1. Introduction
Glioblastoma multiforme (GBM) is the most common and most malignant human brain tumor, with an incidence of 4 out of 100,000 per year [
1]. GBM is extremely invasive and difficult to treat surgically. It is distinguished by severe and aberrant vascularization and high resistance to radiotherapy (RT) and chemotherapy [
1,
2]. The median survival of patients with GBM is about 1 year and only approximately 5% of patients survive no longer than 3 years [
1,
3]. There are no known risk factors directly related to GBM. However, long-term exposure to various carcinogens, including heavy metals and aromatic chemicals has been associated with an increased risk for GBM [
4]. Exposure to these environmental chemicals can either stimulate cell proliferation or lead to cell death, therefore impacting tumor growth [
5]. Phthalates are a group of industrial chemicals that are used as plasticizers, solvents, lubricants, fixatives, and as detergents in personal care products [
6]. Therefore, phthalates can be identified in many widely used industrial consumer products [
7]. The most well-known phthalate is di-ethylhexylphthalate (DEHP). DEHP is a principal component in polyvinyl chloride products frequently found in medical devices [
8] and has the ability to penetrate the blood-brain-barrier.
It is essential to develop novel therapeutic approaches and identify therapeutic targets for advancements in glioblastoma treatment. S100P has been identified as a potential therapeutic target in the treatment of GBM. The family of S100 proteins is comprised of small dimeric constituents of the EF-hand super family of Calcium-binding proteins. Becker
et al. purified and characterized S100P from the placenta [
9]. S100P is a 95-amino acid protein and the gene coding S100P is mapped on the human chromosome 4, at 4p16 [
10]. This particular chromosomal location has been associated with Huntington disease [
11], Wolf–Hirschhorn syndrome [
12,
13], Familial Wolfram syndrome [
12,
14], Crohn’s disease [
15] and cervical cancer [
16]. S100P has been shown to aid in cancer progression through its roles in cell proliferation, survival, angiogenesis, and metastasis [
17]. S100P was absent in normal breast tissue but detected in both typical and atypical hyperplasia as well as
in situ and invasive carcinoma [
18]. Therefore, S100P is highly correlated with tumor progression in breast cancer. S100P expression has also been identified in flat adenomas in the colon [
19]. Moreover, S100P is specifically expressed in cancerous colon tissue [
20], but not in normal colon tissue [
21].
The role of S100P in glioblastoma progression has not yet been investigated. In this study, we examined whether DEHP-induced cell transformation in glioblastoma is mediated through S100P.
2. Experimental Section
2.1. Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), trypsin ethylenediaminetetraacetic acid (EDTA), puromycin, glutamine, penicillin-streptomycin, and culture supplements were purchased from Gibco (Life Technologies, Palto Alto, CA, USA). Propidium iodide (PI), S100P antibody and DEHP were purchased from Sigma-Aldrich, Inc., (St. Louis, MO, USA). Cultrex® 3D spheroid cell invasion assay kit was purchased from Trevigen (Gaithersburg, MD, USA). The kit included 10× spheroid formation extracellular matrix (ECM), 3D culture qualified 96 well spheroid formation plate, and invasion matrix. All other reagents and materials were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
2.2. Cell Culture
Glioblastoma cancer cell line, LN-229, was purchased from American Type Culture Collection (Rockville, MD, USA). The cells were cultured and maintained in DMEM containing 10% fetal bovine serum, 1% penicillin/streptomycin, and 2% glutamine. LN-229 cell lines were grown in BD primaria tissue culture dishes, with dimensions of 100 × 20 mm at 37 °C with 5% CO2 in a humidifier incubator and carried at 2.0 × 106 cells/mL, passaging two to three times weekly as needed. Cells were pelleted by centrifugation at 1,500 rpm for 6 min at 4 °C and resuspended in fresh complete media in tissue culture plates 24 h before use in experiments to avoid any confounding gene expression that might occur because of handling.
2.3. Lentiviral Production and Infection
Lentiviral shRNAs targeting S100P was obtained from Harvard Medical School (Boston, MA, USA). The lentivirus was packaged by co-transfection of human embryonic cells (293T) with the shRNA expression vector, VSV-G (vesicular stomatitis virus-glycoprotein), and delta-VPR (viral protein R) plasmids at the ratio of 1:0.9:0.1, using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Forty-eight hours after transfection, the supernatants containing lentiviral particles were harvested and titering was performed using Hela cells.
2.4. shS100P Infections
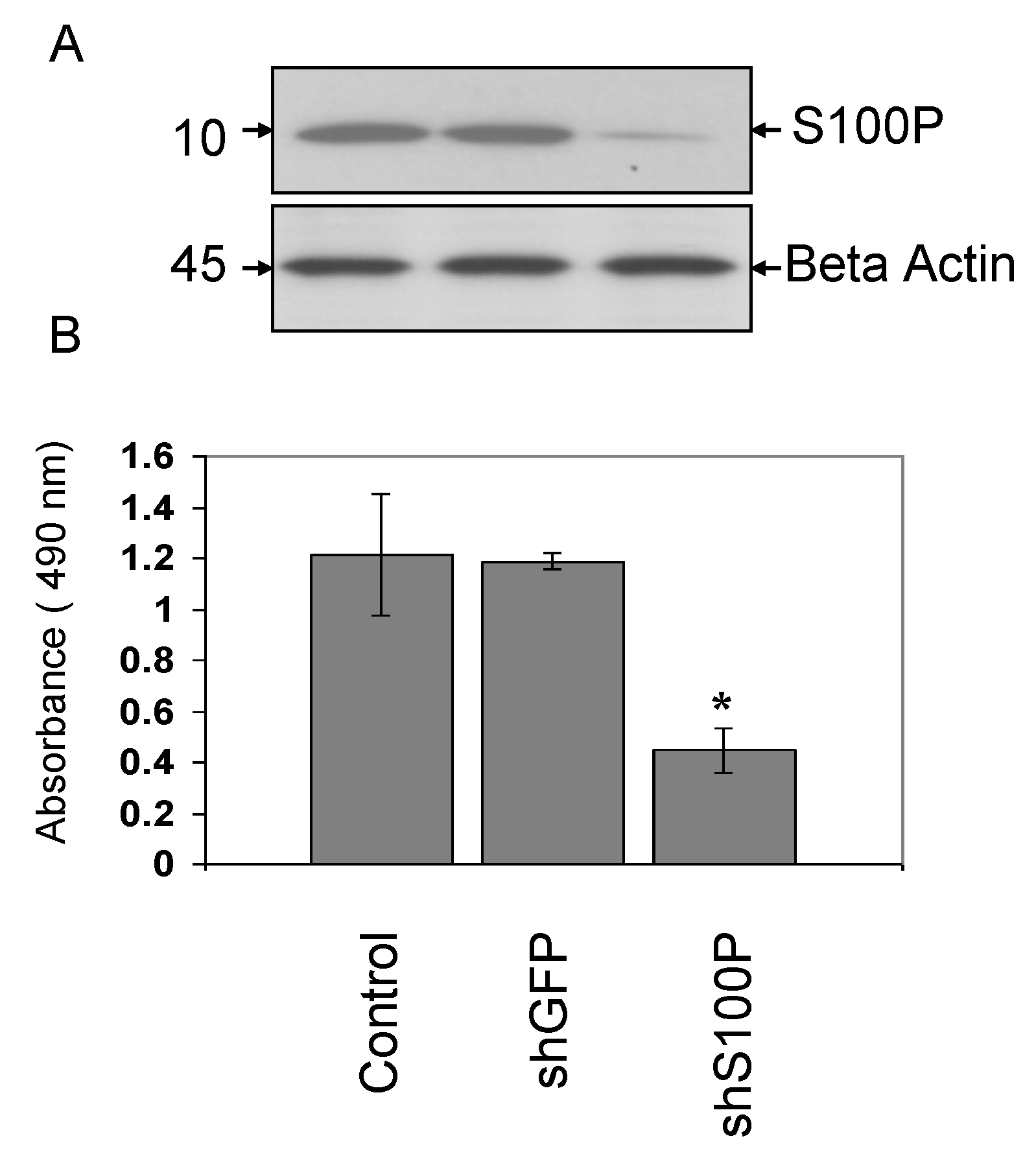
LN-229 cells were plated in 10 cm dishes until 80% confluence. The day of infection, media was removed and replaced with 3 mLs of complete media supplemented with polybrene (8 μg/mL) into each plate. Two hundred and fifty (250) µL of lentivirus were added in each plate and incubated for 24 h. Cells were left to recover from infection for 24 h before initiating selection with puromycin (3 µg/mL) for three days.
2.5. Western Blot Analysis
Western blot analysis was performed. Briefly, cells were harvested and pelleted in an microcentrifuge (1,200 g, 5 min, 4 °C), washed in 1× PBS and resuspended in a cell lysis buffer containing 20 mM Tris (pH 8.0), 0.5% (w/v) Nonidet P-40, 1 mM EDTA, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 mM dithiothreitol, 1 mM PMSF and 0.1 M NaCl. After a 20 min incubation period at 4 °C, supernatants were clarified by centrifugation (8,000 g, 5 min, 4 °C) and their total protein concentration was determined by the Bradford and Lowry method using Bio-Rad Protein Assay reagents in a microtiter assay. Total cellular protein (40 μg) was electrophoresed on a sodium dodecyl sulphate polyacrylamide gel (SDS-PAGE) and then transferred to a polyvinylidine difluoride membrane (GE Healthcare, Little Chalfont, Buckinghamshire, England) by electroblotting overnight in 25 mM Tris (pH 8.3), 192 mM glycine, 20% (v/v) methanol, at 15 V, 100 mA, 4 °C. The membranes were blocked with 10% (w/v) electrophoresis-grade biotin-depleted non-fat dry milk (Bio-Rad Laboratories Inc., Hercules, CA, USA) in 1× PBS, rinsed in PBS, probed with monoclonal rabbit anti- S100P at a 1:250 dilution and washed 3× in PBS. The secondary antibody, anti-rabbit whole IgG, used at 1:5,000 dilution (Transduction Laboratories San Diego, CA, USA) for one hour at room temperature. The protein bands were then visualized using an enhanced chemiluminescence (ECL) detection system (GE Healthcare, Little Chalfont, Buckinghamshire, England).
2.6. Cell Proliferation Assay
Cell proliferation was indirectly assessed with a colorimetric, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay obtained from Promega (Madison, WI, USA). Cells (100 µL, number = 5,000) were plated on 96 well plates (Thermo Fisher Scientific, Waltham, MA, USA) and infected with shS100P and/or treated with 2.5 μg/µL DEHP. After 24 h of incubation, DMEM medium was removed and followed by the addition of 20 µL of MTS solution to each well. The 96 well plates were placed in an incubator at 37 °C in 5% CO2. The absorbance of the solution was measured at 490 nm in one hour increments for three hours using a spectrophotometer (Bio-Rad Model 550; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
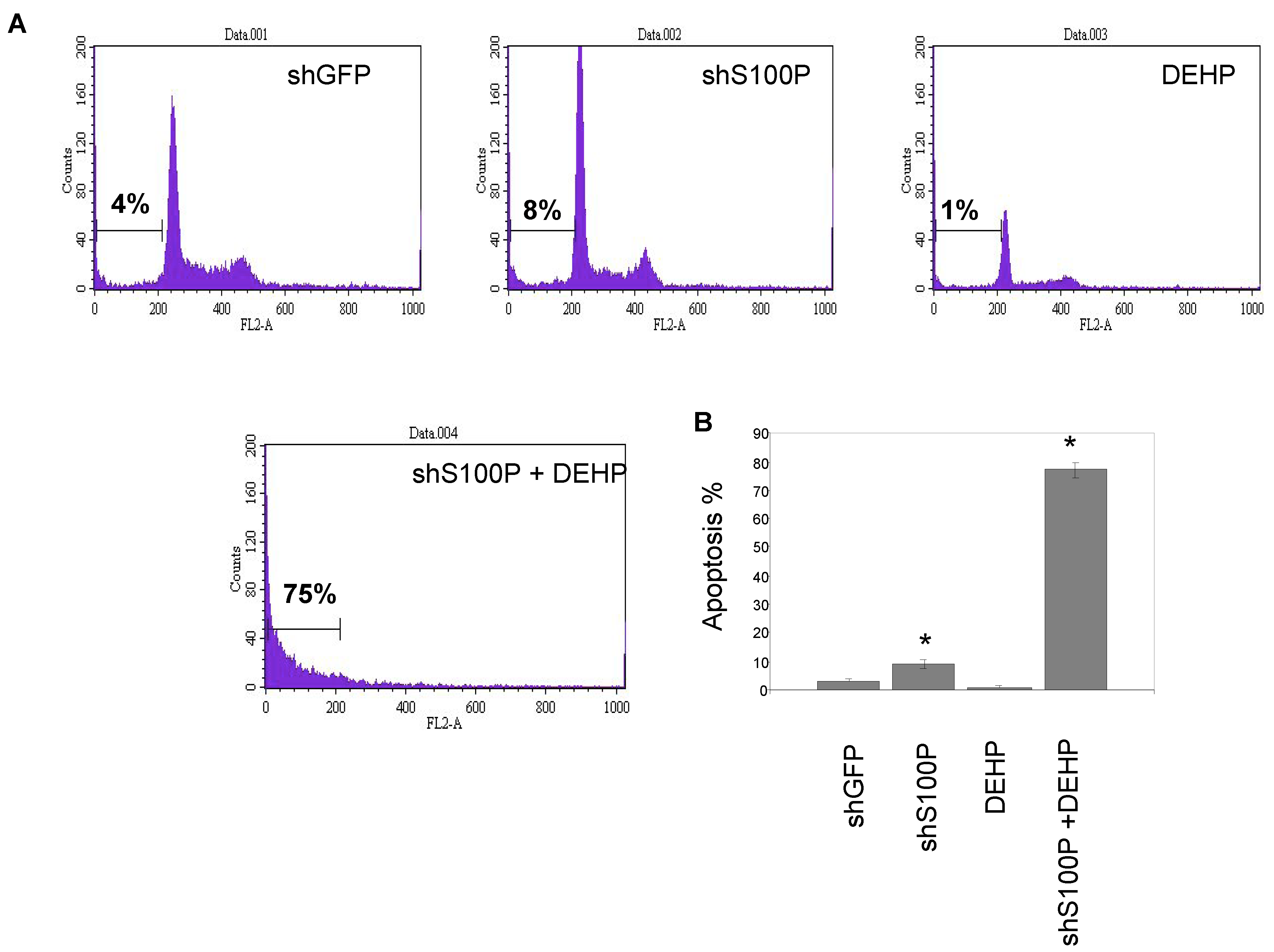
2.7. Sub G1 Apoptosis Assay
Flow cytometry was performed to assess sub G1 DNA content in an entire cell population of LN-229 cells (2 × 105 cells/mL) were seeded in 24-well plates and cultured for 24 h prior to treatment. Cells were then treated with 2.5 µg/µL DEHP and incubated for 24 h. Cells were harvested, washed once with 1XPBS and suspended in 400 µL propidium iodide (PI) solution (propidium iodide 50 µg/mL, 0.1% sodium citrate and 0.1% Triton-X 100). The sub G1 DNA content was determined using a Becton Dickinson Flow Cytometer (Becton-Dickinson, San Jose, CA, USA).
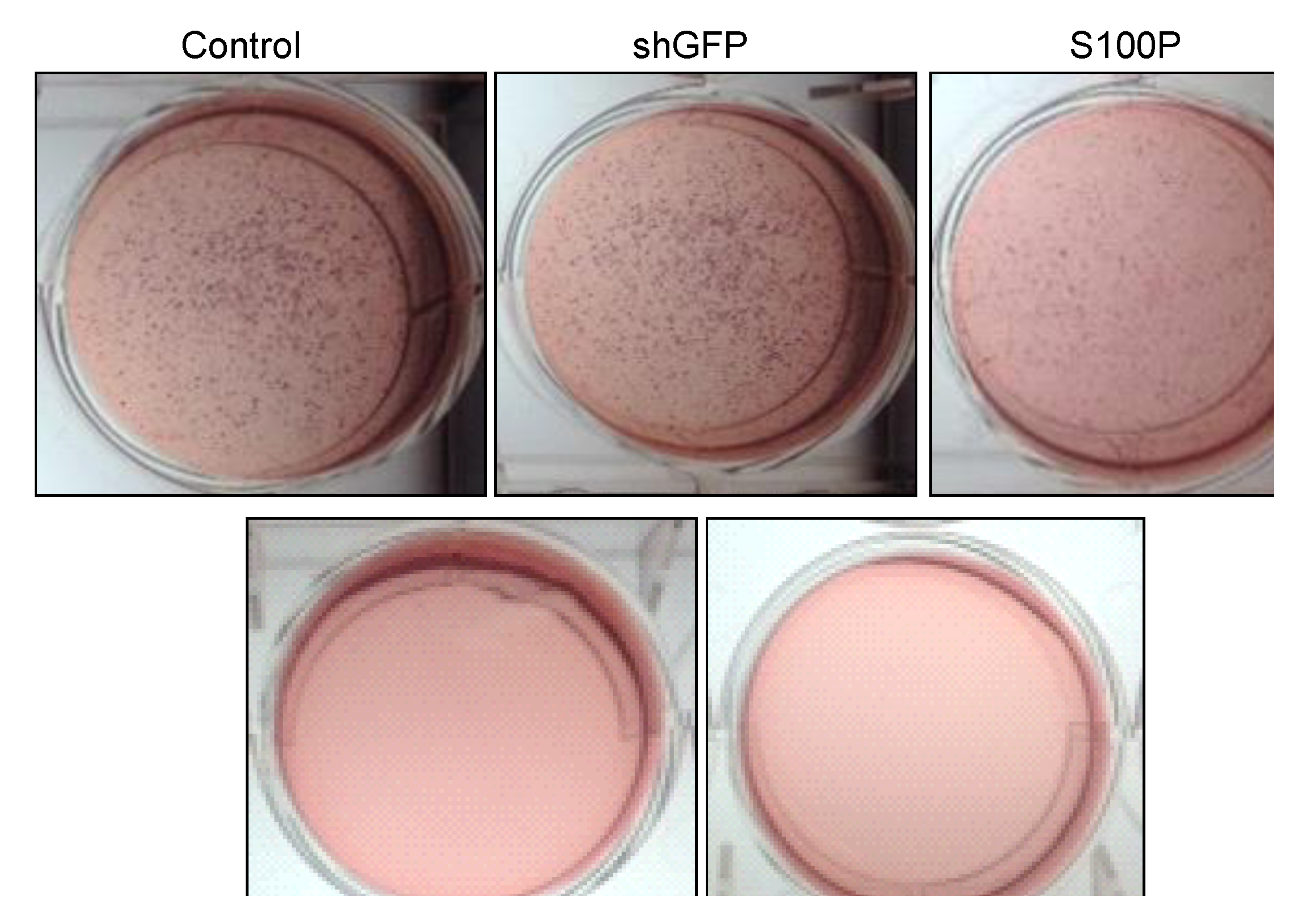
2.8. Soft Agar Assay
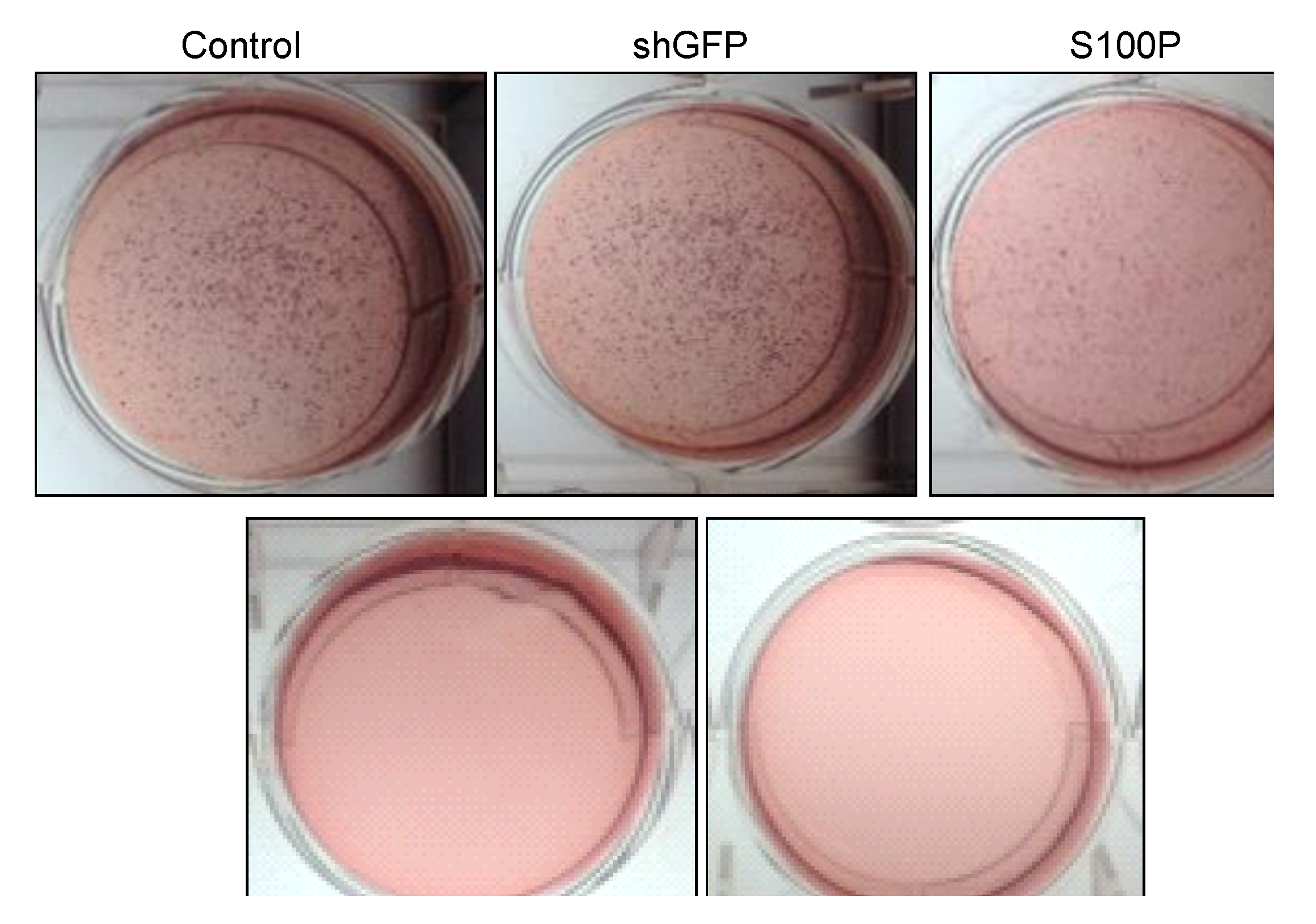
Anchorage independent growth was measured using a soft agar assay. Experiments were carried out on 6-well plates. This assay is comprised of two layers. The bottom layer of agar was prepared first by dissolving 3.2 g of powdered agarose in 200 mL (1.6% agarose) of double distilled water and boiling this solution for 10 min. The bottom agar consisted of 12 mL of 1.6% agarose, and 3 mL FBS. Components for the bottom agar were mixed and 2 mL was added to each well avoiding bubbles and placed in 4 ºC for 15 min to allow agar to solidify. Plates were then incubated (37 °C, 5% CO2) overnight. The top agar was made by mixing 2 mL 2× DMEM, 2 mL 1.0% agarose (2.0 g of agarose in 200 mL of double distilled water boiled for 10 min), 0.5 mL FBS, 0.5 mL DMEM with FBS and penicillin/streptomycin. Untreated and treated (80 µL DEHP; 2.5 µg/µL in 3 mL DMEM) cells (number = 50,000) were added to the mixture and mixed well. Two mL of the top agar mixture was added to each well containing bottom agar. Plates were placed at 4 °C for 5 min as the agar solidified. Plates were then returned to the incubator (37 °C, 5% CO2) for 3 weeks. Two drops of DMEM with FBS and penicillin/streptomycin were added to each plate every two to three days to keep the plates moist over the three-week incubation period. After 3 weeks, plates were then stained with p-iodonitrotetrazolium violet (Santa Cruz, Dallas, TX, USA) in DMSO. Plates were returned to 37 °C incubator overnight. Pictures were then taken.
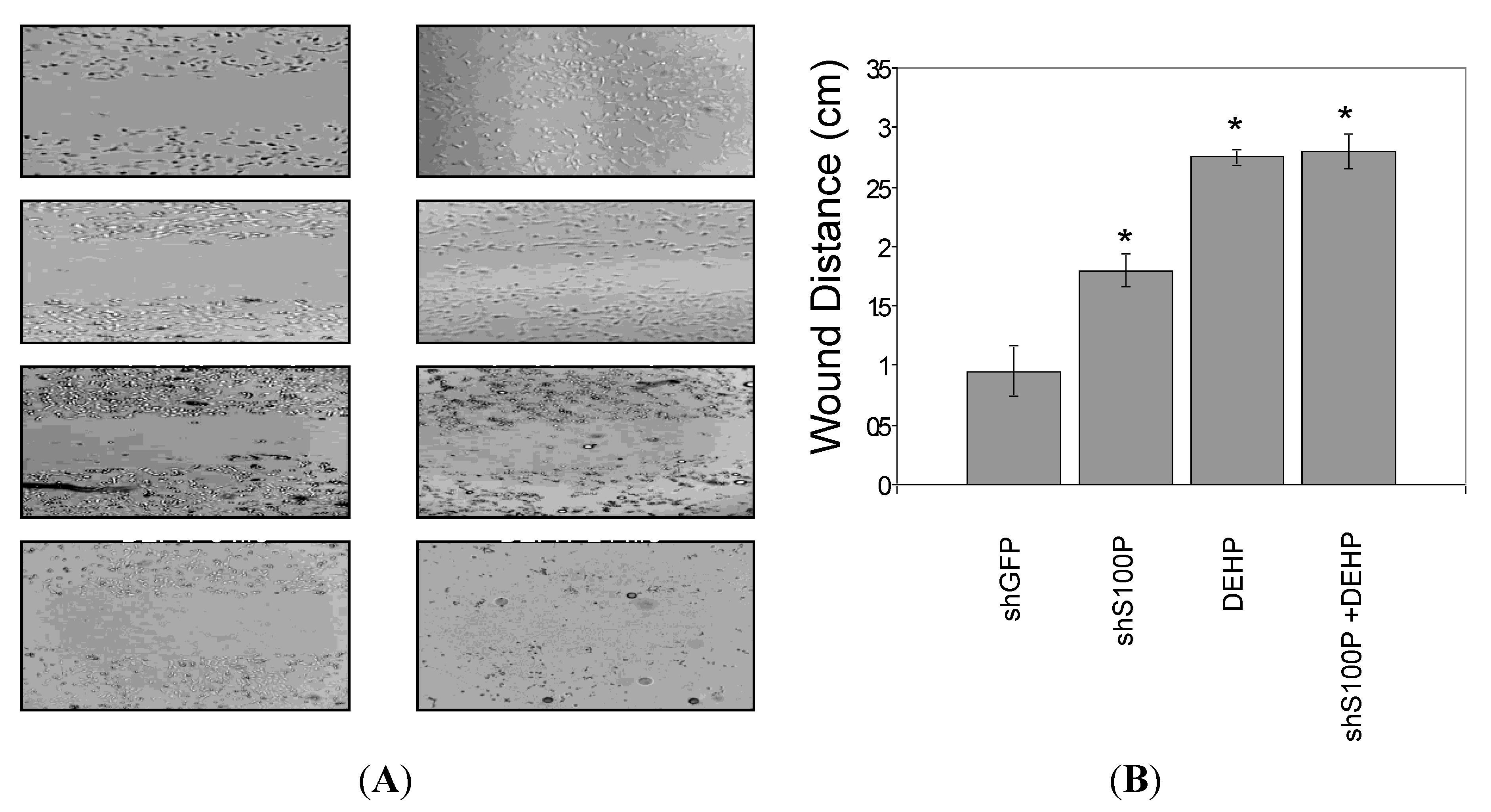
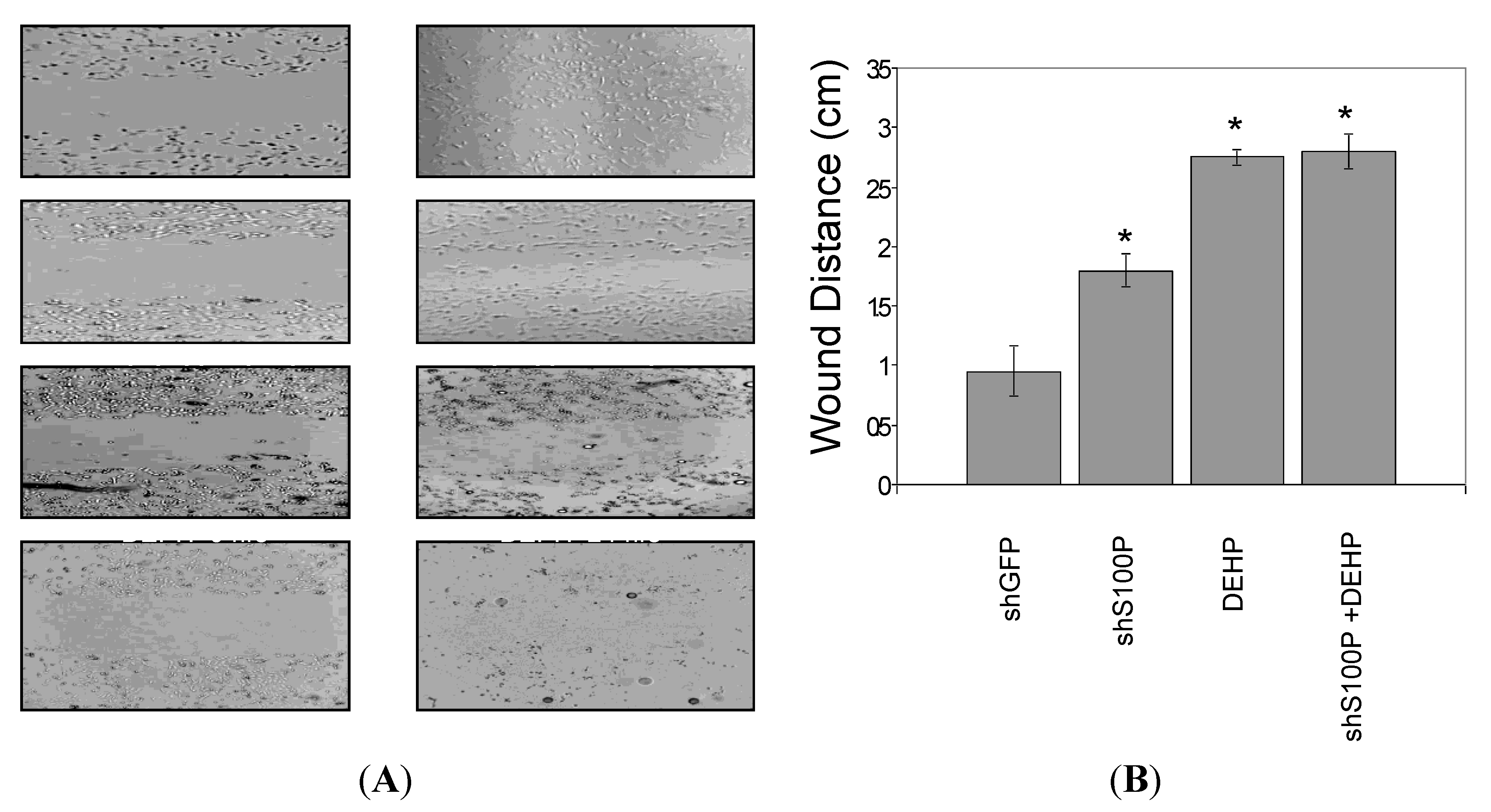
2.9. Wound Scratch Assay
Cell migration was assessed utilizing the wound scratch assay. LN-229 cells were harvested and seeded (1 × 106) on a six well plate and cultured for 24 h. Media was aspirated from each well and a scratch was made on the cell mononlayer with a 200 µL pipette tip. Each well was photographed to assess the scratch and thereafter treated with DEHP. The 6-well plate was incubated at 37 °C for an additional 24 h after which each well was photographed.
2.10. 3D Spheroid BME Cell Invasion Assay
3D Spheroid BME Cell Invasion Assay was performed on LN-229 cells. Cells were cultured at 80% confluence and then treated with DEHP and incubated at 37 °C for 24 h. Cells for each condition were harvested and resuspended in spheroid formation ECM. This mixture was comprised of 5 µL spheroid formation ECM, 15 µL of DMEM with FBS and penicillin/streptomycin. Fifty microliters of cell suspension were added per well to the 3D culture qualified 96-well spheroid formation plate and centrifuged at 200 g for 3 min at room temperature and then incubated at 37 °C for 72 h to promote spheroid formation. Fifty microliters of the invasion matrix was added to each well in the 3D culture qualified 96-well spheroid formation plate. The spheroid formation plate was centrifuged at 300 g at 4 °C for 5 min to eliminate bubbles and position spheroids within the invasion matrix towards the center of the well. The spheroid formation plate was then transferred to the incubator at 37 °C for one hour to promote gel formation. After one hour, 100 µL of DMEM with FBS and penicillin/streptomycin was added to each well. The spheroid formation plate was incubated at 37 °C for 3 to 7 days, and spheroids were photographed in each well every two days.
2.11. Statistical Analyses
Statistical significance was evaluated using one-way ANOVA with multiple comparisons and Student’s t-test for two group comparison. The data are reported as mean ± SEM and a value of p < 0.05 was considered statistically significant.
4. Discussion
There is an imperative need to understand the mechanisms responsible for the aggressiveness and treatment resistance of glioblastoma multiforme. This study addresses the connection between cancer and exposure to toxic DEHP substances in the environment. It is estimated that as many as two-thirds of all cancer cases are attributed to environmental causes. This environmental chemical may be present in the air, water, food, and workplace. Because of the complex interplay of many factors, it is not possible to predict whether DEHP’s environmental exposure and genetics will cause a particular person to develop cancer. Some studies in women have found that exposure to phthalates have been linked to disrupted thyroid hormone levels, increased levels of oxidative stress, and illnesses such as endometriosis and breast cancer. However, DEHP mechanisms are still not completely understood. In this study, we hypothesized that DEHP modulates cell migration, invasion and anchorage independent growth through targeting S100P in LN-229 glioblastoma cells.
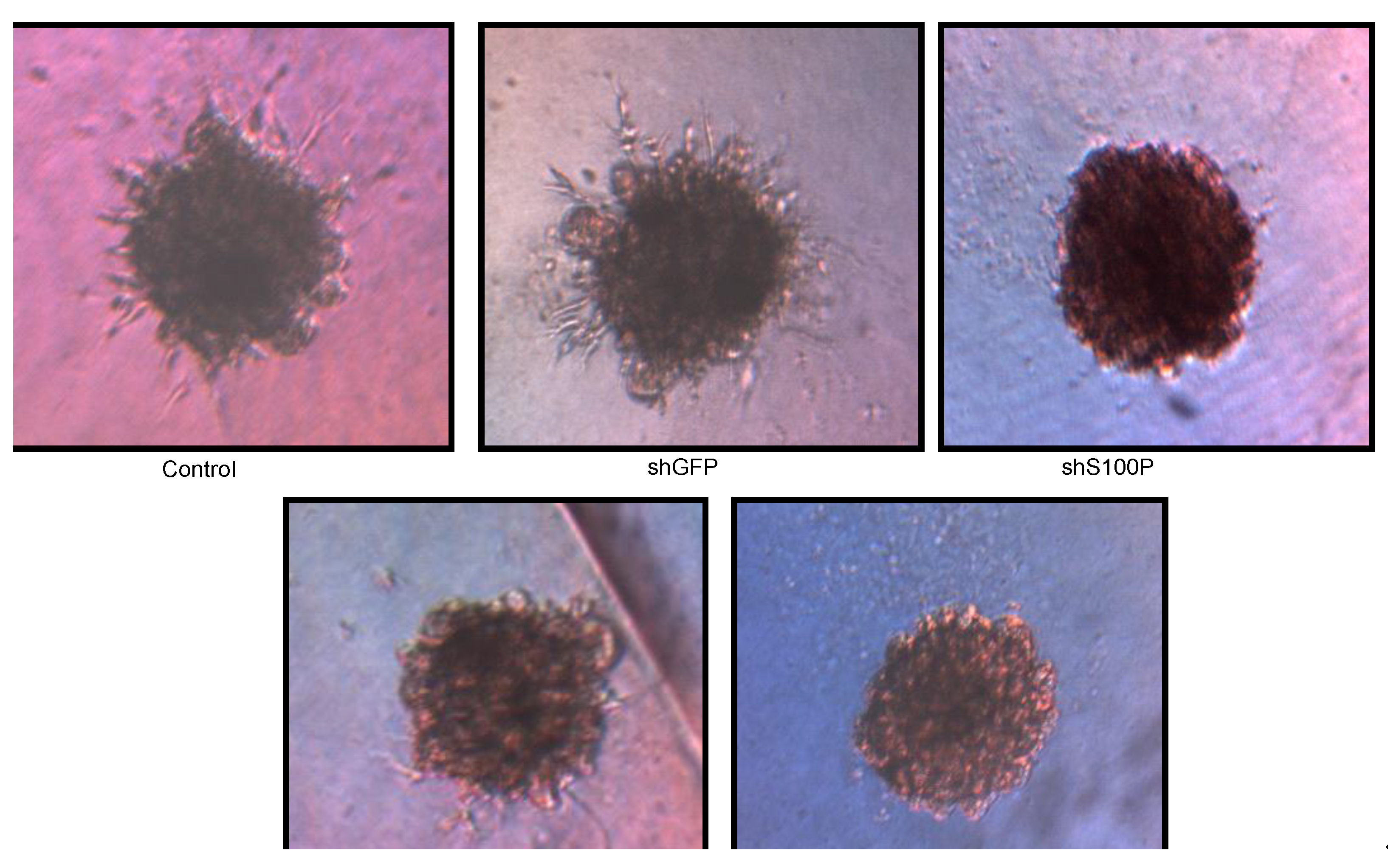
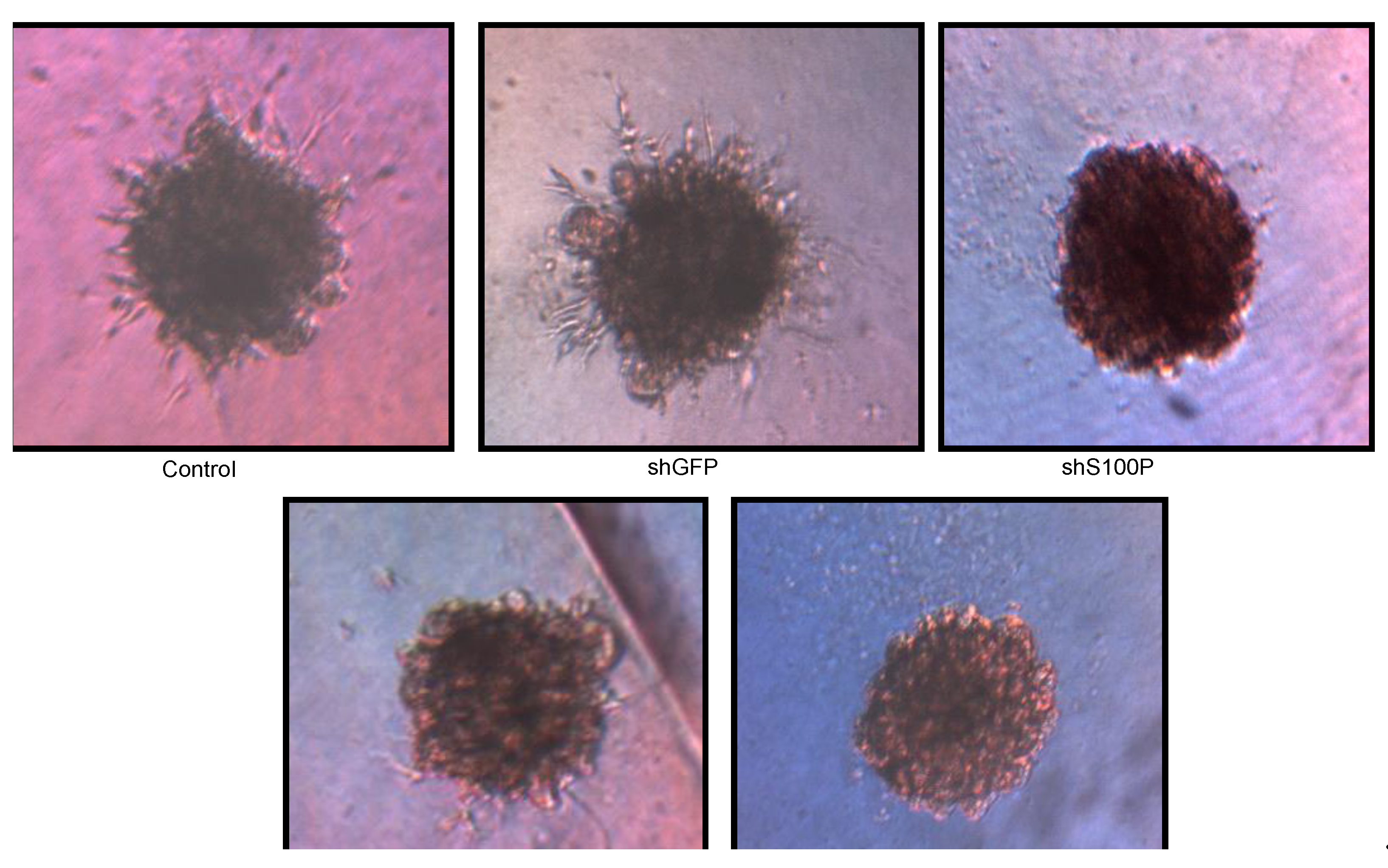
Figure 6.
Representative micrographs of spheroid colony formation by LN-229 cells treated with DEHP alone and in combination with S100P knockdown. Microphotographs were taken after seven days of incubation.
Figure 6.
Representative micrographs of spheroid colony formation by LN-229 cells treated with DEHP alone and in combination with S100P knockdown. Microphotographs were taken after seven days of incubation.
S100P proteins have been found in a variety of tumors and are associated with metastasis, making them a key interest in cancer research. Overexpression of S100P has been demonstrated in several forms of cancer, including pancreas, breast, prostate, lung, and colon. However, the functional role of S100P in glioblastoma has not been elucidated. Consequently, we suppressed S100P expression in glioblastoma cancer cells using lentiviral knockdown to investigate its role in glioblastoma. Knockdown assays were performed by lentiviral infection due to its benefits over other gene therapy methods, including its ability to infect both dividing and non-dividing cells with high efficiency [
22] and to achieve long-term stable expression of the transgene as well as its low immunogenicity [
21]. Silencing is more specific than overexpression for determining the role of a factor in cell biology because it avoids problems associated with overexpression [
23]. The expression of S100P in glioblastoma cell lines was knocked down using shS100P. In this study, we demonstrate that knockdown of S100P expression significantly inhibited cell proliferation and anchorage independent growth in LN-229 cells. A similar study of S100P knockdown in colon cancer cells also exhibited proliferation rates lower in the knockdown cells when compared to colon cancer cells expressing S100P [
21]. Furthermore, in an investigation of the role of S100P in pancreatic cancer, one study revealed that cells with siRNA-silenced S100P expression grew at a significantly reduced rate compared with control siRNA-expressing cells [
23]. We have shown that DEHP has the potential to cause cell transformation and it significantly inhibits cell proliferation in the presence and absence of S100P. Overexpression studies of S100P in prostate cancer reported anchorage independent growth in soft agar [
24]. To determine the role of S100P on anchorage independent growth in glioblastoma cells, we examined colony formation after blocking the expression of S100P. We found that suppressing S100P expression in glioblastoma cells inhibited anchorage independent growth when compared to uninfected and shGFP cells. These results are complementary to a colon cancer study that silenced S100P by RNAi and revealed significant reductions in colony formation of cells infected with shS100P [
21]. We also observed the loss of colony formation in anchorage independent growth in LN-229 cells treated with DEHP alone and in the absence of S100P.
Furthermore, we analyzed the effects of suppressing S100P expression on migration and invasion of LN-229 glioblastoma cells. Our study revealed that blocking S100P expression inhibited glioblastoma cell migration and invasion. Our results are in line with a study that silenced S100P in pancreatic cancer [
23] as well as another study that silenced S100P expression in colon cancer [
21] as suppressing S100P expression also reduced the rate of migration and invasion in pancreatic and colon cancer.
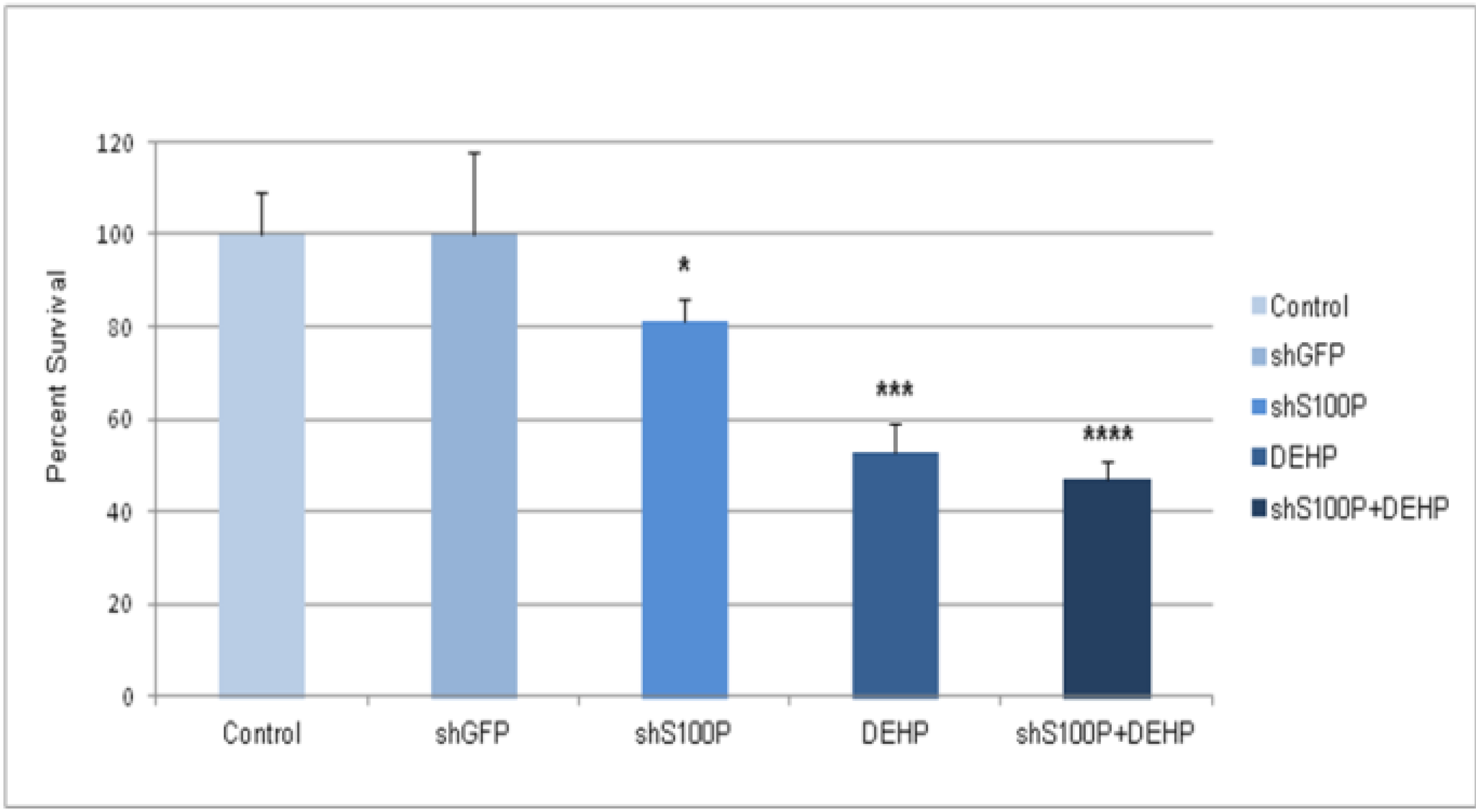
Exposure of DEHP on LN-229 cells caused a significant decrease in cell migration by inducing cell death. LN-229 cells were extremely sensitive to the toxic effects of DEHP. Moreover, the resultant of shS100P and DEHP exposure are not additive implicating the predominant effects of DEHP on cell migration. Van Meir
et al. (1994) [
25] showed that LN-229 glioblastoma cells have low expression levels of
p53. Thus, we sought to determine whether S100P expression is involved in inducing apoptosis in the first growth phase of the cell cycle; our data revealed a substantial increase cell death in shS100P LN-229 cells treated with DEHP. Additionally, we demonstrated that suppressing S100P significantly inhibited spheroid expansion in LN-229 cells as compared to the control uninfected and shGFP cells. We observed that shS100Pcells treated with DEHP showed greater inhibition of spheroid expansion in LN-229; these findings were further supported by the results of a spheroid proliferation assay which revealed the same effect. We also extended the current study to examine the effect of DEHP treatment on glioblastoma cells in the presence and absence S100P expression. DEHP-induced cell death in LN-229 cells resulted in decreased cell migration.

 and
and 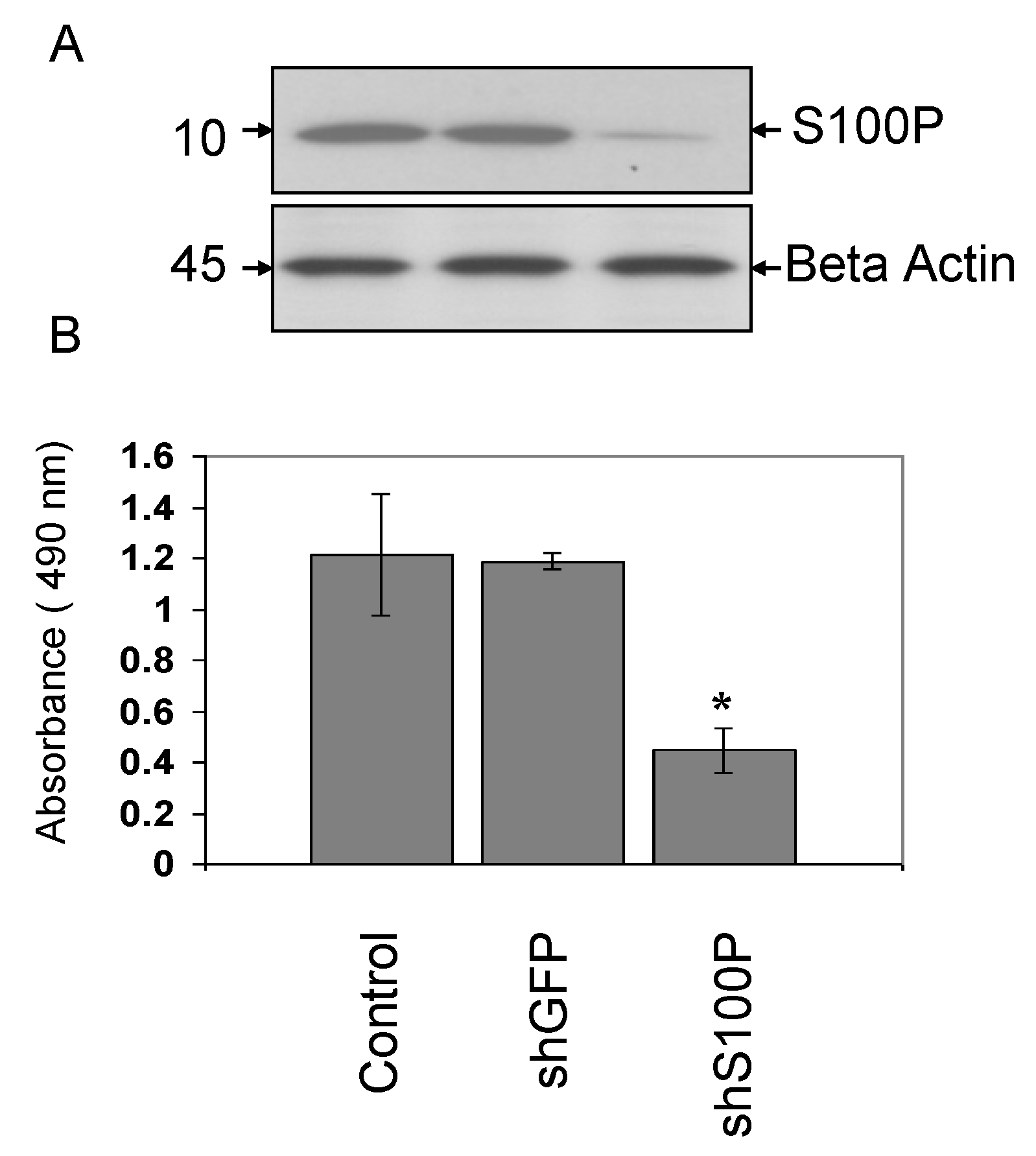{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}