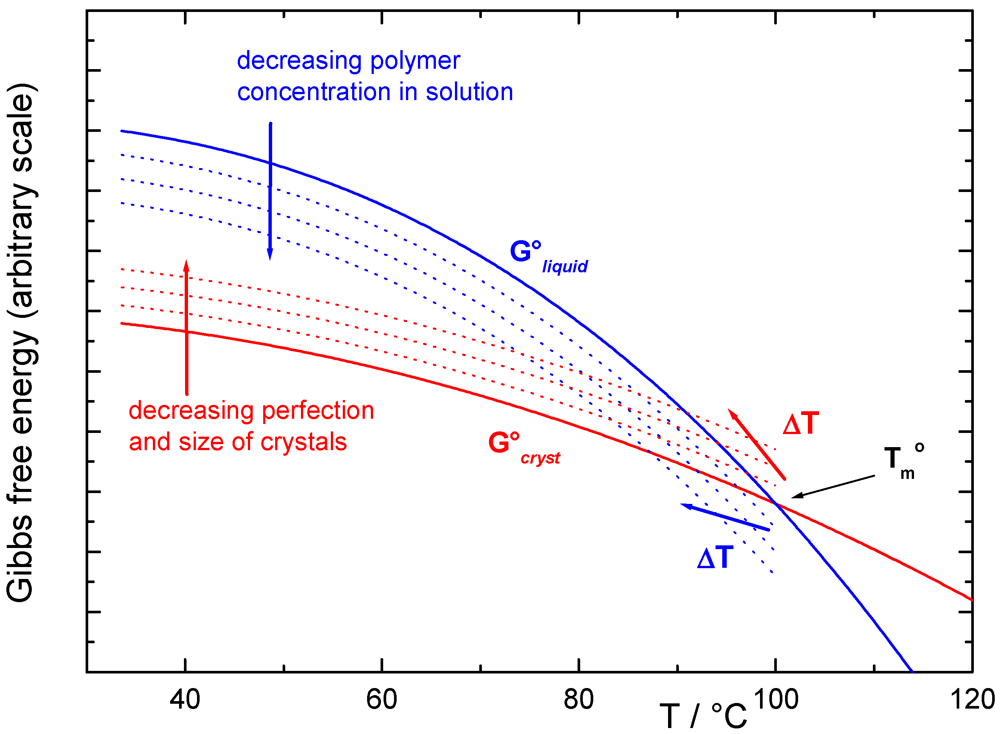
2.2. Thermodynamic Considerations

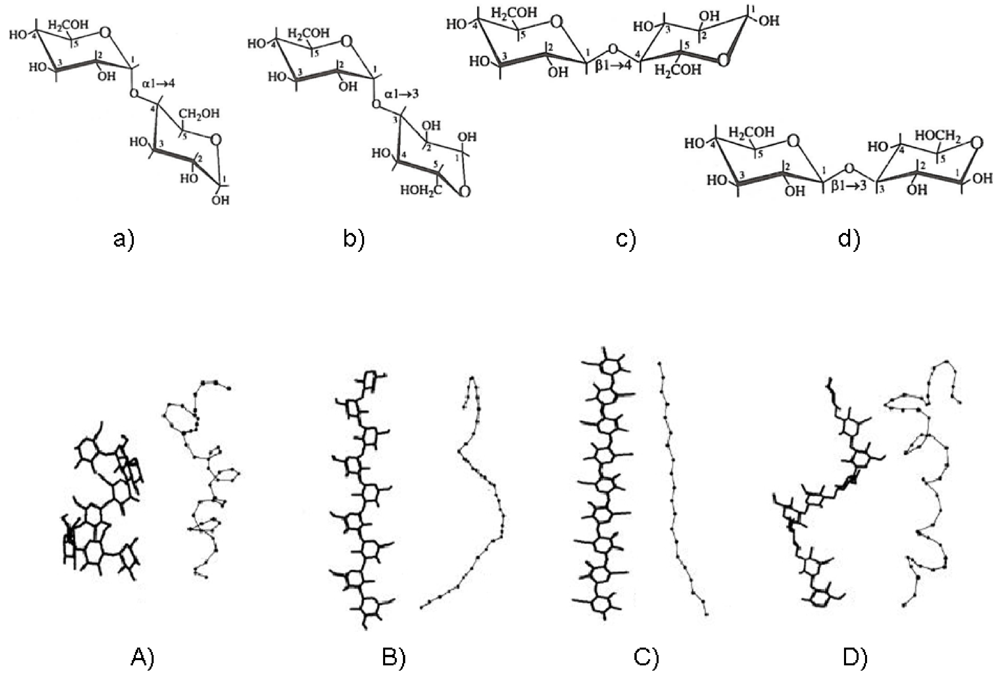
2.3. How Do Polysaccharide Chain Solution Properties Depend on Sugar Monomer Composition?

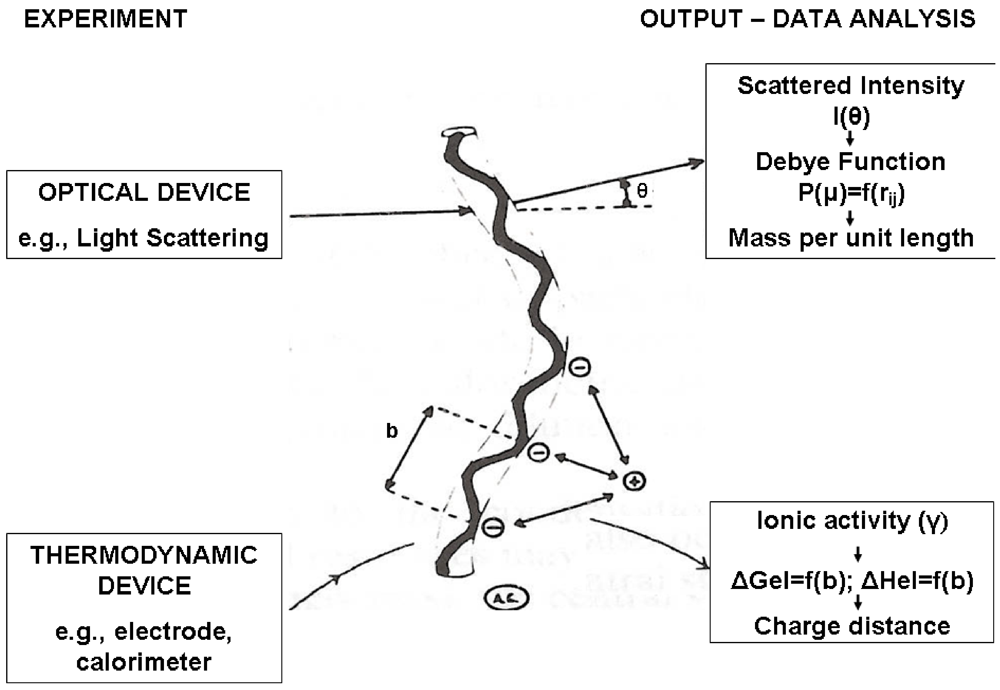
2.4. Solutions of Ionic Polysaccharides

2.5. Marine Polysaccharides: Alginate and Chitosan Solutions
3. Encapsulation in Polysaccharide Hydrogels
3.1. Polysaccharide-Based Hydrogels for Technological Applications
3.2. Pharmaceutical Applications
3.3. Other Applications
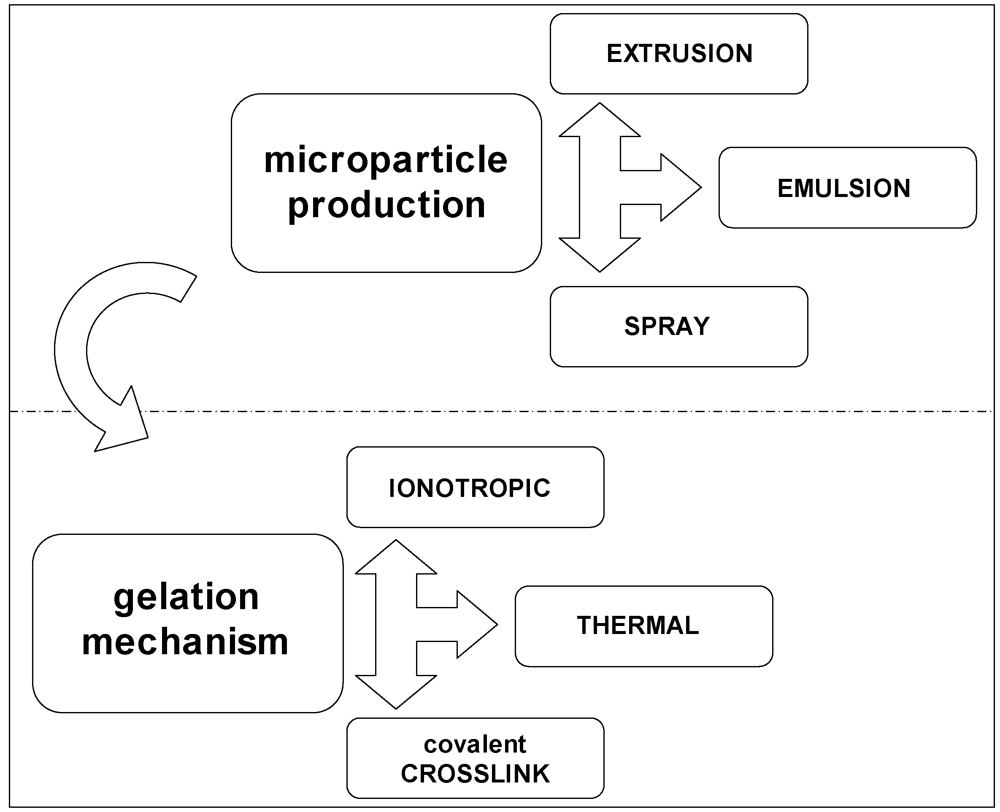
3.4. Methods for Microencapsulation


3.5. Microparticle Production
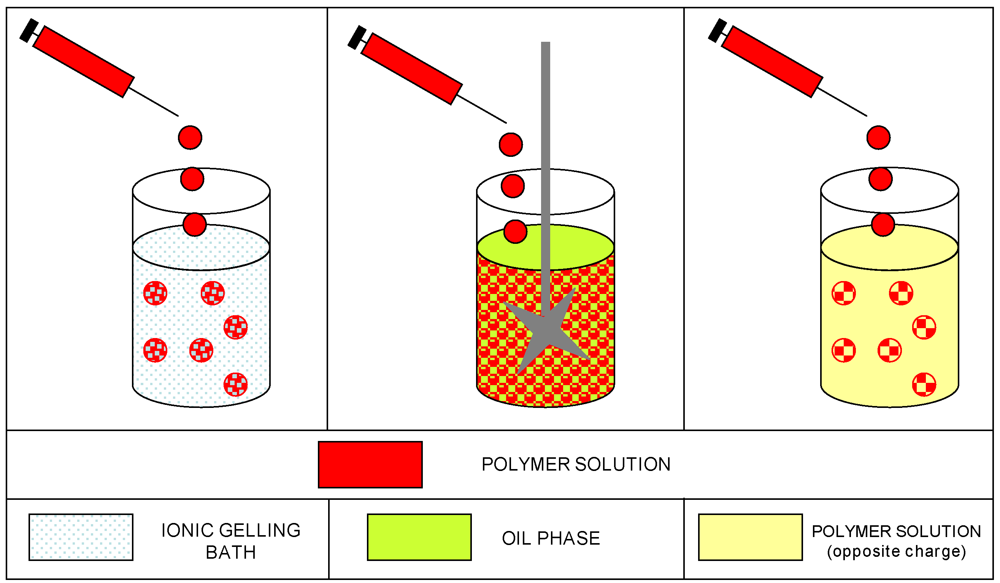
3.6. Gelation Methods
4. Microencapsulation for Fish Vaccination in Aquaculture: A Case Study
4.1. Fish Vaccination in Aquaculture
4.2. Case Study

5. Conclusions
Acknowledgments
- Samples Availability: Available from the authors.
References and Notes
- Donati, I.; Borgogna, M.; Turello, E.; Cesàro, A.; Paoletti, S. Tuning Supramolecular Structuring at the Nanoscale Level: Nonstoichiometric Soluble Complexes in Dilute Mixed Solutions of Alginate and Lactose-Modified Chitosan (Chitlac). Biomacromolecules 2007, 8, 1471–1479. [Google Scholar]
- Cesàro, A.; Bellich, B.; Borgogna, M. Biophysical functionality in polysaccharides: from Lego-blocks to nano-particles. Eur. Biophys. J. 2011, in press. [Google Scholar]
- Woods, R.J.; Tessier, M.B. Computational glycoscience: characterizing the spatial and temporal properties of glycans and glycan-protein complexes. Curr. Opin. Struct. Biol. 2010, 20, 575–583. [Google Scholar]
- Yalpani, M.; Sandford, P.A. Industrial Polysaccharides; Yalpani, M., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; p. 31. [Google Scholar]
- Ross-Murphy, S.B.; Tobitani, A. Hydrocolloids: Physical Chemistry and Industrial Application of Gels, Polysaccharides, and Proteins; Nishinari, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2000; p. 379. [Google Scholar]
- Kawakami, M.; Byrne, K.; Khatri, B.; McLeish, T.C.B.; Radford, S.E.; Smith, D.A. Viscoelastic properties of single polysaccharide molecules determined by analysis of thermally driven oscillations of an atomic force microscope cantilever. Langmuir 2004, 20, 9299–9303. [Google Scholar]
- Khatri, B.S.; Kawakami, M.; Byrne, K.; Smith, D.A.; McLeish, T.C.B. Entropy and barrier-controlled fluctuations determine conformational viscoelasticity of single biomolecules. Biophys. J. 2007, 92, 1825–1835. [Google Scholar]
- Clark, A.; Ross-Murphy, S. Structural and mechanical properties of biopolymer gels. Adv. Polym. Sci. 1987, 83, 57–192. [Google Scholar]
- Makromolekulare Chemie. Macromolecular Symposia; Eichinger, B.E. (Ed.) Wiley-VCH: Weinheim, Germany, 1993; Volume 76, pp. 1–290.
- te Nijenhuis, K. Thermoreversible Networks; Springer: Berlin-Heidelberg, Germany, 1997; Volume 130, p. 1. [Google Scholar]
- Mandelken, L.; Edwards, C.O.; Domszy, R.C.; Davidson, M.W. Microdomains in Polymer Solutions; Dubin, P., Ed.; Plenum Press: New York, NY, USA, 1985; p. 121. [Google Scholar]
- Flory, P.J. Introductory lecture. Faraday Discuss. Chem. Society 1974, 57, 7–18. [Google Scholar]
- Tanaka, F.; Stockmayer, W.H. Thermoreversible gelation with junctions of variable multiplicity. Macromol. Symposia 1994, 81, 171–175. [Google Scholar]
- Xiong, J.Y.; Narayanan, J.; Liu, X.Y.; Chong, T.K.; Chen, S.B.; Chung, T.S. Topology evolution and gelation mechanism of agarose gel. J. Phys. Chem. B 2005, 109, 5638–5643. [Google Scholar]
- Clark, A.; Ross-Murphy, S.B. Biopolymer network assembly: measurement and theory. In Modern Biopolymer Science; Kasapis, S., Norton, I.T., Ubbink, J.B., Eds.; Elsevier: London, UK, 2009; p. 1. [Google Scholar]
- Nordqvist, D.; Vilgis, T. Rheological study of the gelation process of agarose-based solutions. Food Biophys. 2011, 6, 450–460. [Google Scholar]
- Nishinari, K.; Takahashi, R. Interaction in polysaccharide solutions and gels. Curr. Opin. Colloid Interface Sci. 2003, 8, 396–400. [Google Scholar]
- Dai, B.; Matsukawa, S. NMR studies of the gelation mechanism and molecular dynamics in agar solutions. Food Hydrocoll. 2012, 26, 181–186. [Google Scholar]
- Miles, M.J. Developments in Crystalline Polymers; Bassett, D.C., Ed.; Elsevier Applied Science: London, UK, 1988; p. 233. [Google Scholar]
- Guenet, M.J. Factors influencing gelation versus crystallization in cooling polymer solutions. Trends Polym. Sci. 1996, 4, 6–11. [Google Scholar]
- Mandelkern, L. Polymer-diluent mixtures. In Crystallization of Polymers; McGraw-Hill: New York, NY, USA, 1964; Chapter 3. [Google Scholar]
- Keller, A.; Rastogi, S.; Hikosaka, M.H. Polymer crystallisation: Role of metastability and the confluence of thermodynamic and kinetic factors. Macromol. Symposia 1997, 124, 67–81. [Google Scholar]
- Hu, Z.; Lu, X.; Gao, J.; Wang, C. Polymer Gel Nanoparticle Networks. Adv. Mater. 2000, 12, 1173–1176. [Google Scholar]
- Rees, D.A. Polysaccharide Shapes; Chapman and Hall: London, UK, 1977. [Google Scholar]
- Burton, B.A.; Brant, D.A. Comparative flexibility, extension, and conformation of some simple polysaccharide chains. Biopolymers 1983, 22, 1769–1792. [Google Scholar]
- Brant, D.A.; Christ, M.D. Realistic Conformational Modeling of Carbohydrates. In Computer Modeling of Carbohydrate Molecules; French, A.D., Brady, J.W., Eds.; American Chemical Society: Washington, DC, USA, 1990; p. 42. [Google Scholar]
- Kroon-Batenburg, L.M.J.; Kruiskamp, P.H.; Vliegenthart, J.F.G.; Kroon, J. Estimation of the persistence length of polymers by MD simulations on small fragments in solution. Application to cellulose. J. Phys. Chem. B 1997, 101, 8454–8459. [Google Scholar]
- Skovstrup, S.; Hansen, S.G.; Skrydstrup, T.; Schiøtt, B. Conformational flexibility of chitosan: A molecular modeling study. Biomacromolecules 2010, 11, 3196–3207. [Google Scholar]
- Jordan, R.C.; Brant, D.A.; Cesàro, A. A Monte Carlo study of the amylosic chain conformation. Biopolymers 1978, 17, 2617–2632. [Google Scholar]
- Kitamura, S.; Minami, T.; Nakamura, Y.; Isuda, H.; Kobayashi, H.; Mimura, M.; Urakawa, H.; Kajiwara, K.; Ohno, S. Chain dimensions and scattering function of (1→3)-β-D-glucan simulated by the Monte Carlo method. J. Mol. Struct. Theochem 1997, 395-396, 425–435. [Google Scholar]
- Furlan, S.; La Penna, G.; Perico, A.; Cesàro, A. Hyaluronan chain conformation and dynamics. Carbohydr. Res. 2005, 340, 959–970. [Google Scholar]
- Hargittai, I.; Hargittai, M. Molecular structure of hyaluronan: An introduction. Struct. Chem. 2008, 19, 697–717. [Google Scholar]
- Brant, D.A. Shapes and motions of polysaccharide chains. Pure Appl. Chem. 1997, 69, 1885–1892. [Google Scholar]
- Kadkhodaei, M.; Wu, H.; Brant, D.A. Comparison of the conformational dynamics of the (1→4)- and (1→6)-linked a-D-glucans using 13C-NMR relaxation. Biopolymers 1991, 31, 1581–1592. [Google Scholar]
- Brant, D.A.; Liu, H.-S.; Zhu, Z.S. The dependence of glucan conformational dynamics on linkage position and stereochemistry. Carbohydr. Res. 1995, 278, 11–26. [Google Scholar]
- Perico, A.; Mormino, M.; Urbani, R.; Cesàro, A.; Tylianakis, E.; Dais, P.; Brant, D.A. Local dynamics of carbohydrates. 1. dynamics of simple glycans with different chain linkages. J. Phys. Chem. B 1999, 103, 8162–8171. [Google Scholar]
- Landström, J.; Widmalm, G. Glycan flexibility: insights into nanosecond dynamics from a microsecond molecular dynamics simulation explaining an unusual nuclear Overhauser effect. Carbohydr. Res. 2010, 345, 330–333. [Google Scholar]
- Urbani, R.; Cesàro, A. Solvent effects on the unperturbed chain conformation of polysaccharides. Polymer 1991, 32, 3013–3020. [Google Scholar]
- Bertocchi, C.; Navarini, L.; Cesàro, A.; Anastasio, M. Polysaccharides from cyanobacteria. Carbohydr. Polym. 1990, 12, 127–153. [Google Scholar]
- Li, P.; Harding, S.E.; Liu, Z. Cyanobacterial exopolysaccharides: their nature and potential biotechnological applications. Biotechnol. Genet. Eng. Rev. 2001, 18, 375–404. [Google Scholar]
- Sutherland, I.W. Polysaccharides from Microorganisms, Plants and Animals. In Biopolymers; Vandamme, E.J., De Baets, S., Steinbüchel, A., Eds.; Wiley-VCH: Weinheim, Germay, 2002; Volume 5, p. 1. [Google Scholar]
- Straub, P.R.; Brant, D.A. Measurement of preferential solvation of some glucans in mixed solvent systems by gel-permeation chromatography. Biopolymers 1980, 19, 639–653. [Google Scholar]
- Kirschner, K.N.; Woods, R.J. Solvent interactions determine carbohydrate conformation. Proc. Natl. Acad. Sci. USA 2001, 98, 10541–10545. [Google Scholar]
- Almond, A.; Sheenan, J.K. Predicting the molecular shape of polysaccharides from dynamic interactions with water. Glycobiology 2003, 13, 255–264. [Google Scholar]
- Kuttel, M.M.; Naidoo, K.J. Free energy surfaces for the α(1→4)-glycosidic linkage: Implications for polysaccharide solution structure and dynamics. J. Phys. Chem. B 2005, 109, 7468–7474. [Google Scholar]
- Pereira, C.S.; Kony, D.; Baron, R.; Müller, M.; Van Gunsteren, W.F.; Hünenberger, P.H. Conformational and dynamical properties of disaccharides in water: A molecular dynamics study. Biophys. J. 2006, 90, 4337–4344. [Google Scholar]
- Antoniou, E.; Buitrago, C.F.; Tsianou, M.; Alexandridis, P. Solvent effects on polysaccharide conformation. Carbohydr. Polym. 2010, 79, 380–390. [Google Scholar]
- Perić-Hassler, L.; Hansen, H.S.; Baron, R.; Hünenberger, P.H. Conformational properties of glucose-based disaccharides investigated using molecular dynamics simulations with local elevation umbrella sampling. Carbohydr. Res. 2010, 345, 1781–1801. [Google Scholar]
- Brady, J.W.; Schmidt, R.K. The role of hydrogen bonding in carbohydrates: Molecular dynamics simulations of maltose in aqueous solution. J. Phys. Chem. 1993, 97, 958–966. [Google Scholar]
- Ueda, K.; Ueda, T.; Sato, T.; Nakayama, H.; Brady, J.W. The conformational free-energy map for solvated neocarrabiose. Carbohydr. Res. 2004, 339, 1953–1960. [Google Scholar]
- Dogsa, I.; Štrancar, J.; Laggner, P.; Stopar, D. Efficient modeling of polysaccharide conformations based on Small-Angle X-ray Scattering experimental data. Polymer 2008, 49, 1398–1406. [Google Scholar]
- Cesàro, A.; Paoletti, S.; Urbani, R.; Benegas, J. Polyelectrolytic effects in semi-flexible carboxylate polysaccharides. Part 2. Int. J. Biol. Macromol. 1989, 11, 66–72. [Google Scholar]
- Paoletti, S.; Cesàro, A.; Delben, F.; Crescenzi, V.; Rizzo, R. Microdomains in Polymer Solutions; Dubin, P., Ed.; Plenum Press: New York, NY, USA, 1985; p. 159. [Google Scholar]
- Paoletti, S.; Benegas, J.; Cesàro, A.; Manzini, G.; Fogolari, F.; Crescenzi, V. Limiting-laws of polyelectrolyte solutions. Ionic distribution in mixed-valency counterions systems. I: The model. Biophys. Chem. 1991, 41, 73–80. [Google Scholar]
- Manning, G.S. Counterion binding in polyelectrolyte theory. Acc. Chem. Res. 1979, 12, 443–449. [Google Scholar]
- Cesàro, A.; Delben, F.; Paoletti, S. Interaction of divalent cations with polyuronates. J. Chem. Soc. Faraday Trans. 1 1988, 84, 2573–2584. [Google Scholar]
- Paoletti, S.; Delben, F.; Cesàro, A.; Grasdalen, H. Conformational transition of κ-carrageenan in aqueous solution. Macromolecules 1985, 18, 1834–1841. [Google Scholar]
- Smidsrød, O.; Haug, A. Estimation of the relative stiffness of the molecular chain in polyelectrolytes from measurements of viscosity at different ionic strengths. Biopolymers 1971, 10, 1213–1227. [Google Scholar]
- Manning, G.S.; Paoletti, S. Industrial Polysaccharides; Crescenzi, V., Dea, I.C.M., Stivala, S.S., Eds.; Gordon & Breach: New York, NY, USA, 1987; p. 305. [Google Scholar]
- Rotureau, E.; Van Leeuwen, H.P. Kinetics of metal ion binding by polysaccharide colloids. J. Phys. Chem. A 2008, 112, 7177–7184. [Google Scholar]
- Sriamornsak, P.; Kennedy, R.A. Swelling and diffusion studies of calcium polysaccharide gels intended for film coating. Int. J. Pharm. 2008, 358, 205–213. [Google Scholar]
- Kristiansen, K.A.; Schirmer, B.C.; Aachmann, F.L.; Skjåk-Bræk, G.; Draget, K.I.; Christensen, B.E. Novel alginates prepared by independent control of chain stiffness and distribution of G-residues: Structure and gelling properties. Carbohydr. Polym. 2009, 77, 725–735. [Google Scholar]
- Haidara, H.; Vonna, L.; Vidal, L. Unrevealed self-assembly and crystallization structures of Na-alginate, induced by the drying dynamics of wetting films of the aqueous polymer solution. Macromolecules 2010, 43, 2421–2429. [Google Scholar]
- Draget, K.I.; Smidsrød, O.; Skjåk-Bræk, G. Alginates from Algae. In Polysaccharides and Polyamides in the Food Industry. Properties, Production, and Patents; Steinbüchel, A., Rhee, S.K., Eds.; WILEY-VCH: Weinheim, Germany, 2005; p. 1. [Google Scholar]
- Smidsrød, O.; Haug, A. Dependence upon gel-sol state of ionexchange properties of alginates. Acta Chem. Scand. 1972, 26, 2063–2074. [Google Scholar]
- Smidsrød, O. Molecular basis for some physical properties of alginates in the gel state. Faraday Discuss. Chem. Soc. 1974, 57, 263–274. [Google Scholar]
- Kohn, R. Ion binding on polyuronates-alginate and pectin. Pure Appl. Chem. 1975, 42, 371–397. [Google Scholar]
- Grant, G.T.; Morris, E.R.; Rees, D.A.; Smith, P.J.C.; Thom, D. Biological interactions between polysaccharides and divalent cations: The egg-box model. FEBS Lett. 1973, 32, 195–198. [Google Scholar]
- Morris, E.R.; Rees, D.A.; Thom, D.; Boyd, J. Chiroptical and stoichiometric evidence of a specific, primary dimerisation process in alginate gelation. Carbohydr. Res. 1978, 66, 145–154. [Google Scholar]
- Braccini, I.; Pérez, S. Molecular basis of Ca2+-induced gelation in alginates and pectins: The egg-box model revisited. Biomacromolecules 2001, 2, 1089–1096. [Google Scholar]
- Li, L.; Fang, Y.; Vreeker, R.; Appelqvist, I.; Mendes, E. Reexamining the egg-box model in calcium-alginate gels with X-ray diffraction. Biomacromolecules 2007, 8, 464–468. [Google Scholar]
- Sikorski, P.; Mo, F.; Skjåk-Bræk, G.; Stokke, B.T. Evidence for egg-box-compatible interactions in calcium-alginate gels from fiber X-ray diffraction. Biomacromolecules 2007, 8, 2098–2103. [Google Scholar]
- Donati, I.; Paoletti, S. Material properties of alginates. In Alginates: Biology and Applications; Rehm, B.H.A., Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2009; p. 1. [Google Scholar]
- Donati, I.; Cesàro, A.; Paoletti, S. Specific Interactions versus counterion condensation. 1. Nongelling ions/polyuronate systems. Biomacromolecules 2005, 7, 281–287. [Google Scholar]
- Donati, I.; Benegas, J.C.; Cesàro, A.; Paoletti, S. Specific Interactions versus counterion condensation. 2. Theoretical treatment within the counterion condensation theory. Biomacromolecules 2006, 7, 1587–1596. [Google Scholar]
- Donati, I.; Benegas, J.C.; Paoletti, S. Polyelectrolyte study of the calcium-induced chain association of pectate. Biomacromolecules 2006, 7, 3439–3447. [Google Scholar]
- Donati, I.; Holtan, S.; Mørch, Y.A.; Borgogna, M.; Dentini, M.; Skjåk-Bræk, G. New hypothesis on the role of alternating sequences in calcium-alginate gels. Biomacromolecules 2005, 6, 1031–1040. [Google Scholar]
- Vårum, K.M.; Smidsrød, O. Structure-Property Relationship in Chitosans. In Polysaccharides. Structural Diversity and Functional Versatility; Dumitriu, S., Ed.; Marcel Dekker: New York, NY, USA, 2004; p. 625. [Google Scholar]
- Ogawa, K.; Yui, T.; Okuyama, K. Three D structures of chitosan. Int. J. Biol. Macromol. 2004, 34, 1–8. [Google Scholar]
- Signini, R.; Desbrières, J.; Campana Filho, S.P. On the stiffness of chitosan hydrochloride in acid-free aqueous solutions. Carbohydr. Polym. 2000, 43, 351–357. [Google Scholar]
- Lamarque, G.; Lucas, J.-M.; Viton, C.; Domard, A. Physicochemical behavior of homogeneous series of acetylated chitosans in aqueous solution: Role of various structural parameters. Biomacromolecules 2004, 6, 131–142. [Google Scholar]
- Christensen, B.E.; Vold, I.M.N.; Vårum, K.M. Chain stiffness and extension of chitosans and periodate oxidised chitosans studied by size-exclusion chromatography combined with light scattering and viscosity detectors. Carbohydr. Polym. 2008, 74, 559–565. [Google Scholar]
- Ravi Kumar, M.N.V.; Muzzarelli, R.A.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1879. [Google Scholar]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar]
- Lee, K.Y.; Yuk, S.H. Polymeric protein delivery systems. Prog. Polym. Sci. 2007, 32, 669–697. [Google Scholar]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control. Release 2007, 119, 5–24. [Google Scholar]
- D’Ayala, G.; Malinconico, M.; Laurienzo, P. Marine derived polysaccharides for biomedical applications: Chemical modification approaches. Molecules 2008, 13, 2069–2106. [Google Scholar]
- Rinaudo, M. Main properties and current applications of some polysaccharides as biomaterials. Polym. Int. 2008, 57, 397–430. [Google Scholar]
- Laurienzo, P. Marine polysaccharides in pharmaceutical applications: An overview. Mar. Drugs 2010, 8, 2435–2465. [Google Scholar]
- Van Vlierberghe, S.; Dubruel, P.; Schacht, E. Biopolymer-based hydrogels as scaffolds for tissue engineering applications: a review. Biomacromolecules 2011, 12, 1387–1408. [Google Scholar]
- Gombotz, W.R.; Wee, S. Protein release from alginate matrices. Adv. Drug Deliv. Rev. 1998, 31, 267–285. [Google Scholar]
- Faroongsarng, D.; Sukonrat, P. Thermal behavior of water in the selected starch- and cellulose-based polymeric hydrogels. Int. J. Pharm. 2008, 352, 152–158. [Google Scholar]
- Martinsen, A.; Skjåk-Bræk, G.; Smidsrød, O. Alginate as immobilization material: I. Correlation between chemical and physical properties of alginate gel beads. Biotechnol. Bioeng. 1989, 33, 79–89. [Google Scholar]
- Thu, B.; Bruheim, P.; Espevik, T.; Smidsrød, O.; Soon-Shiong, P.; Skjåk-Bræk, G. Alginate polycation microcapsules: II. Some functional properties. Biomaterials 1996, 17, 1069–1079. [Google Scholar]
- Gåserød, O.; Sannes, A.; Skjåk-Bræk, G. Microcapsules of alginate-chitosan. II. A study of capsule stability and permeability. Biomaterials 1999, 20, 773–783. [Google Scholar]
- Takka, F.; Acarturk, S. Calcium alginate microparticles for oral administration: II effect of form ulation factors on drug release and drug entrapment efficiency. J. Microencapsul. 1999, 16, 291–301. [Google Scholar]
- Acarturk, S.; Takka, F. Calcium alginate microparticles for oral administration: I: effect of sodium alginate type on drug release and drug entrapment efficiency. J. Microencapsul. 1999, 16, 275–290. [Google Scholar]
- Vandenberg, G.W.; Drolet, C.; Scott, S.L.; de la Noüe, J. Factors affecting protein release from alginate–chitosan coacervate microcapsules during production and gastric/intestinal simulation. J. Control. Release 2001, 77, 297–307. [Google Scholar]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar]
- Serra, L.; Doménech, J.; Peppas, N.A. Engineering design and molecular dynamics of mucoadhesive drug delivery systems as targeting agents. Eur. J. Pharm. Biopharm. 2009, 71, 519–528. [Google Scholar]
- Bonferoni, M.C.; Sandri, G.; Rossi, S.; Ferrari, F.; Caramella, C. Chitosan and its salts for mucosal and transmucosal delivery. Expert Opin. Drug Deliv. 2009, 6, 923–939. [Google Scholar]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar]
- Oh, J.K.; Lee, D.I.; Park, J.M. Biopolymer-based microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2009, 34, 1261–1282. [Google Scholar]
- He, P.; Davis, S.S.; Illum, L. Chitosan microspheres prepared by spray drying. Int. J. Pharm. 1999, 187, 53–65. [Google Scholar]
- Illum, L.; Jabbal-Gill, I.; Hinchcliffe, M.; Fisher, A.N.; Davis, S.S. Chitosan as a novel nasal delivery system for vaccines. Adv. Drug Deliv. Rev. 2001, 51, 81–96. [Google Scholar]
- George, M.; Abraham, T.E. Polyionic hydrocolloids for the intestinal delivery of protein drugs: Alginate and chitosan—A review. J. Control. Release 2006, 114, 1–14. [Google Scholar]
- Borchard, G. Chitosans for gene delivery. Adv. Drug Deliv. Rev. 2001, 52, 145–150. [Google Scholar]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar]
- Tafaghodi, M.; Sajadi Tabassi, S.A.; Jaafari, M.R. Induction of systemic and mucosal immune responses by intranasal administration of alginate microspheres encapsulated with tetanus toxoid and CpG-ODN. Int. J. Pharm. 2006, 319, 37–43. [Google Scholar]
- Amidi, M.; Mastrobattista, E.; Jiskoot, W.; Hennink, W.E. Chitosan-based delivery systems for protein therapeutics and antigens. Adv. Drug Deliv. Rev. 2010, 62, 59–82. [Google Scholar]
- Wen, Z.-S.; Xu, Y.-L.; Zou, X.-T.; Xu, Z.-R. Chitosan Nanoparticles Act as an Adjuvant to Promote both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice. Mar. Drugs 2011, 9, 1038–1055. [Google Scholar]
- Año, G.; Esquisabel, A.; Pastor, M.; Talavera, A.; Cedré, B.; Fernández, S.; Sifontes, S.; Aranguren, Y.; Falero, G.; García, L.; et al. A new oral vaccine candidate based on the microencapsulation by spray-drying of inactivated Vibrio cholerae. Vaccine 2011, 29, 5758–5764. [Google Scholar]
- Plant, K.P.; Lapatra, S.E. Advances in fish vaccine delivery. Develop. Comp. Immunol. 2011, in press. [Google Scholar]
- Champagne, C.P.; Fustier, P. Microencapsulation for the improved delivery of bioactive compounds into foods. Curr. Opin. Biotechnol. 2007, 18, 184–190. [Google Scholar]
- Chen, L.; Remondetto, G.E.; Subirade, M. Food protein-based materials as nutraceutical delivery systems. Trends Food Sci. Technol. 2006, 17, 272–283. [Google Scholar]
- de Vos, P.; Faas, M.M.; Spasojevic, M.; Sikkema, J. Encapsulation for preservation of functionality and targeted delivery of bioactive food components. Int. Dairy J. 2010, 20, 292–302. [Google Scholar]
- Desai, K.G.H.; Park, H.J. Recent developments in microencapsulation of food ingredients. Dry. Technol. 2005, 23, 1361–1394. [Google Scholar]
- Wandrey, C.; Bartkowiak, A.; Harding, S.E. Materials for Encapsulation. In Encapsulation Technologies for Active Food Ingredients and Food Processing; Zuidam, N.J., Nedović, V., Eds.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar]
- Luo, Y.; Engelmayr, G.; Auguste, D.T.; Ferreira, L.; Karp, J.M.; Saigal, R.; Langer, R. Three Dimensional Scaffolds. In Principles of Tissue Engineering; Lanza, R., Langer, R., Vacanti, J.P., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2007; p. 359. [Google Scholar]
- Brun-Graeppi, A.K.A.S.; Richard, C.; Bessodes, M.; Scherman, D.; Merten, O.-W. Cell microcarriers and microcapsules of stimuli-responsive polymers. J. Control. Release 2011, 149, 209–224. [Google Scholar]
- Smidsrød, O.; Skjåk-Bræk, G. Alginate as immobilization matrix for cells. Trends Biotechnol. 1990, 8, 71–78. [Google Scholar]
- Dulieu, C.; Poncelet, D.; Neufeld, R.J. Encapsulation and Immobilization Techniques. In Cell Encapsulation Technology and Therapeutics; Kühtreiber, W.M., Lanza, R.P., Chick, W.L., Eds.; Birkhäuser: Boston, MA, USA, 1999. [Google Scholar]
- de Vos, P.; Faas, M.M.; Strand, B.; Calafiore, R. Alginate-based microcapsules for immunoisolation of pancreatic islets. Biomaterials 2006, 27, 5603–5617. [Google Scholar]
- Lutolf, M.P.; Gilbert, P.M.; Blau, H.M. Designing materials to direct stem-cell fate. Nature 2009, 462, 433–441. [Google Scholar]
- Hernandez, R.M.; Orive, G.; Murua, A.; Pedraz, J.L. Microcapsules and microcarriers for in situ cell delivery. Adv. Drug Deliv. Rev. 2010, 62, 711–730. [Google Scholar]
- Murua, A.; Portero, A.; Orive, G.; Hernandez, R.M.; de Castro, M.; Pedraz, J.L. Cell microencapsulation technology: Towards clinical application. J. Control. Release 2008, 132, 76–83. [Google Scholar]
- Hunt, N.C.; Grover, L.M. Cell encapsulation using biopolymer gels for regenerative medicine. Biotechnol. Lett. 2010, 32, 733–742. [Google Scholar]
- Janes, K.A.; Calvo, P.; Alonso, M.J. Polysaccharide colloidal particles as delivery systems for macromolecules. Adv. Drug Deliv. Rev. 2001, 47, 83–97. [Google Scholar]
- Gouin, S. Microencapsulation. Trends Food Sci. Technol. 2004, 15, 330–347. [Google Scholar]
- Gharsallaoui, A.; Roudaut, G.; Chambin, O.; Voilley, A.; Saurel, R. Applications of spray-drying in microencapsulation of food ingredients: An overview. Food Res. Int. 2007, 40, 1107–1121. [Google Scholar]
- Cui, J.H.; Goh, J.S.; Park, S.Y.; Kim, P.H.; Lee, B.J. Preparation and physical characterization of alginate microparticles using air atomization method. Drug Develop. Ind. Pharm. 2001, 27, 309–319. [Google Scholar]
- Teunou, E.; Poncelet, D. Rotary disc atomisation for microencapsulation applications-prediction of the particle trajectories. J. Food Eng. 2005, 71, 345–353. [Google Scholar]
- Prüsse, U.; Bilancetti, L.; Bučko, M.; Bugarski, B.; Bukowski, J.; Gemeiner, P.; Massart, B.; Nastruzzi, C.; Nedovic, V.; Poncelet, D.; et al. Comparison of different technologies for alginate beads production. Chem. Pap. 2008, 62, 364–374. [Google Scholar]
- Pinto Reis, C.; Neufeld, R.J.; Ribeiro, A.J.; Veiga, F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 8–21. [Google Scholar]
- Anton, N.; Benoit, J.P.; Saulnier, P. Design and production of nanoparticles formulated from nano-emulsion templates—A review. J. Control. Release 2008, 128, 185–199. [Google Scholar]
- Barbosa-Cánovas, G.V.; Ortega-Rivas, E.; Juliano, P.; Yan, H. Food Powders: Physical Properties, Processing, and Functionality; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Tønnesen, H.H.; Karlsen, J. Alginate in drug delivery systems. Drug Develop. Ind. Pharm. 2002, 28, 621–630. [Google Scholar]
- Zimmermann, H.; Shirley, S.G.; Zimmermann, U. Alginate-based encapsulation of cells: Past, present, and future. Curr. Diabetes Rep. 2007, 7, 314–320. [Google Scholar]
- de Vos, P.; Bučko, M.; Gemeiner, P.; Navrátil, M.; Švitel, J.; Faas, M.; Strand, B.L.; Skjåk-Bræk, G.; Morch, Y.A.; Vikartovská, A.; et al. Multiscale requirements for bioencapsulation in medicine and biotechnology. Biomaterials 2009, 30, 2559–2570. [Google Scholar]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2011. [Google Scholar]
- Gåserød, O.; Smidsrød, O.; Skjåk-Bræk, G. Microcapsules of alginate-chitosan—I: A quantitative study of the interaction between alginate and chitosan. Biomaterials 1998, 19, 1815–1825. [Google Scholar]
- Ribeiro, A.J.; Silva, C.; Ferreira, D.; Veiga, F. Chitosan-reinforced alginate microspheres obtained through the emulsification/internal gelation technique. Eur. J. Pharm. Sci. 2005, 25, 31–40. [Google Scholar]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar]
- Hamman, J.H. Chitosan based polyelectrolyte complexes as potential carrier materials in drug delivery systems. Mar. Drugs 2010, 8, 1305–1322. [Google Scholar]
- Patil, J.S.; Kamalapur, M.V.; Marapur, S.C.; Kadam, D.V. Ionotropic gelation and polyelectrolyte complexation: The novel techniques to design hydrogel particulate sustained, modulated drug delivery system: A review. Digest J. Nanomater. Biostruct. 2010, 5, 241–248. [Google Scholar]
- Batorsky, A.; Liao, J.; Lund, A.W.; Plopper, G.E.; Stegemann, J.P. Encapsulation of adult human mesenchymal stem cells within collagen-agarose microenvironments. Biotechnol. Bioeng. 2005, 92, 492–500. [Google Scholar]
- Kumachev, A.; Greener, J.; Tumarkin, E.; Eiser, E.; Zandstra, P.W.; Kumacheva, E. High-throughput generation of hydrogel microbeads with varying elasticity for cell encapsulation. Biomaterials 2011, 32, 1477–1483. [Google Scholar]
- Jeon, O.; Bouhadir, K.H.; Mansour, J.M.; Alsberg, E. Photocrosslinked alginate hydrogels with tunable biodegradation rates and mechanical properties. Biomaterials 2009, 30, 2724–2734. [Google Scholar]
- Cellesi, F.; Tirelli, N. A new process for cell microencapsulation and other biomaterial applications: Thermal gelation and chemical cross-linking in “tandem”. J. Mater. Sci.: Mater. Med. 2005, 16, 559–565. [Google Scholar]
- Shen, F.; Li, A.A.; Cornelius, R.M.; Cirone, P.; Childs, R.F.; Brash, J.L.; Chang, P.L. Biological properties of photocrosslinked alginate microcapsules. J. Biomed. Mater. Res. Part B 2005, 75, 425–434. [Google Scholar]
- Rokstad, A.M.; Donati, I.; Borgogna, M.; Oberholzer, J.; Strand, B.L.; Espevik, T.; Skjåk-Bræk, G. Cell-compatible covalently reinforced beads obtained from a chemoenzymatically engineered alginate. Biomaterials 2006, 27, 4726–4737. [Google Scholar]
- Rajaonarivony, M.; Vauthier, C.; Couarraze, G.; Puisieux, F.; Couvreur, P. Development of a new drug carrier made from alginate. J. Pharm. Sci. 1993, 82, 912–917. [Google Scholar]
- Skiba, M.; Morvan, C.; Duchene, D.; Puisieux, F.; Wouessidjewe, D. Evaluation of gastrointestinal behaviour in the rat of amphiphilic β-cyclodextrin nanocapsules, loaded with indomethacin. Int. J. Pharm. 1995, 126, 275–279. [Google Scholar]
- Calvo, P.; Remuñan-López, C.; Vila-Jato, J.L.; Alonso, M.J. Chitosan and chitosan/ethylene oxide-propylene oxide block copolymer nanoparticles as novel carriers for proteins and vaccines. Pharm. Res. 1997, 14, 1431–1436. [Google Scholar]
- Riley, T.; Govender, T.; Stolnik, S.; Xiong, C.D.; Garnett, M.C.; Illum, L.; Davis, S.S. Colloidal stability and drug incorporation aspects of micellar-like PLA-PEG nanoparticles. Colloid. Surf. B 1999, 16, 147–159. [Google Scholar]
- Bhavna, M.A.; Sanjula, B.; Javed, A. Patents on nanoparticulate drug delivery systems—A review. Recent Pat. Drug Deliv. Formul. 2008, 2, 83–89. [Google Scholar]
- Wong, T.W. Chitosan and its use in design of insulin delivery system. Recent Pat. Drug Deliv. Formul. 2009, 3, 8–25. [Google Scholar]
- Bawarski, W.E.; Chidlowsky, E.; Bharali, D.J.; Mousa, S.A. Emerging nanopharmaceuticals. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 273–282. [Google Scholar]
- Millotti, G.; Bernkop-Schnürch, A. Nano- and Microparticles in Oral Delivery of Macromolecular Drugs. In Oral Delivery of Macromolecular Drugs; Bernkop-Schnürch, A., Ed.; Springer: New York, NY, USA, 2009; p. 153. [Google Scholar]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: a review on formulation technology, types and applications toward targeted drug delivery. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 9–24. [Google Scholar]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar]
- Phillips, M.A.; Gran, M.L.; Peppas, N.A. Targeted nanodelivery of drugs and diagnostics. Nano Today 2010, 5, 143–159. [Google Scholar]
- Fish Health Management in Aquaculture; Fisheries and Aquaculture Department, Food and Agriculture Organization of the United Nations: Rome, Italy. Available online: www.fao.org/fishery/en (accessed on 27 May 2005).
- Vandenberg, G.W. Oral vaccines for finfish: academic theory or commercial reality? Anim. Health Res. Rev. 2004, 5, 301–304. [Google Scholar]
- Adams, A.; Thompson, K.D. Biotechnology offers revolution to fish health management. Trends Biotechnol. 2006, 24, 201–205. [Google Scholar]
- Sommerset, I.; Krossøy, B.; Biering, E.; Frost, P. Vaccines for fish in aquaculture. Expert Rev. Vaccines 2005, 4, 89–101. [Google Scholar]
- Joosten, P.H.M.; Tiemersma, E.; Threels, A.; Caumartin-Dhieux, C.; Rombout, J.H.W.M. Oral vaccination offish against Vibrio anguillarum using alginate microparticles. Fish Shellfish Immunol. 1997, 7, 471–485. [Google Scholar]
- Toranzo, A.E.; Magariños, B.; Romalde, J.L. A review of the main bacterial fish diseases in mariculture systems. Aquaculture 2005, 246, 37–61. [Google Scholar]
- Research project “Oral vaccine carrier for fish farming of Friuli Venezia Giulia”-Region Friuli Venezia Giulia, Italy.
- Romalde, J.L.; Luzardo-Alvárez, A.; Ravelo, C.; Toranzo, A.E.; Blanco-Méndez, J. Oral immunization using alginate microparticles as a useful strategy for booster vaccination against fish lactoccocosis. Aquaculture 2004, 236, 119–129. [Google Scholar]
- Altun, S.; Kubilay, A.; Ekici, S.; Didinen, B.I.; Diler, O. Oral vaccination against lactococcosis in rainbow trout (Oncorhynchus mykiss) using sodium alginate and poly (lactide-co-glycolide) carrier. Kafkas Univ. Vet. Fak. Derg. 2010, 16, S211–S217. [Google Scholar]
- Bellich, B.; Borgogna, M.; Zorzin, L.; Cocchietto, M.; Blasi, P.; Lapasin, R.; Sava, G.; Cesàro, A. Oral vaccines for aquaculture: development of a vaccine microencapsulation system based on biodegradable polymers. In Third Biotech Workshop—“Drug Delivery Systems For Biotech Products”, Pavia, Italy, 24–25 March 2010; University of Pavia.
- Paolini, A.; Ridolfi, V.; Zezza, D.; Cocchietto, M.; Musa, M.; Pavone, A.; Conte, A.; Giorgetti, G. Vaccination trials of sea bass (Dicentrarchus labrax) against pasteurellosis using oral, intraperitoneal and immersion methods. Vet. Ital. 2005, 41, 137–144. [Google Scholar]
- Zorzin, L.; Cocchietto, M.; Voinovich, D.; Marcuzzi, A.; Filipović-Grčić, J.; Mulloni, C.; Crembiale, G.; Casarsa, C.; Bulla, R.; Sava, G. Lysozyme-containing chitosan-coated alginate microspheres for oral immunisation. J. Drug Deliv. Sci. Technol. 2006, 16, 413–420. [Google Scholar]
- Sava, G. Pharmacological aspects and therapeutic applications of lysozymes. In Lysozyme: Model Enzymes in Biochemistry and Biology; Jolles, P., Ed.; Birkhäuser Verlag: Basel, Switzerland, 1996; p. 433. [Google Scholar]
- Zorzin, L.; Cocchietto, M.; Voinovich, D.; Blasi, P.; Bellich, B.; Borgogna, M.; Sava, G. Production of microparticles by a novel pneumatic -assisted technology: preliminary results. In Proceeding of 17th International Symposium on Microencapsulation, Nagoya, Japan, 29 September–1 October 2009.
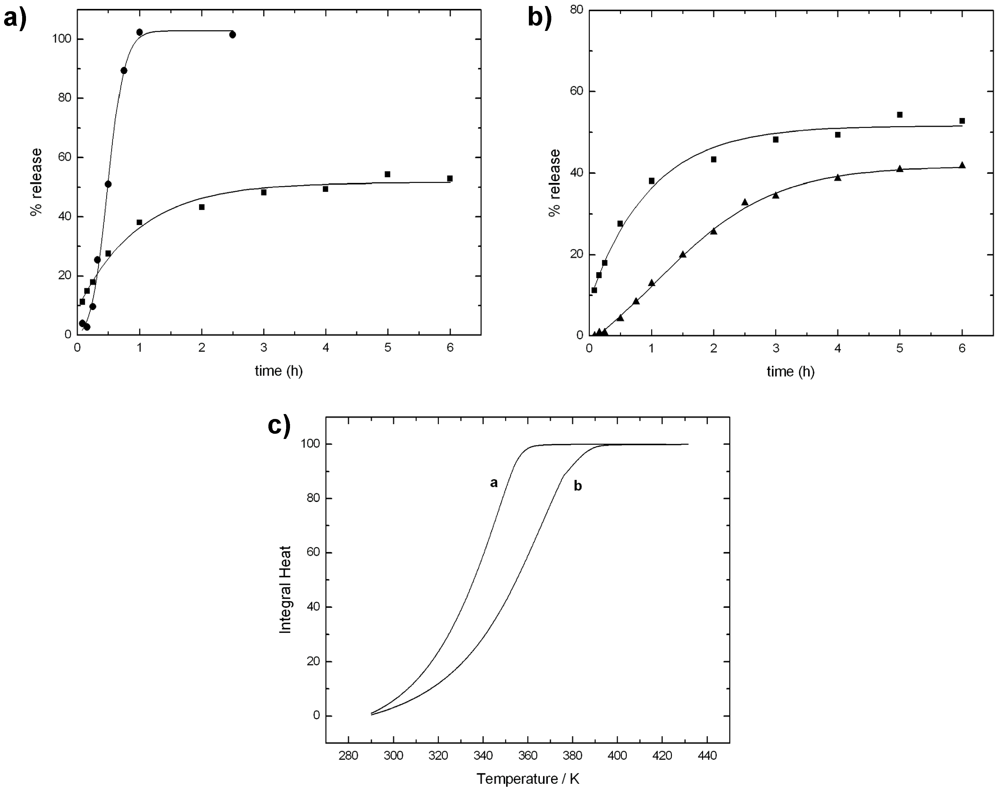
- Bellich, B.; Borgogna, M.; Carnio, D.; Cesàro, A. Thermal behavior of water in micro-particles based on alginate gel. J. Therm. Anal. Calorim. 2009, 97, 871–878. [Google Scholar]
- Borgogna, M.; Bellich, B.; Zorzin, L.; Lapasin, R.; Cesàro, A. Food microencapsulation of bioactive compounds: Rheological and thermal characterisation of non-conventional gelling system. Food Chem. 2010, 122, 416–423. [Google Scholar]
- Bellich, B.; Borgogna, M.; Cok, M.; Cesàro, A. Release Properties of Hydrogels: Water Evaporation from Alginate Gel Beads. Food Biophys. 2011, 6, 259–266. [Google Scholar]
- Bellich, B.; Borgogna, M.; Cok, M.; Cesàro, A. Water evaporation from gel beads: A calorimetric approach to hydrogel matrix release properties. J. Therm. Anal. Calorim. 2011, 103, 81–88. [Google Scholar]
- Martins, S.; Sarmento, B.; Souto, E.B.; Ferreira, D.C. Insulin-loaded alginate microspheres for oral delivery—Effect of polysaccharide reinforcement on physicochemical properties and release profile. Carbohydr. Polym. 2007, 69, 725–731. [Google Scholar]
- Lee, D.W.; Hwang, S.J.; Park, J.B.; Park, H.J. Preparation and release characteristics of polymer-coated and blended alginate microspheres. J. Microencapsul. 2003, 20, 179–192. [Google Scholar]
- Joshi, S.C.; Chen, B. Swelling, Dissolution and Disintegration of HPMC in Aqueous Media. In Proceeding of 13th International Conference on Biomedical Engineering; Lim, C.T., Goh, J.C.H., Eds.; Springer: Berlin Heidelberg, Germany, 2009; 23, p. 1244. [Google Scholar]
- Lin, N.; Huang, J.; Chang, P.R.; Feng, L.; Yu, J. Effect of polysaccharide nanocrystals on structure, properties, and drug release kinetics of alginate-based microspheres. Colloid. Surf. B 2011, 85, 270–279. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Borgogna, M.; Bellich, B.; Cesàro, A. Marine Polysaccharides in Microencapsulation and Application to Aquaculture: “From Sea to Sea”. Mar. Drugs 2011, 9, 2572-2604. https://doi.org/10.3390/md9122572
Borgogna M, Bellich B, Cesàro A. Marine Polysaccharides in Microencapsulation and Application to Aquaculture: “From Sea to Sea”. Marine Drugs. 2011; 9(12):2572-2604. https://doi.org/10.3390/md9122572
Chicago/Turabian StyleBorgogna, Massimiliano, Barbara Bellich, and Attilio Cesàro. 2011. "Marine Polysaccharides in Microencapsulation and Application to Aquaculture: “From Sea to Sea”" Marine Drugs 9, no. 12: 2572-2604. https://doi.org/10.3390/md9122572